

Abstract
T4 phage lysozyme (T4L) is an enzyme that cleaves bacterial cell wall peptidoglycan. Remarkably, the single substitution of the active site Thr26 to a His (T26H) converts T4L from an inverting to a retaining glycoside hydrolase with transglycosylase activity. It has been proposed that T26H–T4L follows a double displacement mechanism with His26 serving as a nucleophile to form a covalent glycosyl‐enzyme intermediate (Kuroki et al., PNAS 1999; 96:8949–8954). To gain further insights into this or alternative mechanisms, we used NMR spectroscopy to measure the acid dissociation constants (pK a values) and/or define the ionization states of the Asp, Glu, His, and Arg residues in the T4L mutant. Most notably, the pK a value of the putative nucleophile His26 is 6.8 ± 0.1, whereas that of the general acid Glu11 is 4.7 ± 0.1. If the proposed mechanism holds true, then T26H–T4L follows a reverse protonation pathway in which only a minor population of the free enzyme is in its catalytically competent ionization state with His26 deprotonated and Glu11 protonated. Our studies also confirm that all arginines in T26H–T4L, including the active site Arg145, are positively charged under neutral pH conditions.
Brief statement
The replacement of a single amino acid changes T4 lysozyme from an inverting to a retaining glycoside hydrolase. Using NMR spectroscopy, we measured the pK a values of the ionizable residues in the active site of this mutant enzyme. Along with previously reported data, these results provide important constraints for understanding the catalytic mechanisms by which the wild‐type and mutant form of T4 lysozyme cleave bacterial peptidoglycan.
Keywords: lysozyme, glycoside hydrolase, pH‐dependent enzymatic mechanism, acid dissociation constant, NMR spectroscopy, hydrogen exchange, deuterium isotope shift, neutron diffraction
Abbreviations
- DAP
diaminopimelic acid
- GH
glycoside hydrolase
- NAG
N‐acetylglucosamine
- NAM
N‐acetylmuramic acid
- pH*
the pH meter reading uncorrected for isotope effects
- pKa
negative logarithm of the acid dissociation constant
- rmsd
root‐mean‐square deviation
- T4L*
cysteine‐free (C54T and C97A) variant of T4 lysozyme
Introduction
Lysozymes are glycoside hydrolases that cleave the β‐1,4‐glycosidic linkage between N‐acetylmuramic acid (NAM) and N‐acetylglucosamine (NAG) of peptidoglycan, a major component of the bacterial cell wall. These enzymes are classified into at least five families by the Carbohydrate Active Enzyme (CAZy) database based on their amino acid sequences.1, 2 Members of a family typically utilize a similar hydrolytic mechanism that leads to either retention or inversion of anomeric carbon stereochemistry at the site of hydrolysis.3, 4
The lysozyme from Enterobacteria phage T4 (T4L) is a member of the GH24 family. Although T4L is an extremely well‐characterized model system for investigating protein folding and dynamics,5 detailed enzymology studies have proven difficult as it acts on a complex glycopeptide substrate6 for which analogs amenable to rigorous kinetic measurements are not readily available. In 1995, Matthews and co‐workers reported that T4L is an inverting glycoside hydrolase.7 This conclusion was based in part on the observation that hydrolysis of a peptidic tetrasaccharide by T4L yielded a disaccharide product with the opposite anomeric stereochemistry to that generated by hen egg white lysozyme, a classic GH22 retaining hydrolase.8 Accordingly, it was proposed that T4L catalyzes a single‐displacement inverting reaction in which a nucleophilic water, activated by the general base Asp20 (with a reported9, 10 pK a value of 3.6), attacks the β‐1,4‐linked anomeric C1 carbon of the peptidic NAM, while the general acid Glu11 (pK a 5.4) concomitantly donates a proton to the leaving aglycone, NAG.
In the X‐ray crystallographic structures of wild‐type T4L, a candidate nucleophilic water is hydrogen bonded between the sidechains of Asp20 and Thr26. As part of an earlier attempt to engineer metal binding sites into T4L, the Matthews group introduced a Glu at position 26. Serendipitously, the T26E mutant was isolated from E. coli with Glu26 covalently bound to a peptidic NAG‐NAM disaccharide via an α‐linkage to the anomeric carbon of a distorted NAM moiety.11 This stable species appeared to be analogous to the glycosyl‐enzyme intermediate formed in the first step of a double‐displacement retaining mechanism. Subsequently, it was found that the T26H mutant is catalytically active, yet produces a hydrolysis product with the opposite anomeric stereochemistry to that yielded by wild‐type T4L.7 Remarkably, a single amino acid substitution at position 26 changes T4L from an inverting to a retaining glycoside hydrolase. Furthermore, unlike the wild‐type enzyme, T26H–T4L also functions as a transglycosylase.12
Based on mutational and X‐ray crystallographic studies, a two‐step double‐displacement mechanism was proposed for T26H–T4L [Fig. 1(A)].7, 12 In the glycosylation step, His26 is postulated to be a nucleophile, forming an α‐linked glycosyl‐enzyme intermediate. This is facilitated by Glu11 serving as a general acid to protonate the leaving aglycone. In the subsequent deglycosylation step, Glu11 functions as a general base to activate either a water (hydrolysis) or carbohydrate (transglycosylation) for nucleophilic displacement of His26 and release of the glycone with a net retention of β‐anomeric stereochemistry.
Figure 1.
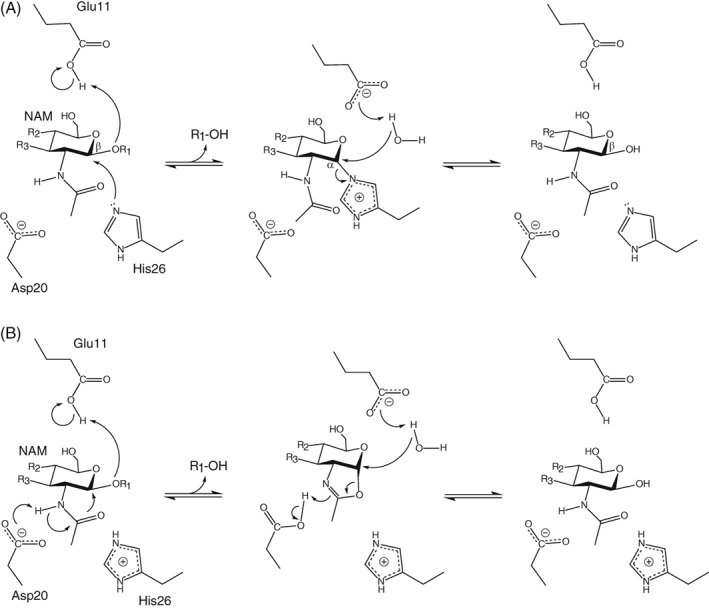
(A) Proposed double‐displacement retaining mechanism for the hydrolysis reaction catalyzed by T26H–T4L.7, 12 (R1 and R2 correspond to NAG; R3 to the peptide‐linked lactyl substituent of NAM.) A minor population of the enzyme is depicted with the hypothesized nucleophile His26 and the general acid/base Glu11 in their catalytically competent protonation states. (B) An alternative mechanism involving substrate‐assisted catalysis. Asp20 is shown as a general base, but could also electrostatically stabilize a charged oxazolinium intermediate without transfer of the N‐acetyl nitrogen‐bonded proton.
An important aspect of understanding the proposed mechanism, or any alternative, for T26H–T4L lies with defining the protonation states of catalytic residues along the reaction pathway. To this end, Kuroki et al.13 used room temperature neutron diffraction to characterize a sample of perdeuterated T26H–T4L*, crystallized in D2O buffer (pD 7.0). In the reported 2.09 Å resolution structure (5XPF.pdb), His26 is predominantly in its neutral state and the carboxyl of Glu11 partially deuterated. This is consistent with the scheme of Figure 1(A). However, Asp20 and nearby Arg145 were also modeled to be neutral. This is perplexing since there are no obvious physicochemical reasons why these active site residues should have such highly perturbed pK a values and unusual sidechain deuteration states at pD ~ 7. Indeed, in the 2.2 Å resolution neutron crystallographic structure of the perdeuterated wild‐type enzyme at cryogenic temperatures (pD 6 – 7, 5VNQ.pdb), all ionizable residues are in their charged forms.14 Inspired by these reports, we used NMR spectroscopy to investigate the protonation states and, when possible, determine the pK a values of the His, Asp, Glu, and Arg residues in T26H–T4L. These measurements provide key insights into the catalytic mechanism and electrostatic properties of this mutant retaining glycoside hydrolase.
