

Abstract
Retinyl palmitate (RP) is frequently used as an ingredient in cosmetics and other retail products. We previously reported that, under UVA light irradiation, RP is facilely decomposed into multiple products, including anhydroretinol (AR) and 5,6-epoxyretinyl palmitate (5,6-epoxy-RP). We also determined that combined treatment of mouse lymphoma cells with RP and UVA irradiation produced a photomutagenic effect. In this study, we evaluated the photomutagenicity of AR and 5,6-epoxy-RP, in L5178Y/Tk+/− mouse lymphoma cells. Treatment of cells with AR or 5,6-epoxy-RP alone at 10 and 25 µg/mL for 4 h did not show a positive mutagenic response. However, because these doses did not induce the required amount of cytotoxicity for mouse lymphoma assay, we are unable to determine whether or not these two compounds are mutagenic. Treatment of cells with 1–25 µg/mL AR or 5,6-epoxy-RP under UVA light (315–400 nm) for 30 min (1.38 mW/cm2) produced a synergistic photomutagenic effect. At 10 µg/mL (37.3 µM) AR with UVA exposure, the mutant frequency (MF) was about 3-fold higher than that for UVA exposure alone, whereas the MF for 25 µg/mL (46.3 µM) of 5,6-epoxy-RP + UVA was approximately 2-fold higher than that for UVA exposure alone. Compared with previous results for RP + UVA treatment, the potency of the induced phototoxicity and photomutagenicity was AR > RP > 5,6-epoxy-RP. To elucidate the underlying photomutagenic mechanism, we examined the loss of heterozygosity (LOH) at four microsatellite loci spanning the entire chromosome 11 for mutants induced by AR or 5,6-epoxy-RP. Most mutants lost the Tk+ allele, and more than 70% of the chromosome damage extended to 38 cM in chromosome length. AR + UVA induced about twice as many mutants that lost all four microsatellite markers from the chromosome 11 carrying the Tk+ allele as RP + UVA or 5,6-epoxy-RP + UVA. These results suggest that two of RP’s photodecomposition products are photomutagenic in mouse lymphoma cells, causing events that affect a large segment of the chromosome.
Introduction
The skin is one of the largest body organs in the human body, and it functions to protect the body from microbial, physical, chemical, and ultraviolet radiation-induced injury (1, 2). One of the essential roles of vitamin A (retinol) is in the maintenance of normal skin function, including the regulation of epidermal cell growth and differentiation (3–6). Retinyl esters account for the major portion (up to 80% or more) of total retinol stored in the skin. Their predominance in skin reflects the importance of retinyl esters for normal skin function. Retinyl palmitate (RP, Figure 1) is one of the retinyl esters endogenously formed in skin (7–11). Also, because RP is thermally more stable than retinol, it is frequently used as an ingredient in retail products, including moisturizing preparations, skin care preparations, lipsticks, suntan gels and preparations, makeup preparations, and bath soaps and detergents (12, 13). However, while people using these products are unavoidably exposed to sunlight, there is limited information on risks associated with use of topically applied RP-containing products and concomitant exposure to sunlight (14).
Figure 1.
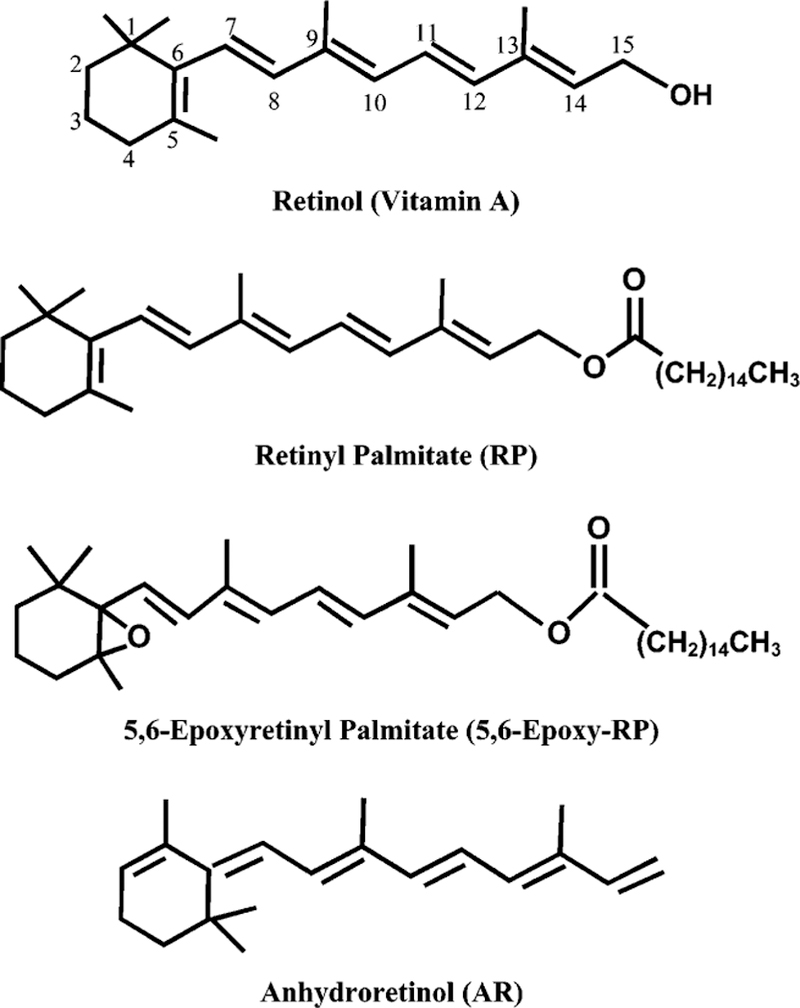
Structures and abbreviations for retinol, retinyl palmitate, 5,6-epoxyretinyl palmitate, and anhydroretinol.
