

Abstract
Antibody–drug conjugates (ADCs) are antibody‐based therapeutics that have proven to be highly effective cancer treatment platforms. They are composed of monoclonal antibodies conjugated with highly potent drugs via chemical linkers. Compared to cysteine‐targeted chemistries, conjugation at native lysine residues can lead to a higher degree of structural heterogeneity, and thus it is important to evaluate the impact of conjugation on antibody conformation. Here, we present a workflow involving native ion mobility (IM)‐MS and gas‐phase unfolding for the structural characterization of lysine‐linked monoclonal antibody (mAb)–biotin conjugates. Following the determination of conjugation states via denaturing Liquid Chromatography‐Mass Spectrometry (LC–MS) measurements, we performed both size exclusion chromatography (SEC) and native IM‐MS measurements in order to compare the structures of biotinylated and unmodified IgG1 molecules. Hydrodynamic radii (Rh) and collision cross‐sectional (CCS) values were insufficient to distinguish the conformational changes in these antibody–biotin conjugates owing to their flexible structures and limited instrument resolution. In contrast, collision induced unfolding (CIU) analyses were able to detect subtle structural and stability differences in the mAb upon biotin conjugation, exhibiting a sensitivity to mAb conjugation that exceeds native MS analysis alone. Destabilization of mAb–biotin conjugates was detected by both CIU and differential scanning calorimetry (DSC) data, suggesting a previously unknown correlation between the two measurement tools. We conclude by discussing the impact of IM‐MS and CIU technologies on the future of ADC development pipelines.
Keywords: biotherapeutics, ion mobility‐mass spectrometry, protein stability, differential scanning calorimetry
Introduction
Antibody–drug conjugates (ADCs) have become a promising class of therapeutics for the treatment of cancer, underscored by the four ADCs currently approved by the US FDA, and the more than 60 ADCs in various clinical trial stages.1, 2, 3, 4 ADCs consist of monoclonal antibodies (mAbs) that are covalently attached to highly potent drugs through chemical linkers comprised of relatively labile bonds. Such conjugation allows for the high selectivity of mAbs to be combined with cytotoxic drugs, achieving discrimination between healthy and diseased tissue in contrast to traditional chemotherapies. While both cysteine and lysine‐targeted chemistries are widely used in ADC generation,5, 6, 7 the latter typically results in a more heterogeneous drug‐to‐antibody ratio (DAR) distribution, often creating therapeutics with increased structural complexity, owing to the large number of native lysine residues in mAb sequences.8, 9, 10
Conjugated species, which differ in terms of their levels and sites of drug incorporation, can potentially exhibit differential structures and pharmacokinetic properties.6 As such, DAR values are considered critical quality attributes of ADCs, necessitating the development of a range of analytical methods for their quantitative evaluation. For example, many separation techniques have been utilized to accurately derive DAR values, such as hydrophobic interaction chromatography (HIC), ion exchange chromatography (IEC), reverse phase liquid chromatography (RPLC), capillary electrophoresis (CE), or capillary isoelectric focusing (cIEF).8, 10, 11, 12 As lysine‐conjugated ADCs typically exhibit greater heterogeneity and poorer chromatographic peak shapes than equivalent cysteine‐modified therapeutics, mass spectrometry (MS) is typically deployed for their DAR assessment.8, 13, 14 Recently, native MS has been increasingly applied for ADC analysis, providing highly accurate DAR values under native conditions, which are especially critical for capturing accurate values produced by cysteine‐linked modification chemistries.7, 15, 16, 17 Moreover, with advances in high‐resolution instrumentation for native MS experiments, DAR values can often be readily extracted from ADCs prepared at physiologically relevant pH.18, 19, 20, 21, 22
In addition to DAR values, it is also of critical importance to assess the impact of conjugation chemistries on mAb higher order structures (HOS). Biophysical assays have been broadly used in the biopharmaceutical industry for assessing such HOS effects in ADCs. For instance, differential scanning calorimetry (DSC) is widely used to assess the thermal stability of mAbs, providing key information used to predict the clinical success of mAb‐based drugs. Nonetheless, only limited information is obtained from such data.23, 24 Recently, MS‐based techniques have emerged as an important class of tools for protein HOS characterization. Among these, hydrogen–deuterium exchange (HDX)‐MS has been used to compare the overall conformation and flexibility of ADCs to their parent mAbs.7, 25 Despite its ability to access localized structure information in large therapeutic proteins, HDX‐MS experiments often take a long time to perform and analyze. Thus, there is a growing need for high‐throughput structural probes of ADC structure as a function of conjugation state and formulation that can operate in both development and quality assessment roles.
The combination of ion mobility (IM) and MS has proven to be a useful tool for the characterization of mAbs and ADCs, by separating such proteins according to their size and recording their ion‐neutral collision cross sections (CCSs) as a means of HOS analysis.15, 26, 27, 28 In an effort to assess antibody structures in greater detail, collision induced unfolding (CIU) experiments can be performed.29 This technology involves the collisional activation of protein ions prior to IM‐MS separation in order to initiate protein unfolding events in the gas‐phase. CIU is capable of differentiating IgG subclasses, detecting minor alterations in mAb glycoforms, assessing stability shifts associated with site‐specific ADCs, and evaluating the comparability of biosimilars.30, 31, 32, 33, 34, 35, 36, 37
In this report, we present a CIU based workflow for the rapid characterization of a human IgG1 mAb conjugated with biotin via its native lysine residues, which we treat as a model ADC system. Despite the high degree of structural similarity across conjugation states revealed by native IM‐MS, our CIU results indicate the presence of subtle structural changes in the mAbs upon biotin conjugation, as revealed by shifts in their overall stabilities and ground state CCSs. Through comparative analysis, we are also able to correlate the differences observed in our CIU data with the DAR values quantified using LC–MS, measured under denaturing conditions. We conclude by discussing the benefits of native IM‐MS and CIU assays for ADC characterization.