Results
T26H–T4L* yielded high‐quality NMR spectra, and thus assignment of the 1H, 13C, and 15N signals from its mainchain and selected sidechain nuclei was straightforward using standard heteronuclear correlation experiments (Fig. S1). A comparison of the 15N‐HSQC spectrum of this mutant with that of pseudo‐wild‐type T4L* revealed moderate 1HN and 15N chemical shift differences for many corresponding amides (Fig. S2). When mapped onto the structure of lysozyme, these amides cluster in the N‐terminal lobe around position 26. The crystallographic co‐ordinates of T4L* (1CX6.pdb) and T26H–T4L* (1QT8.pdb) superimpose closely (0.28 Å rmsd for all atoms), and thus these chemical shift differences likely arise from small conformational changes due to the amino acid substitution, combined with electric field and ring current effects from the charged, aromatic imidazolium sidechain of histidine. These two co‐ordinate files were used to provide the inter‐residue distances cited below.
Histidine pK a and tautomer state determination
The signals from the two histidines in uniformly 13C/15N‐labeled T26H–T4L* were monitored via 13C‐HSQC spectra as the protein was titrated between pH 5 and 11 [Figs. 2(A) and S3]. Fitting the pH‐dependent chemical shifts of the ring 13Cε1, 1Hε1, 13Cδ2, and 1Hδ2 reporter nuclei yielded average pK a values of 6.8 ± 0.1 for His26 and 8.9 ± 0.1 for His31 [Fig. 2(B) and Table 1]. Furthermore, with highly diagnostic 13Cδ2 chemical shifts ~ 127 ppm, the neutral forms of both histidines can be confidently assigned to the less common Nδ1H tautomer.17, 18
Figure 2.
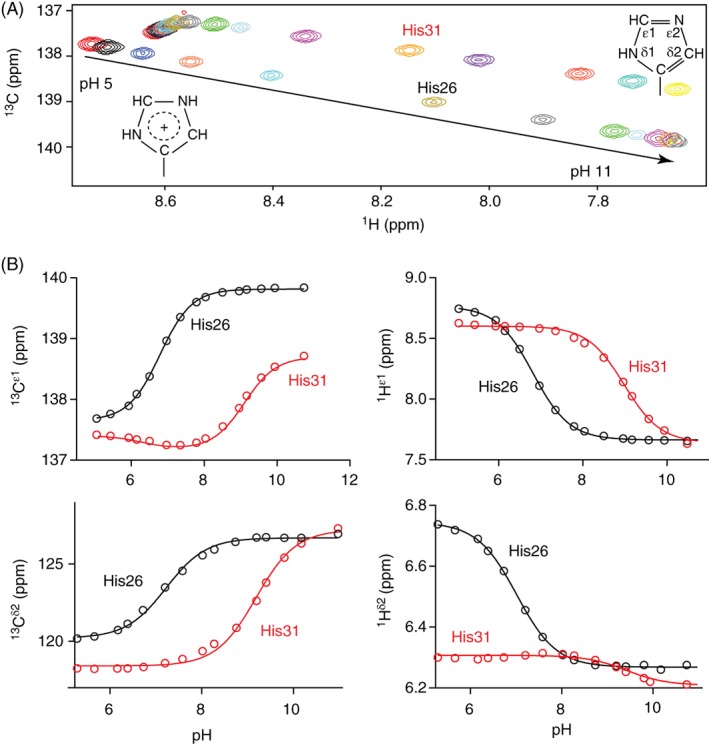
(A) Superimposed 15N‐decoupled 13C‐HSQC spectra of uniformly 13C/15N‐labeled T26H–T4L* showing the 1Hε1–13Cε1 signals of His26 and His31 as the protein was titrated between pH 5 and 11 at 25°C (100 mM KCl, 30 mM potassium phosphate, 5% D2O). Spectra for the 1Hδ2–13Cδ2 signals are provided in Figure S3. (B) Fit titration curves for the 13Cε1, 1Hε1, 13Cδ2, and 1Hδ2 nuclei yielded average pK a values of 6.8 ± 0.1 for His26 and 8.9 ± 0.1 for His31 (Table 1). The biphasic titration curve for the 13Cε1 of His31 has a second fit pK a ~ 6.3 for a small chemical shift change of −0.2 ppm with increasing sample pH that may be attributed to conformational or electrostatic perturbations16 upon deprotonation of nearby (~ 8 Å) His26. The structures of the imidazolium and neutral imidazole Nδ1H tautomer sidechains are shown.
Table 1.
pKa values of T26H–T4L* and wild‐type T4L
| Residue | T26H–T4L*a | T4Lb |
|---|---|---|
| His26 | 6.8 ± 0.1 | |
| His31 | 8.9 ± 0.1 | 9.1 |
| Asp10 | < 0.8c | < 0.5 |
| Asp20 | ~ 1.5d | 3.6 |
| Asp47 | 3.1 ± 0.1 | 3.0 |
| Asp61 | 3.6 ± 0.1 | 3.6 |
| Asp70 | < 0.8c | < 0.5 |
| Asp72 | 3.3 ± 0.1 | 3.5 |
| Asp89 | 4.0 ± 0.1 | 4.0 |
| Asp92 | 2.7 ± 0.1 | 2.5 |
| Asp127 | 3.5 ± 0.1 | 3.5 |
| Asp159 | 3.4 ± 0.1 | 3.5 |
| Glu5 | 3.7 ± 0.1 | |
| Glu11 | 4.7 ± 0.1 | 5.4 |
| Glu22 | 4.3 ± 0.1 | |
| Glu45 | 3.8 ± 0.1 | |
| Glu62 | 2.9 ± 0.1 | 2.8 |
| Glu64 | 4.4 ± 0.1 | |
| Glu108 | 4.2 ± 0.1 | |
| Glu128 | 4.4 ± 0.1 | |
| Arg95 | > 12e | |
| Arg145 | > 11e | |
| Arg148 | > 11e |
Measured herein at 25°C with T26H–T4L* samples initially in 100 mM KCl and 30 mM potassium phosphate.
Measured from the pH‐dependent His 1Hε1 and Asp/Glu 13Cγ/δ chemical shifts of amide deuterated or selectively sidechain labeled wild‐type T4L, respectively, at 10°C and initially in 100 mM KCl and 10 mM phosphate, as reported by Anderson et al.10, 15 The assignments were obtained via single‐site amino acid substitutions.
Estimated upper limit (pK a < pH − 1) as no 13Cγ chemical shift changes due to protonation were seen at the titration endpoint of pH 1.8 [Figs. 3(A,C)].
Fit with a restrained chemical shift change of 3.4 ppm upon deprotonation [Fig. 3(C)].
Estimated lower limit (pK a > pH + 1) as no 15Nε chemical shift changes due to deprotonation were seen up to pH 9.9 for Arg145 and Arg148, and pH 10.7 for Arg95 (Fig. 5).
These results are consistent with the previously reported pK a value of 9.1 for His31 in wild‐type T4L,15 and the X‐ray crystallographic structure of the protein with this histidine donating a hydrogen bond from its protonated ring Nδ1 to the sidechain carboxylate of Asp70 (pK a < 0.8, see below). Similarly, the structure of T26H–T4L* shows that His26 donates a hydrogen bond from its protonated Nδ1 to the buried mainchain carbonyl oxygen of Tyr24, leaving its Nε2 solvent exposed to aid catalysis. Presumably, both hydrogen bonds are maintained when His26 and His31 are Nε2‐deprotonated under alkaline conditions.
The pK a value of an ionizable functional group in a folded protein reflects the pH‐dependent free energy difference between states with that group in its neutral versus charged forms, and hence a complex interplay of entropic and enthalpic terms, including desolvation, hydrogen bonding, and electrostatic interactions with surrounding charged and polar moieties. Dissecting the relative contributions of these terms is challenging experimentally and computationally.19 Nevertheless, it is well accepted that the highly elevated pK a value of His31, compared to that of ~ 6.5 for a histidine in a random coil polypeptide,18 arises in large part from its formation of a very favorable salt‐bridge (i.e. a hydrogen‐bonded ion pair) with ionized Asp70.15 This conclusion is supported by a comparison of the pH‐dependent stability against thermal denaturation of the wild‐type protein versus variants with H31N and/or D70N substitutions.15 In contrast, the relatively unperturbed pK a value of His26 may reflect a balance of unfavorable desolvation of the partially buried imidazolium sidechain and favorable electrostatic interactions with nearby Glu11 (~ 6 Å) and Asp20 (~ 3.5 Å). Based on their pK a values presented below, these latter residues are both predominantly negatively charged around the midpoint pH 6.8 for the deprotonation of positively charged His26.
Aspartic acid pKa determination
A sample of T26H–T4L* selectively labeled with 13Cγ‐aspartic acid was titrated between pH 1.8 and 7.8. The pH‐dependent chemical shifts of the 13Cγ nuclei were monitored via 1D 13C‐NMR spectra [Fig. 3(A)]. The Asp 13Cγ and Glu 13Cδ are part of the carboxylic acid functional group and thus generally reliable reporter nuclei for pK a measurements by NMR spectroscopy.18 Of the 10 Asp residues, seven gave well‐defined titration curves with downfield chemical shift changes of ~ 3.4 ppm upon increasing sample pH [Fig. 3(C)]. Such spectral changes are diagnostic of sidechain deprotonation. Fitting the titration curves yielded the pK a values summarized in Table 1. These values match well those reported previously and discussed in detail for the wild‐type protein.10
Figure 3.
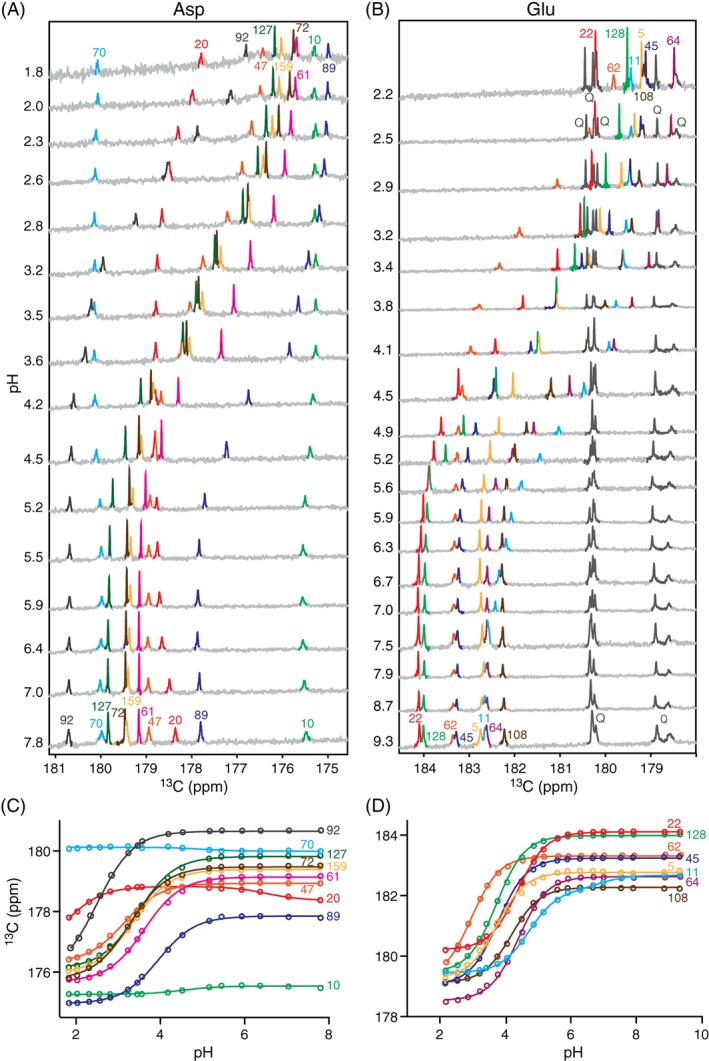
Stacked 13C‐NMR spectra of T26H–T4L* selectively labeled with (A) 13Cγ‐Asp and (B) 13Cδ‐Glu, recorded as a function of sample pH at 25°C (initially 100 mM KCl, 30 mM potassium phosphate, 5% D2O). Due to metabolic interconversion, glutamine (Q) sidechains were also labeled in the latter sample. The titration curves were fit to obtain the (C) Asp and (D) Glu pK a values reported in Table 1. The biphasic titration curve for Asp20 followed one pK a ~ 1.5 with a restrained 13Cγ chemical shift change of 3.4 ppm upon increasing sample pH that is attributed to its own ionization and a second small upfield chemical shift change of −0.5 ppm with pK a ~ 6.6 that likely reflects structural or electrostatic perturbations due to the deprotonation of nearby (~ 3.5 Å) His26. Similarly, the titration curve for Glu11 followed a predominant 13Cδ chemical shift change of 2.7 ppm with pK a 4.7 assigned to its own ionization and a smaller downfield change of 0.5 ppm with pK a ~ 7 that also likely reflects the titration of His26 (~ 6 Å).
In contrast, the 13Cγ chemical shifts of Asp10 and Asp70 did not change markedly over the pH range studied, and that of Asp20 appeared to follow a titration with pK a < 2. However, in the absence of a complete titration curve, it is difficult to infer the ionization state of an Asp residue based on chemical shifts alone. For example, Asp10 has a 13Cγ chemical shift indicative of a protonated carboxylic acid, whereas that of Asp70 corresponds to a deprotonated carboxylate. An alternative approach for distinguishing a carboxylic acid from a carboxylate lies with detecting the presence or absence, respectively, of a 2‐bond isotope shift of ~ 0.23 ppm for the 13Cγ due to a proton versus deuteron on the adjacent oxygen.20, 21, 22 As shown in Figure 4(A), none of the 10 Asp residues exhibited a 13Cγ chemical shift change of this magnitude when T26H–T4L* was transferred from H2O at pH 5.8 to D2O at pH* 5.7. This confirms that all are ionized under these conditions. Furthermore, since no NMR spectroscopic evidence for their protonation was seen even at pH 1.8 in 13C‐NMR monitored titrations, Asp10 and Asp70 are strong acids with pK a values < 0.8 (i.e., from the Henderson–Hasselbalch equation for a simple acid dissociation equilibrium, at pH = pK a + 1, the population ratio of conjugate base to acid is 0.1, which should be detectable). The same conclusions were previously reported for wild‐type T4L (Table 1), as evidenced by the lack of any NMR‐detectable titrations for Asp10 and Asp70 down to pH 1.5, combined with a careful study of the pH‐dependent stability of the protein under acidic conditions.10, 15 These highly reduced pK a values, compared to that of ~ 4 for an aspartic acid in a random coil polypeptide,18 can be rationalized at least in part by the salt‐bridges between Asp10–Arg148 and Asp70–His31 observed by X‐ray crystallography.
Figure 4.
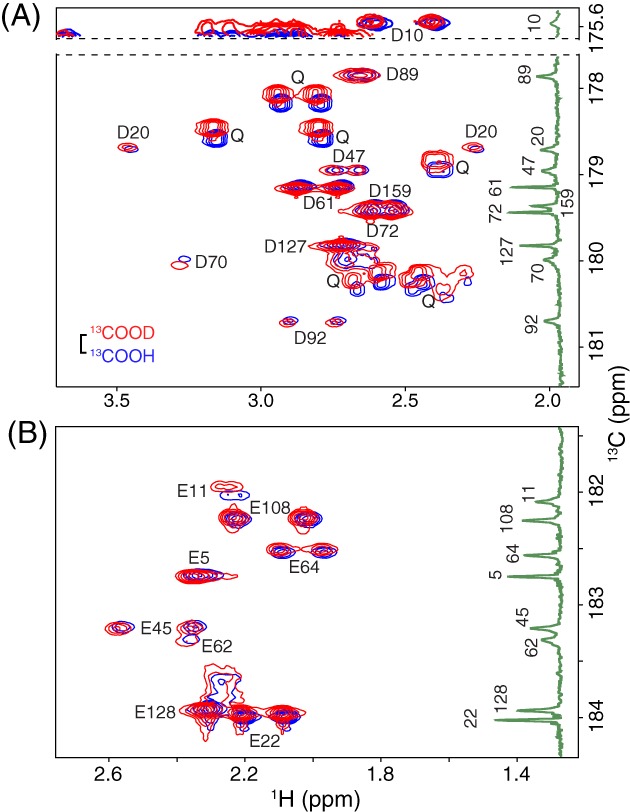
Selected regions of the 2D Hβ/γ 2(Cβ/γ)Cγ/δ spectra of uniformly 15N/13C‐labeled T26H–T4L* in H2O (blue; pH 5.8) and D2O (red; pH* 5.7) NMR sample buffer. The spectra show correlations between (A) the Hββ’ and 13Cγ of Asp and (B) the Hγγ’ and 13Cδ of Glu. The vertical insets are the 1D 13C‐NMR spectra of protein selectively labeled with (A) 13Cγ‐Asp or (B) 13Cδ‐Glu at pH 5.9 (taken from Fig. 3). The scale bar indicates the expected 0.23 ppm isotope shift for a neutral carboxylic acid (13COOH vs. 13COOD). The lack of any significant isotope shifts (< 0.04 ppm in magnitude) demonstrates that the Asp and Glu residues in T26H–T4L* are predominantly ionized under these conditions. Two exceptions are the small 13Cδ shift of 0.08 ppm exhibited by Glu11 (pK a 4.7) that is attributed to a change in its fractional ionization due to the slightly different pH/pH* conditions, and the slight “reversed” isotope shift of Asp70 13Cγ (−0.07 ppm) that may possibly result from protonation versus deuteration of its hydrogen bonded partner His31.22, 23 Also seen in (A) are 15N‐coupled signals from several Gln sidechains, which show expected20, 24 deuterium isotope shifts (13CONH2 vs. 13COND2) of ~ 0.12 ppm. Signals from the Asn sidechains, with similar isotope shifts, are outside of the presented 13C chemical shift windows.
Most important for this study, we conclude that Asp20 has a pK a ~ 1.5 in T26H–T4L* (obtained by fitting the partial titration curve with a restrained chemical shift change of 3.4 ppm due to deprotonation). This is substantially lower than the pK a of 3.6 reported for this residue in the wild‐type protein10 and may at least in part be due to the presence of a positively charged histidine versus neutral threonine at the nearby (~ 3.5 Å) position 26. Parenthetically, in wild‐type T4L, Asp20 shows an anomalously small 13Cγ chemical shift change over the pH range examined.10 Although this limits the confidence that can be placed on assigning the fit pK a value of 3.6 to the deprotonation of this catalytic residue, no other titrations were detected for the 13Cγ of Asp20 between pH 1.5 and 5.5.
Glutamic acid pKa determination
A sample of T26H–T4L* selectively labeled with δ‐13C glutamic acid was also titrated between pH 1.8 and 9.3 [Fig. 3(B,D)]. In 13C‐NMR spectra, all eight Glu residues showed clear titration curves with downfield 13Cγ chemical shift changes of ~ 3.8 ppm diagnostic of deprotonation with increasing sample pH [Fig. 3(B,D)]. Sidechain ionization was confirmed by the absence of a significant deuterium isotope shift for each residue in H2O at pH 5.8 versus D2O at pH* 5.7 [Fig. 4(B)]. Fitting the titration curves yielded the pK a values summarized in Table 1. These results also generally match those reported previously for wild‐type T4L,10 with Glu62 having the lowest pK a value of 2.9, likely due in part to its close proximity to Arg52 (3.2 Å).
Glu11 has a pK a of 4.7 in T26H–T4L*. This is only marginally higher than the pK a of ~ 4.4 for an unperturbed glutamic acid in a random coil polypeptide,18 yet lower than that of 5.4 measured for the wild‐type protein.10 Favorable electrostatic interactions with nearby (~ 6 Å) His26 in the T26H mutant may cause this pK a reduction. However, as noted by Andersen et al.,10 the elevated pK a value of Glu11 in wild‐type T4L is somewhat unexpected given its hydrogen bonding with Arg145, albeit offset by charge repulsion from Asp10 (~ 7 Å) and Asp20 (~ 7 Å). These authors suggested that conformational dynamics of T4L in solution, including a hinge bending motion between its N‐ and C‐lobes, may alter the solvent accessibility and electrostatic environment of active site residues relative to those seen in static crystal structures, thus precluding a simple rationalization of pK a values.
All arginines are N ε protonated at neutral pH
We also sought to determine the protonation states of the arginines in T26H–T4L*. Sidechain 1Hε–15Nε signals from the 13 arginines in both the wild‐type and mutant protein were readily detected and assigned in 15N‐HSQC spectra recorded at pH 5.9 (Fig. 5). This unequivocally demonstrates that under these conditions, all arginines, including Arg145, in both proteins are protonated at the Nε position. However, it is possible that an arginine could be neutral due to the loss of a terminal Hη proton. Therefore, we also assigned the arginine 13Cζ and 15Nη signals of T26H–T4L* at pH 6.5 (Fig. S4). Although caution must be exercised in inferring charge states from chemical shifts, all 13 had 15Nε, 13Cζ, and 15Nη signals highly indicative of a fully protonated guanidinium sidechain (e.g., 84.8 ppm, 159.5 ppm, and 75.0 and ~ 71 ppm, respectively, for Arg145).18, 25 Furthermore, the 13C and 15N NMR signals of corresponding arginines in the T26H–T4L* mutant and in the wild‐type protein are very similar.26, 27, 28 Of these, several, including Arg145, in T4L have been unambiguously demonstrated to be fully protonated at pH 5.5, and hence positively charged, on the basis of 1Hη–15Nη scalar coupling patterns.29
Figure 5.
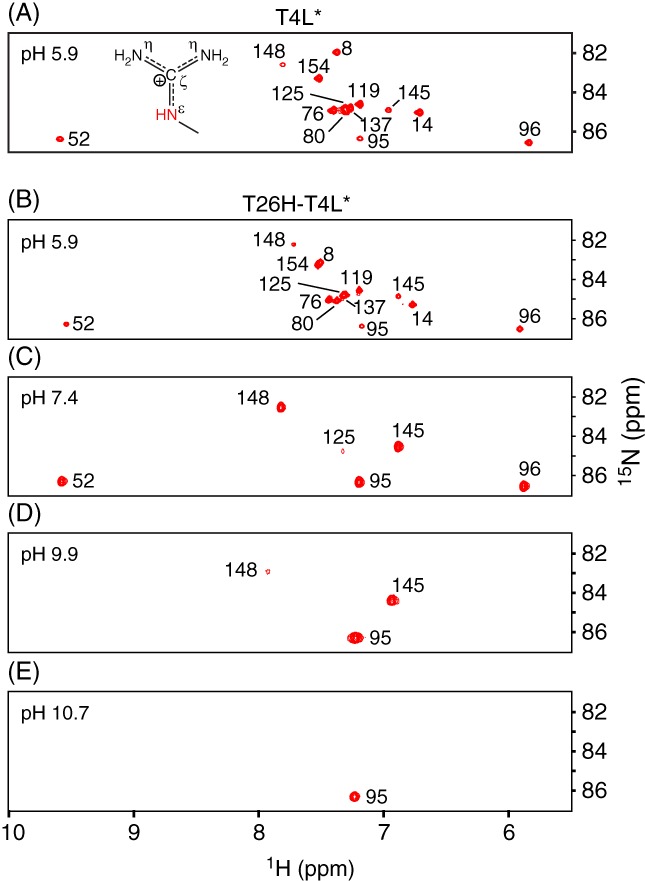
15N‐HSQC spectra showing the arginine guanidinium 1Hε‐15Nε signals from uniformly 15N/13C‐labeled (A) wild‐type T4L* and (B) T26H–T4L* at pH 5.9. The data unambiguously demonstrate that all 13 arginines, including Arg145, are Nε‐protonated in both proteins. Superimposition of these spectra (Fig. S2) reveals small 1Hε–15Nε chemical shift differences between several corresponding arginines of the wild‐type and mutant protein attributable to the T26H substitution and variations in exact sample conditions. (C–E) With increasing pH, the 1Hε–15Nε signals from T26H–T4L* disappeared due to base‐catalyzed hydrogen exchange. Only well‐protected arginines were detected under alkaline conditions. The lack of any significant pH‐dependent 15Nε chemical shift changes shows that the detected Arg95, Arg145, and Arg148 are positively charged under these conditions and hence have high pK a values (Table 1). The small downfield shift in the 1Hε signal of Arg148 followed an apparent pK a of 7.2. There are no ionizable groups proximal to Arg148 with a pK a value in this range and thus we speculate that this chemical shift change reflects altered pH‐dependent interactions with buffer phosphate (pK a2 ~ 7).
We attempted to measure the pK a values of the arginines in T26H–T4L* using 2D Hδ 2(Cδ)Nε‐type experiments.30 However, this was not successful as spectral crowding precluded the confident measurement of pH‐dependent 15Nε chemical shifts. In principle, these reporter shifts, as well as those of the 13Cζ nuclei, could be obtained from a Nε/η–Cζ correlation experiment.29 This was not pursued due to the low sensitivity of such a 13C‐detected approach and signal overlap in the resulting 2D spectra (Fig. S4). Regardless, the unperturbed pK a value of an arginine in a random coil polypeptide is ~ 13.8,18, 25 and it is unlikely that lysozyme would remain folded, or even chemically intact, under the harsh alkaline conditions required to measure complete titration curves.
Alternatively, we simply recorded 15N‐HSQC spectra of T26H–T4L* as a function of sample pH value [Fig. 5(C–E)]. At pH 9.9, the 1Hε–15Nε signals of all but three highly protected arginines (Arg95, Arg145, and Arg148) disappeared due to rapid base‐catalyzed hydrogen exchange with water.31 At pH 10.7, only Arg95 was observed. This protection, which matches the trends in arginine exchange rate constants recently measured for wild‐type T4L*,32 can be attributed at least in part to hydrogen bonds between Arg95 with the mainchain oxygen of Phe153, Arg145 with the sidechain oxygen of Asn101, and Arg148 with the carboxylate of Asp10. Importantly, no significant changes in the 15Nε chemical shifts of these three residues were observed over the pH range examined. Since the 15Nε signal of an arginine moves downfield by ~ 6 ppm upon deprotonation,18, 25 this implies that the pK a values of Arg145 and Arg148 must be > 11, and that of Arg95 > 12 (Table 1).
Discussion
Using NMR spectroscopy, we measured the pK a values of the Asp, Glu, and His residues in T26H–T4L* and confirmed that all arginines are Nε protonated and most certainly charged under neutral pH conditions. Along with previously reported data for T4L, these results provide important constraints for understanding the catalytic mechanisms of the wild‐type and mutant lysozymes.
Inverting mechanism of wild‐type T4L
Wild‐type T4L is an inverting glycoside hydrolase. A reasonable single‐displacement mechanism11 involves Asp20 acting as a general base to activate a nucleophilic water that is also hydrogen bonded to Thr26. This water carries out a nucleophilic substitution, with inversion of stereochemistry, at the anomeric carbon of a peptidic NAM. Concomitantly, Glu11 serves as a proton donor to facilitate the departure of the NAG aglycone.
This mechanism leads to the prediction that T4L should display a bell‐shaped activity versus pH profile, with a pH optimum of ~ 4.5 where Asp20 (pK a 3.6) is predominantly deprotonated to act as a general base and Glu11 (pK a 5.4) predominantly protonated to function as a general acid. In this context (and below), “predicted activity” refers to kcat/Km, the second order rate constant for the reaction of free enzyme and substrate, and neglects likely contributions of pH‐dependent interactions between the charged substrate peptide moiety, lAla‐dGlu‐DAP‐lAla, with an extended binding site on the protein surface.11 The first‐order rate constant for turnover of the Michaelis complex, k cat, will depend upon the pK a values of catalytic residues with the bound substrate, which have yet to be measured. Unfortunately, these fundamental catalytic parameters have not been determined for T4L due to the lack of a well‐defined substrate amenable to detailed kinetic analyses. Although it has been reported that the lytic activity of T4L on chloroform‐treated Escherichia coli cells is maximum at pH ~ 7.3, with sensitivity to ionic strength, amines, and divalent cations,33, 34 numerous factors complicate the mechanistic interpretation of these results.
Retaining mechanism of T26H–T4L with His26 as a nucleophile
Remarkably, the single substitution of Thr26 with a histidine converts T4L from an inverting to a retaining glycoside hydrolase with transglycosylase activity. Based on the classical Koshland double‐displacement mechanism, a plausible pathway [Fig. 1(A)] was proposed for T26H–T4L that involves nucleophilic attack of His26 on the peptidic NAM moiety to form an α‐linked glycosyl‐enzyme intermediate.7, 11, 12 This glycosylation step is facilitated though protonation of the leaving aglycone by the general acid Glu11. In the subsequent deglycosylation step, this same residue serves as a general base to activate either a nucleophilic water (hydrolysis) or carbohydrate (transglycosylation) for His26 displacement and glycone release with overall retention of β‐anomeric stereochemistry. The alternating role of Glu11 as a general acid and base likely results from changes in its environment, and hence pK a values, along the reaction pathway.35 In contrast to the wild‐type enzyme, Asp20 in T26H–T4L would not play any obvious direct catalytic role except perhaps to facilitate substrate binding by hydrogen bonding to the nitrogen of the N‐acetyl group of NAM. Evidence supporting this mechanism includes the observation of a stable peptidic NAG‐NAM moiety α‐linked to Glu26 in T26E–T4L,11 as well as the structural and enzymatic characterization of an extensive series of lysozymes with mutations at residues 11, 20, and 26.7, 12 In particular, the Nε2 of His26 is approximately positioned for nucleophilic attack on a docked substrate, and the T26Q mutant is inactive.
Accepting this proposed mechanism leads to the prediction that the pH‐dependent activity (k cat/K m) of T26H–T4L will follow a bell‐shaped profile with a maximum at pH ~ 5.8. However, the pK a value of the general acid Glu11 (4.7) is lower than that of the postulated nucleophile His26 (6.8). Thus, at this predicted pH optimum, only a small fraction of the protein (~ 1%) will be in the catalytically competent ionization state with Glu11 protonated and His26 deprotonated. Such a “reverse protonation mechanism” is well established (albeit underappreciated) for many enzymes and requires that the low population of suitably ionized residues be offset by their high catalytic efficiency.36 It may not seem intuitive that the pH‐dependence of k cat/K m for a reverse protonation mechanism would reflect the population‐averaged macroscopic pK a values of the catalytic residues measured by NMR spectroscopy.16 That is, due to electrostatic interactions, the microscopic pK a values of proximal Glu11 and His26 will likely differ depending on whether the other is neutral or charged. However, at the predicted pH optimum of ~ 5.8, the major population of T4L with His31 protonated and Glu11 deprotonated will be in a pH‐independent equilibrium with the minor population having the catalytically competent deprotonated His31 and protonated Glu11.
Alternative mechanism for T26H–T4L
Although in principle histidine can serve as a nucleophile for retaining glycoside hydrolases, it is striking that none to date has ever been demonstrated or proposed to play this role in any such enzyme other than T26H–T4L. This prompts the question whether T26H–T4L could follow an alternative catalytic mechanism. One possibility, exemplified by GH20 β‐hexosaminidases,37 involves the nucleophilic participation of the N‐acetyl group of NAM [Fig. 1(B)]. Such participation could become feasible if the T26H mutation results in interactions that are favorable to the requisite alignment of the side chain amide moiety. A particularly attractive feature of this hypothesized mechanism is that we know it is inherently possible for the N‐acetyl moiety to adopt the correct geometry for nucleophilic attack since it does so in other enzymes. In contrast, we do not know with certainty that this is the case for the nitrogen of His26.
In this hypothesized substrate‐assisted catalysis mechanism, Asp20 might act as a general base to abstract a proton from the N‐acetyl nitrogen and assist the formation of a bicyclic oxazoline intermediate. Glu11 would serve as a general acid for aglycone departure and then as a general base for hydrolysis or transglycosylation, with net retention of β‐anomeric stereochemistry. The predicted pH optimum in kcat/Km for such a mechanism would be ~ 3.1 with Asp20 (pK a ~ 1.5) predominantly deprotonated and Glu11 (pK a 4.7) predominantly protonated in the free enzyme. However, with such a low pK a value, Asp20 would be a poor general base and might rather remain negatively charged to stabilize a positively charged oxazolinium intermediate without proton transfer.38 It is also worth considering that, in the Michaelis complex with bound substrate, the pK a values of these catalytic residues could be better tuned for efficient catalysis.
In contrast to the previously proposed mechanism with a histidine‐linked glycosyl‐enzyme intermediate, in this alternative substrate‐assisted pathway Asp20 serves a direct catalytic role whereas that of His26 is not obviously defined. This is conformationally plausible as Asp20 is hydrogen bonded to the N‐acetyl group of NAM in the X‐ray crystallographic structure of glycosylated T26E–T4L,11 yet less consistent with the observations that the T4L–T26Q mutant is inactive and the D20C/T26H double mutant is still active as a retaining glycoside hydrolase.12 However, wild‐type T4L is tolerant to substitutions at position 20, including D20C for which cysteine could serve as a general base.34 Furthermore, the very fact that substituting Thr26 with His changes the stereochemical outcome of peptidogycan hydrolysis suggests an active site plasticity that tips the balance between different catalytic mechanisms. Resolving these mechanisms will require more detailed enzymatic studies with well‐defined substrates.
Arg145 is N ε protonated with a high pK a value
Using NMR spectroscopy, we showed conclusively that the Nε of all arginines, including Arg145, in both wild‐type T4L* and T26H–T4L* are protonated under typical experimental conditions. Also, from chemical shift arguments, the sidechain guanidinium group of this active site residue in the mutant protein is most certainly ionized with a pK a > 11. Although largely buried in the X‐ray crystal structures of both proteins, stabilization of the delocalized positive charge of Arg145 may result from extensive hydrogen bonding between the terminal Nη1 and Nη2 with the carboxylate of Glu11 and the Nη2 and Nε with the sidechain oxygen of Asn101. Considerable debate has centered on the possibility of a deprotonated arginine sidechain in a protein under physiological conditions.25, 39 With an intrinsic pK a value of nearly 14, a substantial energetic penalty would be paid to accommodate a neutral guanidino group. This penalty is on the order of global protein folding energetics, and thus any such species is unlikely to exist except perhaps in a minor population along an enzymatic pathway in which arginine functions as a general base.40
Ionization states determined from neutron crystallography
Neutron crystallography is a well‐established approach for identifying hydrogens in molecules, including proteins. Indeed, the ionization states of the Asp, Glu, His, and Arg residues found herein by NMR spectroscopy are consistent with those determined in a recently published joint neutron and high‐resolution X‐ray diffraction study of perdeuterated T4L* at pD 6–7 (5VNQ.pdb and 5VNR.pdb).14 Thus it somewhat perplexing why in a similar joint diffraction study of perdeuterated T26H–T4L* at pD 7, Arg145 was reported to be Nε deprotonated, Asp20 carboxyl protonated, Glu11 partially (~ 60%) carboxyl protonated, and His26 deprotonated at Nε2 (5XPE.pdb and 5XPF.).13 As we have demonstrated for this protein in solution, Asp20 has a pK a ~ 1.5 and Arg145 is Nε protonated with an inferred pK a > 11. Thus, both are effectively completely charged under physiologically relevant pH conditions. Also, at pH 7, Glu11 (pKa 4.7) and His 31 (pKa 6.8) should be ~ 1% and ~ 40% protonated, respectively. There is no obvious physicochemical reason as to why the substitution of Thr26 to His would dramatically alter the pK a values of these active site residues in T26H–T4L* versus wild‐type T4L*, or why T26H–T4L* would differ between the solution and crystalline states. Although crystal packing or protein and solvent deuteration could explain the smaller discrepancies for Glu11 and His31, it is difficult to imagine how this could lead to such energetically unfavorable changes for Asp20 and Arg145. Unexplained disagreements between results obtained by NMR spectroscopy and neutron diffraction can also be found in the case of photoactive yellow protein.29, 41 This speaks to the challenges in defining protein protonation states and pK a values, and the need to use complementary methods to obtain a comprehensive understanding of the pH‐dependent ionization states of biological macromolecules.
Materials and Methods
Protein expression and purification
The clones encoding T4L* (plasmid 18111) and T26H‐T4L* (plasmid 18251) were obtained from AddGene. T4L* is a cysteine‐free pseudo‐wild‐type lysozyme with the C54T and C97A substitutions. Protein expression and purification followed previously described protocols.42 Briefly, 13C/15N‐ or 15N‐labeled lysozyme was expressed in transformed prototrophic E. coli HMS174 cells. An LB seed culture was used to inoculate M9 media containing 3 g/L 99% 13C6‐glucose and/or 1 g/L 98% 15NH4Cl to an initial OD600 of ~ 0.1. Expression was induced with 1 mM IPTG at OD600 ~ 0.6, and the cells were harvested after ~ 4 h of growth at 37°C by centrifugation.
Samples of T26H–T4L* selectively labeled with 13Cγ‐aspartic acid or 13Cδ‐glutamic acid were expressed in auxotrophic E. coli strains EA1 (aspC, tyrB, asnAB::Tn5) and DL39 (aspC, ilvE, tyrB, avtA::Tn5), respectively.43 The asnAB mutations prevent the metabolic conversion of aspartic acid to asparagine. Unfortunately, a viable strain with a glnA genotype to prevent the conversion of glutamic acid to glutamine was not available. Overnight seed cultures in LB were used to inoculate minimal media containing 500 mg/L 13Cγ‐d/l‐aspartic acid (Cambridge Isotopes) or 800 mg/L 13Cδ‐d/l‐glutamic acid (Cambridge Isotopes), along with all other unlabeled amino acids.10, 44, 45 Cells were grown to an OD600 of ~ 0.6, induced with 1 mM IPTG, and harvested by centrifugation after growth for 4 h at 37°C.
Cells were lysed by homogenization, loaded onto an SP‐Sepharose cation exchange column (GE Healthcare) in 50 mM sodium phosphate, 2 mM EDTA, pH 6.5, and eluted using a linear gradient of NaCl from 0 to 1 M. Samples were further purified by passage through a Superdex 75 size exclusion column in NMR buffer (100 mM KCl, 30 mM potassium phosphate, pH 5.5), and concentrated to ~ 1.5 mM using a 3 kDa MWCO Amicon ultracentrifugal filter. Finally, ~ 5% D2O was added as a lock solvent. Protein concentrations were determined using ultraviolet absorbance (predicted ε280 = 25,440 M−1 cm−1).
NMR spectroscopy
NMR spectra were recorded at 25°C with Bruker Avance III 500, 600, and 850 MHz spectrometers equipped with the xyz‐gradient TCI cryoprobes. Data were processed with NMRpipe46 and analyzed in NMRFAM‐SPARKY47 and Topspin. Standard heteronuclear scalar correlation experiments, including 3D HNCACB, CBCACONH, H(CCO)‐TOCSY‐NH, and C(CCO)‐TOCSY‐NH,48 were automatically interpreted using PINE49 and verified manually to assign relevant backbone and sidechain 1H, 13C, and 15N signals. The sidechain carboxyl signals of Asp/Glu were assigned from a 3D Hβ/γ 2Cβ/γCOγ/δ experiment, which was based on a modified HCACO experiment.50 Carboxyl deuterium isotope shifts were measured from 2D Hβ/γ 2(Cβ/γ)COγ/δ spectra of uniformly 15N/13C‐labeled T26H–T4L* in H2O NMR sample buffer and then after lyophilization and resuspension in an equal volume of D2O. The initial sample pH was 5.8, and the final sample pH* (uncorrected for isotope effects) was 5.7. Histidine imidazole signals were readily identified in conventional and constant time 13C‐HSQC spectra.51, 52 Arginine 15Nε signals were assigned from an HεNεCδ spectrum, recorded using a modified HNCA experiment,53 13Cζ signals from an HεNεCζ spectrum, recorded using a modified HNCO experiment,53 and 15Nη signals from a cross polarization 13C‐detected Nε/η‐Cζ correlation experiment.26, 29 The modifications involved setting the transmitter frequencies for 15Nε (84.5 ppm), 13Cδ (43.5 ppm), and 13Cζ (160 ppm), and lengthening the 1Hε–15Nε de‐phasing/re‐phasing delays to 2.8 ms along with a 1.67 msec 15Nε‐selective 180° pulse.
pKa determination of His, Asp, and Glu
NMR‐monitored pH titrations were recorded at 25°C on a Bruker Avance III 850 MHz spectrometer. The titrations were carried out by transferring the sample between the NMR tube and a 1.5 mL Eppendorf tube and adding small aliquots of NMR buffer with ~ 0.1 M HCl or NaOH. The average pH meter reading, measured before and after spectra acquisition with a ThermoFisher Orion 9110DJWP electrode at room temperature (~ 20°C), was taken as the sample pH value. Histidines in 13C/15N‐labeled T26H–T4L* were monitored between pH 5.0 and 10.7 via 13C‐HSQC spectra. The sidechain carboxyl signals of Asp and Glu in selectively labeled protein samples were monitored between pH 1.8 to 7.8 and 1.8 to 9.8, respectively, via direct detection 1D 13C‐NMR with 1H decoupling during acquisition. The pH‐dependent chemical shifts of individual 13C and 1H nuclei were fit with GraphPad Prism to the Henderson–Hasselbalch equation for one or macroscopic ionization equilibria to obtain corresponding pK a values.16 In the case of each His, results from four reporter nuclei were averaged to obtain the reported pK a values with standard deviations. In the case of Asp and Glu, errors were estimated ± 0.1 as limited by the accuracy in measuring sample pH value.
Conflict of interests
The authors declare no conflicts of interest with respect to the publication of this article.
Supporting information
Appendix S1: Supplementary material
Acknowledgments
We thank Brian Matthews and Rick Dahlquist for helpful discussions. This work was funded through Discovery Grants from the Natural Sciences and Engineering Research Council (NSERC) to S.G.W. and L.P.M., and a UBC Four Year Doctoral Fellowship to J.A.B. Instrument support was provided by the Canada Foundation for Innovation, the British Columbia Knowledge Development Fund, the UBC Blusson Fund, and NSERC.
References
- 1. Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B (2014) The carbohydrate‐active enzymes database (CaZy) in 2013. Nucleic Acids Res 42:D490–D495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wohlkonig A, Huet J, Looze Y, Wintjens R (2010) Structural relationships in the lysozyme superfamily: significant evidence for glycoside hydrolase signature motifs. PLoS One 5:e15388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Davies G, Henrissat B (1995) Structures and mechanisms of glycosyl hydrolases. Structure 3:853–859. [DOI] [PubMed] [Google Scholar]
- 4. Rye CS, Withers SG (2000) Glycosidase mechanisms. Curr Opin Chem Biol 4:573–580. [DOI] [PubMed] [Google Scholar]
- 5. Baase WA, Liu L, Tronrud DE, Matthews BW (2010) Lessons from the lysozyme of phage T4. Protein Sci 19:631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jensen HB, Kleppe G, Schindler M, Mirelman D (1976) The specificity requirements of bacteriophage T4 lysozyme. Eur J Biochem 66:319–325. [DOI] [PubMed] [Google Scholar]
- 7. Kuroki R, Weaver LH, Matthews BW (1995) Structure‐based design of a lysozyme with altered catalytic activity. Nat Struct Biol 2:1007–1011. [DOI] [PubMed] [Google Scholar]
- 8. Vocadlo DJ, Davies GJ, Laine R, Withers SG (2001) Catalysis by hen egg‐white lysozyme proceeds via a covalent intermediate. Nature 412:835–838. [DOI] [PubMed] [Google Scholar]
- 9. Anderson DE (1992) The role of electrostatic interactions in stabilizing T4 lysozyme. PhD Thesis, University of Oregon, Eugene, OR.
- 10. Anderson DE, Lu J, McIntosh L, Dahlquist FW. The folding, stability and dynamics of T4 lysozyme: a perspective using nuclear magnetic resonance In: Clore GM, Groneborn A, Eds. (1993) Topics in Molecular and Structural Biology: NMR of Proteins. London: Macmillan Press; p. 258–304. [Google Scholar]
- 11. Kuroki R, Weaver LH, Matthews BW (1993) A covalent enzyme–substrate intermediate with saccharide distortion in a mutant T4 lysozyme. Science 262:2030–2033. [DOI] [PubMed] [Google Scholar]
- 12. Kuroki R, Weaver LH, Matthews BW (1999) Structural basis of the conversion of T4 lysozyme into a transglycosidase by reengineering the active site. Proc Natl Acad Sci U S A 96:8949–8954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hiromoto T, Meilleur F, Shimizu R, Shibazaki C, Adachi M, Tamada T, Kuroki R (2017) Neutron structure of the T26H mutant of T4 phage lysozyme provides insight into the catalytic activity of the mutant enzyme and how it differs from that of wild type. Protein Sci 26:1953–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li L, Shukla S, Meilleur F, Standaert RF, Pierce J, Myles DAA, Cuneo MJ (2017) Neutron crystallographic studies of T4 lysozyme at cryogenic temperature. Protein Sci 26:2098–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anderson DE, Becktel WJ, Dahlquist FW (1990) pH‐induced denaturation of proteins: a single salt bridge contributes 3‐5 kcal/Mol to the free energy of folding of T4 lysozyme. Biochemistry 29:2403–2408. [DOI] [PubMed] [Google Scholar]
- 16. McIntosh LP, Naito D, Baturin SJ, Okon M, Joshi MD, Nielsen JE (2011) Dissecting electrostatic interactions in Bacillus circulans xylanase through NMR‐monitored pH titrations. J Biomol NMR 51:5–19. [DOI] [PubMed] [Google Scholar]
- 17. Sudmeier JL, Bradshaw EM, Haddad KEC, Day RM, Thalhauser CJ, Bullock PA, Bachovchin WW (2003) Identification of histidine tautomers in proteins by 2D 1H/13Cδ2 one‐bond correlated NMR. J Am Chem Soc 125:8430–8431. [DOI] [PubMed] [Google Scholar]
- 18. Platzer G, Okon M, McIntosh LP (2014) pH‐dependent random coil 1H, 13C, and 15N chemical shifts of the ionizable amino acids: a guide for protein pK a measurements. J Biomol NMR 60:109–129. [DOI] [PubMed] [Google Scholar]
- 19. Bosshard HR, Marti DN, Jelesarov I (2004) Protein stabilization by salt bridges: concepts, experimental approaches and clarification of some misunderstandings. J Mol Recogn 17:1–16. [DOI] [PubMed] [Google Scholar]
- 20. Ladner HK, Led JJ, Grant DM (1975) Deuterium‐isotope effects on 13C chemical‐shifts in amino‐acids and dipeptides. J Magn Reson 20:530–534. [Google Scholar]
- 21. Led JJ, Petersen SB (1979) Deuterium‐isotope effects on 13C chemical‐shifts in selected amino‐acids as function of pH. J Magn Reson 33:603–617. [Google Scholar]
- 22. Wang YX, Freedberg DI, Yamazaki T, Wingfield PT, Stahl SJ, Kaufman JD, Kiso Y, Torchia DA (1996) Solution NMR evidence that the HIV‐1 protease catalytic aspartyl groups have different ionization states in the complex formed with the asymmetric drug KNI‐272. Biochemistry 35:9945–9950. [DOI] [PubMed] [Google Scholar]
- 23. Guo J, Tolstoy PM, Koeppe B, Golubev NS, Denisov GS, Smirnov SN, Limbach HH (2012) Hydrogen bond geometries and proton tautomerism of homoconjugated anions of carboxylic acids studied via H/D isotope effects on 13C NMR chemical shifts. J Phys Chem A 116:11180–11188. [DOI] [PubMed] [Google Scholar]
- 24. Liu A, Wang J, Lu Z, Yao L, Li Y, Yan H (2008) Hydrogen‐bond detection, configuration assignment and rotamer correction of side‐chain amides in large proteins by NMR spectroscopy through protium/deuterium isotope effects. Chembiochem 9:2860–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fitch CA, Platzer G, Okon M, Garcia‐Moreno BE, McIntosh LP (2015) Arginine: its pK a value revisited. Protein Sci 24:752–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Werbeck ND, Kirkpatrick J, Hansen DF (2013) Probing arginine side‐chains and their dynamics with carbon‐detected NMR spectroscopy: application to the 42 kDa human histone deacetylase 8 at high pH. Angew Chem Int Ed Engl 52:3145–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gerecht K, Figueiredo AM, Hansen DF (2017) Determining rotational dynamics of the guanidino group of arginine side chains in proteins by carbon‐detected NMR. Chem Commun (Camb) 53:10062–10065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mackenzie HW, Hansen DF (2017) A 13C‐detected 15N double‐quantum NMR experiment to probe arginine side‐chain guanidinium 15Nη chemical shifts. J Biomol NMR 69:123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yoshimura Y, Oktaviani NA, Yonezawa K, Kamikubo H, Mulder FA (2017) Unambiguous determination of protein arginine ionization states in solution by NMR spectroscopy. Angew Chem Int Ed Engl 56:239–242. [DOI] [PubMed] [Google Scholar]
- 30. Andre I, Linse S, Mulder FA (2007) Residue‐specific pK a determination of lysine and arginine side chains by indirect 15N and 13C NMR spectroscopy: application to apo calmodulin. J Am Chem Soc 129:15805–15813. [DOI] [PubMed] [Google Scholar]
- 31. Henry GD, Sykes BD (1995) Determination of the rotational‐dynamics and pH‐dependence of the hydrogen‐exchange rates of the arginine guanidino group using NMR spectroscopy. J Biomol NMR 6:59–66. [DOI] [PubMed] [Google Scholar]
- 32. Mackenzie HW, Hansen DF (2018) Arginine side‐chain hydrogen exchange: quantifying arginine side‐chain interactions in solution. Chemphyschem 19:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jensen HB, Kleppe K (1972) Effect of ionic strength, pH, amines and divalent cations on the lytic activity of T4 lysozyme. Eur J Biochem 28:116–122. [DOI] [PubMed] [Google Scholar]
- 34. Hardy LW, Poteete AR (1991) Reexamination of the role of asp20 in catalysis by bacteriophage T4 lysozyme. Biochemistry 30:9457–9463. [DOI] [PubMed] [Google Scholar]
- 35. McIntosh LP, Hand G, Johnson PE, Joshi MD, Korner M, Plesniak LA, Ziser L, Wakarchuk WW, Withers SG (1996) The pK a of the general acid/base carboxyl group of a glycosidase cycles during catalysis: a 13C‐NMR study of Bacillus circulans xylanase. Biochemistry 35:9958–9966. [DOI] [PubMed] [Google Scholar]
- 36. Mock WL (1992) Theory of enzymatic reverse‐protonation catalysis. Bioorg Chem 20:377–381. [Google Scholar]
- 37. Mark BL, Vocadlo DJ, Knapp S, Triggs‐Raine BL, Withers SG, James MN (2001) Crystallographic evidence for substrate‐assisted catalysis in a bacterial beta‐hexosaminidase. J Biol Chem 276:10330–10337. [DOI] [PubMed] [Google Scholar]
- 38. Coines J, Alfonso‐Prieto M, Biarnes X, Planas A, Rovira C (2018) Oxazoline or oxazolinium ion? The protonation state and conformation of the reaction intermediate of chitinase enzymes revisited. Chem Eur J. https://onlinelibrary.wiley.com/doi/epdf/10.1002/chem.201803905. [DOI] [PubMed] [Google Scholar]
- 39. Harms MJ, Schlessman JL, Sue GR, Garcia‐Moreno B (2011) Arginine residues at internal positions in a protein are always charged. Proc Natl Acad Sci U S A 108:18954–18959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schlippe YVG, Hedstrom L (2005) A twisted base? The role of arginine in enzyme‐catalyzed proton abstractions. Arch Biochem Biophys 433:266–278. [DOI] [PubMed] [Google Scholar]
- 41. Yonezawa K, Shimizu N, Kurihara K, Yamazaki Y, Kamikubo H, Kataoka M (2017) Neutron crystallography of photoactive yellow protein reveals unusual protonation state of Arg52 in the crystal. Sci Rep 7:9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vallurupalli P, Hansen DF, Lundstrom P, Kay LE (2009) CPMG relaxation dispersion NMR experiments measuring glycine 1Hα and 13Cα chemical shifts in the 'invisible' excited states of proteins. J Biomol NMR 45:45–55. [DOI] [PubMed] [Google Scholar]
- 43. Waugh DS (1996) Genetic tools for selective labeling of proteins with alpha‐15N‐amino acids. J Biomol NMR 8:184–192. [DOI] [PubMed] [Google Scholar]
- 44. Muchmore DC, McIntosh LP, Russell CB, Anderson DE, Dahlquist FW (1989) Expression and nitrogen‐15 labeling of proteins for proton and nitrogen‐15 nuclear magnetic resonance. Methods Enzymol 177:44–73. [DOI] [PubMed] [Google Scholar]
- 45. McIntosh LP, Wand AJ, Lowry DF, Redfield AG, Dahlquist FW (1990) Assignment of the backbone 1H and 15N NMR resonances of bacteriophage T4 lysozyme. Biochemistry 29:6341–6362. [DOI] [PubMed] [Google Scholar]
- 46. Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRpipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293. [DOI] [PubMed] [Google Scholar]
- 47. Lee W, Tonelli M, Markley JL (2015) NMRFAM‐sparky: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31:1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sattler M, Schleucher J, Griesinger C (1999) Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog Nucl Mag Res Spec 34:93–158. [Google Scholar]
- 49. Bahrami A, Assadi AH, Markley JL, Eghbalnia HR (2009) Probabilistic interaction network of evidence algorithm and its application to complete labeling of peak lists from protein NMR spectroscopy. PLoS Comput Biol 5:e1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kay LE, Ikura M, Tschudin R, Bax A (1989) Three‐dimensional triple‐resonance NMR spectroscopy of isotopically enriched proteins. J Magn Reson 89:496–514. [DOI] [PubMed] [Google Scholar]
- 51. Vuister GW, Bax A (1992) Resolution enhancement and spectral editing of uniformly 13C‐enriched proteins by homonuclear broad‐band 13C decoupling. J Magn Reson 98:428–435. [Google Scholar]
- 52. Plesniak LA, Connelly GP, Wakarchuk WW, McIntosh LP (1996) Characterization of a buried neutral histidine residue in Bacillus circulans xylanase: NMR assignments, pH titration, and hydrogen exchange. Protein Sci 5:2319–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kay LE, Ikura M, Tschudin R, Bax A (1990) 3‐Dimensional triple‐resonance NMR‐spectroscopy of isotopically enriched proteins. J Magn Reson 89:496–514. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary material