

Abstract
G protein‐coupled receptors (GPCRs) constitute the largest family of cell surface receptors that mediate numerous cell signaling pathways, and are targets of more than one‐third of clinical drugs. Thanks to the advancement of novel structural biology technologies, high‐resolution structures of GPCRs in complex with their signaling transducers, including G‐protein and arrestin, have been determined. These 3D complex structures have significantly improved our understanding of the molecular mechanism of GPCR signaling and provided a structural basis for signaling‐biased drug discovery targeting GPCRs. Here we summarize structural studies of GPCR signaling complexes with G protein and arrestin using rhodopsin as a model system, and highlight the key features of GPCR conformational states in biased signaling including the sequence motifs of receptor TM6 that determine selective coupling of G proteins, and the phosphorylation codes of GPCRs for arrestin recruitment. We envision the future of GPCR structural biology not only to solve more high‐resolution complex structures but also to show stepwise GPCR signaling complex assembly and disassembly and dynamic process of GPCR signal transduction.
Keywords: Structural biology, G protein‐coupled receptors, G protein, arrestin, GPCR, signaling
Introduction
G protein‐coupled receptors (GPCRs) represent a superfamily of cell surface receptors. They regulate numerous physiological processes and serve as targets of a large number of pharmaceutical drugs for various human diseases.1, 2, 3, 4 When stimulated by extracellular signals, GPCRs couple to intracellular heterotrimeric G proteins, and mediate the exchange of GDP for GTP in the Gα subunits. GTP binding activates the Gα subunit and induces dissociation of the Gα from the GPCR and the dimeric Gβγ subunits. Both the GTP‐bound Gα and the dissociated Gβγ in turn regulate downstream signaling events through binding to downstream effectors.5 G protein‐mediated signaling can be terminated when a GPCR kinase (GRK) is recruited and phosphorylates the GPCR. Receptor phosphorylation promotes the GPCR to bind to arrestin, initiating receptor internalization that leads to receptor recycling or degradation, or redirecting the signaling to arrestin‐mediated pathways.
Up to now, over one hundred GPCR structures have been solved in either agonist‐ or antagonist‐bound conformations (http://gpcrdb.org), leading to accumulated understanding of the structural mechanism of ligand‐induced conformational changes in GPCRs. Structure determination of GPCRs in complex with their signal transducers, however, is still challenging due to the transient and dynamic interactions of GPCRs with their signaling partners.
The crystal structure of the β2‐adrenergic receptor (β2AR) in complex with the stimulatory Gs protein was determined in 2011, with the help of a stabilizing nanobody (Nb35) that binds to the G protein.6 This complex structure has demonstrated that an activated GPCR recruits Gs protein by binding to its Gα subunit through the cytoplasmic portion of the transmembrane helical domain (TMD) of the receptor. The interface between GPCR and G protein consists of two parts. The first interface is formed between the transmembrane helices (TM) 5 and 6 and the intracellular loop (ICL) 3 of the receptor (β2AR TM5‐ICL3‐TM6), and the Gα C‐terminal domain, specifically the C‐terminal helices (α) 4 and 5 and the β‐strand (β) 6 of the Ras‐like domain of the Gα subunit (Gα α4‐β6‐α5). The second interface is formed between ICL2 of the receptor and the Gα C‐terminal end of the N‐terminal helix (αN), the central β‐sheet, and α5 of the Gs α subunit. The same principal GPCR–G protein interface was observed in a crystal structure of the adenosine A2A receptor bound to an engineered mini‐Gs protein, which consists solely of the Ras‐like GTPase domain of the G protein.7 The first GPCR–arrestin complex structure, rhodopsin in complex with visual arrestin, was solved by our group.8, 9 This is so far the only high‐resolution GPCR–arrestin complex structure that shows the overall GPCR–arrestin assembly, and thus provides important insight into structural understanding of GPCR signaling mediated by arrestin.`
Since 2017, we have witnessed a tremendous progress in GPCR structural biology using cryo‐EM. The first two cryo‐EM GPCR complex structures were determined for glucagon‐like peptide 1 receptor (GLP1R) and calcitonin receptor (CTR), both in complex with the stimulatory Gs protein.10, 11 Entering 2018, a cryo‐EM structure of rhodopsin in complex with Gi, a homologue of the G protein transducin, and three other structures, μ‐opioid receptor (μOR) –Gi complex, adenosine A1 receptor (A1R)‐Gi complex, and serotonin 5‐HT1bR in complex with Go, have been reported.12, 13, 14, 15 Most recently, a crystal structure of bovine rhodopsin in complex with a mini‐Go that includes the Ras domain of Gαo subunit, was published.16
Rhodopsin is a prototypical GPCR that has served as a model system for structural and biological studies of the whole superfamily of GPCRs. It plays a key role as a photoreceptor in the rod cells of the retina, responsible for converting photons into chemical signals that stimulate biological processes in the nervous system to perceive light. Rhodopsin adopts an inactive conformation when bound to its inverse agonist 11‐cis retinal. Light‐induced isomerization of 11‐cis retinal to all‐trans retinal triggers conformational changes in rhodopsin that facilitate the opening of an intracellular pocket of the 7TM domain of the receptor. This pocket accommodates the binding of the G protein transducin, a visual‐specific G protein belonging to the Gi subgroup that is responsible for photon perception in humans and animals. Upon rhodopsin activation, the GTP‐bound α subunit of transducin activates cGMP phosphodiesterase, leading to cGMP hydrolysis and the closing of cGMP‐gated cation channels. This causes the hyperpolarization of the photoreceptor cell, and the build‐up of a charge difference across the membrane of the cells to send an electrical impulse to the brain through adjacent nerve cells for photon perception.
The crystal structure of ground‐state bovine rhodopsin in complex with the reverse agonist 11‐cis‐retinal was the first high‐resolution GPCR structure solved by X‐ray crystallography.17, 18, 19, 20 Later, crystal structures have been published for rhodopsin and its retinal‐free form opsin in active conformations, with or without the binding of a peptide derived from the C‐terminal helix α5 of the α subunit of G protein transducin (Gt).21, 22, 23, 24, 25 The overall structural information of how rhodopsin recruits its downstream signaling partners, the G protein transducin and visual arrestin, however, had not been available until the structures of rhodopsin–visual arrestin and rhodopsin‐Gi complexes were determined.8, 9, 26 As rhodopsin serves as a prototype for the GPCR family, the discovery of the structural information of these complexes has significantly impacted the understanding of the signaling mechanisms of the entire GPCR superfamily.
During the revision of this manuscript, Glukhova et al. published a review paper that summarized recently reported GPCR‐G protein complex structures.27 By comparing all available GPCR‐G protein complex structures, they proposed general rules for GPCR‐G protein engagement, and identified receptor regions that are selectively involved in binding to Gs or Gi/o. In this review, we focus on the structural studies of the rhodopsin‐Gi and rhodopsin–visual arrestin complexes. Based on these structures, we discuss the underlying molecular mechanisms for the recruitment and activation of the receptor's signaling partners, G protein and visual arrestin, and G protein‐ and arrestin‐mediated signaling by rhodopsin and other GPCRs.
GPCR–G Protein Interaction
Rhodopsin–Gi protein interaction
G proteins are signal transducers that play a key role in regulating GPCR signal transduction.28 G proteins are composed of three different subunits: α, β, and γ. The α subunit contains a Ras‐like GTPase domain and an α‐helical domain (AHD), with a guanine nucleotide (GDP or GTP) binding site between the two domains. The Gβ and Gγ subunits tightly bind to each other as a dimeric protein complex. G proteins are classified into four subfamilies, Gs, Gi/o, Gq/11, and G12/13, based on sequence homology of their Gα subunit. Each G protein subfamily contains members that play distinct roles in GPCR signaling transduction.29 While there is so far no published GPCR structures in complex with Gq/11 or G12/13 proteins, GPCR structures in complex with Gs (Gs group), and Gi and Go (both belong to the Gi group) have been reported. The Gs proteins stimulate the production of cAMP second messengers by activating adenylyl cyclase, while the Gi/o family members reduce intracellular cAMP levels through repressing adenylyl cyclase activity or activating phosphodiesterases.30
Gi belongs to the same G protein family as transducin, and functions exchangeably with transducin in cellular and biochemical in vitro assays.31, 32, 33 Thus, the 3D structure of activated rhodopsin in complex with Gi is a good model for the binding mode of G protein transducin with rhodopsin and provides structural insights of photon perception through rhodopsin–G protein signaling.
