

Abstract
Vaccinia‐related kinase 1 (VRK1), a serine/threonine mitotic kinase, is widely over‐expressed in dividing cells and regarded as a cancer drug target primarily due to its function as an early response gene in cell proliferation. However, the mechanism of VRK1 phosphorylation and substrate activation is not well understood. More importantly even the molecular basis of VRK1 interaction with its cofactor, adenosine triphosphate (ATP), is unavailable to‐date. As designing specific inhibitors remains to be the major challenge in kinase research, such a molecular understanding will enable us to design ATP‐competitive specific inhibitors of VRK1. Here we report the molecular characterization of VRK1 in complex with AMP‐PNP, a non‐hydrolyzable ATP‐analog, using NMR titration followed by the co‐crystal structure determined upto 2.07 Å resolution. We also carried out the structural comparison of the AMP‐PNP bound‐form with its apo and inhibitor‐bound counterparts, which has enabled us to present our rationale toward designing VRK1‐specific inhibitors.
Keywords: kinase, mitotic kinase, vaccinia‐related kinase, VRK1, adenosine triphosphate, ATP
Short abstract
PDB Code(s): 6AC9
Introduction
Protein kinases phosphorylate signaling proteins in cellular pathways to regulate essential biological activities. Primarily, they catalyze the transfer of a phosphate group from ATP to serine, threonine, or tyrosine residues of substrate proteins and induce a conformational change to activate the protein. The human genome consists of ~518 protein kinases,1 making it one of the most represented protein families, with many of them involved in regulating cell division.2 Thus, kinase inhibitors serve as the most sought out drug targets, mainly for cancer, evident from the ~37 FDA approved kinase inhibitors available in the market to‐date.3 Among the kinase superfamily, the mitotic kinases belong to a class of kinases which can regulate the cell‐cycle entry, exit, checkpoint, and cytokinesis, deemed to be important for the growth and development of an organism.4 To‐date, members of the cyclin‐dependent kinase family are the well‐studied mitotic regulators.4, 5, 6 Members of the Polo, Aurora and NIMA mitotic kinases have also been studied for their involvement in cell cycle regulation.4, 5, 6 A recent addition to this list is the human vaccinia‐related kinase 1 (VRK1) highlighted for its role in regulating cell‐cycle progression7 and chromatin condensation.8, 9 VRKs are a family of Ser/Thr kinases, which belong to the casein kinase (CK1) group, consisting of three paralogous members: VRK1, VRK2, and VRK3.10 VRK1 is mainly found in the nucleus, whereas VRK2 is found in ER membranes and the nuclear envelope. Although catalytic domain structures of VRK family reveal typical kinase fold, only VRK1 and 2 are catalytically active VRK's,10 while VRK3 is catalytically inactive, which is attributable to its non‐canonical active site topology11 (Supporting Information Fig. S1). In particular, the extra carboxyl‐terminal tail of VRK1 orients toward its catalytic site and forms multiple interactions that are critical for catalysis and structural stability.12 Interestingly, its catalytic activity on histone H3 is oscillated during cell cycle and is regulated by macroH2A1, a histone variant.13 VRK1 is discovered as an early response gene and is one of the important factors for the initiation of cell division process,7, 14, 15 reflected on its overexpression in many cancerous tissues.16 Recent studies have shown direct correlation of poor clinical outcomes of breast cancer patients was associated with high expression levels of VRK117 suggesting it as a promising cancer drug target.16, 17, 18, 19 VRK1 can phosphorylate itself (auto‐phosphorylate) as well as its interacting substrate proteins. During mitosis, VRK1 can phosphorylate (a) barrier‐to‐autointegration factor (BAF) to regulate the morphology of nuclear envelope20; (b) H3's N‐terminal tail to control chromatin condensation8; and (c) p53 which helps to stabilize DNA damage regulation.21 Hence, dysregulation of VRK1 during mitosis can result in high cell proliferation. Recent studies led to the discovery of small molecule VRK1 inhibitors from natural products, such as luteolin,22 brazilin,23 and ursolic acid.24 An extensive thermal shift screening using a well‐known kinase library, followed by crystal structure determination, revealed the first molecular basis of the VRK1 inhibitors, ASC24 (PDBID 3OP5), BI‐D1870 (PDBID 5UVF), and GW297361X (PDBID 5UKF), providing cues toward possible VRK1 selective inhibitor design.25 While these inhibitors were shown to recognize the ATP binding pocket, their specificity as ATP‐competitive or ATP‐mimetic VRK1 inhibitors remains elusive. It has been shown that VRK1 shows poor selectivity toward known kinase inhibitors,26, 27, 28 indicating that there is a scope for identifying VRK1‐specific inhibitors which could also have low promiscuity with other kinases. In this direction, we feel the lack of ATP‐bound VRK1 structure cripples the efforts toward identifying ATP‐competitive inhibitors “specific” to VRK1. The structural information delineating the underlying molecular interaction of VRK1 with ATP could facilitate the design of selective ligands for VRK1. To this end, we report here the NMR titration study to analyze the interaction of VRK1 with a non‐hydrolyzable ATP‐analog, AMP‐PNP, followed by the crystal structure of VRK1–AMP‐PNP complex highlighting its structural characteristics and comparison with other VRK1 structures.