We have been interested in studying the possible adverse effects exerted by topical application of RP-containing cream products on sun-exposed skin (14, 15). We previously reported that photoirradiation of RP by UVA light resulted in 14 photodecomposition products, including 5,6-epoxyretinyl palmitate (5,6-epoxy-RP) and anhydroretinol (AR, Figure 1) (16). The study indicated that the photodecomposition of RP to 5,6-epoxy-RP is mediated by a light-initiated free radical chain reaction and that AR is formed through an ionic photodissociation mechanism. In addition, photoirradiation of RP, 5,6-epoxy-RP, and AR with UVA light in the presence of methyl linoleate resulted in lipid peroxide (methyl linoleate hydroperoxide) formation. This reaction was inhibited by dithiothreitol (DTT), NaN3, and superoxide dismutase (SOD), which suggests that photoirradiation of RP, 5,6-epoxy-RP, and AR by UVA light generated reactive oxygen species (ROS) that resulted in lipid (methyl linoleate) peroxidation. We demonstrated that lipid peroxidation photosensitized by RP was inhibited by NaN3 and enhanced by the presence of D2O (16), which suggests that singlet oxygen is involved in this process. Subsequent studies employing the electron spin resonance (ESR) spin-trap technique provided direct evidence that photoirradiation of RP by UVA light generates ROS (singlet oxygen and superoxide) that initiate lipid peroxidation (17).
Results from in vitro studies using the Comet assay suggest that RP, AR, and 5,6-epoxy-RP photosensitize DNA, leading to damage and cytotoxicity (18). RP, AR, and 5,6-epoxy-RP, however, are not mutagenic or photomutagenic in Salmonella typhimurium tester strains (16). We have demonstrated that treatment of mouse lymphoma cells with RP and concomitant exposure to UVA light results in mutations, mainly via loss of heterozygosity (LOH). These results suggest that RP under UVA light is photomutagenic to mouse lymphoma cells through a clastogenic mode but not through a point mutation mode (19). As a continuation of the study on the mechanism by which RP and its photodecomposition products, AR and 5,6-epoxy-RP, lead to genotoxicity, in the present study we investigate the photomutagenic potential of AR or 5,6-epoxy-RP, using L5178Y/ Tk+/− mouse lymphoma cells and UVA light, and also the underlying mechanism of mutation by LOH analysis.
Experimental Procedures
Materials.
RP was purchased from Sigma Chemical Co. (St. Louis, MO). AR and 5,6-epoxy-RP were prepared as previously described (16, 17). Fischer’s medium was purchased from Quality Biological Inc. (Gaithersburg, MD), and all cell culture supplies were purchased from Invitrogen Life Technologies (Carlsbad, CA). PCR Master Mix was purchased from Promega Company (Madison, WI). The primers used for detection of LOH at the Tk locus and the D11Mit42, D11Mit29, and D11Mit74 loci were purchased from Invitrogen Life Technologies.
UVA Light Source.
The UVA light box was custom-made with four UVA lamps (National Biologics, Twinsburg, OH) (16, 17, 19). The irradiance of the light box was determined by use of an Optronics OL754 spectroradiometer (Optronics Laboratories, Orlando, FL), and the light dose was routinely measured with a Solar Light PMA-2110 UVA detector (Solar Light Inc., Philadelphia, PA). The maximum emission of the UVA light box was 350–352 nm with the following spectral distribution: UVA (315–400 nm), 98.93%; UVB (280–315 nm), 1.07%; and UVC (250–280 nm), <0.0001%.
Cells and Culture Conditions.
The L5178Y/Tk+/− 3.7.2C mouse lymphoma cell line was utilized for the mutation assay. Cells were grown according to the methods described by Chen and Moore (20). Briefly, the basic medium was Fischer’s medium for leukemic cells of mice with L-glutamine supplemented with pluronic F68 (0.1%), sodium pyruvate (1 mM), penicillin (100 units/mL), and streptomycin (100 µg/mL). The treatment medium (F5p), growth medium (F10p), and cloning medium (F20p) were the basic medium supplemented with 5%, 10%, and 20% heat-inactivated horse serum, respectively. The cultures were maintained in a humidified incubator with 5% CO2 in air at 37 °C.
Cell Treatment with AR or 5,6-Epoxy-RP in the Absence of Light Irradiation.
The AR and 5,6-epoxy-RP working solutions (100×) were prepared just prior to use by dissolving with anhydrous dimethyl sulfoxide (DMSO). The cells were suspended in 100mm diameter tissue culture dishes at a concentration of 6 × 106 cells in 10 mL of treatment medium. Aliquots (100 µL) of the AR or 5,6-epoxy-RP working solutions were added to give final concentrations of 10 or 25 µg/mL, and the cells were incubated for4h at 37 °C. In all cases, including the solvent controls (DMSO only) and positive controls [0.1 µg/mL 4-nitroquinoline-1-oxide (4-NQO)], the final concentration of DMSO in the medium was 1%.
Cell Treatment with AR or 5,6-Epoxy-RP and UVA Light.
Cells were treated with different concentrations of AR or 5,6-epoxy-RP (1–25 µg/mL) and exposed to 2.48 J/cm2 UVA light during a period of 30 min (e.g., 1.38 mW/cm2). The treated cultures were then incubated at 37 °C (without UVA light irradiation) for an additional 3.5 h. After treatment, the cells were centrifuged, washed once with fresh medium, and then resuspended in growth medium at a density of 3 × 105 cells/mL in 25 cm2 cell culture flasks to begin the 2-day phenotypic expression.
Tk Microwell Mutation Assay.