Materials and Methods
Biotinylation of mAb
The mAb–biotin conjugates were prepared as previously described by reacting the IgG1 mAb with 5, 10, 15, 20, and 30 equivalents of EZ‐Link sulfo‐NHS‐LC‐Biotin (Pierce).22
Denaturing LC/MS
LC–MS measurements were performed on an Agilent 6230 TOF LC/MS system with a 1290 Infinity LC system as previously described.22
Native ion mobility‐mass spectrometry (IM‐MS) and collision induced unfolding (CIU)
Native IM‐MS data were acquired either on a traveling wave ion mobility (TWIM) mass spectrometer, Synapt G2 HDMS instrument, or on a Synapt HDMS instrument modified with an RF‐confining drift cell. For the CCS measurements on TWIMS instrument, samples were buffer exchanged into 100 mM ammonium acetate using Bio‐rad microspin column with 6 kDa MWCO. The nESI voltage was set at 1.1–1.3 kV, and sampling cone was set at 60 V. The traveling‐wave ion mobility separator was operated at a pressure of ~2.5 mbar with wave height and wave velocity set at 14 V and 300 m/s, respectively. IM wave velocity and wave height were optimized to achieve higher accuracy in CCS measurements with a bit trade off in the arrival time resolution. To derive the CCS(Ω) values, protein drift times measured in the TWIM device were calibrated using the protein standards concanavalin A (Con A), alcohol dehydrogenase (ADH), and pyruvate kinase (PK). The ToF‐MS was operated over the m/z range of 1000–10,000 at a pressure of 1.5 × 10−6 mbar. For CCS measurements carried out using the RF‐confining drift tube instrument, samples were infused at a concentration of 8 μM. The instrument was operated in positive ESI mode and instrument parameters were set as described previously.38 CIU experiments were performed in the trap region prior to IM separation on a Synapt G2 HDMS instrument as previously described. The antibody ions at desired charge state were selected by the high m/z transmission quadrupole. The trap collision voltage was applied on the selected protein ions incrementally ramping from 5 V to 200 V by 5 V. The ion mobility traveling wave height was set to 40 V and the wave velocity was set to 600 m/s.
MS data analysis
Denaturing LC/MS data was extracted and deconvoluted using MassHunter Qualitative Analysis Software (Agilent, Santa Clara, CA). Native mass spectra were calibrated externally using a solution of cesium iodide (100 mg/mL) and processed with Masslynx V4.1 software (Waters, Milford, MA). CIU data was extracted using TWIMExtract39 and analyzed using the CIUSuite40 to generate CIU plots and perform CIU comparisons. Root‐mean‐square deviation (RMSD) values were computed from a pixel‐to‐pixel comparison of two CIU fingerprints. In order to analyze the IM peak widths in CIU data, IM arrival time distributions (ATDs) observed at each collision voltage were modeled as a sum of Gaussian components. Components were added sequentially until the goodness of fit (r 2) exceeded 0.99, allowing optimized fitting without over‐fitting. CIU features in the dataset are detected by grouping observed arrival time peaks that are present across multiple collision voltages. The tolerance (allowed deviation) from a median drift time was three drift bins, and the minimum number of collision voltages required for feature fitting was three (or 10 V range). Following feature detection, the transition region between features is fitted to a logistic (generalized sigmoid) function. The logistic function parameters describe the lower and upper asymptotes (centroid drift times of the features before and after the transition), the growth rate or steepness of the transition, and the midpoint voltage, which we term the “CIU‐50” value.
Differential scanning calorimetry (DSC)
DSC experiments were performed on a Nano DSC instrument (TA Instrument, New Castle, DE). Samples were buffer exchanged into 100 mM ammonium acetate, pH 7.1 buffer and diluted to ~0.5 mg/mL. Accurate concentration was determined from UV absorbance at 280 nm. 300 μL of the protein solution was loaded to the capillary sample cell while the reference cell contained ammonium acetate buffer. The chamber was pressurized to 3 atm and the temperature ramped from 25°C to 95°C at 1°C/min heating rate. The recorded DSC thermograms were baseline subtracted and subjected to a multi‐component Gaussian fitting in the NanoAnalyze software (TA Instrument, New Castle, DE). The temperatures for three major transitions were extracted from the fitted Gaussian models, relating to the unfolding of CH2, Fab, and CH3 domains. Replicates were not available for each sample. Thus, we used a conservative value of 1°C as the cutoff limit for evaluating the significance of the differences observed in melt temperatures.
Results and Discussion
Determining DAR values from denaturing LC–MS measurements
The conjugation of one biotin to the lysine residue yields a mass addition of 339.5 Da. After the conjugation reaction, the parent mAb and the resulting ADCs models were subjected to denaturing LC–MS analysis to assess both the distribution of attached biotins produced, as well as their DAR values (Supporting Information Fig. S1). Under denaturing MS conditions, the resolution of different glycoforms (G0F, G1F, and G2F) is achieved for the parent mAb. However, the charge state distributions for biotinylated mAbs overlap significantly as more than 10 eqv. conjugation events are achieved, producing ADC mimics of increasing heterogeneity. As expected, individual biotin conjugation states cannot readily be differentiated from antibody glycoforms in LC–MS data acquired under denaturing conditions. To obtain accurate biotin‐to‐antibody ratios, protein samples were treated with PNGase F under native conditions in order to remove all N‐linked glycans and reduce the heterogeneity of our model ADCs. Denaturing LC–MS data acquired for the deglycosylated samples reveals primarily a single peak for the unconjugated deglycosylated mAb (Fig. 1). Experimental masses calculated for deglycosylated biotinylated antibodies range from 145.1 kDa to 152.2 kDa, comprising mAb species with 0–21 biotins attached. Average DAR values were calculated using the deconvoluted mass peak areas recorded in these experiments. Based on these results, reacting IgG 1 with 5 eqv., 10 eqv., 15 eqv., 20 eqv., and 30 eqv. of biotin under the conditions described yielded mAb‐biotin conjugates with average DAR values of 2.5, 4.6, 7.1, 9.5, and 14.3, respectively.
Figure 1.

(A) Deconvoluted mass spectrum for deglycosylated parent mAb recorded from denaturing LC–MS analysis. One main species is observed with one low intensity species corresponding to glycated mAb (asterisk labeled). (B–F) Zero charge mass spectra for biotinylated mAbs under same experiment conditions. Average DAR for each mAb‐biotin sample is calculated based on the peak area.