Results and Discussion
NMR titration of VRK1 with AMP‐PNP
The binding of AMP‐PNP on VRK1 was examined by performing a NMR study on a two‐dimensional 1H–15N TROSY spectroscopy (Fig. 1). The residues undergoing chemical shift perturbations upon AMP‐PNP titration were picked and analyzed, showing that the backbone amides of residues Gln45, Gly46, Gly49, Val 70, Lys71, Glu73, Phe81, Glu83, Leu84, Lys85, and Gln88 displayed chemical shift perturbations, while the residues Gly47, Cys50, and Val69 completely disappeared upon AMP‐PNP titration [Fig. 1(B) and (C)]. These perturbed residues are located on the highly flexible P‐loop, β3 strand, and αC helix. Residues Asp132, Arg133, and Phe134 of DRF motif, also known as the hinge region, along with the neighboring residues, Met131 and Gly135 also undergo chemical shift perturbations [Fig. 1(B) and (C)], indicating their roles in interacting with AMP‐PNP. Apart from these, residues His175, Gly176, Asp177, Ile178, Lys179, and Ser181 on the β6 strand (the catalytic loop region) and Asp197, Tyr198, and Gly199 of DYG motif, showed perturbations or disappearance upon AMP‐PNP binding [Fig. 1(B) and (C)]. The NMR titration studies apart from helping us to identify the perturbed residues on the canonical ATP binding pocket of VRK1 [Fig. 1(C)] also serves as a platform for fragment screening in future.
Figure 1.
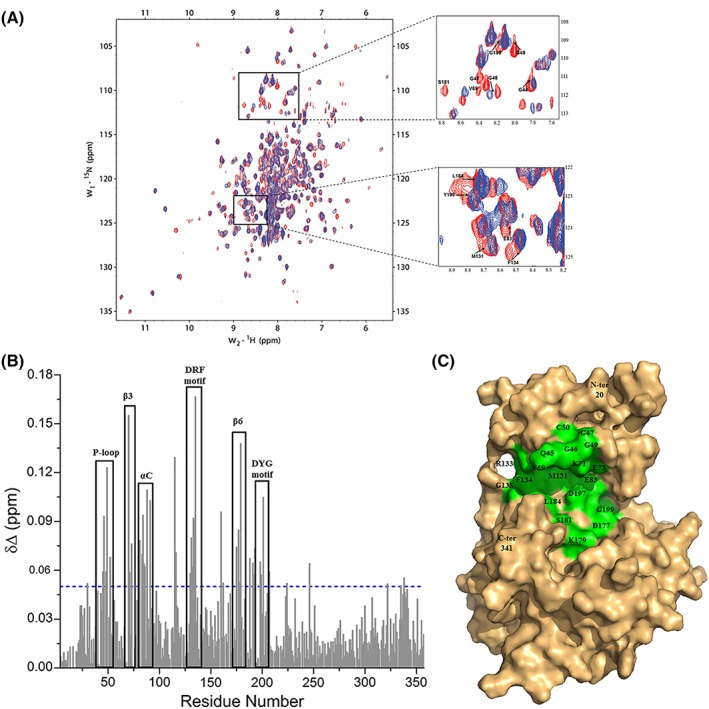
The interaction between VRK1 and AMP‐PNP, an ATP analog characterized using NMR titration. (A) The overlay of 2D 1H–15N TROSY spectra of VRK1 in free state (red) with that of VRK1 in the presence of AMP‐PNP at a molar ratio of 1:5 (VRK1:AMP‐PNP; blue). A closer view of a few regions showing chemical shift perturbations or NMR signal disappearance upon the addition of AMP‐PNP are displayed as insets on the right. The direction of the shifts from free VRK1 (in red) toward AMP‐PNP bound form is indicated by black arrows and the residues are labeled for reference. (B) The chemical shift perturbation plot showing the regions of VRK1 affected upon the addition of AMP‐PNP, shown in the black box, and numbered according to the secondary structure elements of VRK1. The blue broken line represents the chemical shift perturbation (δΔ) threshold value set at 0.05. (C) The surface representation of VRK1 (PDB ID 2LAV), displaying residues 20 to 341 (in pale orange), with residues affected by the addition of AMP‐PNP labeled and colored in green.
Crystal structure of VRK1–AMP‐PNP
To understand the atomic‐level interactions, the crystal structure of VRK1–AMP‐PNP complex was determined to a resolution of 2.07 Å. The crystals belonged to the orthorhombic space group, P212121, with four molecules in the asymmetric unit, with one AMP‐PNP along with a magnesium ion bound to each chain [Fig. 2(A)]. Overall, the VRK1–AMP‐PNP complex structure revealed an active site topology in ATP‐bound conformation of typical Ser/Thr kinases in which AMP‐PNP molecule docks its adenosine moiety into the active site pocket, while the phosphate group end orients toward the P‐loop. The AMP‐PNP is stabilized by a network of hydrogen bonds and non‐bonded interactions [Fig. 2(B) and Table 1], and the electron density map revealed that all its atoms are completely resolved [Fig. 2(C)]. Notable hydrogen bonds are those made by the adenosine nitrogen atoms with Asp132 and Phe134 backbone atoms (part of DRF hinge motif). One of the phosphate oxygen is hydrogen bonded to the Asp197 side chain (part of the DYG motif), while another phosphate oxygen forms a hydrogen bond with Lys71 side‐chain. In addition to this, the ribose sugar oxygen forms a hydrogen bond with Ser181 in Chain D, while not in others. Several non‐bonded interactions made by the adenosine and ribose rings with residues Ile51, Val69, Phe134, and Leu184, locks the adenosine end of AMP‐PNP within the active site. Residues from the DRF motif interact with adenine ring, while the DYG motif interacts with phosphate groups, bridged by the magnesium ion. AMP‐PNP adopts a similar orientation in the three chains (A, B, C) while there is a slight difference in that bound to the D‐chain. A closer look revealed the position of the bound magnesium is identical in the three chains, forming ionic interactions (~2.8 Å) with the neighboring atoms, while that bound to D‐chain is co‐ordinated (~2.1 Å) by the neighboring residues [Fig. 2(B) and Table 1]. In addition, a few water molecules stabilize the AMP‐PNP and magnesium by forming hydrogen bonded interactions [Fig. 2(B) and Table I]. It is worthwhile to mention that AMP‐PNP is a non‐hydrolyzable ATP analog expected to possess subtle changes to the mode of interactions made by ATP binding, nonetheless ATP analogs29 are being widely used for trapping the structural snapshot mimicking ATP binding.