Mutant selection was performed as described previously (20). Briefly, the cells were counted and the densities were adjusted with fresh medium at approximately 1 and 2 days following exposure. For mutant enumeration, trifluorothymidine (TFT, 3 µg/mL) was added to the cells in cloning medium. Cells were seeded into four 96-well flat-bottom microtiter plates, 200 µL/well for a final density of 2000 cells/well. For the determination of plating efficiency, approximately 1.6 cells were aliquoted in 200 µL/well into two 96-well flat-bottom microtiter plates. All plates were incubated at 37 °C in a humidified incubator with 5% CO2 in air. After 11 days of incubation, colonies were counted and mutant colonies were categorized as small or large. Small colonies are defined as those smaller than 25% of the well diameter. Mutant frequencies (MFs) were calculated by use of the Poisson distribution. Cytotoxicity was measured via relative total growth (RTG), which includes a measure of growth during treatment, expression, and cloning (20).
Tk Mutant Evaluation for LOH at the Thymidine Kinase (Tk1) and Three Other Microsatellite Loci Spanning the Entire Chromosome 11.
Mutant clones were directly taken from TFT selection plates. Forty-eight large and 48 small mutant colonies resulting from treatment with 10 µg/mL AR + UVA or 25 µg/mL 5,6-epoxy-RP + UVA were analyzed. The mutant cells were washed once with phosphate-buffered saline (PBS) by centrifugation, and cell pellets were quickly frozen and stored at −80 °C. Genomic DNA was extracted by digesting the cells in lysis buffer [10 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 1% (v/v) Triton X-100, and 1% (v/v) Tween 20] with 200 µg/mL proteinase K at 60 °C for 90 min, followed by inactivation of proteinase K at 95 °C for 10 min. For polymerase chain reaction (PCR) analysis of LOH at Tk and other loci (D11Mit42, D11Mit29, and D11Mit74 loci), the amplification reactions were carried out in a total volume of 20 µL by use of 2× PCR Master Mix and pairs of primers described previously (21). The thermal cycling conditions were as follows: initial incubation at 94 °C for 3 min; 40 cycles of 94 °C denaturation for 30 s, 55 °C annealing for 30 s, and 72 °C extension for 30 s; and a final extension at 72 °C for 7 min. The amplification products were scored for the presence of one band (indicating LOH) or two bands (retention of heterozygosity at the given locus) after 2% agarose gel electrophoresis.
Data Analyses.
The data evaluation criteria developed by the Mouse Lymphoma Assay Expert Workgroup of the International Workgroup for Genotoxicity Tests (IWGT) were used to determine whether a response was positive or negative (22). The recommendation for the determination of a positive test chemical response includes both the requirement that the response exceeds a defined value (the global evaluation factor) and that there also is a positive dose-response. To evaluate the differences in induced mutant frequency between the treatment groups, model fitting and comparisons were done with the lm library in the public domain R software program (23).
Results
Photocytotoxicity and Photomutagenicity of AR or 5,6-Epoxy-RP.
Our previous study showed that combined exposure to RP and UVA light is photomutagenic in mouse lymphoma cells (19). In the present study, under similar experimental conditions, the photomutagenicity of two RP photodecomposition products, AR and 5,6-epoxy-RP (Figure 1), exposed to UVA light was examined. First, cells were treated with AR or 5,6-epoxy-RP at concentrations of 10 or 25 µg/ mL for 4 h without UVA light irradiation. Then, the photomutagenicity was determined by treating cells with AR or 5,6-epoxy-RP (1–25 µg/mL) and UVA at a total light dose of 2.48 J/cm2 (1.38 mW/cm2 for 30 min). The results from two experiments are presented in Table 1. Treatments with AR or 5,6-epoxy-RP alone (10 and 25 µg/mL) for 4 h caused little or no cytotoxicity and did not give a positive mutagenic response. Because we did not have sufficient chemical samples to evaluate doses higher than 25 µg/mL. That is, we could not attain sufficient cytotoxicity [RTG between 20% and 10% (20)]. Therefore, we were unable to determine whether or not AR and 5,6-epoxy-RP are mutagenic without UVA exposure.
Table 1.
Toxicity and Mutagenicity of AR and 5,6-Epoxy-RP with UVA in L5178Y/Tk+/− Mouse Lymphoma Cells
| experiment 1 |
experiment 2 |
|||||
|---|---|---|---|---|---|---|
| treatment | dose (µg/mL) | concn (µM) | mutant frequencya (×10−6) | rel total growth (%) | mutant frequencya (×10−6) | rel total growth (%) |
| control | b | 55 (20/35) | 100 | 96 (24/72) | 100 | |
| AR | 10 | 37.3 | 78 (29/49) | 99 | 119 (32/87) | 93 |
| AR | 25 | 93.3 | 77 (38/39) | 74 | 127 (44/83) | 68 |
| AR + UVAc | 0 | 0 | 153 (63/90) | 77 | 147 (38/109) | 70 |
| AR + UVA | 1 | 3.7 | 212 (106/106) | 50 | 171 (75/96) | 68 |
| AR + UVA | 2.5 | 9.3 | 284 (136/148) | 40 | 212 (104/108) | 50 |
| AR + UVA | 5 | 18.7 | 318 (150/168) | 23 | 261 (140/121) | 33 |
| AR + UVA | 7.5 | 28.0 | NDd | NDd | 396 (220/176) | 23 |
| AR + UVA | 10 | 37.3 | 453 (298/155) | 10 | 448 (294/154) | 11 |
| 4-NQOe | 0.1 | 614 (245/368) | 70 | 611 (278/332) | 69 | |
| control | b | 53 (25/28) | 100 | 101 (33/68) | 100 | |
| 5,6-epoxy-RP | 10 | 18.5 | 45 (21/24) | 95 | 99 (30/69) | 104 |
| 5,6-epoxy-RP | 25 | 46.3 | 61 (28/33) | 95 | 89 (30/59) | 87 |
| 5,6-epoxy-RP +UVAc | 0 | 0 | 141 (69/72) | 68 | 179 (61/118) | 78 |
| 5,6-epoxy-RP +UVA | 1 | 1.9 | 161 (62/99) | 67 | 187 (71/116) | 69 |
| 5,6-epoxy-RP +UVA | 5 | 9.3 | 179 (52/127) | 66 | 197 (80/117) | 72 |
| 5,6-epoxy-RP +UVA | 10 | 18.5 | 184 (91/93) | 70 | 184 (79/105) | 76 |
| 5,6-epoxy-RP +UVA | 15 | 27.8 | 182 (73/109) | 64 | 196 (72/124) | 73 |
| 5,6-epoxy-RP +UVA | 20 | 37.0 | 187 (84/103) | 45 | 239 (96/143) | 55 |
| 5,6-epoxy-RP +UVA | 25 | 46.3 | 237 (108/129) | 39 | 276 (127/149) | 47 |
| 4-NQOe | 0.1 | 599 (256/343) | 59 | 598 (305/293) | 54 | |
Numbers in parentheses denote the mutant frequencies for small/large colonies.
In the control group, cells were treated with 1% DMSO only.
Cells were concomitantly exposed to different concentrations of compounds (AR or 5,6-epoxy-RP) and 1.38 mW/cm2 UVA irradiation for 30 min.
Not determined.
4-NQO was used as a positive control. Abbreviation: concn, concentration; rel, relative.
In contrast, treatment of cells with various concentrations (1−25 µg/mL) of AR or 5,6-epoxy-RP concomitantly exposed to UVA (2.48 J/cm2) resulted in dose-dependent positive photocytotoxic and photomutagenic responses (Table 1). Because the 10 µg/mL dose of AR resulted in RTG values of 10% and 11% in the two experiments, the MFs for doses higher than 10 µg/mL AR concomitantly with UVA light exposure were not determined. Within the dose range studied, 5,6-epoxy-RP exhibited lower photocytotoxicity and photomutagenicity compared to AR (Table 1 and Figure 2). Considering that the molecular weight of AR is about half that of 5,6-epoxy-RP, the photomutagenicity of AR is about 2–3-fold higher than that of 5,6-epoxy-RP assayed at the same molar concentration. When results obtained for AR + UVA and 5,6-epoxy-RP + UVA are compared with our previous results obtained for RP + UVA, it is seen that all treatments resulted in dose-dependent increases in photomutagenicity (Figure 2). The slopes of these linear regressions were 8.0 for AR + UVA, 5.5 for RP + UVA, and 1.6 for 5,6-epoxy-RP + UVA (R2 = 0.986, 0.992, and 0.824, respectively). The following relative potencies were observed: AR > RP > 5,6-epoxy-RP. There were significant differences for paired comparisons of these regression coefficients (P < 0.001).
Figure 2.
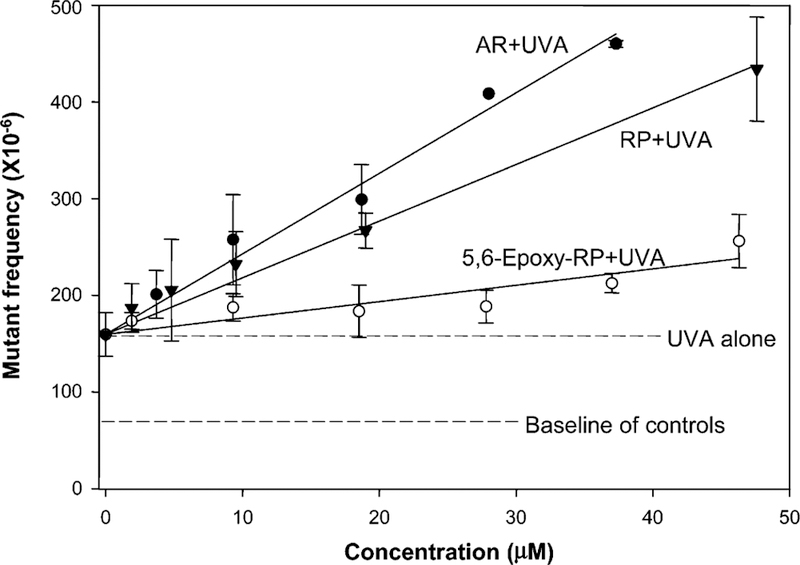
Comparison of photomutagenicity of RP (▼), AR (●), and 5,6-epoxy-RP (O) in mouse lymphoma cells. The cells were treated with different concentrations of compounds in conjunction with UVA irradiation of 2.48 J/cm2 (1.38 mW/cm2 for 30 min). The data points for AR + UVA and 5,6-epoxy-RP + UVA represent the mean of two independent experiments from Table 1. The RP + UVA data were from our previous results (19). UVA alone was the mean of mutant frequencies generated from all experiments with UVA treatment alone. The data for RP, AR, and 5,6-epoxy-RP were adjusted according to the mean of UVA alone as the origin of regression lines.
Analysis of Mutants of AR or 5,6-Epoxy-RP with UVA for LOH.
DNA samples isolated from 48 large and 48 small mutant colonies from cultures treated with 10 µg/mL AR + UVA or 25 µg/mL 5,6-epoxy-RP + UVA were analyzed for LOH. As was previously done for mutants from RP-treated cultures (19), LOH was evaluated by allele-specific PCR with four microsatellite loci (the Tk1 locus, D11Mit42, D11Mit29, and D11Mit74) spanning the entire chromosome 11 (Table 2). More than 92% of the mutants from the two treatment groups (AR + UVA and 5,6-epoxy-RP + UVA) lost heterozygosity at the Tk1 locus. The different types of mutations are shown in Figure 3. For comparison we have included the microsatellite mutants previously obtained for control, UVA alone, and RP + UVA (19). The most common type of mutation for the AR + UVA exposure (51%) was LOH involving all four microsatellite makers from chromosome 11 carrying the Tk+ allele, whereas the major type of mutation for 5,6-epoxy-RP + UVA exposure (45%) was LOH extending to D11Mit29, an alternation involving approximately half of the chromosome. Approximately 70−80% of the mutants in the present study showed LOH extending to 38 cM (D11Mit29), which was 2−3 fold higher than that of RP + UVA (Figure 3).
Table 2.
LOH at Different Loci along Chromosome 11 in 48 Large- and 48 Small-Colony Tk Mutants from Cells Concomitantly Treated with AR + UVA or 5,6-Epoxy−RP + UVA
| AR + UVA |
5,6-epoxy-RP + UVA |
||||
|---|---|---|---|---|---|
| locus | position (cM)a | no. in large colonies (%) | no. in small colonies (%) | no. in large colonies (%) | no. in small colonies (%) |
| D11Mit74 | 0 | 33 (69) | 20 (42) | 11 (23) | 14 (29) |
| D11Mit29 | 40 | 40 (83) | 39 (81) | 34 (71) | 34 (71) |
| D11Mit42 | 72 | 45 (94) | 44 (92) | 42 (88) | 43 (90) |
| Tk | 78 | 47 (98) | 46 (96) | 44 (92) | 47 (98) |
Locus position in centimorgans (cM) is the distance to the top of chromosome 11
Figure 3.
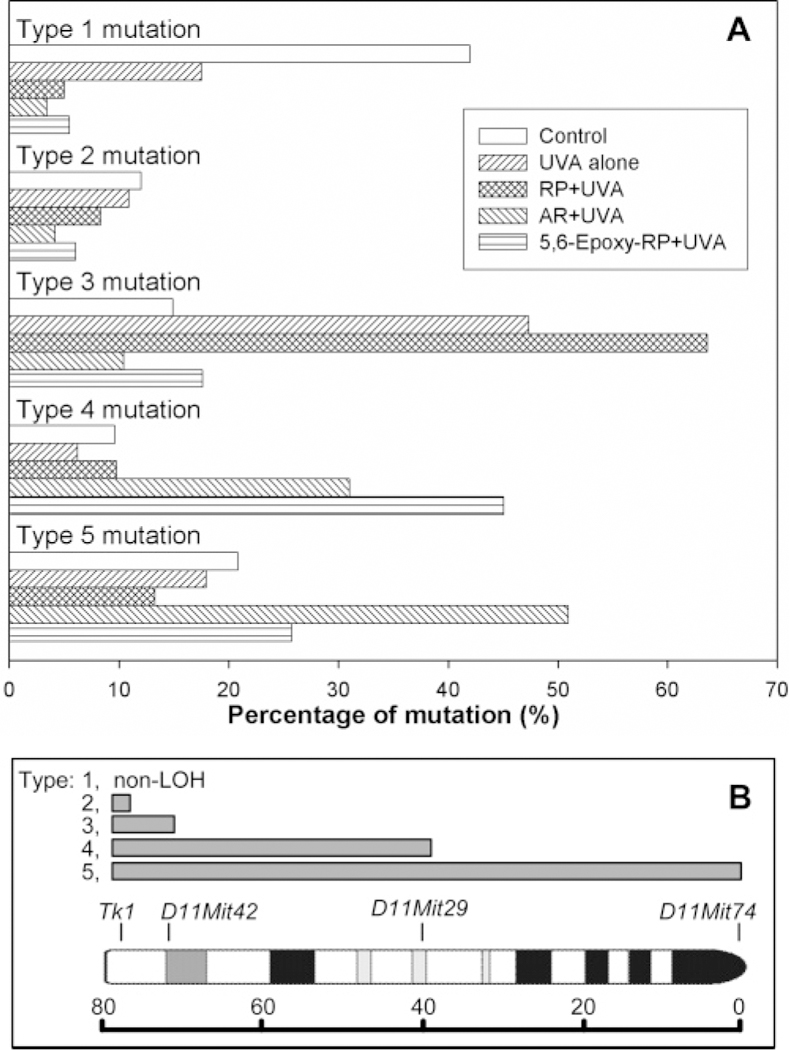
(A) Comparison of the percentage of mutational types for all (large and small) colonies produced in mouse lymphoma cells treated with control, UVA, RP + UVA, AR + UVA, and 5,6-epoxy-RP + UVA. The data for control, UVA alone, and RP + UVA are from our previous study (19). (B) Different types of mutations shown by histograms indicating the range of LOH, at the same scale as used in the ideogram of mouse chromosome 11. The loci that were analyzed for LOH (Tk1, D11Mit42, D11Mit29, and D11Mit74) are marked. The ruler in centimorgans indicates the distance from the top of the chromosome. Type 1 mutation, non-LOH; type 2, LOH at Tk locus only; type 3, LOH extending to D11Mit42 (about 6 cM); type 4, LOH extending to D11Mit29 (about 38 cM); and type 5, LOH extending to the top of chromosome 11.
Discussion
We have previously demonstrated that photoirradiation of RP by UVA generates photodecomposition products, produces ROS, and elicits toxicological responses, including lipid peroxidation, photocytotoxicity, DNA damage, and photomutagenicity (16–19). RP is photomutgenic through a clastogenic mode of action, and more than 95% of the mutants showed chromosome 11 LOH (19). LOH is the loss of the remaining normal allele of a heterozygous locus, resulting in either hemizygous or homozygous status for the deleterious allele. It can be indicative for a deletion or other mutational event within the normal allele. To explore further the underlying mechanisms by which photoirradiation of RP by UVA light elicits phototoxicity and photomutagenicity, we investigated whether two of RP’s decomposition products, AR and 5,6-epoxy-RP, are phototoxic and photomutagenic to mouse lymphoma cells by analysis of the induced MFs and LOH.
When mouse lymphoma cells were exposed to UVA light irradiation, both AR and 5,6-epoxy-RP were phototoxic and photomutagenic (Table 1). Furthermore, RP, AR, and 5,6-epoxy-RP all were photomutagenic by causing LOH involving chromosome 11 (Figure 3). These results indicate that their photomutagenicities are through a clastogenic mode-of-action (MOA) rather than a point mutation MOA. It is worth noting that AR and 5,6-epoxy-RP are produced from photoirradiation of RP through different mechanistic pathways. Our previous study showed that 5,6-epoxy-RP is formed through a free radical chain reaction, and AR is generated through an ionic photoirradiation mechanism (16). We observed previously that RP, 5,6-epoxy-RP, and AR are not mutagenic and photomutagenic in Salmonella typhimurium tester strains, indicating that these compounds do not generate point mutations with or without UVA light irradiation (16). Consequently, our overall mechanistic studies provide evidence that photoirradiation of RP by UVA results in photomutagenicity through multiple activation pathways and that the mode of action is not the induction of point mutations but rather chromosomal mutations.
The proposed activation pathways leading to chromosome mutations from photoirradiation of RP and its two photodecomposition products, 5,6-epoxy-RP and AR, by UVA light is shown in Figure 4. From our studies, it appears that AR and 5,6-epoxy-RP may be at least partially responsible for the phototoxicity of RP. Photoirradiation of RP with UVA light results in the formation of photodecomposition products through three distinct mechanisms: a UVA-initiated free radical mechanism (5,6-epoxy-RP), an ionic photodissociation mechanism (AR), and energy absorption (excited RP) (16, 17). As with other photodynamic sensitizers, upon photoirradiation by UVA light, these compounds absorb light energy and act as photosensitizers by transferring energy to molecular oxygen to form singlet oxygen (1O2), and by transferring electrons to molecular oxygen to form ROS such as superoxide radical anion (− •O2) (17). ROS may then damage nucleic acids and proteins, which may result in immediate functional consequences or mutations. ROS also target lipids, leading to lipid peroxidation (16, 17). Singh et al. (21) reported that the lipid peroxidation product 4-hydroxy-2-nonenal (4-HNE) is mutagenic in mouse lymphoma cells, and the major type of mutation is LOH extending to D11Mit42. This type of mutation is similar to that found in our previous study for the RP + UVA treatment group (19). In this study, the range of DNA damage induced by 5,6-epoxy-RP + UVA or AR + UVA treatments appeared more extensive than for RP + UVA, resulting in even the entire loss of the chromosome 11 carrying the Tk+ allele for part of the mutants. Thus, as shown in Figure 4, we propose that, upon UVA irradiation of RP, 5,6-epoxy-RP, or AR, the resulting lipid peroxidation products can lead to chromosome mutations.
Figure 4.
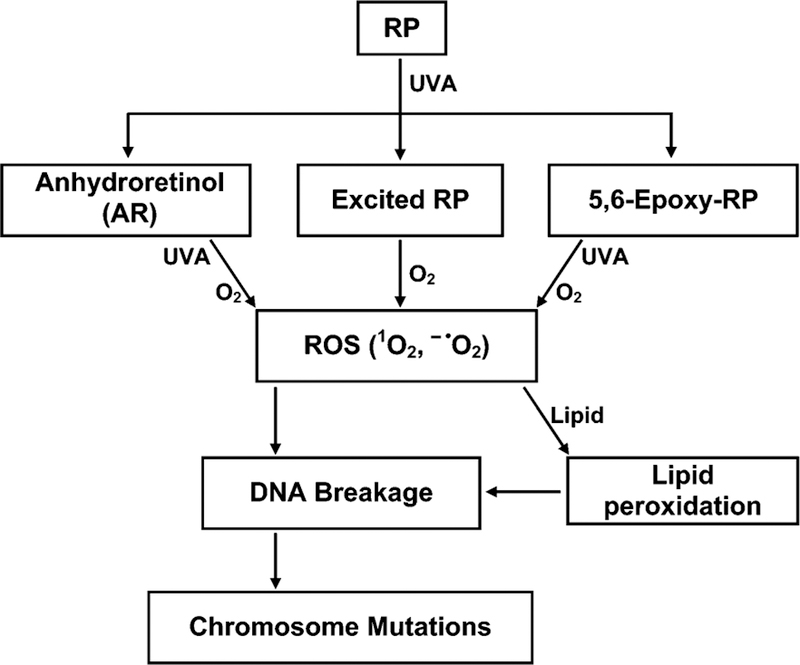
Proposed activation pathways leading to chromosome mutations induced by photoirradiation of RP and two of its photodecomposition products, AR and 5,6-epoxy-RP.
It is significant to note that RP, 5,6-epoxy-RP, and AR elicit phototoxicity and photomutagenicity with different potencies (Table 1 and Figure 2). The potency of the induced phototoxicity and photomutagenicity follows the order AR > RP > 5,6-epoxy-RP. The potency of AR may be explained by its shorter length and the fact that it is more lipophilic than RP and 5,6-epoxy-RP (Figure 1). Therefore it may readily diffuse across the plasma membrane and enter the cell where it is reactive. Another possible reason for the observed difference in potencies may be explained by the chemical differences. AR, RP, and 5,6-epoxy-RP have their maximum UV−visible absorption at 330−390, 320−330, and 300−325 nm, respectively (16). These absorbance maxima are all within the UVA light range (315−400 nm), and thus all the compounds studied can be excited by UVA light and subsequently interact with molecular oxygen. This capability was demonstrated on our previous study. Upon UVA light irradiation, RP, 5,6-epoxy-RP, and AR all can generate ROS (singlet oxygen and superoxide) (16, 17). AR, RP, and 5,6-epoxy-RP have six, five, and four conjugated double bonds, respectively. As such, the ease of absorbing the UVA light energy by these compounds should be in the order AR > RP > 5,6-epoxy-RP, following the same order of induction of phototoxicity and photomutagenicity.