Native IM‐MS analysis reveals similar native gas‐phase structures for antibody–biotin conjugates
Although LC–MS is a well‐validated method to determine DAR values for lysine‐linked ADCs, it is unable to assess native mAb structures upon conjugation. To obtain more structural information, we analyzed the biotinylated mAbs by IM‐MS under native‐like solution conditions. Figure 2(A) displays the mass spectra for glycosylated ADCs, occupying a much narrower charge state envelope (from 21+ to 26+) when compared to typical denaturing MS data. As highlighted in the mass spectra, signals for the 23+ charge state shift to higher m/z values when more biotin equivalents are added for the conjugation reaction, indicating increased mAb molecular mass values. In order to preserve compact mAb structures in the gas phase we limited the amount of activation experienced by mAb ions in our experiments. These conditions result in broader features in the resulting mass spectra, produced through the non‐specific binding of buffer components to mAbs which are carried into the gas phase, and make resolution of either glycoforms or biotin conjugation states impossible. Thus, for the data shown in Figure 2, the intact masses of detected mAbs were calculated based on the recorded centroid m/z values of the observed charge states. Our measurements indicate an average M w of 148.92 ± 0.06 kDa for the parent mAb which increases linearly as a function of equivalents of biotin added for ADC model production, terminating in an intact mass of 154.02 ± 0.16 kDa, recorded for the sample containing 30 equivalents of biotin [Fig. 2(C), blue]. Relative average mass differences between the biotinylated mAbs and the parent mAb were calculated in order to estimate the average DAR values for each mAb‐biotin sample, producing DARs of 1.8, 4.8, 6.5, 8.2, and 15.0, in agreement with our LC–MS DAR evaluation.
Figure 2.

Overlay of the native MS spectra for (A) glycosylated and (B) deglycosylated mAb‐biotins. The 23+ charge state of the parent mAb is highlighted by the blue dashed line. (C) the average masses are calculated and plotted against the number of biotin equivalents reacted with the mAb. The peak width or mass resolution is shown as dashed line. (D) CCS values in nitrogen derived from TWIMS measurements are shown for each charge state of each sample.
To better resolve these conjugates by native IM‐MS, we analyzed deglycosylated ADC models as discussed above, and observed narrower peak MS widths as expected [Fig. 2(B)]. These data produced measured intact ADC masses ranging from 145.58 ± 0.05 kDa to 150.23 ± 0.17 kDa [Fig. 2(C), orange] and DAR values of 2.1, 4.5, 6.7, 9.6, and 13.7 for samples produced from reactions using increasing equivalents of biotin. We note that the DAR values extracted from native MS are systematically lower than those obtained by LC–MS, and importantly, if the peak widths or mass resolving power of our native MS data are taken into account [dashed line in Fig. 2(C)], the discrimination between some of the mAb–biotins states is challenging to accomplish at high confidence.
In order to characterize the gas‐phase structures of mAb‐biotin conjugates produced under native‐like conditions and evaluate the ability of IM‐MS alone to differentiate our group of ADC mimics, a comprehensive series of CCS measurements were carried out. The CCS values for each charge state of the ADC samples in nitrogen acquired by TWIM are shown in Figure 2(D). CCS values averaged over all observed charge states range from 76.2 ± 0.8 nm2 to 77.8 ± 1.0 nm2 for glycosylated mAbs, and from 75.0 ± 0.7 nm2 to 76.2 ± 0.6 nm2 for their deglycosylated analogues. IM analyses were also performed on an RF‐confining drift tube device containing helium (Tables 1 and Supporting Information Table S1). Overall, measured CCS values for ADC models produced from reaction conditions containing 30 biotin equivalents differ by ~2% when compared with unmodified IgG1 ions, indicating that only minor changes in global mAb structure take place upon biotin conjugation. The absolute sizes of the parent mAbs and the biotinylated mAbs in solution were measured by SEC‐MALS/QELS (size‐exclusion chromatography equipped with multi‐angle light scattering and quasi‐elastic light scattering detectors) and reported as hydrodynamic radius (Rh) values (Tables 1 and Supporting Information Table S1). Similarly, all the ADC mimics have indistinguishable Rh values in solution. We note a structural compaction of ~22% for both parent mAb and mAb–biotin conjugates in the gas phase, when comparing experimental CCS values with the CCS values derived from R h using the equation described by Hewitt et al.,41 consistent with previous reports.26, 27 Taken together, native IM‐MS analysis results suggest the presence of similarly compact gas‐phase structures for the mAb–biotin conjugates compared to the unmodified mAb.
Table 1.
Traveling Wave Ion Mobility (TWIM) Derived CCS Values in Nitrogen, RF‐Confining Drift Cell Measured CCS Values in Helium, and SEC‐MALS/QELS Determined Hydrodynamic Radius (for full list, see Supporting Information Table S1)
| ΩN2 (nm2) | ΩHe (nm2) | Rh (nm) | |
|---|---|---|---|
| Glycosylated | |||
| Parent mAb | 76.2 ± 0.8 | 70.32 ± 0.28 | 5.06 ± 0.1 |
| mAb + 30 eqv. biotin | 77.8 ± 1.0 | 71.66 ± 0.48 | 5.10 ± 0.1 |
| Deglycosylated | |||
| Parent mAb | 75.0 ± 0.7 | 69.34 ± 0.57 | 5.04 ± 0.1 |
| mAb + 30 eqv. biotin | 76.2 ± 0.6 | 71.17 ± 0.23 | 5.13 ± 0.1 |
aCCS values are averaged from all charge states.
Thermal stabilities of mAb–biotin conjugates in solution
In order to evaluate the thermal stabilities of our ADC models in solution, we utilized DSC to record melt temperatures for glycosylated and deglycosylated mAbs in the absence of biotinylation. As shown in Figure 3(A), we detect a shift in the first mAb transition temperature, with the other two transition temperatures remaining essentially unchanged. Specifically, the first melt temperature we observe is apparently decreased from 73.8°C to 67.5°C upon deglycosylation, indicating altered CH2 domain structure upon glycan removal, in line with previous literature reports.42, 43
Figure 3.