Figure 2.
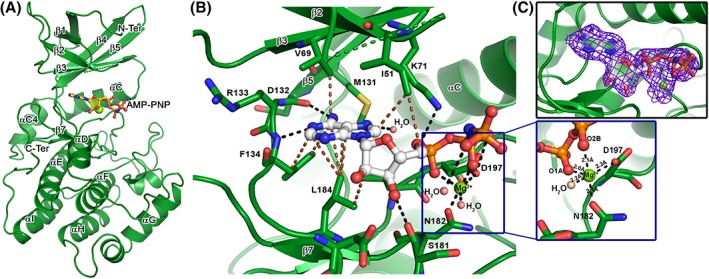
Crystal structure of VRK1–AMP‐PNP complex. (A) The cartoon representation of VRK1 bound to AMP‐PNP, in ball and stick mode, and a magnesium ion (Mg), shown as a green sphere. The major secondary structure elements are labeled for reference. (B) The interactions made by AMP‐PNP (shown in ball and stick), with the active residues of VRK1 (shown in stick mode). The hydrogen bonds and non‐polar interactions are shown as black and brown broken lines, respectively. The Mg ion (in green sphere) form a coordination geometry with AMP‐PNP phosphate groups and nearby Asp197 and Asn182 (shown as inset). (C) The 2Fo–Fc electron density map, contoured at 1σ cut‐off, showing density cover for all the atoms of AMP‐PNP and the co‐ordinated Mg ion.
Table 1.
Interactions made by AMP‐PNP (ANP) with VRK1. It is consolidated for all the four chains, while the magnesium coordination distances refer to Chain D only. For sake of comparison inhibitor (8E1) interactions with VRK1 (PDB ID 5UKF) are also provided
| Hydrogen bonds | |||||
|---|---|---|---|---|---|
| VRK1–ANP | VRK1‐8E1 | ||||
| ANP Atom | VRK1/ Mg2+ Atoms | D–A distance (Å) | 8E1 atom | VRK1 atoms | D–A distance (Å) |
| ANP N6 | Asp 132 O | 2.8 | 8E1 N2 | Asp 132 O | 3.1 |
| ANP O2A | Lys 71 NZ | 3.4 | 8E1 O | Phe 134 N | 2.9 |
| ANP O2B | Asp 197 OD1 | 3.0 | 8E1 N3 | Asp 137 N | 3.1 |
| ANP N1 | Phe 134 N | 2.8 | |||
| ANP O3’ | Ser 181 O | 2.9 | |||
| ANP O1A | Mg2+ | 2.8 | |||
| ANP O3A | Mg2+ | 3.2 | |||
| ANP N7 | H2O O | 2.8 | |||
| ANP O3’ | H2O O | 3.3 | |||
| Mg coordination (in Chain D alone) | |||||
| Mg2+ | ANP O1A | 2.1 | |||
| Mg2+ | ANP O2B | 2.0 | |||
| Mg2+ | Asp 197 OD1 | 2.3 | |||
| Mg2+ | Asn 182 OD1 | 2.1 | |||
| Mg2+ | H2O O | 2.2 | |||
| Non‐polar contacts | |||||
| VRK1‐ANP | VRK1‐8E1 | ||||
| ANP Atom | VRK1 Residues | 8E1 Atom | VRK1 Residues | ||
| ANP C2 | Phe134, Leu184 | 8E1 C | Phe134, Gly135 | ||
| ANP C4 | Leu184 | 8E1 C1 | Gly135 | ||
| ANP C5 | Leu184 | 8E1 C2 | Gly135 | ||
| ANP C6 | Val69, Asp132, Phe134, Leu184 | 8E1 C5 | Gly135 | ||
| ANP C8 | Ile51 | 8E1 C6 | Leu184 | ||
| ANP C5’ | Ile51 | 8E1 C7 | Leu184 | ||
| ANP C2’ | Leu184 | 8E1 C9 | Val69, Phe134 | ||
| ANP PA | Ile51 | 8E1 C10 | Met131, Phe134 | ||
| ANP O1A | Ile51, K71 | 8E1 C14 | Phe48 | ||
| ANP O2B | Asp197 | 8E1 C15 | Phe134 | ||
| ANP O2’ | Leu184 | 8E1 S | Phe48, Ile51 | ||
| ANP O4’ | Ile51 | 8E1 O | Val69, Arg133, Phe134 | ||
| ANP O5’ | Ile51 | 8E1 N1 | Val196 | ||
| ANP N1 | Asp132, Arg133, Phe134 | 8E1 N2 | Val69, Asp132, Arg133, Phe134 | ||
| ANP N6 | Val69, Met131, Asp132, Phe134 | 8E1 N3 | Ser136, Asp137, Lys140 | ||
Structural comparison of VRK1–AMP‐PNP with apo and inhibitor counterparts
The superposition of VRK1–AMP‐PNP complex on the apo structure12 (PDB ID 2LAV) and VRK1–inhibitor complex25 (PDB ID 5UKF) revealed a root‐mean‐square deviation (RMSD) of 1.70 Å (270 equivalent Cα atoms) and 0.56 Å (243 equivalent Cα atoms), respectively, indicating that the ligand bound structures are similar to each other compared to the apo form (Fig. S2). For the comparison studies, we used only the apo structure's representative model from its NMR ensemble. The formation of the salt‐bridge between Lys71 and Glu83, a primary feature of active