The L5178Y mouse lymphoma assay has been widely used for short-term mutagenicity bioassay (24, 25). This assay is capable of determining whether chemicals can induce either or both point mutations and chromosomal mutations (26, 27). This assay is particularly useful for evaluating the ability of mutagens to induce a wide variety of mutational events, because it detects not only intragenic events (mainly point mutations) but also LOH including Tk gene loss, karyotypically visible deletions, and rearrangements of the Tk+-bearing chromosome 11b (28–30).
In recent years, assessing the photogenotoxic potential of a compound has become an issue for certain drugs and cosmetic products. It has been considered that concomitant exposure of the cells with test compound and irradiation would constitute an appropriate general approach that can be used in screening assays (31). The present study of 5,6-epoxy-RP and AR and the previous study of RP suggest that the mouse lymphoma assay has utility for investigating photomutagenicity and, following LOH analysis of the mutants, it also can be used for establishing a mode of action for mutant induction.
Acknowledgment.
We thank Mr. Wayne G. Wamer (Center for Food Safety and Applied Nutrition, FDA) for helpful discussions, comments, and criticisms. We also thank Drs. Chen-An Tsai (Institute of Statistical Science, Taiwan) and Jianjun Zhang (University of Arkansas for Medical Sciences) for their help with statistical tests. This research was partially supported by appointment (L.C.) to the Postgraduate Research Program at the National Center for Toxicological Research, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and the U.S. Food and Drug Administration. The views presented in this article do not necessarily reflect those of the Food and Drug Administration.
References
- (1).Mukhtar H (1995) Skin Cancer: Mechanisms and Human Relevance CRC Press: Boca Raton, FL. [Google Scholar]
- (2).Ahmad N, and Mukhtar H (2004) Toxicology of the skin: new and emerging concepts. Toxicol. Appl. Pharm 195, 265–266. [Google Scholar]
- (3).International Agency for Research on Cancer (IARC). (1998) Handbooks of Cancer Prevention, Vitamin A Vol. 3, Lyon, France. [Google Scholar]
- (4).Idson B (1990) Vitamins in cosmetics, an update I. Overview and Vitamin A. Drug Cosmet. Ind 146, 26–91. [Google Scholar]
- (5).Tee ES (1992) Carotenoids and retinoids in human nutrition. Crit. Rev. Food Sci. Nutr 31, 103–163. [DOI] [PubMed] [Google Scholar]
- (6).Ries G, and Hess R (1999) Retinol: Safety considerations for its use in cosmetics products. J. Toxicol.-Cutan. Ocul 18, 169–185. [Google Scholar]
- (7).Torma H, Brunnberg L, and Vahlquist A (1987) Age-related variations in acyl-CoA:retinol acyltransferase activity and vitamin A concentration in the liver and epidermis of hairless mice. Biochim. Biophys. Acta 921, 254–258. [DOI] [PubMed] [Google Scholar]
- (8).Sorg O, Tran C, Carrau P, Didierjean L, and Saurat JH (1999) Retinol and retinyl ester epidermal pools are not identically sensitive to UVB irradiation and anti-oxidant protective effect. Dermatology 199, 302–307. [DOI] [PubMed] [Google Scholar]
- (9).Sklan D (1987) Vitamin A in human nutrition. Prog. Food Nutr. Sci 11, 39–55. [PubMed] [Google Scholar]
- (10).Kang S, Duell EA, Fisher GJ, Datta SC, Wang ZQ, Reddy AP, Tavakkol A, Yi JY, Griffiths CE, Elder JT, et al. (1995) Application of retinol to human skin in vivo induces epidermal hyperplasia and cellular retinoid binding proteins characteristic of retinoic acid but without measurable retinoic acid levels or irritation. J. Invest. Dermatol 105, 549–556. [DOI] [PubMed] [Google Scholar]
- (11).Yan J, Xia Q, Webb P, Warbritton AR, Wamer WG, Howard PC, Boudreau M, and Fu PP (2006) Levels of retinyl palmitate and retinol in stratum corneum, epidermis, and dermis of female SKH-1 mice. Toxicol. Ind. Health 22, 103–112. [DOI] [PubMed] [Google Scholar]
- (12).FDA (2000) Data from FDA’s voluntary cosmetics registration program
- (13).Cosmetic, Toiletry and Fragrance Association (CTFA). (1999) In International Cosmetic Ingredient Dictionary and Handbook, 8th ed. (Wenninger J, Canterbery RC, and McEwen GN Jr., Eds.) p 1279, CTFA, Washington, DC. [Google Scholar]
- (14).Fu PP, Howard PC, Culp SG, Xia Q, Webb PJ, Blankenship LR, Wamer WG, and Bucher JR (2002) Do topically applied skin creams containing retinyl palmitate affect the photocarcinogenecity of simulated solar light? J. Food Drug Anal 10, 262–268. [Google Scholar]
- (15).Fu PP, Cherng SH, Coop L, Xia Q, Culp SJ, Tolleson WH, Wamer WG, and Howard PC (2003) Photoreaction, phototoxicity, and photocarcinogenicity of retinoids. J. Environ. Sci. Health C: Environ. Carcinog. Ecotoxicol. Rev C21, 165–197. [DOI] [PubMed] [Google Scholar]
- (16).Cherng SH, Xia Q, Blankenship LR, Freeman JP, Wamer WG, Howard PC, and Fu PP (2005) Photodecomposition of retinyl palmitate in ethanol by UVA light-formation of photodecomposition products, reactive oxygen species, and lipid peroxides. Chem. Res. Toxicol 18, 129–138. [DOI] [PubMed] [Google Scholar]
- (17).Xia Q, Yin JJ, Cherng SH, Wamer WG, Boudreau M, Howard PC, and Fu PP (2006) UVA photoirradiation of retinyl palmitate–formation of singlet oxygen and superoxide, and their role in induction of lipid peroxidation. Toxicol. Lett 163, 30–43. [DOI] [PubMed] [Google Scholar]
- (18).Yan J, Xia Q, Cherng SH, Wamer WG, Howard PC, Yu H, and Fu PP (2005) Photo-induced DNA damage and photocytotoxicity of retinyl palmitate and its photodecomposition products. Toxicol. Ind. Health 21, 167–175. [DOI] [PubMed] [Google Scholar]
- (19).Mei N, Xia Q, Chen L, Moore MM, Fu PP, and Chen T (2005) Photomutagenicity of retinyl palmitate by ultraviolet A irradiation in mouse lymphoma cells. Toxicol. Sci 88, 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chen T and Moore MM (2004) Screening for chemical mutagens using the mouse lymphoma assay. In Optimization in Drug Discovery: In-vitro Methods (Yan Z, and Caldwell GW, Eds.) pp 337–352, Humana Press, Totowa, NJ. [Google Scholar]
- (21).Singh SP, Chen T, Chen L, Mei N, McLain E, Samokyszyn V, Thaden JJ, Moore MM, and Zimniak P (2005) Mutagenic effects of 4-hydroxynonenal triacetate, a chemically protected form of the lipid peroxidation product 4-hydroxynonenal, as assayed in L5178Y/Tk+/− mouse lymphoma cells. J. Pharmacol. Exp. Ther 313, 855–861. [DOI] [PubMed] [Google Scholar]
- (22).Moore MM, Honma M, Clements J, Bolcsfoldi G, Burlinson B, Cifone M, Clarke J, Delongchamp R, Durward R, Fellows M, Gollapudi B, Hou S, Jenkinson P, Lloyd M, Majeska J, Myhr B, O’Donovan M, Omori T, Riach C, San R, Stankowski LF Jr., Thakur AK, Van Goethem F, Wakuri S, and Yoshimura I (2006) Mouse lymphoma thymidine kinase gene mutation assay: Follow-up meeting of the international workshop on Genotoxicity testing, Aberdeen, Scotland, 2003. Assay acceptance criteria, positive controls, and data evaluation. Environ. Mol. Mutagen 47, 1–5. [DOI] [PubMed] [Google Scholar]
- (23).R project for statistical computing (http://www.r-project.org).
- (24).Dearfield KL, Auletta AE, Cimino MC, and Moore MM (1991) Considerations in the U.S. Environmental Protection Agency’s testing approach for mutagenicity. Mutat. Res 258, 259–283. [DOI] [PubMed] [Google Scholar]
- (25).Muller L, Kikuchi Y, Probst G, Schechtman L, Shimada H, Sofuni T, and Tweats D (1999) ICH-harmonised guidances on genotoxicity testing of pharmaceuticals: evolution, reasoning and impact. Mutat. Res 436, 195–225. [DOI] [PubMed] [Google Scholar]
- (26).Moore MM, Amtower A, Doerr C, Brock KH, and Dearfield KL (1987) Mutagenicity and clastogenicity of acrylamide in L5178Y mouse lymphoma cells. Environ. Mutagen 9, 261–267. [DOI] [PubMed] [Google Scholar]
- (27).Moore MM, Honma M, Clements J, Bolcsfoldi G, Cifone M, Delongchamp R, Fellows M, Gollapudi B, Jenkinson P, Kirby P, Kirchner S, Muster W, Myhr B, O’Donovan M, Oliver J, Omori T, Ouldelhkim MC, Pant K, Preston R, Riach C, San R, Stankowski LF Jr., Thakur A, Wakuri S, and Yoshimura I (2003) Mouse lymphoma thymidine kinase gene mutation assay: International Workshop on Genotoxicity Tests Workgroup report,–Plymouth, U.K., 2002. Mutat. Res 540, 127–140. [DOI] [PubMed] [Google Scholar]
- (28).Applegate ML, Moore MM, Broder CB, Burrell A, Juhn G, Kasweck KL, Lin PF, Wadhams A, and Hozier JC (1990) Molecular dissection of mutations at the heterozygous thymidine kinase locus in mouse lymphoma cells. Proc. Natl. Acad. Sci. U.S.A 87, 51–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Chen T, Harrington-Brock K, and Moore MM (2002) Mutant frequencies and loss of heterozygosity induced by N-ethyl-N-nitrosourea in the thymidine kinase gene of L5178Y/Tk+/−−3.7.2C mouse lymphoma cells. Mutagenesis 17, 105–109. [DOI] [PubMed] [Google Scholar]
- (30).Clements J (2000) The mouse lymphoma assay. Mutat. Res 455, 97–110. [DOI] [PubMed] [Google Scholar]
- (31).Gocke E, Muller L, Guzzie PJ, Brendler-Schwaab S, Bulera S, Chignell CF, Henderson LM, Jacobs A, Murli H, Snyder RD, and Tanaka N (2000) Considerations on photochemical genotoxicity: report of the International Workshop on Genotoxicity Test Procedures Working Group. Environ. Mol. Mutagen 35, 173–184. [DOI] [PubMed] [Google Scholar]