While the receptors in the GPCR–Gs complexes of known structures also interact with Gβγ, the interface between rhodopsin and the Gi is exclusively maintained by the Gα subunit. The major interface between rhodopsin and Gαi is formed between the very C‐terminus of the Gα subunit (Residues 337–350 of α5 and Residues 351–354 of the adjacent C‐terminal loop) and the intracellular TM pocket of rhodopsin [Fig. 1(A,B)]. The interface is largely mediated by hydrophobic interaction through G protein residues I344, L348, and C351 on α5, and L353 and F354 on the C‐terminal loop, with their side chains inserting into the intracellular pocket of the receptor, interacting with residues L72 and L76 on TM2, V138 and V139 on TM3, L226 and V230 on TM5, A246, V250, M253, and Y257 on TM6, and M309 at the turn between TM7 and Helix 8 [Fig. 1(B)]. In addition, we observed two sets of electrostatic interactions: between the carboxyl group of the last residue of Gα, F354, and Rho K311 at the N‐terminus of Helix 8; and polar associations between carbonyl oxygens of the C‐terminal loop of the Gα protein and amide nitrogens from M309, N310, and K311 of the receptor [Fig. 1(B)]. These polar interactions, noted as “capping interactions”, serve as one of the distinguished features for GPCR‐Gi or ‐Go coupling, are not observed in GPCR–Gs complexes.26
Figure 1.

Rhodopsin–Gi assembly. (A) Two views of the rhodopsin‐Gi protein‐Fab complex structure (human rhodopsin with human Gαi/rat Gβ1/Bovine Gγ2; PDB code: 6CMO). Rhodopsin is colored in dark green, Gαi Ras domain in magenta, Gαi α helical domain (AHD) in red, Gβ in blue, Gγ in yellow, and the Fab in gray. The key structure interface elements of Gαi with rhodopsin, α5 and αN, are labeled.(B) Structure interface between the rhodopsin TM pocket and α5 of Gαi. The key interface residues are labeled; hydrophobic residues in the rhodopsin TM pocket that interact with α5 of Gαi are depicted in gray surface presentation. The same color code is used as in panel A.
Specificity of GPCR–G protein interaction: Gs versus Gi/o
The rhodopsin–Gi complex structure, together with other GPCR–G protein complex structures6, 10, 11, 12, 13, 14, 15 allows us to study molecular mechanisms of the recruitment and activation of different G protein subfamilies. We observed similar overall complex assemblies of GPCRs with Gi or Gs proteins, with a conserved major interface between the α5 helices of Gα and the intracellular binding pockets of the receptors including helices TM3, TM5, TM6, TM7, and Helix 8. These GPCR–G protein complexes, however, also exhibit interesting details that differ in their receptor–G protein interfaces, particularly in α5 helices of G proteins with TM6, TM7 and Helix 8 of the cytoplasmic TM pockets of the receptors. Some of these differences are G protein subfamily‐dependent and are likely relevant to their distinct functions in G protein mediated signaling.
Rhodopsin and β2AR represent well‐studied class A GPCRs that bind to Gi and Gs, respectively. In their inactive states, both receptors adopt similar closed conformations with their cytoplasmic ends of TM helices and Helix 8 in nearly identical positions. Receptor activation induces conformational changes in the TM domains of the receptors, resulting in different active conformations, particularly in cytoplasmic ends of TM6 and TM7, and the turn between TM7 and Helix 8, for differentially accommodating Gi and Gs [Fig. 2(A,B)]. Alignment of rhodopsin‐Gi with β2AR‐Gs structures shows that the intracellular end of the TM6 of the Gs‐bound β2AR is tilted outward by approximately 8 Å compared with that of the Gi‐bound rhodopsin, which is relatively straight. Correspondingly, the α5 helix of β2AR‐bound Gs is inserted upward in the TM pocket and tilted towards the intracellular end of the TM6 of the receptor. We observed that the α5 helix of rhodopsin‐bound Gi is positioned about 3.5 Å away from that of β2AR‐bound Gs in the TM pocket, and the two α5 helices orient differently, with a 20° angle between each other, when rhodopsin and β2AR are aligned [Fig. 2(A,B)].
Figure 2.
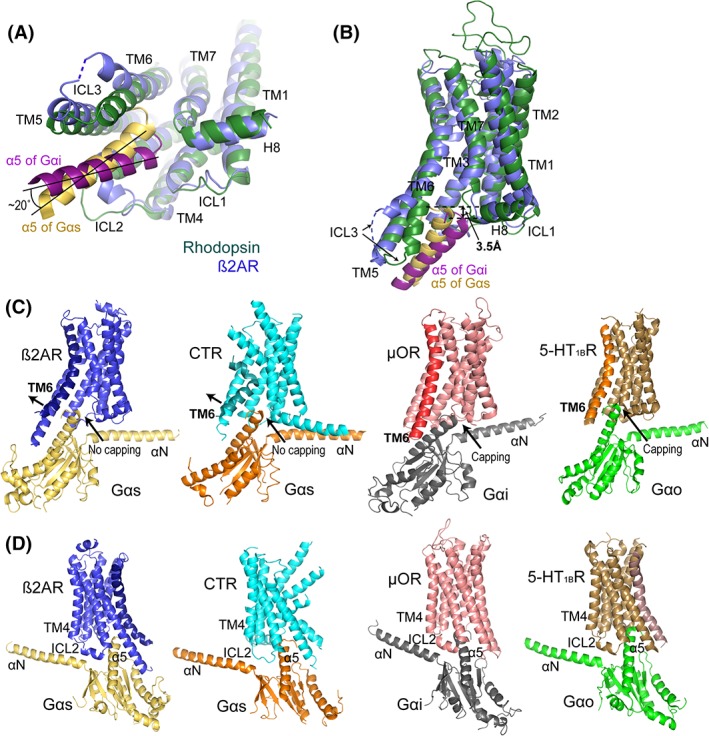
Specificity of GPCR–G protein interaction. (A) The orientations of α5 of Gαi (magenta) and Gαs (yellow) in their complexes with rhodopsin (dark green) and β2AR (blue), respectively. Rhodopsin‐bound Gi is about 20° twisted in a clockwise direction compared with β2AR‐bound Gs when viewed from the intracellular end of the receptors. (B) The α5 of Gαs in the β2AR‐Gs complex is upward‐positioned and closer to TM6 of the receptor than that of Gi in rhodopsin‐Gi complexes. The α5 of Gi positioned about 3.5 Å away from that of Gs in the TM pocket. The color code is the same as in Panel A. (C) GPCR–G protein complex structures. From left to right: β2AR‐Gs (PDB: 3SN6), CTR‐Gs (PDB: 5UZ7), μOR‐Gi (PDB: 6DDE), and 5‐HT1BR‐Go (PDB: 6G79). Key structural elements of the complexes are labeled. TM6 of each of the receptor is highlighted by darker colors. (D) Different orientation of the structures shown in Panel C to highlight the ICL2 conformations.
The different conformations of TM6 between Gi‐ and Gs‐coupling receptors and their distinct binding to α5 of those two G proteins are also observed in other available GPCR–G protein complex structures. Gs‐coupling receptors exhibit strongly bent TM6 and more widely opened TM pockets, while Gi‐ or Go‐coupling receptors show relatively straight TM6 helices and narrowly opened intracellular TM pockets. Correspondingly, α5 of Gαs is tilted up to maintain a close interaction with the outward bent TM6 of a Gs‐coupling receptor, compared with that of Gi or Go, which is positioned relatively further down in the TM pocket, matching the relatively straight TM6 of a Gi‐ or Go‐coupling receptor [Fig. 2(C,D)].
The conformational differences in TM6 helices of Gi‐ or Gs‐coupling GPCRs have been verified by all‐atom mollified adaptive biasing potential (mABP) simulation. The biased simulations indicated that the intracellular ends of TM6 helices of Gi‐coupling rhodopsin and μOR were considerably less dynamic and remained in a more straight conformation, whereas those of Gs‐coupling β2AR and A2AR favored an outward tilted conformation.26
Sequence analysis revealed distinct patterns of polar/positively charged and hydrophobic residues as well as secondary structure‐destabilizing glycines at six TM6 positions TM6.31/34/35/36/38/42, of Class A GPCRs. These patterns are likely related to the different conformations of the intracellular ends of TM6 helices of GPCRs for differentially coupling Gs or Gi proteins [Fig. 3(A,B)]. Positions TM6.31/34/35 are located at the cytosolic surface of the bilayer membrane, which differ between Gs‐ and Gi−/Go‐coupling receptors [Fig. 3(C,D)]. Gi‐coupling receptors exhibit patterns of polar or positively charged residues at TM6.31 and TM6.35, which can stabilize a straight conformation of the intracellular ends of TM6 helices among the negatively charged lipid heads or being exposed to cytosol, while Gs‐coupling receptors show high probability of hydrophobic patterns at TM6.31 and TM6.34 to favor an outward movement of the intracellular ends of TM6 and allow those hydrophobic residues to bury inside the lipid layers of the membrane [Fig. 3(A–D)]. Position TM6.36 is at the back side of TM6. This position has a high probability of hydrophobic residues in Gi‐coupling receptors, but is occupied by Thr/Ser in 72% of the Gs‐coupling receptors. Thr or Ser at this position can form hydrogen bonds with the carbonyl oxygen of the residue that precedes the Ser/Thr by four positions (i‐4), which stabilizes the bent conformation of the TM6 of a Gs‐coupling receptor [Fig. 3(A–D)]. We also observed a highly conserved Gly at TM6.42 of Gs‐coupling GPCRs. This Gly at TM6.42 of Gs‐couplers allows more flexibility for the TM6 of those receptors to adopt an outwardly tilted conformation26 [Fig. 3(A–D)]. Based on these observations, we derived a TM6 sequence motif of ΦXXΦXT/SXXXXXG (where Φ is a hydrophobic residue, X is any residue, T is threonine, S is serine and G is glycine) from TM6 residues 6.31 through 6.42 for Gs‐coupling receptors. Correspondingly, a TM6 sequence motif of θXXXθΦXΦXXXX (where θ is a polar residue) is derived for Gi‐coupled receptors [Fig. 3(C,D)].