kinases, was stronger in the presence of AMP‐PNP (at 2.8 Å) in comparison to the inhibitor bound form (3.7 Å), where the apo structure has a distance of 7.9 Å [Fig. 3(A)]. In addition, the distance between Glu83 and Tyr198 (of DYG) (7.2 Å) indicated that the α‐C helix adopts an “out‐like” conformation in the apo form, against the ligand bound conformation (a distance of 4.7 Å), which adopts an ‘in’ conformation.30 The conformation of the P‐loop from “Open” to “Close” was evident from the apo and inhibitor bound forms, respectively [Fig. 3(B)]. Although we could not trace electron density for the P‐loop residues in the AMP‐PNP bound form, the orientation of the residues on either side of this missing loop mimic that of the inhibitor bound conformation. We speculate that even in the AMP‐PNP bound form the P‐loop might adopt a “Close‐conformation” [Fig. 3(B)], though this needs further investigation. It was concluded that the P‐loop is a highly flexible region in all VRK1 crystal structures and could be traced in only one of chains in the inhibitor complex (Chain A of PDB ID 5UKF).25 Nonetheless, our NMR titration shows that the residues in the P‐loop (Gln45, Gly45, Gly46, and Gly49) are affected upon AMP‐PNP binding, validating their interaction. The DYG [Fig. 3(C)] and DRF [Fig. 3(D)] motifs are similar in the AMP‐PNP and inhibitor complexes compared with the apo form, in which a few side‐chains adopt different orientations. In all three structures, the “DYG‐in” orientation is observed. The orientation of the Met131, next to the DRF motif, also exhibits flexibility depending on the occupancy of the active site pocket. In the apo form and AMP‐PNP complex, Met131 protrudes toward the pocket, as opposed to the inhibitor bound complex, forming non‐bonded interactions with the ligand. It is evident that the inhibitor which docks deeper into the pocket compared to AMP‐PNP relocates the Met131 further away in comparison to the AMP‐PNP complex [Fig. 3(E)]. A similar flexibility was also observed when comparing the VRK2 apo and inhibitor complexes. It is to be mentioned that these active site Met residues are well‐conserved among the Ser/Thr kinases31 and more insights are required to explore their functional role in VRK1.
Figure 3.
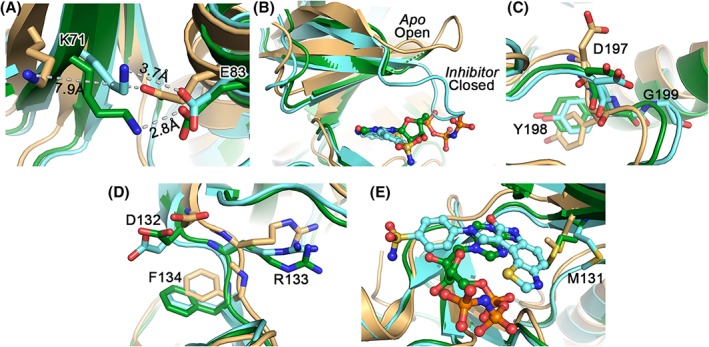
Comparison of the VRK1–AMP‐PNP complex (green) with the apo (pale orange) and inhibitor‐bound (cyan) structures, highlighting the major signature motifs. (A) the salt bridge formed between the K71 and E83 (in stick mode) can be clearly visible in the AMP‐PNP (2.8 Å) and inhibitor (3.7 Å) bound structures, but not in the apo form (7.9 Å). (B) The P‐loop conformation adopts an open form in the apo structure, while is closed in inhibitor‐bound structure. As this region cannot be traced in the AMP‐PNP structure, it needs to be seen if it resembles a close‐conformation. The residues of the DYG (C) and DRF (D) motifs are also similar in the ligand‐bound conformations in comparison to the apo form. (E) Met131, shown in stick mode, can be observed to adopt a flexible orientation dependent on the nature of bound ligand, with the inhibitor pushing it the farthest from the active site pocket as this docks deeper. In (B) and (E), the ligands are shown in ball and stick mode for reference.