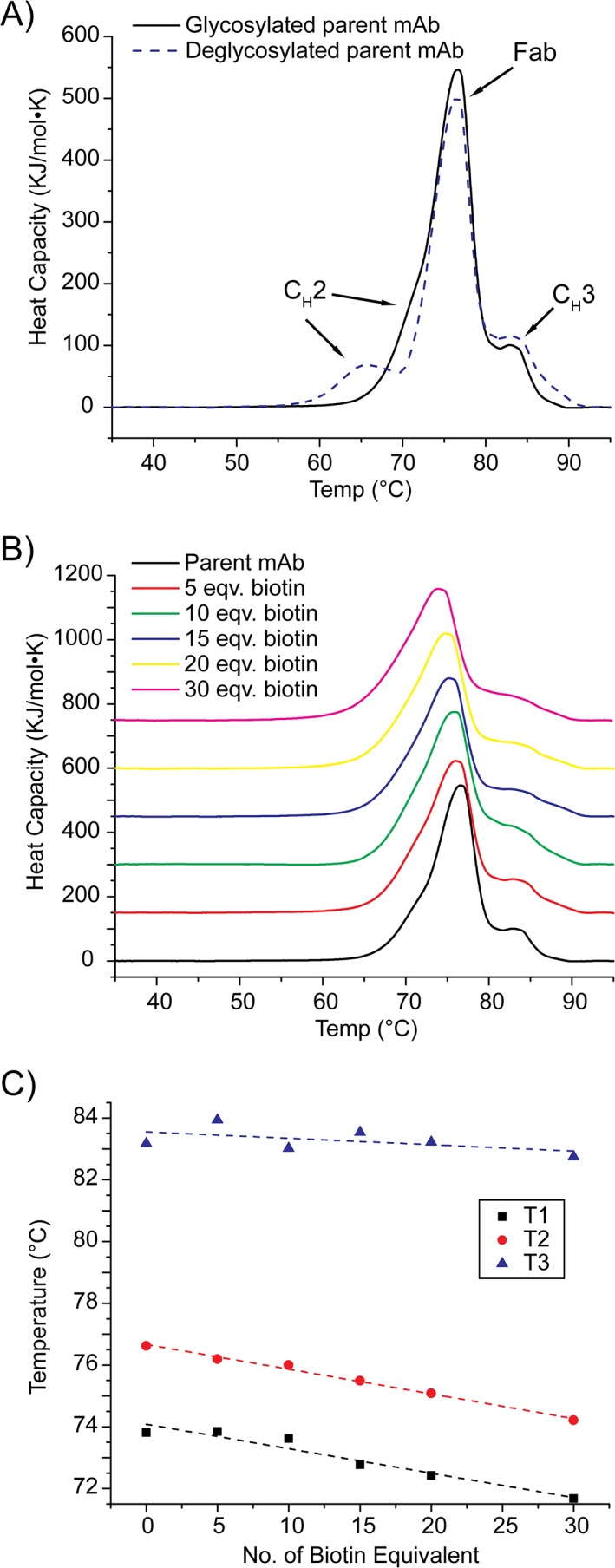
(A) Overlay of the baseline subtracted DSC thermograms recorded for the control mAbs with or without N‐linked glycosylation. (B) Overlay of the baseline subtracted DSC thermograms for the biotinylated mAbs with N‐linked glycosylation. (C) The transition temperatures extracted from the DSC thermograms plotted as a function of biotin equivalents reacted with the mAb.
Following these control experiments, we further compared DSC results acquired for ADC models prepared using a range of biotin equivalents with those acquired for the parent mAb [Fig. 3(B)]. While biotinylated mAbs also unfold to produce three distinct melting transitions, we observe differences in their absolute values that are correlated with expected levels of biotinylation for these samples. We observe a linear decrease in Fab melt temperature as a greater number of biotins are conjugated to the target mAb, shifting from 76.6°C to 74.2°C, whereas the unfolding temperature we observe for the CH2 domain remains unchanged at ~73.8°C until more than 15 equivalents of biotins are used for the conjugation reaction. For deglycosylated ADC models, the observed melt temperatures also shift as greater numbers of biotins are conjugated (Supporting Information Fig. S2). In contrast to glycosylated mAbs, the higher‐temperature transitions shift to lower temperatures as a function of biotinylation for our deglycosylated ADC models. Overall, it is interesting to note that biotinylated mAbs are destabilized upon biotin conjugation. Despite this, the level of destabilization we detect is minimal and only detectable by DSC with confidence when comparing mAbs conjugated with greater than 15 biotin equivalents.
CIU fingerprints reveal significant stability shifts upon biotin conjugation
As our native IM‐MS and classical DSC exhibited poor sensitivities to biotin conjugation within IgG1 mAbs, we performed CIU analysis in an effort to capture structure and stability changes that remain too subtle to detect using other approaches. Figure 4(A–F) shows averaged CIU fingerprints for the 25+ mAb ions reacted with 0–30 equivalents of biotin. We observe three unfolded features for the parent IgG1 mAb in addition to its ground state conformer family, with centroid IM drift times ranging from ~7.7 ms to ~12.6 ms. These same intermediate states are observed throughout our 25+ ADC model CIU dataset. While CIU data recorded for each biotinylated mAb appears similar to the control experiments, detailed quantitative analysis of the complete fingerprints recorded reveals shifts in mAb stability that are highly correlated with the extent of expected protein conjugation.
Figure 4.
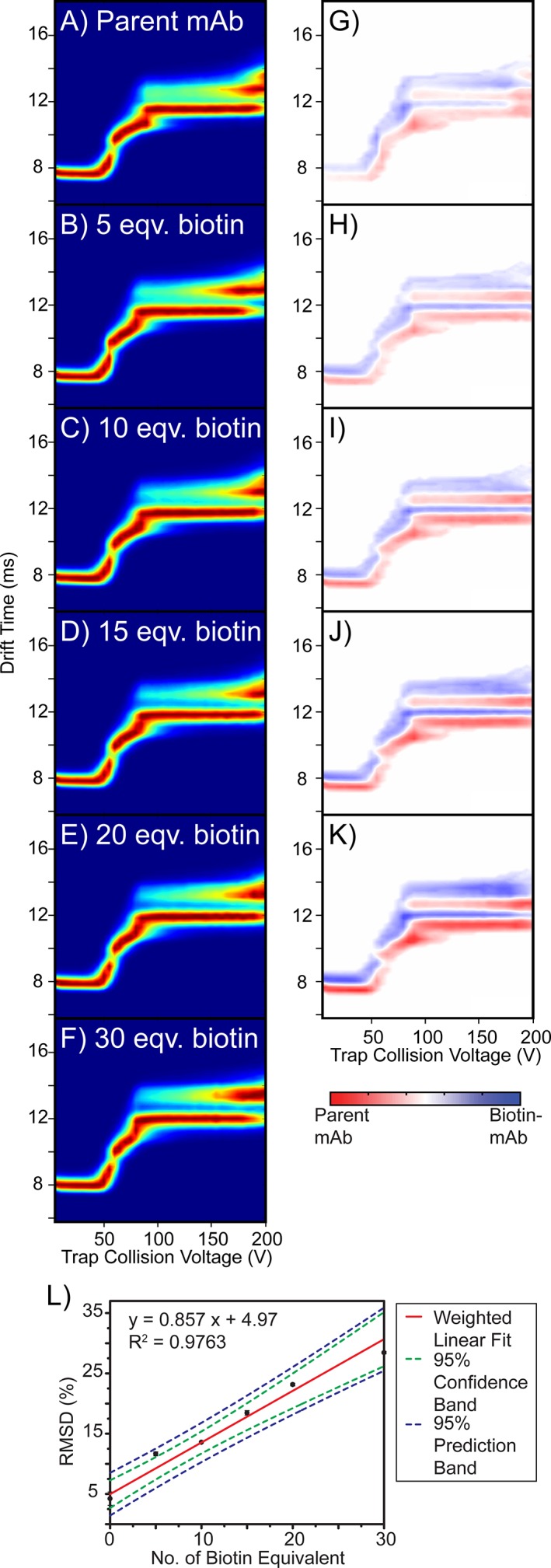
(A–F) Triplicate averaged CIU fingerprints for the parent mAb and the mAb–biotin conjugates. (G–K) CIU difference plots generated by subtracting the fingerprint of the biotinylated mAb from that of the parent mAb. (L) RMSD values plotted as a function of biotin equivalent used in the conjugation reactions. Each data point represents the average value from triplicate data, with the standard deviation shown as the error bar. A weighted linear regression was performed for data fitting (red line). 95% confidence interval (green dashed line) and 95% prediction interval (blue dashed line) were computed.