Figure 3.

Enrichment of distinct motifs on TM6 of Gs or Gi/o protein‐coupling GPCRs. GPCRDB generic numbering system is used in this figure (http://gpcrdb.org). (A) Sequence alignment of TM6 motifs of Gi−/Go‐coupling and Gs‐coupling GPCRs shows distinct motifs for coupling Gi or Gs subtypes. (B) Relative probability of polar/positively charged, hydrophobic or specific type (Gly) residues for Gi/o (n = 76) and Gs (n = 25) coupling receptors. (C and D) TM6 sequence motifs of GPCRs for coupling Gs (C) or Gi/o (D) proteins and representative Gs or Gi/o‐coupling GPCR structures with distinct patterns of polar/positively charged (red), hydrophobic (green), or specific type (Gly, black) residues at TM6.31/34/35/36/38/42 positions. Dashed lines indicate the cytoplasmic surface of the bilayer membrane. Φ, hydrophobic residue; θ, polar or positively charged residue; T, threonine; S, serine; G, glycine; and X, any residue. (E) Sequence alignment of TM6 motifs of class B GPCRs shows motifs for coupling Gs protein. (F and G) TM6 sequence motifs of CTR (F) and GLP1R (G) for coupling Gs protein. Labeled are distinct patterns of T/S (red), hydrophobic (green), or specific residues (Gly and Pro, black) on TM6.
While class A GPCRs can couple both Gs and Gi/o subfamilies, Class B GPCRs predominantly recruit Gs protein for signal transduction. The available Gs‐bound Class B GPCR structures displayed conserved outwardly bent TM6 helices similar to those of Gs‐coupling Class A receptors. The TM6 sequences of the Class B GPCRs show distinct conserved patterns, which, however, are different from those of Class A GPCRs [Fig. 3(E–G)].
We observed a pronounced kink in TM6 in the structures of Gs‐bound CTR and GLP1R at the region of TM6.47–50. This corresponds to conserved residues P6.47 and G6.50 on TM6 of these Class B receptors, which break TM6 at this region and lead to outward bending of both extracellular and intracellular ends of the TM6 in these Class B GPCRs [Fig. 3(E–G)]. The kink in the same area of Class A GPCRs, however, is not so pronounced because Classes A GPCRs lack a proline in this region [Fig. 3(A–C)].
A conserved Residue T occurs at TM6.42 of CTR, CRFR1, and CRFR2, and Residues ST at TM6.41/42 of other Class B receptors, which resemble the residue T/S at TM6.36 of Gs‐coupling class A GPCRs [Fig. 3(A,E)]. We observed that T6.42 of CTR and S6.41 of GLP1R form hydrogen bonds with the carbonyl oxygen of the residues that precede the S or T by four positions [Fig. 3(F,G)]. Residue T6.42 of Class B GPCRs has been identified as a key residue of a conserved H/E/T/Y polar core network that is important for maintaining inactive conformation of Class B GPCRs. Mutations that disrupt the polar core induce constitutive GPCR activity.34 This finding was later validated by Gs‐bound Class B GPCR cryo‐EM structures that displayed rearranged polar core with T6.42 dissociated from the polar core network, and pronouncedly bent TM6 in these activated class B GPCRs [Fig. 3(F,G)].
We observed hydrophobic patterns at TM6.34/36/38/39 of GIPR, GLP1R, and GLR, at positions of TM6.35/36/38/39 of CTR and PACR, and TM6.35/38/39 of most other Class B GPCRs, which may correspond to the hydrophobic motif of TM6.31/34 of the Gs‐coupling class A GPCRs that allows the bending of TM6 in activated conformation of these receptors [Fig. 3(C,E)]. Actually, L6.36 and V6.39 of CTR, and C6.36, L6.38 and A6.39 of GLP1R are at the intracellular membrane surface based on the structural models of their Gs complexes [Fig. 3(F,G)].
The C‐termini of Gαs are tilted away from the TM pocket core in Gs‐coupling GPCRs. In contrast, the C‐termini of Gαi and Gαo are positioned close to TM7 and Helix 8 of the receptors, which allows them to form “capping interactions” with the residues at the turn between TM7 and Helix 8 of the receptors. “Capping interactions” are not observed (or very weak) between β2AR, CTR and GLP1R, and the Gαs subunit due to the large distance (>6 Å) between the C‐terminus of α5 of Gαs and TM7 of the receptors [Figs. 1(B) and 2(C)]. The difference in capping interactions could serve as a hallmark that helps to distinguish Gi‐coupling receptors from Gs‐coupling receptors.
When coupling to G proteins, GPCRs exhibit variable conformations in their ICL2. The ICL2 of rhodopsin, CTR, and GLP1R display a loop that forms a few polar interactions with αN of the Gα subunit. The ICL2 of μOR, β2AR, and 5‐HT1BR, however, adopt a short helix that forms not only polar interactions with αN but also hydrophobic interactions with α5 of the Gα subunit [Figs. 1(A) and 2(D)]. This indicates that the ICL2 of a GPCR can adopt either a helix to form a larger hydrophobic interface with both αN and α5 of the G protein α subunit, or a complete loop that forms polar interactions with only αN of the G protein. These differences do not seem to be G protein subtype‐specific, but are likely dependent upon the structural features of both sides of the receptor–G protein interface of the complex.
Structural dynamics of the α‐helical domain of the G proteins
We observed significant conformational changes in the Gαi subunit of rhodopsin‐bound Gi compared with that of the GDP bound inactive G protein conformation (PDB code: 1GP2) [Fig. 4(A,B)]. Most dramatically, the α‐helical domain (AHD) is swung away about 55° from the Ras domain along the two loops that connect the two domains, whereas in the inactive Gi protein α subunit, the Ras domain and the AHD are close to each other to form a nucleotide binding pocket. Receptor association thus leads to the opening of the nucleotide binding pocket, the rearrangement of the Ras domain, the dissociation of the GDP, and the increased dynamics of the AHD of the Gα subunit [Fig. 4(B)]. The separation of the AHD from the Ras domain and the release of GDP allow the Gα subunit to bind to GTP, which has higher binding affinity and a much higher cellular concentration than GDP. GTP binding leads to the re‐closure of the AHD domain on the Ras domain and Gα‐mediated signaling. GTP hydrolysis through the intrinsic G protein GTPase activity completes the G protein cycle. The mechanism of the GPCR‐mediated nucleotide exchange cycle seems to be highly conserved between Gi and Gs, which has been the subject of an excellent earlier review.35 Below we discuss the AHD dynamics in the rhodopsin‐Gi complex.
Figure 4.
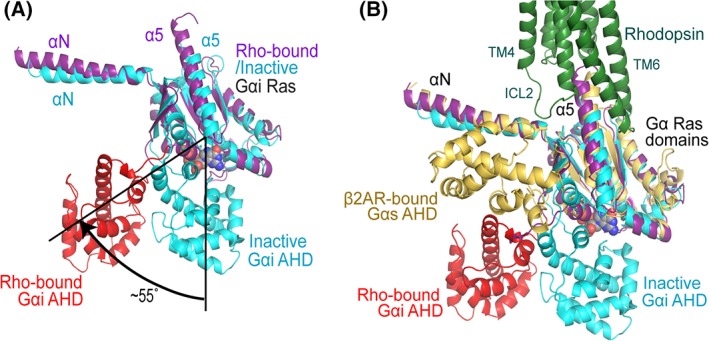
Structural dynamics of the α‐helical domain of G proteins. (A) The α‐helical domain (AHD, red) of the Gαi subunit is 55° tilted away from the Ras domain (magenta) compared with its inactive GDP bound conformation (blue), in which AHD and Ras domain are close to each other to form the nucleotide binding pocket. (B) The comparison of the AHD of Gαi in rhodopsin‐bound conformation with that of Gαs in β2AR‐bound conformation. Gαs is colored in yellow; the same color code as in Panel A is used for rhodopsin‐bound and inactive Gαi subunits.
In the rhodopsin‐bound Gi protein, the displaced AHD is located close to the Gβ subunit, interacting with the Gβ subunit through a salt bridge (between E116 of Gi and R134 and R137 of the Gβ subunit) and a few hydrogen bonding interactions. While the Gβ subunit interaction likely stabilized the AHD position, it is also possible that this position of the AHD is due to the binding of the Fab antibody fragment in the complex structures because we observed a few polar interactions between AHD and Fab [Fig. 1(A)].
Displacement of the AHD was first prominently observed in the β2AR‐Gs structure, in which the AHD was even further moved away from the Ras domain [Fig. 4(B)] and, in contrast to the GPCR‐Gi structures, also associates with the Gβ subunit of the Gs.
Due to the involvement of Fab and nanobody in the solved complex structures, we cannot determine the physiological positions of the AHD of the receptor‐bound G proteins. However, it is obvious that in the nucleotide‐free state the AHD is very dynamic and can adopt variable positions relative to the Ras domain. The positions of the AHDs observed in the Rho‐Gi and β2AR‐Gs structures are likely two possible positions that occur during the dynamic GPCR‐mediated G protein recruitment and signaling [Fig. 4(A,B)].
GPCR–Arrestin Interaction
The rhodopsin–arrestin assembly
While several GPCR–G protein complex structures have been published, rhodopsin–visual arrestin is so far the only available fully‐engaged atomic resolution GPCR–arrestin complex structure, which was determined by femtosecond X‐ray free electron laser (XFEL) crystallography.8, 9 The crystal structure of rhodopsin in complex with visual arrestin displayed an asymmetric rhodopsin–arrestin assembly, with the finger loop of arrestin interacting with the cytoplasmic pocket of the TM domain of rhodopsin [Fig. 5(A)]. From a view along the cross‐section of the cell membrane and the short arrestin axis (perpendicular to the plane of the figure), the arrestin N‐ and C‐domains extend beyond the width of the rhodopsin TM domain by about 20 Å and 40 Å, respectively. Relative to the cell membrane, arrestin is tilted in the rhodopsin‐arrestin assembly with its long axis forming an ~70° angle with the central axis of the rhodopsin TM domain. As a consequence, the C‐edge loops of the arrestin C–domain interact with the bilayer of the cell membrane to anchor the C‐edge to the membrane [Fig. 5(A)]. The direct interaction of this C‐edge tip with the membrane has been directly proven by in vitro biochemical assays and computational modeling.9, 36
Figure 5.
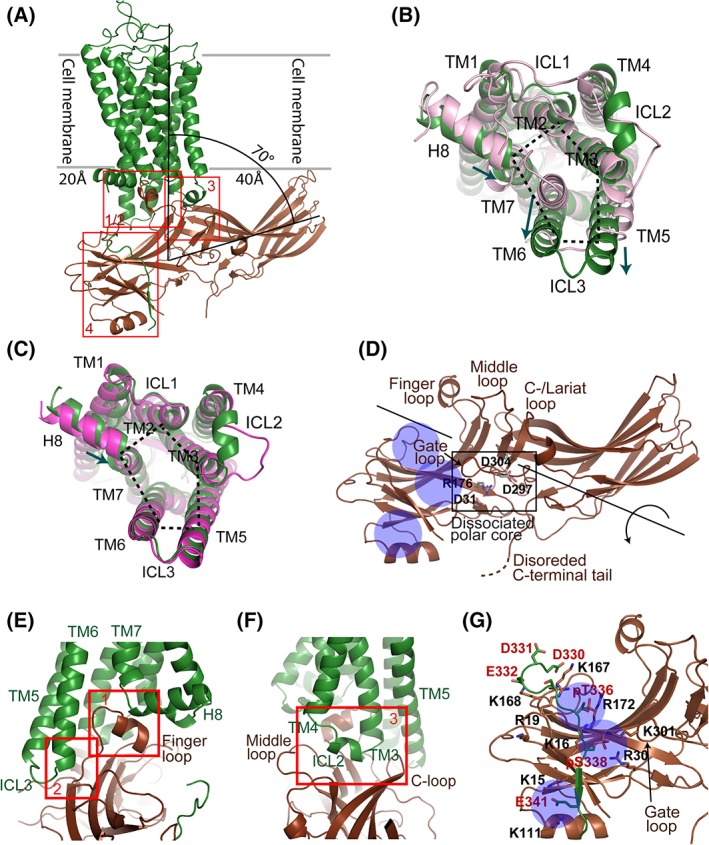
Rhodopsin–visual arrestin interaction. (A) Rhodopsin–visual arrestin complex structure (human rhodopsin with mouse visual arrestin; PDB code: 5W0P). Rhodopsin is colored in dark green, and arrestin in dark brown. Red boxes indicate rhodopsin–arrestin interface patches. (B) Comparison of arrestin‐bound rhodopsin (dark green) with the inactive state of the receptor (pink). The inward shift of the intracellular side of TM7 and Helix 8 of arrestin‐bound rhodopsin is indicated by the small arrow. The outward tilt of TM6 and the extension of TM5 enlarge the intracellular pocket in the TM domain of rhodopsin (indicated with dotted lines) for arrestin binding. (C) Comparison of arrestin‐bound (dark green) with Gi‐bound (red) conformations of rhodopsin. The inward shift of the intracellular side of TM7 and Helix 8 of arrestin‐bound rhodopsin is indicated by the small arrow. The intracellular pocket in the TM domain of arrestin‐bound rhodopsin is similar to that of Gi‐bound rhodopsin, which is indicated by dotted lines. (D) Structural conformation of rhodopsin‐bound visual arrestin with its C‐terminal tail disordered. The structure features a dissociated polar core, a gate loop that is shifted to the N‐domain, and an opened cleft between the N‐ and C‐domains at the central crest region of arrestin that is resulted from a 20° rotation of the domains against each other. The positive surface on the N‐domain is exposed and accessible for receptor binding. (E and F) Rhodopsin–visual arrestin interface patches. Interfaces are formed between 1) the arrestin finger loop and the TM pocket as well as the turn between TM7 and Helix 8 of rhodopsin (E); 2) between the loop and the β‐strand following the finger loop of arrestin and TM5, TM6 and ICL3 of rhodopsin (E); and 3) between arrestin crest loops (middle loop and C‐loop) and rhodopsin ICL2 (F). (G) Rhodopsin–visual arrestin N–C interaction. Rhodopsin C‐terminal tail Residues K339 through E341 form an intermolecular β‐sheet with βI of arrestin. The phosphate groups of pT336 and pS338, and the side chain of E341 of the rhodopsin C‐terminal tail interact with the three positively charged pockets (indicated by blue circles) of the arrestin N‐domain. Key residues of the interface are labeled.
Structure features of arrestin‐bound rhodopsin
Compared with its basal state, arrestin‐bound rhodopsin is in an activated conformation that features an outward tilted TM6 and ICL3 and a C‐terminally extended TM5. These changes expanded the intracellular pocket together with TM2, TM3, and TM7 for accommodating arrestin binding [Fig. 5(B,C)]. These conformational changes are similar to those observed in G protein‐bound rhodopsin and other GPCRs [Fig. 5(C)]. The intracellular side of TM7 and the N‐terminus of Helix 8 of the receptor shifted slightly inward comparing to both the inactive rhodopsin and the G protein bound rhodopsin, in order to better interact with the finger loop of arrestin in arrestin‐bound rhodopsin [Fig. 5(B,C)].
Another important structural change is in the C‐terminal tail of rhodopsin, which in the structure was phosphorylated at residues T336 and S338 [Fig. 5(G)]. The phosphate groups together with negatively charged residues of the C‐terminal tail of rhodopsin form an intermolecular charge interaction network with positively charged residues of the arrestin N‐domain that largely contribute to arrestin recruitment by the receptor.
Structure features of rhodopsin‐bound arrestin
Rhodopsin‐bound arrestin is in an activated conformation, whose structural features were first revealed by the crystal structure of the visual arrestin splice variant p4437, 38 and by the crystal structure of β‐arrestin in complex with a phosphorylated vasopressin 2 receptor (V2R) C‐tail peptide.39 The activated arrestin has an open cleft at the central crest region between the N and C domains, which is resulted from a 20° rotation of the two domains relative to each other. This rotation is caused by the breakage of the central polar core and other inter‐domain hydrophobic interactions that maintain the basal conformation of arrestin40 [Fig. 5(D)]. The open cleft and the rearranged receptor‐binding loops (finger loop, middle loop, C−/lariat loop, and back loop) are important structural features of an activated arrestin for binding to the intracellular TM pocket of a GPCR [Fig. 5(D)].
We observed in the rhodopsin–visual arrestin complex that the C‐terminal tail of arrestin is disordered, and the position of the arrestin C‐terminal tail in the inactive basal state is occupied by the phosphorylated C‐terminal tail of rhodopsin [Fig. 5(D)]. Structural and biochemical evidences have shown that the release of the arrestin C‐terminal tail from the intramolecular N‐C lock allows a cascade of conformational changes in arrestin, including the breakage of the polar core, the formation of three positively charged pockets on the surface of the arrestin N‐domain for binding to the phosphorylated C‐terminal tail of rhodopsin, and the rotation of the N‐ and C‐domains relative to each other, to finally transform arrestin into a pre‐activated conformation that is ready to associate with a receptor37, 38, 41 [Fig. 5(D)].
Rhodopsin–arrestin interface
The rhodopsin–arrestin complex is maintained by a multi‐patched interface, which consists of four distinct interface patches [red boxes in Fig. 5(A,E,F)]. The most noticeable interface patch is formed between the arrestin finger loop (D72 through L78) and the cytoplasmic pocket of the TM domain of rhodopsin. The key arrestin finger loop residues D72 through M76 interact with residues N310 through Q312 from the intracellular end of TM7 and the N‐terminus of Helix 8 of the receptor. Finger loop residues M76 and L78 are positioned in the receptor's hydrophobic TM pocket surrounded by TM3, TM5 and TM6 of the receptor [Patch 1 in Fig. 5(E)]. The finding that the finger loop interacts with the residues at the turn between TM7 and Helix 8 is in agreement with biological data that TM7 and Helix 8 are essential for GPCR desensitization and arrestin‐mediated signaling.42 Following the finger loop is a β‐strand (R81–F86) that interacts with residues from ICL3 and the cytoplasmic ends of TM5 and TM6 of rhodopsin [Patch 2 in Fig. 5(E)].
The ICL2 of rhodopsin, which adopts a one‐turn helix in the activated receptor, inserts into the cleft between the top β‐sheets of the N‐ and C‐domains of arrestin that is resulted from the rotation of the two domains against each other. Specifically, the arrestin middle loop (P135 through S143) and C‐loop (V249‐S253) associate at each side of the ICL2 helix, and arrestin residues R292 and G293 of loop 17–18 (a long loop between β‐strands XVII and XVIII of the C‐domain) at the bottom of the cleft associate with the bottom surface of the ICL2 helix of rhodopsin [Fig. 5(F)]. This is an important interface patch specific for binding to arrestin by a GPCR. Interestingly, in the rhodopsin–Gi complex, ICL2 lacks the helical turn and only associates with the Gα N‐terminal helix through a few hydrogen bonds.
The key interface patch for arrestin recruitment is the intermolecular N–C interaction between the phosphorylated C‐terminal tail of rhodopsin and the N domain of arrestin [Fig. 5(G)]. The rhodopsin C‐terminal residues K339 through T342 adopt a β‐strand that forms an intermolecular β‐sheet with the N‐terminal β‐strand (V12–V17) of arrestin. The residues N‐terminal to the rhodopsin C‐tail β‐strand, D330 through S338, form a turn (D330‐A333) positioned alongside the loop following the N‐terminal β‐strand of arrestin. They are followed by a loop region (334–338) connecting the turn with the C‐terminal β‐strand of rhodopsin, which harbors phosphate groups on Residues T336 and S338 [Fig. 5A,G]. This interface patch is maintained by the hydrogen bonds between the intermolecular β‐strands and, more importantly, the charge interactions between the phosphate groups at T336 and S338 and the negatively charged E341 of rhodopsin with three positively charged pockets (noted as A, B, and C) on the surface of the arrestin N‐domain. The pockets are formed by three groups of basic residues: K16, R19, and R172 (Pocket A), K16, R30, and K301 (Pocket B), and K15 and K111 (Pocket C) [Fig. 5(G)]. It is interesting to notice that all positively charged residues that form the receptor binding pockets are from the arrestin N‐domain, except residue K301, a key residue of pocket B, that is located on the tip of the gate loop from C‐domain of arrestin [Fig. 5(G)].
GPCR phosphorylation code and arrestin activation and recruitment by a GPCR
GPCR phosphorylation code
The phosphorylation of the receptor C‐tail and the charge interaction between the phosphorylated receptor and arrestin are essential for the intermolecular interaction and arrestin recruitment by the receptor.43 The rhodopsin–arrestin complex structure suggested a phosphorylation code for the arrangement of phosphorylated and negatively charged receptor residues to occupy the three positively charged pockets of the arrestin N‐domain. Accordingly, the phosphorylated T336 and S338 together with the negatively charged E341 represent the pattern PXPXXP/E/D (P for phosphate group, E for glutamic acid, D for aspartic acid, and X for other residues) [Fig. 5(G)]. A second phosphorylation code with the pattern PXXPXXP/E/D is also compatible with the position of the positively charged surface pockets. This pattern is found in the phosphorylated C‐terminal peptide of V2R in complex with β‐arrestin‐1. The three phosphorylated residues (pS357, pT360, and pS363) of the V2R C‐terminal peptide were recognized by β‐arrestin‐1 through three positively charged pockets that correspond to those on the N‐domain of visual arrestin in the rhodopsin–visual arrestin complex.39, 44 Sequence searches indicate that the majority of GPCRs contain at least one phosphorylation code (http://tools.vai.org/phoscofinder/). Furthermore, phosphorylation codes are also found in non‐GPCR membrane proteins, including ion channels, transporters, and receptor tyrosine kinases, implying that phosphorylation codes might also play roles in arrestin‐mediated internalization of non‐GPCR membrane proteins. The discovery of phosphorylation codes provides understanding of the molecular mechanism of arrestin activation and recruitment by GPCRs, which we will discuss in the following paragraphs.
Structure features of basal state arrestin
Substantial biochemical evidence supports that the first step of arrestin recruitment is the electrostatic interaction between a C‐tail‐phosphorylated receptor and an arrestin that may still be in a basal conformation.43 Compared with the receptor‐bound state, basal state arrestin has its own C‐terminal tail bound to the N‐domain, which locks it in an inactive conformation (intramolecular N‐C lock). The C‐terminal residues L374 through E378 (F375 through E379 for bovine rhodopsin) form a parallel β‐sheet with the first β‐strand of arrestin, with its L374/V375/F376 (Bovine F375/V376/F377) motif associating with hydrophobic residues from β1 (I13 and Y26; Bovine I12 and Y25) and Helix 1 (L104, L108 and L112; Bovine L103, L107, and L111) of the arrestin N‐domain.
This region of the arrestin C‐terminal tail blocks a large part of its positive surface (Pockets B and C) for receptor binding (Fig. 6). Residue R381 (Bovine R382) on a loop region following the β‐strand of the C‐terminal tail is a key residue involved in the polar core network, which is formed together with charge residues from both the N‐domain (R30, D31, and R176, or R29, D30, and R175 for bovine arrestin) and the C‐domain gate loop (D297 and D304, or D296 and D303 for bovine arrestin) of arrestin. The polar core is required to maintain the basal conformation of arrestin by keeping the two domains of arrestin from rotating against each other, which is a key step of arrestin activation. The polar core further blocks the gate loop from shifting to the N‐domain through a charge interaction of its Residue K301 (K300 for bovine) with the phosphorylated receptor C‐tail (Fig. 6).
Figure 6.

Basal conformation of visual arrestin (bovine arrestin, PDB code: 1CF1). The arrestin C‐terminal tail interacts with the N‐domain, thus blocking the positively charged pockets B and C on the N‐domain that are required for receptor C‐tail interaction. The positively charged pocket A of the arrestin N‐domain (circled), however, is still solvent‐exposed for binding to the phosphorylated receptor C‐tail. Arrestin N‐domain is colored in blue, and C‐domain in brown. Black square: polar core network.
Mechanism of arrestin activation and recruitment by a receptor
The crystal structure of the truncated p44 splice variant of visual arrestin indicates that truncation of the arrestin C‐terminal tail results in conformational changes from its basal state to a pre‐activated conformation for binding to rhodopsin. Those conformational changes include the rotation of the N‐ and C‐domains against each other and the shifting of the gate loop to the N‐domain. The mechanism of activation of full length visual arrestin, however, had not been clearly identified until the structures of the rhodopsin–visual arrestin and the β‐arrestin1‐V2R peptide complexes were solved.
The basal state arrestin structure shows that the positively charged pockets B and C of the N‐domain are blocked by arrestin's own C‐terminal tail, while the pocket A (K16, R19, and R172, may also include K167) remains solvent‐exposed and ready for electrostatic interaction with a phosphate group from a receptor C‐terminal tail (Fig. 6). Arrestin activation and recruitment by a GPCR is initiated when a phosphorylated receptor C‐terminal tail binds to the positively charged pocket A of arrestin through the first phosphate group of its phosphorylation code. With its first phosphate group attached to the N‐domain, the phosphorylated C‐tail can displace the arrestin C‐terminal tail and break the intramolecular N‐C lock, and bind with its second and third phosphate groups (or E/D) to the positively charged pockets B and C. The dissociation of the arrestin C‐terminal tail de‐represses arrestin by disrupting the polar core network, allowing a cascade of conformational changes that transform arrestin from basal state to activated state. These changes include the shifting of the gate loop and its residue K301 to the N‐domain to join the positively charged network (K301 is a residue of Pocket B) for binding to the second phosphate group on the receptor C‐tail, a 20° rotation of the two domains relative to each other, and conformational changes in the loops at the central crest of arrestin, which are required for the formation of a fully‐engaged receptor–arrestin complex.
The interaction of the arrestin N‐domain with a phosphorylated receptor C‐tail is more stable than that with its own C‐terminal tail due to the formation of an extensive electrostatic network between the arrestin N‐domain and the phosphorylated receptor C‐tail. Arrestin activation by the phosphorylated receptor C‐tail is, therefore, energy‐releasing and thermodynamically favorable [Fig. 5(G)].
The first phosphate group is a key residue for arrestin activation and recruitment by a GPCR. While the second and the third phosphate groups of a phosphorylation code may not be as important as the first phosphate group, it is likely that the dissociation of the arrestin C‐tail and the following arrestin activation would not happen if both the second and the third phosphate groups are missing in a phosphorylation code [Figs. 5(G) and 6]. Receptors without a phosphorylation site at their C‐terminal tails cannot initiate arrestin activation, but may form a weak complex with a pre‐activated arrestin through only the core interaction interface. The signaling and relevant physiological functions of those GPCRs are still to be investigated.45, 46, 47
There can be a few different receptor–arrestin assemblies during arrestin activation and arrestin‐mediated GPCR signaling, some of which could be visualized by negative stain EM imaging.48 An initial receptor‐arrestin complex conformation may form when the first phosphate group of the receptor C‐tail binds to the positively charged pocket A of arrestin that is likely still in a basal state. The second complex conformation appears when the arrestin C‐terminal tail is displaced, the polar core is dissociated, and the whole interface patch between receptor C‐tail and arrestin N‐domain is formed. This is likely a tail‐engaged complex conformation as visualized by negative stain EM and in the crystal structure of β‐arrestin1 in complex with the phosphorylated C‐terminal peptide of V2R.39, 48 The tail‐engaged conformation may either be an intermediate state that can readily transition to a high‐affinity fully‐engaged signaling complex, form a super‐complex with a G protein, or serve as a signaling conformation that leads to distinct signaling pathways.45, 49 When the conformational changes in both arrestin domains and the central crest loop occur, the interface patches between the TM domain of the receptor and the arrestin crest loops may rapidly form, leading to the formation of the fully‐engaged complex conformation [Fig. 5(A)]. Biochemical evidence has suggested that those two different receptor–arrestin complex states may result in different arrestin‐mediated signaling consequences.49, 50
Humans express only four arrestins, among which the two visual arrestins are specialized to couple to visual receptors, while the two β‐arrestins serve about 800 non‐visual GPCRs.51 These two β‐arrestins demonstrate a high versatility but low specificity to GPCRs. Arrestin‐mediated GPCR signaling starts from arrestin activation and recruitment by the GPCR, which is largely regulated by the phosphorylation code on the receptor's C‐terminal tail. The specificity of GPCR signaling, therefore, is dependent on distinct Ser/Thr patterns on the receptor's C‐terminal tail due to the recruitment of a specific GRK to phosphorylate the receptor C‐tail.
GPCR‐Biased Conformations for the Recruitment of G Protein or Arrestin
While most receptors and agonists activate both G proteins and arrestins, certain receptors and agonists signal preferentially through either G proteins or arrestins (biased receptors or biased agonists). Based on structural and biological data, biased agonists induce GPCR conformations that more favorably recruit either G proteins for G protein‐mediated signaling, or recruit GRK for receptor phosphorylation followed by arrestin‐mediated signaling. All‐trans retinal is the balanced agonist of rhodopsin, which can regulate either G protein and/or arrestin signaling. Comparing the structures of rhodopsin in their G protein‐bound state versus arrestin‐bound state allows the identification of structural features that favor the binding to either G protein or arrestin.
The TM pocket of rhodopsin is opened when the receptor is activated by photon‐induced isomerization of 11‐cis retinal to all trans retinal. The open pocket that binds to Gi protein does not show significant differences from that that binds to visual arrestin. Due to the hydrophobic nature of the overall TM pocket, the core interactions between rhodopsin's TM pocket and either G protein or arrestin are predominantly maintained by hydrophobic associations. The α5 C‐terminus of Gi and the finger loop of the arrestin can be nicely overlaid when they bind to the intracellular pocket of the receptor [Fig. 7(A,B)]. The only difference is that rhodopsin residues on the turn between TM7 and Helix 8, which form an important interface with the arrestin finger loop, form relatively weak capping interactions with the C‐terminus of the α5 helix of the G protein [Fig. 7(A,B)]. This is consistent with an earlier observation that TM7/Helix 8 (in addition to TM6) is a key region that determines biased signaling in β2AR.42
Figure 7.
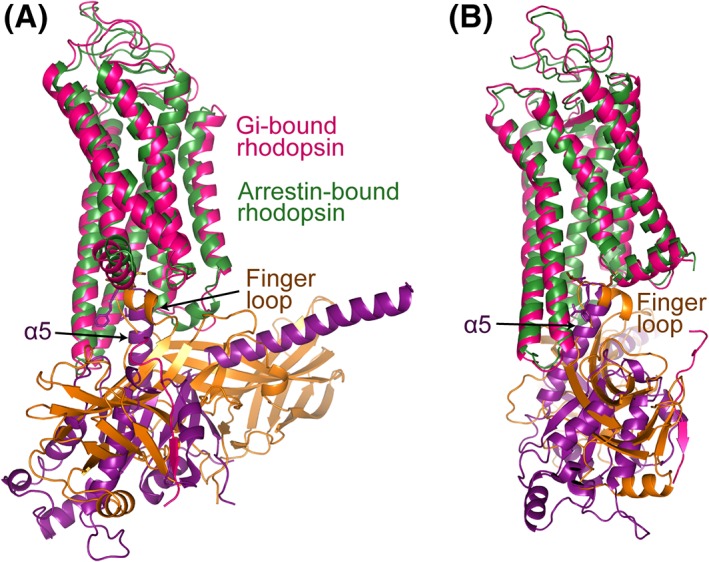
Comparison of the interfaces of Gαi and visual arrestin with rhodopsin. (A) Gαi α5 can be closely superpositioned with the arrestin finger loop when Gi‐bound rhodopsin is aligned with arrestin‐bound rhodopsin. Gi‐bound rhodopsin is colored in red, arrestin‐bound rhodopsin in dark green, Gαi in magenta, and visual arrestin in brown. (B) A view with 90° rotation about the vertical axis.
While ICL2 of rhodopsin forms a loop that weakly associates with a few residues from αN of Gαi in the rhodopsin–Gi complex, it adopts a short helix to provide an important interface with arrestin in the rhodopsin–visual arrestin complex [Figs. 2(C) and 5(F)]. This interface is hydrophobic with rhodopsin Residues P142, M143, and F146 from the ICL2 helix positioned in the hydrophobic cleft at the central crest between the two domains, associating with the hydrophobic Residues F66, Y68, L133, and C144 on the top β‐sheet, and Residues V248, L250, and Y256 of the lariat loop of arrestin [Fig. 2(C,F)].
Based on the rhodopsin–visual arrestin complex structure, a helical ICL2 seems to be required for a GPCR to bind to arrestin. The conformation of the ICL2, however, is not conserved in reported GPCR–G protein complex structures. While ICL2 can adopt helices in both the β2AR‐Gs and μOR–Gi complex structures, it formed loops in the two Class B GPCR–Gs complex structures.12 It is likely that the conformation of the receptor ICL2 in a GPCR–G protein complex is not defined by G protein subtype, but the distinct GPCR–G protein interface.
In addition, receptor C‐tail phosphorylation by a GRK is a key step of the transition from G protein‐mediated signaling to GPCR internalization and/or arrestin‐mediated signaling, and thus a specific structural feature for arrestin recruitment. It has been shown that GRK can directly interact with the TMD pocket of GPCRs and this interaction is required for GRK to phosphorylate receptors. Thus, a specific conformation of GPCR for GRK recognition would play a key role for subsequent arrestin recruitment and signaling.52, 53
Future Outlook
Recent technological advancements in structural biology, including the development of XFEL crystallography and single particle cryo‐EM, have led to the determination of multiple GPCR signaling complex structures with G proteins and arrestins. Those structures have revealed structural information of binding and activation of G proteins and arrestins by GPCRs, and provided insights into the molecular mechanisms of GPCR signaling pathways mediated by these two transducers. Specifically, these structural studies highlight the distinct conformational states of the GPCR signaling complexes, the sequence motifs that determine the selective coupling of G proteins by GPCRs, and the phosphorylation codes for arrestin recruitment. Together, these structures illustrate that specificity of GPCR signaling is not only determined by a specific GPCR conformational states but also determined by short protein motifs within the GPCR sequences such as the TM6 sequence motif or the C‐terminal phosphorylation codes.
Beyond the above GPCR signaling complexes, a high‐resolution structure of a GPCR in complex with a GRK is still needed to fill the knowledge gap of GPCR phosphorylation, which is crucial for the transition from G protein‐mediated signaling to arrestin‐mediated pathways. While GPCR complex structures with both Gs and Gi proteins are available, complex structures with other G protein subtypes, such as Gq and G12/13, are still needed as their physiological functions differ from those of Gs and Gi. Gq‐ or G12/13‐coupling GPCRs are expected to have similar G protein complex assemblies as those of Gs‐ or Gi‐coupling receptors, but will likely show distinct TM6 sequence motifs and specific conformations for binding to the α5 helices of those G proteins, which remain unknown in the absence of the awaited structural analysis of GPCRs in complex with Gq and G12/13.
Non‐visual β‐arrestins are key regulators of signaling pathways of hundreds of non‐rhodopsin GPCRs, thus high‐resolution complex structures of β‐arrestins with full length non‐rhodopsin GPCRs, are important for understanding the mechanism of GPCR signaling and designing therapeutic treatments targeting GPCR signaling pathways.
Because GPCR signaling is involved in many complicated physiological functions, their signaling complexes with transducers are highly dynamic. Structures that catch static snapshots of the dynamic process of GPCR signaling, therefore, provide only limited information that is not enough to display the complete process of the signaling pathways. We are expecting further advancement of structural biology, including the development of cryo‐EM tomography and time‐resolved single particle XFEL imaging, to shed additional light on the dynamic assembly and disassembly of GPCR signaling complexes and to improve our understanding of signaling mechanisms of GPCRs.
Acknowledgments
This work was supported in part by the National Institutes of Health Grants DK071662 (to H. E. X.), and GM102545 and GM129436 (to K. M.). American Asthma Foundation (to H. E. X.), Jay and Betty Van Andel Foundation, Ministry of Science and Technology (China) Grants 2012ZX09301001 and 2012CB910403, 2013CB910600, XDB08020303, and 2013ZX09507001 (to H. E. X.). We thank Parker W. de Waal for manuscript editing and figure preparation.
H. Eric Xu is the winner of the 2016 Hans Neurath Award.
Contributor Information
X. Edward Zhou, Email: edward.zhou@vai.org
H. Eric Xu, Email: eric.xu@vai.org
References
- 1. Hall RA, Premont RT, Lefkowitz RJ (1999) Heptahelical receptor signaling: beyond the G protein paradigm. J Cell Biol 145:927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pierce KL, Premont RT, Lefkowitz RJ (2002) Seven‐transmembrane receptors. Nat Rev Mol Cell Biol 3:639–650. [DOI] [PubMed] [Google Scholar]
- 3. Ritter SL, Hall RA (2009) Fine‐tuning of GPCR activity by receptor‐interacting proteins. Nat Rev Mol Cell Biol 10:819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bjarnadottir TK, Gloriam DE, Hellstrand SH, Kristiansson H, Fredriksson R, Schioth HB (2006) Comprehensive repertoire and phylogenetic analysis of the G protein‐coupled receptors in human and mouse. Genomics 88:263–273. [DOI] [PubMed] [Google Scholar]
- 5. Hamm HE (1998) The many faces of G protein signaling. J Biol Chem 273:669–672. [DOI] [PubMed] [Google Scholar]
- 6. Rasmussen SFG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah STA, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK (2011) Crystal structure of the beta2 adrenergic receptor–Gs protein complex. Nature 477:549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carpenter B, Nehme R, Warne T, Leslie AG, Tate CG (2016) Structure of the adenosine A(2A) receptor bound to an engineered G protein. Nature 536:104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, Barty A, White TA, Yefanov O, Han GW, Xu Q, de Waal PW, Ke J, Tan MHE, Zhang C, Moeller A, West GM, Pascal BD, Van Eps N, Caro LN, Vishnivetskiy SA, Lee RJ, Suino‐Powell KM, Gu X, Pal K, Ma J, Zhi X, Boutet S, Williams GJ, Messerschmidt M, Gati C, Zatsepin NA, Wang D, James D, Basu S, Roy‐Chowdhury S, Conrad CE, Coe J, Liu H, Lisova S, Kupitz C, Grotjohann I, Fromme R, Jiang Y, Tan M, Yang H, Li J, Wang M, Zheng Z, Li D, Howe N, Zhao Y, Standfuss J, Diederichs K, Dong Y, Potter CS, Carragher B, Caffrey M, Jiang H, Chapman HN, Spence JCH, Fromme P, Weierstall U, Ernst OP, Katritch V, Gurevich VV, Griffin PR, Hubbell WL, Stevens RC, Cherezov Y, Melcher K, Xu HE (2015) Crystal structure of rhodopsin bound to arrestin by femtosecond X‐ray laser. Nature 523:561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou XE, He Y, de Waal PW, Gao X, Kang Y, Van Eps N, Yin Y, Pal K, Goswami D, White TA, Barty A, Latorraco NR, Chapman HN, Hubbell WL, Dror RO, Stevens RC, Cherezov V, Gurevich VV, Griffin PR, Ernst OP, Melcher K, Xu HE (2017) Identification of phosphorylation codes for arrestin recruitment by G protein‐coupled receptors. Cell 170:457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liang YL, Khoshouei M, Radjainia M, Zhang Y, Glukhova A, Tarrasch J, Thal DM, Furness SGB, Christopoulos G, Coudrat T, Danev R, Baumeister W, Miller LJ, Christopoulos A, Kobilka BK, Wootten D, Skiniotis G, Sexton PM (2017) Phase‐plate cryo‐EM structure of a class B GPCR‐G‐protein complex. Nature 546:118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Y, Sun B, Feng D, Hu H, Chu M, Qu Q, Tarrasch JT, Li S, Sun Kobilka T, Kobilka BK, Skiniotis G (2017) Cryo‐EM structure of the activated GLP‐1 receptor in complex with a G protein. Nature 546:248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Koehl A, Hu H, Maeda S, Zhang Y, Qu Q, Paggi JM, Latorraca NR, Hilger D, Dawson R, Matile H, Schertler GFX, Granier S, Weis WI, Dror RO, Manglik A, Skiniotis G, Kobilka BK (2018) Structure of the micro‐opioid receptor‐Gi protein complex. Nature 558:547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Draper‐Joyce CJ, Khoshouei M, Thal DM, Liang Y‐L, Nguyen ATN, Furness SGB, Venugopal H, Baltos J‐A, Plitzko JM, Danev R, Baumeister W, May LT, Wootten D, Sexton PM, Glukhova A, Christopoulos A (2018) Structure of the adenosine‐bound human adenosine A1 receptor–Gi complex. Nature 558:559–563. [DOI] [PubMed] [Google Scholar]
- 14. Garcia‐Nafria J, Nehme R, Edwards PC, Tate CG (2018) Cryo‐EM structure of the serotonin 5‐HT1B receptor coupled to heterotrimeric Go. Nature 558:620–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liang YL, Khoshouei M, Glukhova A, Furness SGB, Zhao P, Clydesdale L, Koole C, Truong TT, Thal DM, Lei S, Radjainia M, Danev R, Baumeister W, Wang M‐W, Miller LJ, Christopoulos A, Sexton PM, Wootten D (2018) Phase‐plate cryo‐EM structure of a biased agonist‐bound human GLP‐1 receptor‐Gs complex. Nature 555:121–125. [DOI] [PubMed] [Google Scholar]
- 16. Tsai CJ, Pamula F, Nehme R, Muhle J, Weinert T, Flock T, Nogly P, Edwards PC, Carpenter B, Gruhl T, Ma P, Deupi X, Standfuss J, Tate CG, Schertler GFX (2018) Crystal structure of rhodopsin in complex with a mini‐Go sheds light on the principles of G protein selectivity. Sci Adv 4:eaat7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M (2000) Crystal structure of rhodopsin: a G protein‐coupled receptor. Science 289:739–745. [DOI] [PubMed] [Google Scholar]
- 18. Li J, Edwards PC, Burghammer M, Villa C, Schertler GF (2004) Structure of bovine rhodopsin in a trigonal crystal form. J Mol Biol 343:1409–1438. [DOI] [PubMed] [Google Scholar]
- 19. Standfuss J, Xie G, Edwards PC, Burghammer M, Oprian DD, Schertler GF (2007) Crystal structure of a thermally stable rhodopsin mutant. J Mol Biol 372:1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V (2004) The retinal conformation and its environment in rhodopsin in light of a new 2.2 A crystal structure. J Mol Biol 342:571–583. [DOI] [PubMed] [Google Scholar]
- 21. Salom D, Lodowski DT, Stenkamp RE, Le Trong I, Golczak M, Jastrzebska B, Harris T, Ballesteros JA, Palczewski K (2006) Crystal structure of a photoactivated deprotonated intermediate of rhodopsin. Proc Natl Acad Sci USA 103:16123–16128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Standfuss J, Edwards PC, D'Antona A, Fransen M, Xie G, Oprian DD, Schertler GF (2011) The structural basis of agonist‐induced activation in constitutively active rhodopsin. Nature 471:656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP (2008) Crystal structure of the ligand‐free G‐protein‐coupled receptor opsin. Nature 454:183–187. [DOI] [PubMed] [Google Scholar]
- 24. Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP (2008) Crystal structure of opsin in its G‐protein‐interacting conformation. Nature 455:497–502. [DOI] [PubMed] [Google Scholar]
- 25. Choe HW, Kim YJ, Park JH, Morizumi T, Pai EF, Krauss N, Hofmann KP, Scheerer P, Ernst OP (2011) Crystal structure of metarhodopsin II. Nature 471:651–655. [DOI] [PubMed] [Google Scholar]
- 26. Kang Y, Kuybeda O, de Waal PW, Mukherjee S, Van Eps N, Dutka P, Zhou XE, Bartesaghi A, Erramilli S, Morizumi T, Gu X, Yin Y, Liu P, Jiang Y, Meng X, Zhao G, Melcher K, Ernst OP, Kossiakoff AA, Subramaniam S, Xu HE (2018) Cryo‐EM structure of human rhodopsin bound to an inhibitory G protein. Nature 558:553–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Glukhova A, Draper‐Joyce CJ, Sunahara RK, Christopoulos A, Wootten D, Sexton PM (2018) Rules of engagement: GPCRs and G proteins. ACS Pharm Transl Sci . 10.1021/acsptsci.8b00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oldham WM, Hamm HE (2008) Heterotrimeric G protein activation by G‐protein‐coupled receptors. Nat Rev Mol Cell Biol 9:60–71. [DOI] [PubMed] [Google Scholar]
- 29. Cabrera‐Vera TM, Vanhauwe J, Thomas TO, Medkova M, Preininger A, Mazzoni MR, Hamm HE (2003) Insights into G protein structure, function, and regulation. Endocr Rev 24:765–781. [DOI] [PubMed] [Google Scholar]
- 30. Neves SR, Ram PT, Iyengar R (2002) G protein pathways. Science 296:1636–1639. [DOI] [PubMed] [Google Scholar]
- 31. Gutierrez DV, Mark MD, Masseck O, Maejima T, Kuckelsberg D, Hyde RA, Krause M, Kruse W, Herlitze S (2011) Optogenetic control of motor coordination by Gi/o protein‐coupled vertebrate rhodopsin in cerebellar Purkinje cells. J Biol Chem 286:25848–25858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kaya AI, Lokits AD, Gilbert JA, Iverson TM, Meiler J, Hamm HE (2016) A conserved hydrophobic core in Galphai1 regulates G protein activation and release from activated receptor. J Biol Chem 291:19674–19686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maeda S, Sun D, Singhal A, Foggetta M, Schmid G, Standfuss J, Hennig M, Dawson RJ, Veprintsev DB, Schertler GF (2014) Crystallization scale preparation of a stable GPCR signaling complex between constitutively active rhodopsin and G‐protein. PLoS One 9:e98714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yin Y, de Waal PW, He Y, Zhao LH, Yang D, Cai X, Jiang Y, Melcher K, Wang MW, Xu HE (2017) Rearrangement of a polar core provides a conserved mechanism for constitutive activation of class B G protein‐coupled receptors. J Biol Chem 292:9865–9881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mahoney JP, Sunahara RK (2016) Mechanistic insights into GPCR–G protein interactions. Curr Opin Struct Biol 41:247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lally CC, Bauer B, Selent J, Sommer ME (2017) C‐edge loops of arrestin function as a membrane anchor. Nat Commun 8:14258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Granzin J, Cousin A, Weirauch M, Schlesinger R, Buldt G, Batra‐Safferling R (2012) Crystal structure of p44, a constitutively active splice variant of visual arrestin. J Mol Biol 416:611–618. [DOI] [PubMed] [Google Scholar]
- 38. Kim YJ, Hofmann KP, Ernst OP, Scheerer P, Choe HW, Sommer ME (2013) Crystal structure of pre‐activated arrestin p44. Nature 497:142–146. [DOI] [PubMed] [Google Scholar]
- 39. Shukla AK, Manglik A, Kruse AC, Xiao K, Reis RI, Tseng WC, Staus DP, Hilger D, Uysal S, Huang LY, Paduch M, Tripathi‐Shukla P, Koide A, Koide S, Weis WI, Kossiakoff AA, Kobilka BK, Lefkowitz RJ (2013) Structure of active beta‐arrestin‐1 bound to a G‐protein‐coupled receptor phosphopeptide. Nature 497:137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vishnivetskiy SA, Lee RJ, Zhou XE, Franz A, Xu Q, Xu HE, Gurevich VV (2017) Functional role of the three conserved cysteines in the N domain of visual arrestin‐1. J Biol Chem 292:12496–12502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ostermaier MK, Peterhans C, Jaussi R, Deupi X, Standfuss J (2014) Functional map of arrestin‐1 at single amino acid resolution. Proc Natl Acad Sci USA 111:1825–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu JJ, Horst R, Katritch V, Stevens RC, Wuthrich K (2012) Biased signaling pathways in beta2‐adrenergic receptor characterized by 19F‐NMR. Science 335:1106–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gurevich VV, Gurevich EV (2006) The structural basis of arrestin‐mediated regulation of G‐protein‐coupled receptors. Pharmacol Ther 110:465–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhou XE, Melcher K, Xu HE (2017) Understanding the GPCR biased signaling through G protein and arrestin complex structures. Curr Opin Struct Biol 45:150–159. [DOI] [PubMed] [Google Scholar]
- 45. Thomsen ARB, Plouffe B, Cahill TJ 3rd, Shukla AK, Tarrasch JT, Dosey AM, Kahsai AW, Strachan RT, Pani B, Mahoney JP, Huang L, Breton B, Heydenreich FM, Sunahara RK, Skiniotis G, Bouvier M, Lefkowitz RJ (2016) GPCR‐G protein‐β‐arrestin super‐complex mediates sustained G protein signaling. Cell 166:907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Latorraca NR, Wang JK, Bauer B, Townshend RJL, Hollingsworth SA, Olivieri JE, Xu HE, Sommer ME, Dror RO (2018) Molecular mechanism of GPCR‐mediated arrestin activation. Nature 557:452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Eichel K, Jullie D, Barsi‐Rhyne B, Latorraca NR, Masureel M, Sibarita JB, Dror RO, von Zastrow M (2018) Catalytic activation of beta‐arrestin by GPCRs. Nature 557:381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shukla AK, Westfield GH, Xiao K, Reis RI, Huang LY, Tripathi‐Shukla P, Qian J, Li S, Blanc A, Oleskie AN, Dosey AM, Su M, Liang C‐R, Gu L‐L, Shan J‐M, Chen X, Hanna R, Choi M, Yao XJ, Klink BU, Kahsai AW, Sidhu SS, Koide S, Penczek PA, Kossiakoff AA, Woods VL Jr, Kobilka BK, Skiniotis G, Lefkowitz RJ (2014) Visualization of arrestin recruitment by a G‐protein‐coupled receptor. Nature 512:218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kumari P, Srivastava A, Banerjee R, Ghosh E, Gupta P, Ranjan R, Chen X, Gupta B, Gupta C, Jaiman D, Shukla AK (2016) Functional competence of a partially engaged GPCR‐beta‐arrestin complex. Nat Commun 7:13416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cahill TJ 3rd, Thomsen AR, Tarrasch JT, Plouffe B, Nguyen AH, Yang F, Huang LY, Kahsai AW, Bassoni DL, Gavino BJ, Lamerdin JE, Triest S, Shukla AK, Berger B, Little J IV, Antar A, Blanc A, Qu C‐X, Chen X, Kawakami K, Inoue A, Aoki J, Steyaert J, Sun J‐P, Bouvier M, Skiniotis G, Lefkowitz RJ (2017) Distinct conformations of GPCR‐beta‐arrestin complexes mediate desensitization, signaling, and endocytosis. Proc Natl Acad Sci USA 114:2562–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gurevich VV, Gurevich EV (2017) Molecular mechanisms of GPCR signaling: a structural perspective. Int J Mol Sci 18:E2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. He Y, Gao X, Goswami D, Hou L, Pal K, Yin Y, Zhao G, Ernst OP, Griffin P, Melcher K, Xu HE (2017) Molecular assembly of rhodopsin with G protein‐coupled receptor kinases. Cell Res 27:728–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Komolov KE, Du Y, Duc NM, Betz RM, Rodrigues J, Leib RD, Patra D, Skiniotis G, Adams CM, Dror RO, Chung KY, Kobilka BK, Benovic JL (2017) Structural and functional analysis of a β2‐adrenergic receptor complex with GRK5. Cell 169:407–421. [DOI] [PMC free article] [PubMed] [Google Scholar]