Structural insights into VRK1 inhibitor design
A comparison of the inhibitor (GW297361X – PDB ID 5UKF) and AMP‐PNP‐bound VRK1 structures enabled us to provide structural insights into designing specific inhibitors for VRK1 (Fig. 4 and Table 1). (a) The thiazole moiety in the inhibitor is inserted deeper into the active site pocket, in comparison to the adenosine ring, a conductive characteristic of good ATP‐competitive inhibitor [Fig. 4(A) and (B)]. A Sulfur‐Π interaction with Phe48 has been reported to stabilize this region in one of the chains in the inhibitor bound structures.25 The nitrogen atom in thiazole ring forms a hydrogen bond with a water molecule, but not with any neighboring residues [Fig. 4(D)], the nearest Asp197 (DYG motif) backbone is 4.2 Å away. Thus, modifications to this thiazole nitrogen which can form hydrogen bonds with Asp197 could enhance binding affinity of the inhibitor. (b) The oxindole ring of the inhibitor mimics the interactions of the adenosine in AMP‐PNP, making hydrogen bonds with Asp132 and Phe134, and non‐polar interactions with Ile51 (Fig. 4 and Table 1). (c) The benzene sulfonamide moiety in the inhibitor is stabilized by fewer interactions in comparison to the equivalent ribose and phosphate groups in AMP‐PNP (Fig. 4 and Table I). Modifications to this region, which can enhance interactions with Asp197 and Ser181, similar to AMP‐PNP [Fig. 4(C)], might provide a better affinity to the inhibitor. Furthermore, based on the KLIFS database30 a search for kinases with similar interaction patterns, using a cut‐off of 0.65, resulted in six kinases. A structure‐based sequence alignment32 of all these kinases with VRK members, along with the prototypical protein kinase A (PKA‐C) was carried out to identify unique VRK1 residues (Fig. S1). Variations in the hinge region residues were observed, but the structure reveals that the interactions with the ligand are mainly governed by the backbone chain atoms from this region. The conserved Arg in the HRD motif is converted to Gly in VRK, and might have consequences in substrate protein recognition rather than inhibitor design. Interestingly, among the ligand binding residues, Ser181 in VRK1 is unique in comparison to other kinases, except ERK. Serine specific warheads could also be utilized toward designing VRK1 specific inhibitors, as this residue is unique among the VRK family (Fig. S1). Although the comparison provides a structural perspective, in future, it would be necessary to study the enzyme kinetics of these inhibitors and AMP‐PNP, lacking till‐date, to further enhance our understanding into VRK1 activity and specificity. In conclusion, as VRK1 is regarded as an important cancer drug target due to its enhanced expression levels in many cancer tissues, the molecular insights on the VRK1 interaction with AMP‐PNP presented here, along with the earlier inhibitor complexes,25 could be used to develop a pharmacophore model followed by fragment screening, toward identifying effective VRK1‐specific inhibitors.
Figure 4.
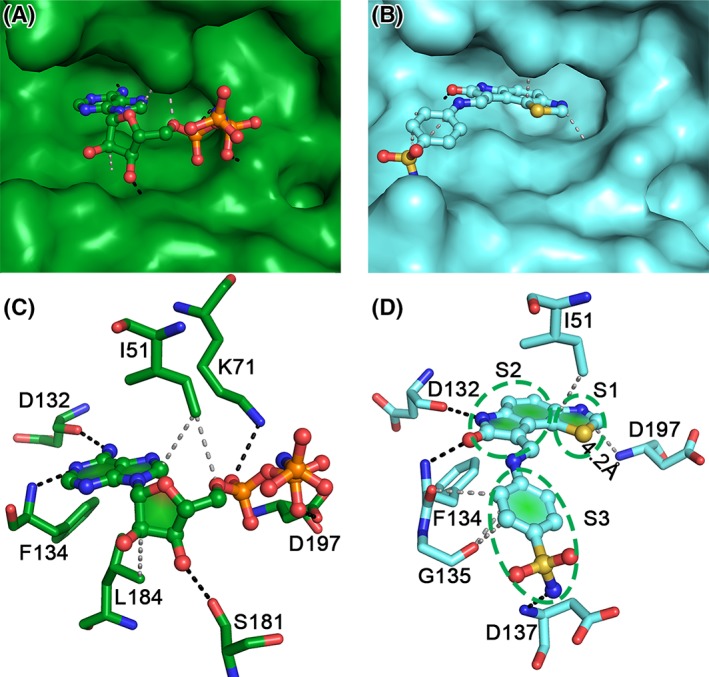
Comparison of AMP‐PNP (green) and inhibitor (GW297361X; cyan) bound VRK1 structures. Surface representation showing the docking of AMP‐PNP (A) and inhibitor (B) in the VRK1‐active site pocket. The P‐loop has been removed in the inhibitor bound structure (B) for clarity. It is visible that the inhibitor (B) sits deeper in comparison to AMP‐PNP (A). The hydrogen bonds and non‐polar contacts made by AMP‐PNP (C) and the inhibitor (D) are indicated by black and gray broken lines, respectively. The interactions can be categorized based on the three moieties (denoted by S1, S2, and S3) in the inhibitor. The thiazole moiety (S1) nitrogen is ~4 Å from the Asp197 backbone (B), while an equivalent region in AMP‐PNP is missing. The oxindole ring (S2) mimics the interactions made by the adenosine ring in AMP‐PNP, with Ile51, Asp132, and Phe134. The benzene sulfonamide moiety (S3) of the inhibitor is replaced by the ribose and phosphate groups in AMP‐PNP and is stabilized by more interactions than the inhibitor.
Materials and Methods
Protein expression and purification
The 15N‐labeled VRK1Δ35 for NMR titration study was prepared as previously reported.12 The codon optimized human VRK1 gene (Residues 1–364) with 11 surface Lys and Glu residues mutated to Ala, to enable crystallization as previously reported,25 was synthesized from GenScript and subcloned into pETSUMO vector. The resulting pETSUMO‐VRK1 was expressed and purified similar to the protocol described previously,12, 33 with a slight modification to the protein induction temperature (16°C, overnight).
NMR titration
2D 1H–15N transverse relaxation optimized spectroscopy (TROSY) NMR experiment was employed to study protein‐ligand interactions in solution. For this, 15N‐labeled VRK1Δ35 (0.2 mM) was titrated with AMP‐PNP. Data were acquired at 25°C in the presence of excess amount of ligand (1 mM) on a Bruker 700 MHz NMR spectrometer as previously described.34 The spectra acquired were processed through Topspin version 2.1 (Bruker BioSpin) and analyzed using the NMRFAM Sparky.35 The chemical shift perturbation was calculated using the formula .
Crystallization and X‐ray diffraction
VRK1, 25 mg/mL in 20 mM Tris buffer (pH 7.5), 150 mM NaCl, and 1 mM DTT, was incubated with Adenylyl‐imidodiphosphate (AMP‐PNP) (Sigma‐Aldrich, USA), in a molar ratio of 1:6 and supplemented with 10 mM of MgCl2, overnight at 4°C prior to crystallization using the hanging‐vapor diffusion method. The best crystals of VRK1–AMP‐PNP complex appeared in 27.5% w/v PEG 3350, 0.2 M of ammonium sulfate and 0.1 M of HEPES (pH 7.0) after 12–15 days at 18°C, with 2 μL of protein complex mixed with 1 μL of the reservoir solution equilibrated against 1 mL of reservoir solution. The crystals were cryo‐protected with 15% glycerol supplemented to the reservoir solution and diffraction data were collected at the National Synchrotron Radiation Research Center (Hsinchu, Taiwan) at 100 K on beamline, TPS05A using a MX300HS detector. Three datasets from a single crystal were collected.
Structure determination
The images were indexed and integrated using iMosflm,36 after which they were merged and scaled by AIMLESS37; both software's were part of the CCP4 suite of programs.38 A molecular replacement solution was obtained using the program BALBES,39 followed by structure refinement using BUSTER40 and map fitting using COOT.41 The AMP‐PNP electron density, accompanied by a magnesium ion, in the active site was clearly observed in each of the four chains in the asymmetric unit. Apart from this, PEG, sulfate, and chloride ions, from the crystallization solution were also observed. In addition, water molecules were identified using the Fo–Fc and 2Fo–Fc electron density maps contoured at 3.0 and 1.0 σ cut‐offs, respectively. The Ramachandran statistics were tabulated using Molprobity.42 The protein ligand interactions were analyzed using LigPlot43 and manual inspection. All figures were prepared using PyMOL (DeLano Scientific, Palo Alto, CA, USA). The statistical analysis for data collection and refinement was summarized in Table 2.
Table 2.
X‐ray crystallographic data reduction and refinement statistics for VRK1 in complex with AMP‐PNP
| Data reduction | |
|---|---|
| Wavelength (Å) | 0.9998 |
| Space group | P 21 21 21 |
| Cell dimensions | |
| a; b; c (Å) | 92.59; 96.84; 192.52 |
| α = β = γ (°) | 90 |
| Resolution (Å) | 68.00–2.07 (2.11–2.07)a |
| R merge | 0.106 (0.701) |
| R pim | 0.032 (0.215) |
| Unique reflections | 106113 (5210) |
| Mean [(I)/σ(I)] | 21.3 (5.7) |
| Completeness | 100 (100) |
| Multiplicity | 22.4 (22.5) |
| CC1/2 | 0.999 (0.968) |
| Refinement | |
| Number of reflections | 105361 |
| Resolution (Å) | 20.00–2.07 |
| R‐value | 0.186 |
| R‐free | 0.223 |
| No. of atoms | |
| Total/VRK1 atoms/AMP‐PNP atoms/water molecules/Hetatoms | 11,080/9986/124/889/81 |
| Mean B‐value (Å2) | |
| Total/VRK1 atoms/AMP‐PNP atoms/water molecules/Hetatoms | 41.2/39.9/76.5/47.0/80.3 |
| R.m.s.d. from ideal values | |
| Bond lengths (Å) | 0.010 |
| Bond angles (°) | 1.00 |
| Ramachandran statistics (%) | |
| Preferred regions | 96.9 |
| Allowed regions | 3.1 |
| Outliers | 0 |
Values in parentheses refer to the corresponding values of the highest‐resolution shell.
Protein Data Bank deposition
Co‐ordinates and structure factors for VRK1–AMP‐PNP complex have been deposited in the Protein Data Bank with accession number 6AC9.
Supporting information
Figure S1: Sequence alignment of VRK members with other kinases. The secondary structure elements corresponding to VRK1 are shown above and labeled accordingly. The alignment is shown only for the region covering the ligand binding residues. The canonical kinase motifs are shown within green boxes and labelled below, corresponding to VRK1. The AMP‐PNP interacting residues in VRK1 are indicated by blue colored stars. Both these motifs and interacting residues, indicate the VRK3 is a non‐canonical kinase. In addition, the location of the unique Ser181 in VRK1 is indicated by a black arrow. It could be observed that this is mutated to Ala and Glu in VRK2 and VRK3, respectively. This Ser181 can be an ideal hotspot to achieve specificity for VRK1.
Figure S2: Cartoon representation of the superposition of apo (pale orange), AMP‐PNP (green) and inhibitor (cyan) bound VRK1 structures. It can be observed that the ligand bound structures are similar in comparison to the apo form. The important motifs are colored uniquely to indicate their locations in the structure. The P‐loop (red), Met131 (yellow), DRF motif (marine blue), DYG (purple), Lys71 and Glu83 (dark blue).
Acknowledgments
The authors thank the National Synchrotron Radiation Research Center (NSRRC) and their staff at beamline TPS05A for help with data collection. The NSRRC is a national user facility supported by the National Science Council of Taiwan, ROC; the Synchrotron Radiation Protein Crystallography Facility at NSRRC is supported by the National Research Program for Genomic Medicine. This work was supported by the Ministry of Education Singapore AcRF Tier 1 (RG137/17).
Contributor Information
Sreekanth Rajan, Email: srajan@ntu.edu.sg.
Ho Sup Yoon, Email: hsyoon@ntu.edu.sg.
References
- 1. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S (2002) The protein kinase complement of the human genome. Science 298:1912–1934. [DOI] [PubMed] [Google Scholar]
- 2. Ma HT, Poon RY (2011) How protein kinases co‐ordinate mitosis in animal cells. Biochem J 435:17–31. [DOI] [PubMed] [Google Scholar]
- 3. Bhullar KS, Lagaron NO, McGowan EM, Parmar I, Jha A, Hubbard BP, Rupasinghe HPV (2018) Kinase‐targeted cancer therapies: progress, challenges and future directions. Mol Cancer 17:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nigg EA (2001) Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol 2:21–32. [DOI] [PubMed] [Google Scholar]
- 5. Welburn JPI, Jeyaprakash AA (2018) Mechanisms of mitotic kinase regulation: a structural perspective. Front Cell Dev Biol 6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bayliss R, Fry A, Haq T, Yeoh S (2012) On the molecular mechanisms of mitotic kinase activation. Open Biol 2:120136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Valbuena A, Lopez‐Sanchez I, Lazo PA (2008) Human VRK1 is an early response gene and its loss causes a block in cell cycle progression. PLoS One 3:e1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kang TH, Park DY, Choi YH, Kim KJ, Yoon HS, Kim KT (2007) Mitotic histone H3 phosphorylation by vaccinia‐related kinase 1 in mammalian cells. Mol Cell Biol 27:8533–8546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moura DS, Campillo‐Marcos I, Vazquez‐Cedeira M, Lazo PA (2018) VRK1 and AURKB form a complex that cross inhibit their kinase activity and the phosphorylation of histone H3 in the progression of mitosis. Cell Mol Life Sci 75:2591–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nichols RJ, Traktman P (2004) Characterization of three paralogous members of the Mammalian vaccinia related kinase family. J Biol Chem 279:7934–7946. [DOI] [PubMed] [Google Scholar]
- 11. Scheeff ED, Eswaran J, Bunkoczi G, Knapp S, Manning G (2009) Structure of the pseudokinase VRK3 reveals a degraded catalytic site, a highly conserved kinase fold, and a putative regulatory binding site. Structure 17:128–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shin J, Chakraborty G, Bharatham N, Kang C, Tochio N, Koshiba S, Kigawa T, Kim W, Kim KT, Yoon HS (2011) NMR solution structure of human vaccinia‐related kinase 1 (VRK1) reveals the C‐terminal tail essential for its structural stability and autocatalytic activity. J Biol Chem 286:22131–22138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim W, Chakraborty G, Kim S, Shin J, Park CH, Jeong MW, Bharatham N, Yoon HS, Kim KT (2012) Macro histone H2A1.2 (macroH2A1) protein suppresses mitotic kinase VRK1 during interphase. J Biol Chem 287:5278–5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schober CS, Aydiner F, Booth CJ, Seli E, Reinke V (2011) The kinase VRK1 is required for normal meiotic progression in mammalian oogenesis. Mech Dev 128:178–190. [DOI] [PubMed] [Google Scholar]
- 15. Valbuena A, Sanz‐Garcia M, Lopez‐Sanchez I, Vega FM, Lazo PA (2011) Roles of VRK1 as a new player in the control of biological processes required for cell division. Cell Signal 23:1267–1272. [DOI] [PubMed] [Google Scholar]
- 16. Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, Sanli K, von Feilitzen K, Oksvold P, Lundberg E, Hober S, Nilsson P, Mattsson J, Schwenk JM, Brunnstrom H, Glimelius B, Sjoblom T, Edqvist PH, Djureinovic D, Micke P, Lindskog C, Mardinoglu A, Ponten F (2017) A pathology atlas of the human cancer transcriptome. Science 357:eaan2507. [DOI] [PubMed] [Google Scholar]
- 17. Molitor TP, Traktman P (2013) Molecular genetic analysis of VRK1 in mammary epithelial cells: depletion slows proliferation in vitro and tumor growth and metastasis in vivo. Oncogene 2:e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Salzano M, Vazquez‐Cedeira M, Sanz‐Garcia M, Valbuena A, Blanco S, Fernandez IF, Lazo PA (2014) Vaccinia‐related kinase 1 (VRK1) confers resistance to DNA‐damaging agents in human breast cancer by affecting DNA damage response. Oncotarget 5:1770–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Campillo‐Marcos I, Lazo PA (2018) Implication of the VRK1 chromatin kinase in the signaling responses to DNA damage: a therapeutic target? Cell Mol Life Sci 75:2375–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gorjanacz M, Klerkx EP, Galy V, Santarella R, Lopez‐Iglesias C, Askjaer P, Mattaj IW (2007) Caenorhabditis elegans BAF‐1 and its kinase VRK‐1 participate directly in post‐mitotic nuclear envelope assembly. EMBO J 26:132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vega FM, Sevilla A, Lazo PA (2004) p53 stabilization and accumulation induced by human vaccinia‐related kinase 1. Mol Cell Biol 24:10366–10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim YS, Kim SH, Shin J, Harikishore A, Lim JK, Jung Y, Lyu HN, Baek NI, Choi KY, Yoon HS, Kim KT (2014) Luteolin suppresses cancer cell proliferation by targeting vaccinia‐related kinase 1. PLoS One 9:e109655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim SH, Lyu HN, Kim YS, Jeon YH, Kim W, Kim S, Lim JK, Lee HW, Baek NI, Choi KY, Lee J, Kim KT (2015) Brazilin isolated from Caesalpinia sappan suppresses nuclear envelope reassembly by inhibiting barrier‐to‐autointegration factor phosphorylation. J Pharmacol Exp Ther 352:175–184. [DOI] [PubMed] [Google Scholar]
- 24. Kim SH, Ryu HG, Lee J, Shin J, Harikishore A, Jung HY, Kim YS, Lyu HN, Oh E, Baek NI, Choi KY, Yoon HS, Kim KT (2015) Ursolic acid exerts anti‐cancer activity by suppressing vaccinia‐related kinase 1‐mediated damage repair in lung cancer cells. Sci Rep 5:14570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Counago RM, Allerston CK, Savitsky P, Azevedo H, Godoi PH, Wells CI, Mascarello A, de Souza Gama FH, Massirer KB, Zuercher WJ, Guimaraes CRW, Gileadi O (2017) Structural characterization of human Vaccinia‐related kinases (VRK) bound to small‐molecule inhibitors identifies different P‐loop conformations. Sci Rep 7:7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fedorov O, Marsden B, Pogacic V, Rellos P, Muller S, Bullock AN, Schwaller J, Sundstrom M, Knapp S (2007) A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc Natl Acad Sci U S A 104:20523–20528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vazquez‐Cedeira M, Barcia‐Sanjurjo I, Sanz‐Garcia M, Barcia R, Lazo PA (2011) Differential inhibitor sensitivity between human kinases VRK1 and VRK2. PLoS One 6:e23235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR (2011) Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol 29:1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bagshaw C (2001) ATP analogues at a glance. J Cell Sci 114:459–460. [DOI] [PubMed] [Google Scholar]
- 30. Kooistra AJ, Kanev GK, van Linden OP, Leurs R, de Esch IJ, de Graaf C (2016) KLIFS: a structural kinase–ligand interaction database. Nucleic Acids Res 44:D365–D371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Veredas FJ, Cantón FR, Aledo JC (2017) Methionine residues around phosphorylation sites are preferentially oxidized in vivo under stress conditions. Sci Rep 7:40403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Di Tommaso P, Moretti S, Xenarios I, Orobitg M, Montanyola A, Chang JM, Taly JF, Notredame C (2011) T‐coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res 39:W13–W17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rajan S, Choi M, Nguyen QT, Ye H, Liu W, Toh HT, Kang C, Kamariah N, Li C, Huang H, White C, Baek K, Gruber G, Yoon HS (2015) Structural transition in Bcl‐xL and its potential association with mitochondrial calcium ion transport. Sci Rep 5:10609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pervushin K, Riek R, Wider G, Wuthrich K (1997) Attenuated T2 relaxation by mutual cancellation of dipole–dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci U S A 94:12366–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee W, Tonelli M, Markley JL (2015) NMRFAM‐SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31:1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG (2011) iMOSFLM: a new graphical interface for diffraction‐image processing with MOSFLM. Acta Cryst D67:271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Evans PR, Murshudov GN (2013) How good are my data and what is the resolution? Acta Cryst D69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Cryst D67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Long F, Vagin AA, Young P, Murshudov GN (2008) BALBES: a molecular‐replacement pipeline. Acta Cryst D64:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Smart OS, Womack TO, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrhein C, Bricogne G (2012) Exploiting structure similarity in refinement: automated NCS and target‐structure restraints in BUSTER. Acta Cryst D68:368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 42. Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Cryst D66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wallace AC, Laskowski RA, Thornton JM (1995) LIGPLOT: a program to generate schematic diagrams of protein‐ligand interactions. Protein Eng 8:127–134. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Sequence alignment of VRK members with other kinases. The secondary structure elements corresponding to VRK1 are shown above and labeled accordingly. The alignment is shown only for the region covering the ligand binding residues. The canonical kinase motifs are shown within green boxes and labelled below, corresponding to VRK1. The AMP‐PNP interacting residues in VRK1 are indicated by blue colored stars. Both these motifs and interacting residues, indicate the VRK3 is a non‐canonical kinase. In addition, the location of the unique Ser181 in VRK1 is indicated by a black arrow. It could be observed that this is mutated to Ala and Glu in VRK2 and VRK3, respectively. This Ser181 can be an ideal hotspot to achieve specificity for VRK1.
Figure S2: Cartoon representation of the superposition of apo (pale orange), AMP‐PNP (green) and inhibitor (cyan) bound VRK1 structures. It can be observed that the ligand bound structures are similar in comparison to the apo form. The important motifs are colored uniquely to indicate their locations in the structure. The P‐loop (red), Met131 (yellow), DRF motif (marine blue), DYG (purple), Lys71 and Glu83 (dark blue).