To perform the abovementioned quantitative analysis of our CIU data, we generated a series of difference plots by comparing averaged CIU fingerprints for the mAb–biotin conjugates with that of the parent mAb [Fig. 4(G–K)]. A color scale is used to denote the magnitude of the differences in signal intensities detected for each comparison, with deeper colors representing the greater signals detected for either the dominated parent mAb (red) or the selected ADC mimic (blue). As shown in Figure 4(G–K), the minor differences observed in our ground state CCS values are carried forward across all voltages probed in our experiment. To quantify the sensitivity of our CIU analysis to small changes in mAb conjugation state, we plot total RMSD values, extracted from pixel‐by‐pixel comparisons between CIU fingerprints acquired for ADC models and control mAbs, where larger RMSD values suggest higher degrees of dissimilarity between the two CIU fingerprints. We observe that the CIU RMSD and the number of biotin equivalents used for the conjugation reaction are strongly correlated to a linear function, with a sufficiently steep slope and narrow error bars so that quantification of stability shifts produced through eight biotin equivalents during mAb conjugation is possible with high confidence [Fig. 4(L)]. Importantly, using CIU RMSD analysis, we are able to globally quantify the structural differences between the biotinylated mAbs versus their parent mAb, even in cases where only small numbers of biotin have been bound.
CIU width and stability analyses further differentiate conjugated mAbs
In an effort to further differentiate ADC samples using CIU, we chose to perform comprehensive analyses that track shifts in TWIM peak widths and stabilities observed in our CIU fingerprints. The width of the ATD recorded in an IM separation is attributable to multiple factors including ion diffusion, the length of the initial ion pulse, space charge, reaction chemistry, and any conformational heterogeneity that manifests on a timescale faster than the IM separation.44, 45, 46, 47 Most IM experiments are designed to minimize peak width influencers other than those associated with diffusional and conformational broadening. As such, if one assumes that diffusional broadening will be roughly equivalent for all mAb species studied here, dramatic changes in the full width at half‐maximum (FWHM) of the TWIM ATDs result from differences in mAb flexibility and structural polydispersity. In previous reports, broader IM ATDs have been observed for mAbs when compared to other proteins of similar size, suggesting mAbs display a greater than average level of structural polydispersity in the gas phase.26, 27
To pursue a TWIM width analysis for our ADC CIU datasets, we utilized multi‐modal Gaussian fitting to analyze IM datasets acquired for all mAb samples at each collision voltage recorded. Centroid drift time (DT) and FWHM values for each fitted Gaussian distribution were determined and plotted as a function of trap collision voltage (Supporting Information Fig. S3). As shown in Supporting Information Figure S3(A), and discussed above, all six mAbs exhibit nearly identical ground state DT values, with the minor CCS differences observed likely attributable to the extra mass associated with biotinylation. For example, the centroid value for maximally conjugated mAb ATDs recorded prior to activation is ~0.33 ms longer when compared with control mAb data, representing a 4% increase. Such relatively minor differences, however, are amplified during CIU, producing differences in TWIM ATD centroids as large as 6% for extended mAb states populated at higher activation voltages.
Supporting Information Figure S3(B) compares the TWIM widths recorded for ADC models with those observed for the parent mAb. Highly similar FWHM values are observed for the ground state arrival time distributions at collision voltages lower than 40 V, suggesting that ADC mimics comprise structures with similar degrees native flexibility and heterogeneity. We detect significantly increased FWHM values at collision voltages between 50 and 70 V, corresponding with the initial CIU transition observed. Furthermore, the TWIM width analysis at higher voltages indicates that different levels of structural heterogeneity appear in the unfolding intermediates populated during CIU of the biotinylated mAbs studied here. Taking the IM data collected at a collision voltage of 180 V as an example, two states are observed, with the state at longer DT exhibiting FWHM and IM resolution trends that are highly correlated with the amount of biotin used during conjugation. Conversely, the IM width and resolution of the state that appears at shorter drift times exhibit opposite trends with biotinylation [Supporting Information Fig. S3(D) and (E)]. The correlation detected in the width analysis serves as a partial explanation for the discrimination observed through the RMSD analysis shown in Figure 4(L), which is clearly governed by the accumulated differences in the CIU response across the whole activation range.
To further mine our ADC CIU data for quantitative differences associated with biotin conjugation, we performed a CIU‐50 analysis to track the activation voltages at which half of the ion population transitions from one CIU conformer state to another. Two major CIU transitions were examined in our analysis: the initial transition that occurs at voltages around 57 V, and a higher‐energy transition that appears at collision voltages between 78.3 V and 87.8 V showing apparent correlations (Supporting Information Table S2). For example, Figure 5 illustrates that while an averaged collision voltage of 87.8 V is required to transition half of the control mAb population from the first CIU intermediate observed to the second, progressively lower voltages are needed to achieve this unfolding transition as mAb conjugation increases in a linear fashion. Although this trend is a long way from ideal and can be less obvious for different sample batches [Supporting Information Fig. S3(F)], it is the first time that we observe a trend in the gas‐phase unfolding energies that is similar to the correlation observed in the melt temperatures measured by DSC. This relationship strongly suggests that future CIU data for similar systems may be used to estimate mAb melting temperatures in solution.
Figure 5.
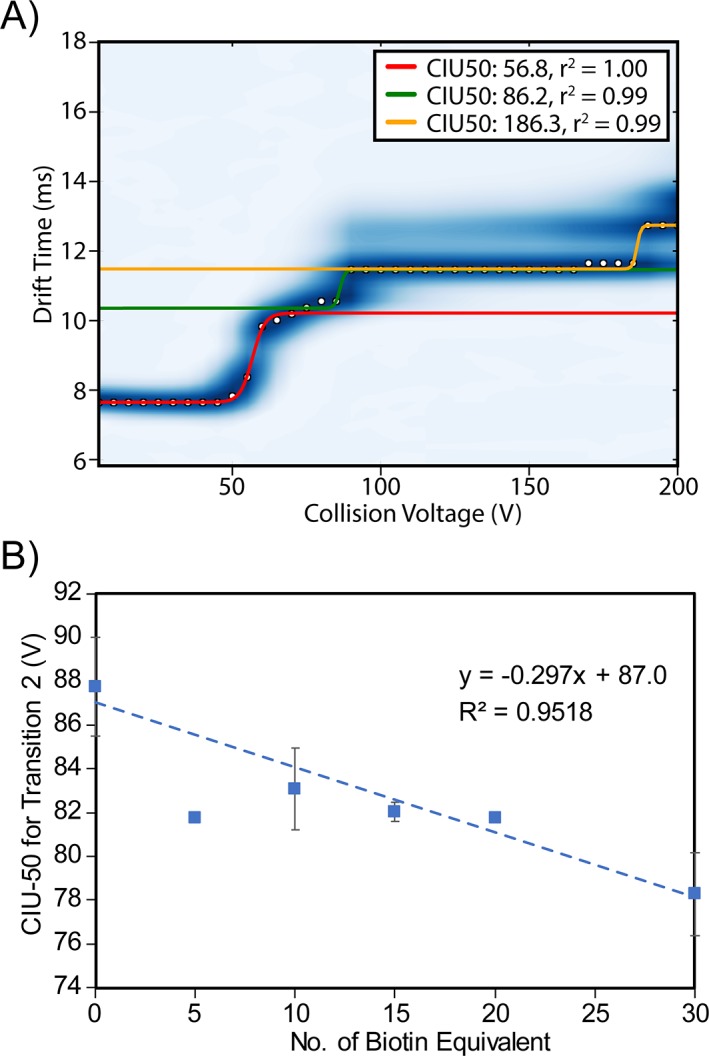
(A) A representative CIU fingerprint for the parent mAb with three gas‐phase unfolding transitions fitted. Our analysis is focused on the first two transitions, highlighted as red and green lines respectively. (B) Collision voltage required for the second CIU transition plotted against the number of biotin equivalent. Each data point is the average value from triplicate data. An initial linear regression was performed and the studentized deleted residuals (r i) were calculated to detect outliers. Data point for 5 eqv. biotin was identified as an outlier, as |r| = 3.732 > t 0.975 (df = 3). The trendline shown in this figure was fitted based on the dataset omitting the outlier.
Conclusions
This work presents a thorough structural characterization of ADC‐like models using a battery of techniques, including denaturing LC–MS, DSC, SEC, native IM‐MS, and CIU analysis. Our data suggest that DAR values for these IgG1 mAb–biotin conjugates can be estimated based on their average intact masses measured by a Q‐ToF mass spectrometer under native conditions, but that native MS remains relatively insensitive to small changes in biotin conjugation level. Similarly, IM measurements performed on native like ions, DSC measurements, and SEC data all struggle to differentiate conjugated mAbs. In contrast, however, CIU data demonstrate the ability to detect stability shifts associated with relatively small numbers (DAR = 8) of bound biotin, while mirroring the structural trends observed in other, lower throughput data types. The sensitivity of CIU to the conjugation levels of our ADC mimics exceeds those observed in our native MS data by a factor of two. As such, our data strongly indicate that CIU has the potential to play a synergistic role in ADC development and selection efforts, by providing a means to acquire conjugation‐dependent gas‐phase stability shift information in a rapid manner that is maximally sensitive to small changes to the target therapeutic.
Our CIU analysis includes RMSD, CIU‐50, and peak width values, all of which are able to individually discriminate between ADC mimics. This report represents the first example of such multi‐parameter CIU data analysis and presents exciting possibilities for future software development efforts. Interestingly, an analysis of CIU‐50 from our data reveals mAb gas‐phase stability values that decrease in a fashion similar to what is observed for these same samples by DSC. While this correlation is far from perfect, our analysis indicates a high confidence level associated with this trend, and our data present the first evidence of such a relationship between CIU and DSC data. At one level, this correlation provides experimental evidence indicating a strong solution‐phase memory effect for a portion of the mAb structure in the gas phase. This result stands in stark contrast to low‐energy CCS‐values obtained for mAbs, which appear to correspond to significantly collapsed versions of their native states, complicating their use in downstream mAb HOS assessments.26, 27 At another level, by converting single IM peaks into detailed fingerprints composed of many resolved features, CIU offers the potential to produce significantly richer structural datasets for mAbs than is currently possible using IM‐MS alone, and is thus poised to propel this technology into a role complimentary to DSC assays in the development of future biopharmaceuticals.48, 49
Supporting information
Appendix S1: Supporting information
Acknowledgments
We gratefully acknowledge support from the National Science Foundation Division of Chemistry under Grant nos. 1253384 and 1808541 (with co‐funding from the Division of Molecular and Cellular Biosciences). The authors further thank Daniel Polasky, Sugyan Dixit and Sarah Fantin for their help in developing some of the CIU data analysis proceedures used in this report.
Brandon Ruotolo is the winner of the 2018 Protein Science Young Investigator Award.
References
- 1. Chari RVJ, Miller ML, Widdison WC (2014) Antibody–drug conjugates: an emerging concept in cancer therapy. Angew Chem Int Ed Engl 53:3796–3827. [DOI] [PubMed] [Google Scholar]
- 2. Lambert JM, Morris CQ (2017) Antibody–drug conjugates (ADCs) for personalized treatment of solid tumors: a review. Adv Ther 34:1015–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beck A, Goetsch L, Dumontet C, Corvaïa N (2017) Strategies and challenges for the next generation of antibody–drug conjugates. Nat Rev Drug Discov 10:345–352. [DOI] [PubMed] [Google Scholar]
- 4. Diamantis N, Banerji U (2016) Antibody–drug conjugates – an emerging class of cancer treatment. Br J Cancer 114:362–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Panowski S, Bhakta S, Raab H, Polakis P, Junutula JR (2014) Site‐specific antibody drug conjugates for cancer therapy. MAbs 6:34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hamblett KJ, Senter PD, Chace DF, Sun MMC, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, Zabinski RF, Meyer DL, Francisco JA (2004) Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res 10:7063–7070. [DOI] [PubMed] [Google Scholar]
- 7. Valliere‐Douglass JF, Hengel SM, Pan LY (2015) Approaches to interchain cysteine‐linked adc characterization by mass spectrometry. Mol Pharm 12:1774–1783. [DOI] [PubMed] [Google Scholar]
- 8. Redman EA, Mellors JS, Starkey JA, Ramsey JM (2016) Characterization of intact antibody drug conjugate variants using microfluidic capillary electrophoresis‐mass spectrometry. Anal Chem 88:2220–2226. [DOI] [PubMed] [Google Scholar]
- 9. Kim MT, Chen Y, Marhoul J, Jacobson F (2014) Statistical modeling of the drug load distribution on trastuzumab emtansine (Kadcyla), a lysine‐linked antibody drug conjugate. Bioconjug Chem 25:1223–1232. [DOI] [PubMed] [Google Scholar]
- 10. Luo Q, Chung HH, Borths C, Janson M, Wen J, Joubert MK, Wypych J (2016) Structural characterization of a monoclonal antibody–maytansinoid immunoconjugate. Anal Chem 88:695–702. [DOI] [PubMed] [Google Scholar]
- 11. Ouyang J. Drug‐to‐antibody ratio (DAR) and drug load distribution by hydrophobic interaction chromatography and reversed phase high‐performance liquid chromatography. Totowa, NJ: Humana Press, 2013;p. 275–283. [DOI] [PubMed] [Google Scholar]
- 12. Bobály B, Fleury‐Souverain S, Beck A, Veuthey J‐L, Guillarme D, Fekete S (2018) Current possibilities of liquid chromatography for the characterization of antibody–drug conjugates. J Pharm Biomed Anal 147:493–505. [DOI] [PubMed] [Google Scholar]
- 13. Lazar AC, Wang L, Blättler W a, Amphlett G, Lambert JM, Zhang W (2005) Analysis of the composition of immunoconjugates using size‐exclusion chromatography coupled to mass spectrometry. Rapid Commun Mass Spectrom 19:1806–1814. [DOI] [PubMed] [Google Scholar]
- 14. Wakankar A, Chen Y, Gokarn Y, Jacobson FS (2011) Analytical methods for physicochemical characterization of antibody drug conjugates. MAbs 3:161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Debaene F, Bœuf A, Wagner‐Rousset E, Colas O, Ayoub D, Corvaïa N, Van Dorsselaer A, Beck A, Cianférani S (2014) Innovative native MS methodologies for antibody drug conjugate characterization: high resolution native MS and IM‐MS for average DAR and DAR distribution assessment. Anal Chem 86:10674–10683. [DOI] [PubMed] [Google Scholar]
- 16. Hengel SM, Sanderson R, Valliere‐Douglass J, Nicholas N, Leiske C, Alley SC (2014) Measurement of in vivo drug load distribution of cysteine‐linked antibody–drug conjugates using microscale liquid chromatography mass spectrometry. Anal Chem 86:3420–3425. [DOI] [PubMed] [Google Scholar]
- 17. Valliere‐Douglass JF, McFee WA, Salas‐Solano O (2012) Native intact mass determination of antibodies conjugated with monomethyl auristatin E and F at interchain cysteine residues. Anal Chem 84:2843–2849. [DOI] [PubMed] [Google Scholar]
- 18. Rose RJ, Damoc E, Denisov E, Makarov A, Heck AJR (2012) High‐sensitivity Orbitrap mass analysis of intact macromolecular assemblies. Nat Methods 9:1084–1086. [DOI] [PubMed] [Google Scholar]
- 19. Belov ME, Damoc E, Denisov E, Compton PD, Horning S, Makarov AA, Kelleher NL (2013) From protein complexes to subunit backbone fragments: a multi‐stage approach to native mass spectrometry. Anal Chem 85:11163–11173. [DOI] [PubMed] [Google Scholar]
- 20. Dyachenko A, Wang G, Belov M, Makarov A, de Jong RN, van den Bremer ETJ, Parren PWHI, Heck AJR (2015) Tandem native mass‐spectrometry on antibody–drug conjugates and submillion Da antibody–antigen protein assemblies on an Orbitrap EMR equipped with a high‐mass quadrupole mass selector. Anal Chem 87:6095–6102. [DOI] [PubMed] [Google Scholar]
- 21. Yang Y, Wang G, Song T, Lebrilla CB, Heck AJR (2017) Resolving the micro‐heterogeneity and structural integrity of monoclonal antibodies by hybrid mass spectrometric approaches. MAbs 9:638–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Campuzano IDG, Netirojjanakul C, Nshanian M, Lippens JL, Kilgour DPA, Van Orden S, Loo JA (2018) Native‐MS analysis of monoclonal antibody conjugates by Fourier transform ion cyclotron resonance mass spectrometry. Anal Chem 90:745–751. [DOI] [PubMed] [Google Scholar]
- 23. Acchione M, Kwon H, Jochheim CM, Atkins WM (2012) Impact of linker and conjugation chemistry on antigen binding, Fc receptor binding and thermal stability of model antibody–drug conjugates. MAbs 4:362–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wakankar AA, Feeney MB, Rivera J, Chen Y, Kim M, Sharma VK, Wang YJ (2010) Physicochemical stability of the antibody–drug conjugate trastuzumab‐DM1: changes due to modification and conjugation processes. Bioconjug Chem 21:1588–1595. [DOI] [PubMed] [Google Scholar]
- 25. Pan LY, Salas‐Solano O, Valliere‐Douglass JF (2014) Conformation and dynamics of interchain cysteine‐linked antibody–drug conjugates as revealed by hydrogen/deuterium exchange mass spectrometry. Anal Chem 86:2657–2664. [DOI] [PubMed] [Google Scholar]
- 26. Pacholarz KJ, Porrini M, R A G, Burnley RJ, Taylor RJ, Henry AJ, Barran PE (2014) Dynamics of intact immunoglobulin G explored by drift‐tube ion‐mobility mass spectrometry and molecular modeling. Angew Chem Int Ed Engl 53:7765–7769. [DOI] [PubMed] [Google Scholar]
- 27. Campuzano IDG, Larriba C, Bagal D, Schnier PD. Ion mobility and mass spectrometry measurements of the humanized IgGk NIST monoclonal antibody Washington, DC: Oxford University Press, 2015. ACS symposium series. Volume 1202; p. 75–112. [Google Scholar]
- 28. Ehkirch A, D'Atri V, Rouviere F, Hernandez‐Alba O, Goyon A, Colas O, Sarrut M, Beck A, Guillarme D, Heinisch S, Cianferani S (2018) An anline four‐dimensional HIC×SEC‐IM×MS methodology for proof‐of‐concept characterization of antibody drug conjugates. Anal Chem 90:1578–1586. [DOI] [PubMed] [Google Scholar]
- 29. Dixit SM, Polasky DA, Ruotolo BT (2018) Collision induced unfolding of isolated proteins in the gas phase: past, present, and future. Curr Opin Chem Biol 42:93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tian Y, Han L, Buckner AC, Ruotolo BT (2015) Collision induced unfolding of intact antibodies: rapid characterization of disulfide bonding patterns, glycosylation, and structures. Anal Chem 87:11509–11515. [DOI] [PubMed] [Google Scholar]
- 31. Tian Y, Ruotolo BT (2018) Collision induced unfolding detects subtle differences in intact antibody glycoforms and associated fragments. Int J Mass Spectrom 425:1–9. [Google Scholar]
- 32. Pisupati K, Tian Y, Okbazghi S, Benet A, Ackermann R, Ford M, Saveliev S, Hosfield CM, Urh M, Carlson E, Becker C, Tolbert TJ, Schwendeman SP, Ruotolo BT, Schwendeman A (2017) A multidimensional analytical comparison of Remicade and the biosimilar Remsima. Anal Chem 89:4838–4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ferguson CN, Gucinski‐Ruth AC (2016) Evaluation of ion mobility‐mass spectrometry for comparative analysis of monoclonal antibodies. J Am Soc Mass Spectrom 27:822–833. [DOI] [PubMed] [Google Scholar]
- 34. Huang Y, Salinas ND, Chen E, Tolia NH, Gross ML (2017) Native mass spectrometry, ion mobility, and collision‐induced unfolding categorize malaria antigen/antibody binding. J Am Soc Mass Spectrom 28:2515–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Watanabe Y, Vasiljevic S, Allen JD, Seabright GE, Duyvesteyn HME, Doores KJ, Crispin M, Struwe WB (2018) Signature of antibody domain exchange by native mass spectrometry and collision‐inducing unfolding. Anal Chem 90:7325–7331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hernandez‐Alba O, Wagner‐Rousset E, Beck A, Cianferani S (2018) Native mass spectrometry, ion mobility, and collision‐induced unfolding for conformational characterization of IgG4 monoclonal antibodies. Anal Chem 90:8865–8872. [DOI] [PubMed] [Google Scholar]
- 37. Botzanowski T, Erb S, Hernandez‐Alba O, Ehkirch A, Colas O, Wagner‐Rousset E, Rabuka D, Beck A, Drake PM, Cianférani S (2017) Insights from native mass spectrometry approaches for top‐ and middle‐level characterization of site‐specific antibody–drug conjugates. MAbs 9:801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bush MF, Hall Z, Giles K, Hoyes J, Robinson CV, Ruotolo BT (2010) Collision cross sections of proteins and their complexes: a calibration framework and database for gas‐phase structural biology. Anal Chem 82:9557–9565. [DOI] [PubMed] [Google Scholar]
- 39. Haynes SE, Polasky DA, Dixit SM, Majmudar JD, Neeson K, Ruotolo BT, Martin BR (2017) Variable‐velocity traveling‐wave ion mobility separation enhances peak capacity for data‐independent acquisition proteomics. Anal Chem 89:5669–5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Eschweiler JD, Rabuck‐Gibbons JN, Tian Y, Ruotolo BT (2015) CIUSuite: a quantitative analysis package for collision induced unfolding measurements of gas‐phase protein ions. Anal Chem 87:11516–11522. [DOI] [PubMed] [Google Scholar]
- 41. Hewitt D, Marklund E, Scott DJ, Robinson CV, Borysik AJ (2014) A hydrodynamic comparison of solution and gas phase proteins and their complexes. J Phys Chem B 118:51. [DOI] [PubMed] [Google Scholar]
- 42. Zheng K, Bantog C, Bayer R (2011) The impact of glycosylation on monoclonal antibody conformation and stability. MAbs 3:568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pawlowski JW, Bajardi‐Taccioli A, Houde D, Feschenko M, Carlage T, Kaltashov IA (2018) Influence of glycan modification on IgG1 biochemical and biophysical properties. J Pharm Biomed Anal 151:133–144. [DOI] [PubMed] [Google Scholar]
- 44. Wu C, Siems WF, Asbury GR, Hill HH (1998) Electrospray ionization high‐resolution ion mobility spectrometry−mass spectrometry. Anal Chem 70:4929–4938. [DOI] [PubMed] [Google Scholar]
- 45. Wyttenbach T, von Helden G, Bowers MT (1996) Gas‐phase conformation of biological molecules: Bradykinin. J Am Chem Soc 118:8355–8364. [Google Scholar]
- 46. Shvartsburg AA, Smith RD (2008) Fundamentals of traveling wave ion mobility spectrometry. Anal Chem 80:9689–9699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhong Y, Hyung S‐J, Ruotolo BT (2011) Characterizing the resolution and accuracy of a second‐generation traveling‐wave ion mobility separator for biomolecular ions. Analyst 136:3534–3541. [DOI] [PubMed] [Google Scholar]
- 48. Campuzano IDG, Lippens JL (2018) Ion mobility in the pharmaceutical industry: an established biophysical technique or still niche? Curr Opin Chem Biol 42:147–159. [DOI] [PubMed] [Google Scholar]
- 49. Tian Y, Ruotolo BT (2018) The growing role of structural mass spectrometry in the discovery and development of therapeutic antibodies. Analyst 143:2459–2468. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting information