

To the editor,
Presenilin 1 gene (PSEN1)-linked Alzheimer’s disease with cotton wool plaques (CWP-AD) is a rare variant that is often clinically characterized by dementia with spastic paraparesis [4]. CWPs are eosinophilic, round, large, non-cored, and Aβ-positive plaques with minor dystrophic neurites. The clinical heterogeneity of this variant remains unclear. Here, we describe an autopsy case of CWP-AD with a novel PSEN1 mutation that showed slowly progressive cognitive and motor disturbances from the mid-20s with a very long disease duration of about 30 years.
The proband was a right-handed Japanese woman who initially presented with a decrease of calculation ability at age 25. Her brother initially exhibited slurred speech and bradykinesia at 39 years. Subsequently, parkinsonism, alien hand sign, and dementia developed. He died at 48 years. The proband’s mother, uncle, and maternal grandfather showed dementia and/or gait disturbance, and all died at 48 years. None of these relatives was genetically examined.
The proband exhibited muscle weakness of both hands and forgetfulness at 26 years. Clumsiness in the right upper and lower extremities developed at 30 years. She first went to the department of neurology at a general hospital and was suspected as having AD with parkinsonism. Dysarthria and dysphagia emerged at 34 and 35 years, respectively. Eleven years after the onset, she was admitted to the department of neurology at a university hospital. Neurological examination revealed limitation of upward and lateral gaze, bilaterally increased tendon reflex in all four extremities, bilaterally positive Babinski sign, spastic paraparesis, akinesia, and rigidity of the neck and four extremities. Parkinsonism was unresponsive to L-dopa treatment. She scored 20/30 points on the Mini-Mental State Examination [5], and on the WAIS-Revised, she obtained a verbal IQ score of 69, performance IQ score of 46, and full-scale IQ score of 54. Baseline blood and cerebrospinal fluid examinations were normal. She could walk without support until 37 years old. Brain MRI at 40 years demonstrated diffuse cerebral atrophy (Fig. 1a-d). At 42 years, she needed tube feeding due to dysphagia. 99mTc-ECD single-photon emission computed tomography (SPECT) at 44 years disclosed hypoperfusion in the posterior part of the cingulate gyrus, precuneus, and parieto-occipital cortices (Fig. 2). Brain MRI at 54 years showed cerebral atrophy with severe dilatation of the ventricles (Fig. 1e and f). She died of respiratory failure at age 54 after a disease duration of 29 years. No respiratory support was given throughout the course. Her final neurological diagnosis was unclassifiable dementia.
Fig. 1.

MR images of the present case. a T1-weighted horizontal, b T2-weighted horizontal, c T1-weighted coronal, and d T1-weighted sagittal images at 40 years. Evident bilateral atrophy of the hippocampus and symmetric white matter atrophy with occipital predominance are noted. The width of the corpus callosum is reduced (c). The brain stem doesn’t show evident atrophy (d). e T1-weighted horizontal and f T2-weighted horizontal images at 54 years. Diffuse cortical atrophy is progressed. The symmetric dilatation of the lateral ventricles becomes evident, suggesting the potential complication of idiopathic normal pressure hydrocephalus in addition to the atrophy of the white matter
Fig. 2.

99mTc-ECD SPECT images at age 44. The cerebral blood flow in the bilateral posterior cingulate gyri, precuneus, and parietal and occipital cortices is reduced with left-side predominance
The brain weighed 895 g before fixation. Macroscopically, severe atrophy in the neocortex (Fig. 3a-c) and marked depigmentation in the substantia nigra (Fig. 3d) and locus coeruleus (Fig. 3e) were noted. The pyramidal tract at the level of the medulla oblongata was atrophic (Fig. 3f). Histopathologically, abundant CWPs were noted throughout the cerebral cortex (Fig. 4a-g, Table 1). Neuritic plaques with dense amyloid cores were hardly noted in any region. Abundant Aβ deposits were noted in the cerebellum (Figs. 4h, 5c, and d) and spinal gray matter (Fig. 6e and f). Aβ42 rather than Aβ40 was predominantly accumulated in CWPs and cerebellar Aβ plaques (Fig. 5a-d). Remarkable cerebral amyloid angiopathy was also noted, although it was hardly related to CWPs spatially (Figs. 4c, g, and 5). The distributions of Aβ deposits and neurofibrillary changes were classified as Thal phase 5 [24] and Braak stage VI [1]. Viewing Congo red-stained sections with polarized light did not demonstrate apple green birefringence in CWPs (Fig. 7a and b). Neuronal loss associated with the proliferation of GFAP-positive astrocytes and Iba1-positive microglias was remarkable in the cerebral cortex and basal ganglia (Figs. 4a, 7c, d, and Table 1). Loss of Betz cells in the motor cortex (Fig. 6a) and degeneration of the pyramidal tract (Fig. 6b-d) were evident. Motor neurons in the spinal anterior horns and hypoglossal nuclei were spared in number (Fig. 6d). α-Synuclein-positive Lewy bodies were extensively distributed, corresponding to diffuse neocortical type Lewy body disease [13] and Braak Parkinson’s disease stage 5 [2]. Pigmented neurons in the substantia nigra were severely reduced in number (Fig. 8a-d). TDP-43-positive neurocytoplasmic inclusions, intranuclear inclusions, and short neurites were noted in the limbic region and temporal cortex, corresponding to Josephs stage III (Fig. 8e-g) [9]. No argyrophilic grain, tufted astrocyte, astrocytic plaque, FUS pathology, p62-positive inclusion in the cerebellar dentate nucleus, 1C2-positive inclusion, or pathological 3F4-positive lesion was noted.
Fig. 3.
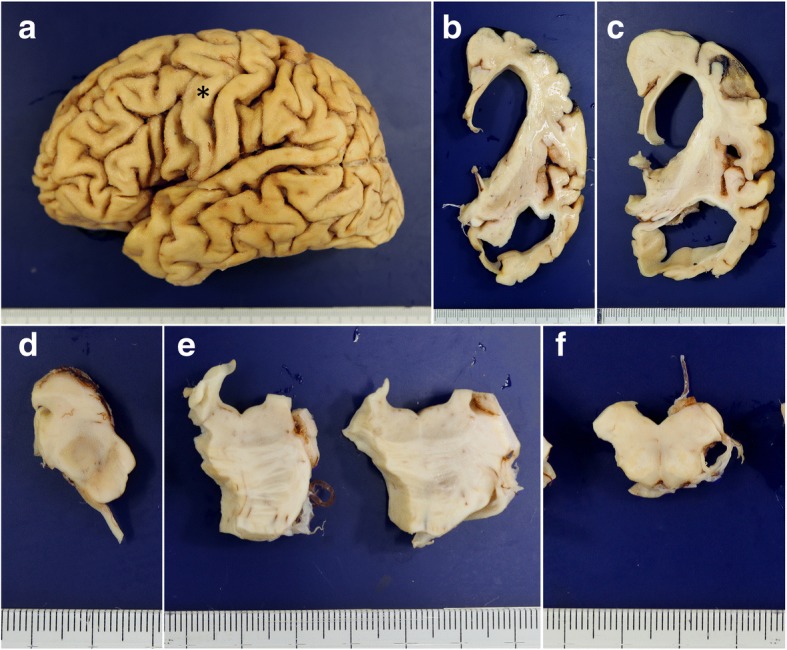
Macroscopic findings of the present case. a Lateral view of the left hemisphere. Severe diffuse atrophy including the precentral gyrus (an asterisk) is seen. b On a coronal section, severe atrophy is evident in the cortex and white matter of the frontal and temporal lobes. The basal ganglia also show severe atrophy. The width of the corpus callosum is severely reduced. c Remarkable atrophy of the hippocampus and parahippocampal gyrus. d Severe depigmentation in the substantia nigra. e Depigmentation in the locus coeruleus. f Severe atrophy in the pyramidal tract at the level of the medulla oblongata
Fig. 4.

Histopathological findings of the present case. a, b Numerous eosinophilic, round, non-cored, and large cotton wool plaques in the insula cortex. The diameter of the plaques is often 100 μm or over. The rarefaction in the neuropil is also remarkable. Hematoxylin-eosin stain. c Aβ-positive abundant CWPs in all cortical layers. The inferior frontal gyrus. 12B2 immunohistochemistry. d A CWP showing a mass effect on around myelinated fibers in the neuropil. The inferior frontal gyrus. Klüver-Barrera stain. e Modified Bielschowsky silver stain showed heterogeneous argyrophilia but no clear amyloid core in a CWP. The middle frontal gyrus. f Gallyas silver stain shows only weak argyrophilia of homogeneous material composing a CWP. The inferior frontal gyrus. g A CWP strongly stained with an anti-Aβ antibody. The inferior frontal gyrus. 12B2 immunohistochemistry. h Aβ deposits in the dentate nucleus in the cerebellum. 12B2 immunohistochemistry. Scale bars = a 100 μm, b 30 μm, c 100 μm, d, e, f, g 30 μm, h 40 μm
Table 1.
Distribution of lesions in the present case
| Neuronal loss/gliosis | Aβ deposits/CWPs | NFTs | Lewy bodies/Lewy neurites | TDP-43 positive NCIs/short neurites | |
|---|---|---|---|---|---|
| Primary motor cortex | +++ | +++++/+++ | ++++ | −/− | −/− |
| Superior frontal gyrus | +++ | +++++/+++++ | ++++ | −/− | −/− |
| Middle frontal gyrus | +++ | +++++/++++ | ++++ | −/− | −/− |
| Inferior frontal gyrus | +++ | +++++/++++ | ++++ | −/− | −/− |
| Frontal white matter | +++ | ++/− | – | −/− | −/− |
| Superior temporal gyrus | +++ | +++++/+++++ | ++++ | −/− | −/− |
| Middle temporal gyrus | +++ | +++++/+++++ | ++++ | −/− | −/− |
| Inferior temporal gyrus | +++ | +++++/+++++ | ++++ | ++/++ | +/+ |
| Lateral occipitotemporal gyrus | +++ | +++++/+++++ | ++++ | +/++ | ++/+ |
| Temporal white matter | +++ | ++++/− | – | −/− | −/− |
| Parietal cortex | +++ | +++++/++++ | ++++ | −/− | −/− |
| Parietal white matter | +++ | ++++/− | – | −/− | −/− |
| Occipital cortex | +++ | ++++/+++ | ++++ | −/− | −/− |
| Occipital white matter | +++ | ++++/− | – | −/− | −/− |
| Entorhinal cortex | +++ | +++++/++++ | ++++ | +++/++++ | ++++/− |
| Hippocampal CA1 | +++ | +++++/++++ | ++++ | +/− | ++++/− |
| Amygdala | + | +++++/++++ | +++++ | ++++/++++ | ++++/++ |
| Caudate nucleus | +++ | +++++/+++++ | ++++ | −/− | −/− |
| Putamen | +++ | +++++/+++++ | +++++ | −/− | −/− |
| Globus pallidus | +++ | ++/− | ++++ | −/− | −/− |
| Subthalamic nucleus | n.a. | n.a. | n.a. | n.a. | n.a. |
| Oculomotor nucleus | – | −/− | ++ | −/− | −/− |
| Substantia nigra | +++ | +/− | ++++ | −/+ | −/− |
| Frontopontine tract | – | +/− | – | −/− | −/− |
| Corticospinal tract | |||||
| at level of cerebral peduncle | +++ | −/− | – | −/− | −/− |
| at level of pons | +++ | −/− | – | −/− | −/− |
| at level of medulla oblongata | +++ | −/− | – | −/− | −/− |
| Locus coeruleus | ++ | +/− | ++ | +/− | −/− |
| Pontine nucleus | – | ++/− | +++ | −/− | −/− |
| Superior cerebellar peduncle | – | −/− | – | −/− | −/− |
| Transverse pontine fibers | – | −/− | – | −/− | −/− |
| Dorsal vagal nucleus | + | +/− | ++ | −/− | −/− |
| Hypoglossal nucleus | – | +/− | – | −/− | −/− |
| Inferior olivary nucleus | – | +/+ | – | −/− | −/− |
| Cerebellum | |||||
| Molecular layer | + | ++++/− | – | −/− | −/− |
| Purkinje cell layer | – | ++/− | – | −/− | −/− |
| Granular layer | – | ++++/− | – | −/− | −/− |
| Dentate nucleus | – | +++++/− | +++ | −/− | −/− |
| White matter | ++ | +/− | – | −/− | −/− |
| Spinal cord | |||||
| Anterior horn | – | ++/− | – | −/− | −/− |
| Corticospinal tract | +++ | +/− | – | −/− | −/− |
Neuronal loss -: none, +: mild, ++: moderate, +++: severe. The severity of the degeneration of the tract was evaluated with the following system using hematoxylin-eosin and Klüver-Barrera stains: - (no degeneration), neither loss of myelin nor glial proliferation was found; + (mild degeneration), slight myelin loss and gliosis without atrophy of the tract; ++ (moderate degeneration), evident myelin loss and gliosis with slight atrophy of the tract; +++ (severe degeneration), evident myelin loss and gliosis with severe atrophy of the tract. Aβ deposits and cotton wool plaques (CWPs) were semiquantitatively evaluated with the following staging system using 12B2 immunohistochemistry: -, no lesion; ±, one lesion in each anatomical region; +, one lesion per ×200 visual field; ++, 2 to 10 lesions per × 200 visual field; +++, 11 to 20 lesions per × 200 visual field; ++++, 21 to 50 lesions per × 200 visual field; +++++, 51 or more lesions per × 200 visual field. AT-8 positive neurofibrillary tangles (NFTs), Lewy body, Lewy neurites, TDP-43 positive neuronal cytoplasmic inclusions (NCIs), and TDP-43 positive short neurites were semiquantitatively evaluated with the following staging system using AT8, pSyn#64, and pS409/410–2 immunohistochemistry, respectively: -, no lesion; +, one lesion in the anatomical region; ++, two to four lesions in the anatomical lesion but less than one lesion per × 200 visual field; +++, one lesion per × 200 visual field; ++++, 2 to 10 lesions per × 200 visual field; +++++, over 11 lesions per × 200 visual field. n.a., not available
Fig. 5.

Aβ42 and Aβ40 immunohistochemistry on serial sections from the temporal cortex and cerebellar cortex. a, b Serial sections from the inferior temporal cortex. Aβ42 (a) rather than Aβ40 (b) is predominantly accumulated in CWPs as well as cerebral amyloid angiopathy. c, d Serial sections from the cerebellar cortex. Aβ42 (c) rather than Aβ40 (d) is predominantly accumulated in the cerebellar cortex, while both are almost equally accumulated in cerebral amyloid angiopathy. Scale bars = a, b 100 μm, c, d 30 μm
Fig. 6.

Degeneration of upper motor neurons. a Loss of Betz cells. (Arrows) A few remaining Betz cells. (Arrowheads) Numerous unstained CWPs that tend to be densely distributed in the deep cortical layers. b Degeneration with evident atrophy of the pyramidal tract at the level of the medulla oblongata. c, d Degeneration in the lateral tract in the thoracic (c) and lumbar cords (d). The anterior horn cells are well preserved in number (d). e Remarkable Aβ deposits in the anterior horns in the lumbar cord, while no Aβ deposit is seen in the corticospinal tract. f A high power view of Aβ deposits in the anterior horn in the lumbar cord. Scale bars = a 300 μm, b-e 1 mm, f 50 μm. a-d Klüver-Barrera stain. e, f 12B2 immunohistochemistry
Fig. 7.

Absence of congophilia of CWPs with glial proliferation. a, b Cotton wool plaques (CWPs) on section stained with Congo red. Observation with polarized light demonstrates apple green birefringence in the amyloid angiopathy but not CWPs (b). The insular cortex. c, d GFAP-positive astrocytes (c) and Iba1-positive microglias (d) surrounding CWPs. The inferior temporal gyrus. All scale bars = 50 μm
Fig. 8.

Pathological findings in the substantia nigra and limbic system. a Severe loss of pigmented neurons in the substantia nigra. The corticospinal tract is also severely degenerated. Klüver-Barrera stain. b Glial proliferation with free melanin in the substantia nigra. Hematoxylin-eosin stain. c Phosphorylated tau-positive dystrophic neurites and NFTs in the substantia nigra. AT8 immunohistochemistry. d Phosphorylated α-synuclein-positive Lewy neurites in the substantia nigra. Psyn#64 immunohistochemistry. (e, f) Phosphorylated TDP-43-positive neurocytoplasmic inclusions in the hippocampal dentate gyrus (e) and occipitotemporal gyrus (f). pS409/410–2 immunohistochemistry. g An intranuclear inclusion immunopositive for phosphorylated TDP-43. The occipitotemporal gyrus. pS409/410–2 immunohistochemistry. Scale bars = a 1 mm, b, c 50 μm, d-f 40 μm, g 5 μm
Mutational analysis of coding exons and flanking intronic sequences of APP, PSEN1, and PSEN2 using frozen brain tissue demonstrated a novel c.1249G > A mutation (p.Gly417Ser) in exon 12 of PSEN1 in the proband (Fig. 9a). The mutation is not present in the ExAC database (http://exac.broadinstitute.org) or jMorp database (https://jmorp.megabank.tohoku.ac.jp). There was no other mutation in APP or PSEN2. The APOE genotype was 3*4. A functional assay by establishing N2a cells that stably express the PS1 wild-type or p.G417S mutant [7] and sandwich ELISA demonstrated that the expression of PSEN1 p.G417S resulted in increases in the Aβ42 and Aβ42/40 ratio, which were significantly higher than those in wild-type-expressing cells, suggesting that the mutation was likely to be causative in the present case (Fig. 9b. See details of methods in Additional files 1 and 2 [7]).
Fig. 9.

Detection of novel PSEN1 mutation and functional assay. a Direct sequencing of PSEN1 exon 12 of the patient demonstrated a novel mutation of c. 1249G > A indicated by arrow, resulted in a missense mutation of p.Gly417Ser. This mutation is predicted as probably damaging with a score of 0.979 by Polyphen-2 and a CADD score of 29.6. b The level of Aβ42 and the ratio of Aβ42/40 were significantly increased in the media of cells stably expressing mutant PS1 of p.G417S compared with those of wild-type. Data were plotted as mean ± SEM (n = 3). **P < 0.01
The most noteworthy clinical features in the present case were the young age at onset and very long disease duration. It was reported that the mean age at onset in 564 patients with PSEN1-linked AD was 43.3 ± 8.6 years, and that the interquartile range of the disease duration was five to 11 years [19]. In the previous 27 PSEN1-linked CWP-AD cases with sufficient clinical information that we reviewed (Table 2 [3, 4, 6, 8, 10, 12, 14, 15, 17, 18, 20–23]), the age at onset ranged from 29 to 58 years (mean: 45.4 ± 8.5 years), and the disease duration was from 3 to 20 years (mean: 9.9 ± 5.8 years) (Fig. 10a–c). Spastic paraparesis and parkinsonism were described in 15 (55.6%) and 6 cases (22.2%), respectively. As far as we know, the present case showed the youngest age at onset and the longest disease duration. In some CWP-AD cases, neurons surrounding CWPs tend to be spared in number and inflammatory changes are often relatively mild [4, 11, 25, 26]. However, our case showed remarkable neuronal loss with tissue rarefaction in the cerebral cortex. It may be explained by the extremely long disease duration. Interestingly, we recently found a poster abstract in which two siblings having dementia, spastic paraplegia, and the same PSEN1 mutation was reported [16]. Although the information was limited, the ages at onset in these siblings were 32 and 36 years, respectively. To our knowledge, these individuals were not included in our pedigree. As shown in Fig. 10, the ages at onset in these clinical cases, like that in our case, are relatively young among previously reported CWP-AD cases. What factors besides mutations affect the age at onset and speeds of tissue degeneration and clinical progression in CWP-AD cases remain unclear. However, clinicians should be aware at least that the differential diagnosis of slowly progressive cognitive decline with spasticity and parkinsonism in young adults includes CWP-AD.
Table 2.
Clinical and pathological features in the present case (case 1) and previously reported PSEN1-linked CWP-AD cases (cases 2–28)
| Case | Sex | Age at onset (y) | Age at death (y) | Duration (y) | Initial symptoms | Spastic paraparesis | Parkinsonism | Brain weight (g) | CWPs | Lewy body disease | PSEN1 mutation | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | f | 25 | 54 | 29 | cognitive impairment | + | + | 895 | + | diffuse | G417S | Present case |
| 2 | n.d. | 29 | n.d. | n.d. | spastic paraparesis | + | n.d. | n.d. | + | n.d. | P436Q | Houlden H et al. [6] |
| 3 | m | 30s | 48 | n.a. | disorientation | – | n.d. | n.d. | + | n.d. | L420R | Shrimpton AE et al. (II:1) [20] |
| 4 | f | 31 | 43 | 12 | forgetfulness | – | + | 1170 | + | – | L420R | Niwa A et al. [14] |
| 5 | m | 34 | 39 | 5 | morbid jealousy | – | n.d. | 1550 | + | n.d | deletion of exon 9 sequence from PSEN1 transcripts | Brooks WS et al. (EOFAD-2 IV:45) [3] |
| 6 | m | 34 | 52 | 18 | parkinsonism | – | + | 1150 | + | diffuse | in-frame 3bp ACC deletion in exon 12 | Ishikawa A et al. [8] |
| 7 | n.d. | 36 | n.d. | n.d. | spastic paraparesis | + | n.d. | n.d. | + | n.d. | Δ83,84IM | Houlden H et al. [6] |
| 8 | f | 37 | 50 | 13 | cognitive dysfunction, parkinsonism | + | + | 740 | + | limbic | G217D | Takao M et al. (case III-2) [23] |
| 9 | f | 40 | biopsy | n.a. | cognitive impairment | + | n.d. | biopsy | + | n.d. | E280G | O’Riordan S et al. (patient 3) [17] |
| 10 | f | 41 | 45 | 4 | cognitive decline | – | n.d. | 992 | + | n.d. | G- > T exon 9 splice acceptor mutation | Brooks WS et al. (EOFAD-3 II:10) [3] |
| 11 | m | 41 | 46 | 5 | dementia | n.d. | n.d. | n.d. | + | n.d. | deletion of exon 9 | Smith MJ et al. (case II:12) [22] |
| 12 | m | 42 | 46 | 4 | antiflexion gait | + | + | 1150 | + | – | G217D | Takao M et al. (case III-1) [23] |
| 13 | f | 46 | 52 | 6 | memory loss and disorientation | – | n.d. | 1144 | + | n.d. | G217R | Norton JB et al. (case 4:4) [15] |
| 14 | m | 46 | 65 | 19 | depression | + | + | 1100 | + | – | P264L | Martikinen P et al. (case 3) [12] |
| 15 | f | 47 | 51 | 4 | cognitive decline | + | n.d. | n.d. | + | n.d. | G- > T exon 9 splice acceptor mutation | Brooks WS et al. (EOFAD-3 II:8) [3] |
| 16 | m | 47 | 67 | 20 | spasticity and weakness in legs | + | n.d. | 1110 | + | – | E280G | Sinha N et al. [21] |
| 17 | f | 50 | 53 | 3 | spastic paraparesis | + | n.d. | n.d. | + | n.d. | deletion of exon 9 | Smith MJ et al. (case III:9) [22] |
| 18 | f | 50 | 60 | 10 | cognitive decline | – | n.d. | 918 | + | n.d. | deletion of exon 9 sequence from PSEN1 transcripts | Brooks WS et al. (EOFAD-2 III:18) [3] |
| 19 | f | 51 | 68 | 17 | memory impairment | – | + | 1050 | + | – | P264L | Martikinen P et al. (case 1) [12] |
| 20 | m | 52 | 56 | 4 | cognitive decline | – | n.d. | 910 | + | n.d. | deletion of exon 9 sequence from PSEN1 transcripts | Brooks WS et al. (EOFAD-2 IV:23) [3] |
| 21 | m | 52 | 56 | 4 | dementia | n.d. | n.d. | n.d. | + | n.d. | deletion of exon 9 | Smith MJ et al. (case II:7) [22] |
| 22 | m | 52 | 67 | 15 | memory difficulty and weakness in both legs | + | n.d. | 890 | + | n.d. | E280Q | Rogaeva E et al. [18] |
| 23 | f | 54 | 63 | 9 | dementia and spastic paraparesis | + | n.d. | n.d. | + | n.d. | deletion of exon 9 | Smith MJ et al. case (III:7) [22] |
| 24 | m | 54 | 64 | 10 | dementia | + | n.d. | 1360 | + | n.d. | deletion of exon 9 | Crook R et al. (case III:15) [4] |
| 25 | m | 55 | 61 | 6 | back pain, stiffness of legs | + | n.d. | n.d. | + | n.d. | deletion of exon 9 splice site | Crook R et al. (patient III:9) [4] |
| 26 | m | 57 | 69 | 12 | dementia | + | n.d. | 1075 | + | n.d. | deletion of exon 9 | Crook R et al. (case III:14) [4] |
| 27 | m | 58 | 75 | 17 | memory impairment | + | – | 1320 | + | – | P264L | Martikinen P et al. (case 2) [12] |
| 28 | f | n.d. | 60 | n.a. | n.d. | – | n.d. | n.d. | + | n.d. | L271V | Kwok JB et al. (III:28) [10] |
Fig. 10.
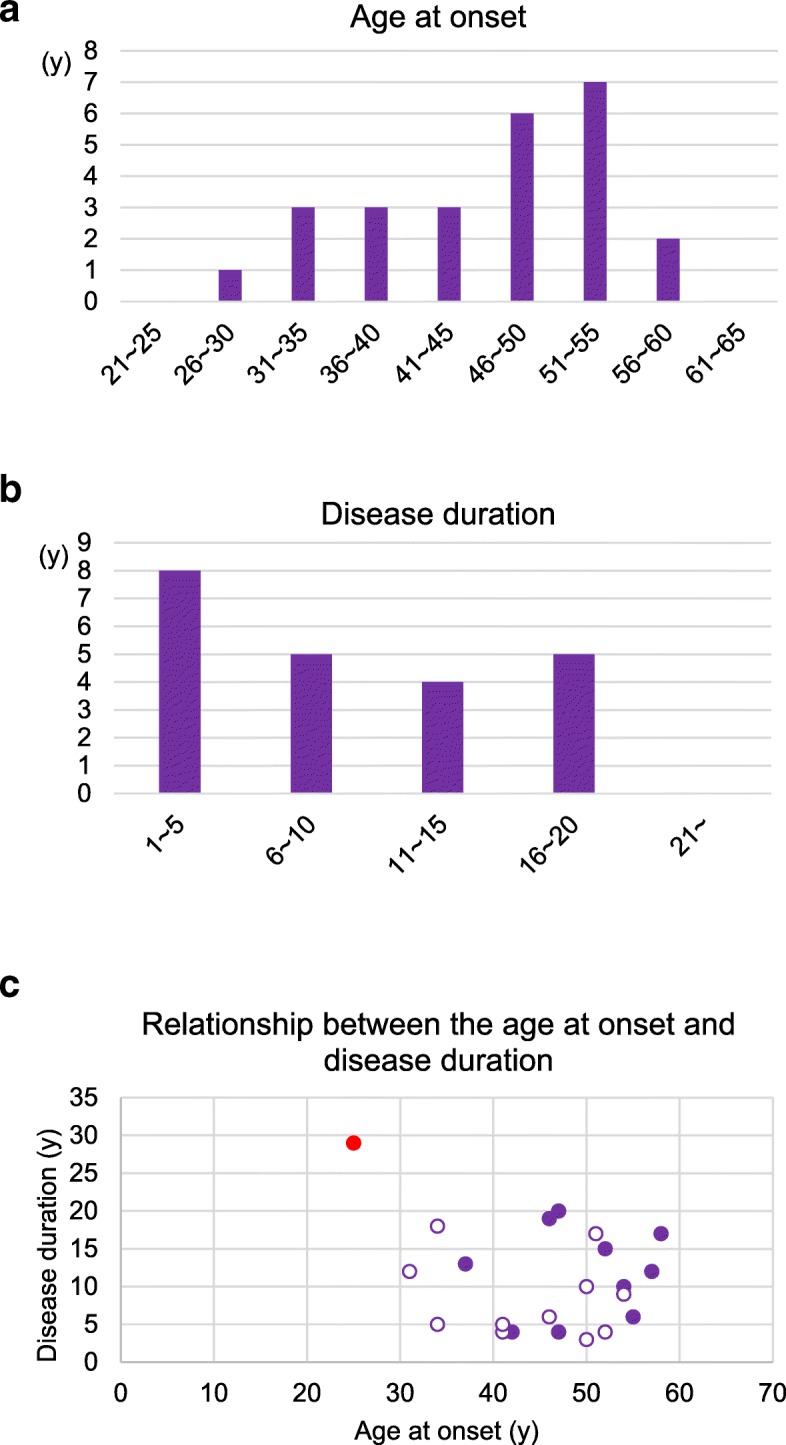
Distribution of the age at onset and disease duration in previously reported CWP-AD cases. a The age at onset in previously reported CWP-AD cases due to PSEN1 mutations. b The disease duration in previously reported CWP-AD cases due to PSEN1 mutations. c The relationship between the age at onset and disease duration in the present case and previously reported CWP-AD. The data of cases 1 to 28 were extracted from the references cited in Table 2. No significant correlation between the age at onset and disease duration was demonstrated by Spearman rank order correlation analysis when all cases whose disease duration was available were examined (ρ = − 0.101, p = 0.65) or when only previous cases were examined (ρ = 0.058, p = 0.80). These findings suggest that young age at onset is not necessarily a factor that predicts rapid progression or short disease duration in CWP-AD cases. Red solid circle: the present case, purple solid circle: previously reported CWP-AD cases with spastic paraparesis, purple open circle: previously reported CWP-AD cases without spastic paraparesis
Additional files
Details of methods. (DOCX 20 kb)
Antibodies used in this study. (DOCX 24 kb)
Acknowledgements
We thank Mses. Y. Matsuo and M. Onbe for their technical assistance.
Funding
This work was supported by Grants-in-Aid for Scientific Research (C) from the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT KAKENHI Grant No. 15K09867, 18K07559), Grants-in-Aid from the Research Committee of CNS Degenerative Diseases and Research on Dementia from the Ministry of Health, Labour and Welfare of Japan (H29-Nanchi-Ippan-033), an Intramural Research Grant for Neurological and Psychiatric Disorders from National Center of Neurology and Psychiatry (NCNP) (27–6-2, 30–8), grants from the Strategic Research Program for Brain Sciences from Japan Agency for Medical Research and Development (AMED, JP18dm0107109, JP18kk0205009, JP18kk0205009), and grants from Zikei Institute of Psychiatry.
Availability of data and materials
Not applicable.
Abbreviations
- AD
Alzheimer’s disease
- CWP
Cotton wool plaque
- CWP-AD
Alzheimer’s disease with cotton wool plaques
- PSEN1
Presenilin 1 gene
Authors’ contributions
TM: data collection, pathological studies, and drafting and revising manuscript. OY: data collection, pathological studies, and critical revision of the manuscript for intellectual content. TH: data collection, pathological studies, and revising manuscript. TI: genetic analysis and revising manuscript. BZ: genetic analysis and revising manuscript. STa: pathological studies and revising manuscript. STe: study supervision and critical revision of the manuscript for intellectual content. NY: study supervision and critical revision of the manuscript for intellectual content. All authors read and approved the final manuscript.
Ethics approval
Autopsy and gene analysis were carried out after written informed consent was obtained from family members, and all experiments in this study were approved by the ethical committees of the Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences (RIN1626–260), Niigata University (H28–870), and National Hospital Organization Minami-Okayama Medical Center (H29–65).
Consent for publication
Family members have consented to publication.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Tomoko Miki, Email: tomokomoreaux@gmail.com.
Osamu Yokota, Email: oyokota1@yahoo.co.jp.
Takashi Haraguchi, Email: haraguchit1@yahoo.co.jp.
Takeshi Ikeuchi, Email: ikeuchi@bri.niigata-u.ac.jp.
Bin Zhu, Email: n17m610a@mail.cc.niigata-u.ac.jp.
Shintaro Takenoshita, Email: shintaro.take@gmail.com.
Seishi Terada, Email: terada@okayama-u.ac.jp.
Norihito Yamada, Email: nyamada@okayama-u.ac.jp.
References
- 1.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/S0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 3.Brooks WS, Kwok JB, Kril JJ, Broe GA, Blumbergs PC, Tannenberg AE, et al. Alzheimer’s disease with spastic paraparesis and ‘cotton wool’ plaques: two pedigrees with PS-1 exon 9 deletions. Brain. 2003;126:783–791. doi: 10.1093/brain/awg084. [DOI] [PubMed] [Google Scholar]
- 4.Crook R, Verkkoniemi A, Perez-Tur J, Mehta N, Baker M, Houlden H, et al. A variant of Alzheimer’s disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nat Med. 1998;4:452–455. doi: 10.1038/nm0498-452. [DOI] [PubMed] [Google Scholar]
- 5.Folstein MF, Folstein SE, McHugh PR. Mini-mental state: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 6.Houlden H, Baker M, McGowan E, Lewis P, Hutton M, Crook R, et al. Variant Alzheimer’s disease with spastic paraparesis and cotton wool plaques is caused by PS-1 mutations that lead to exceptionally high amyloid-beta concentrations. Ann Neurol. 2000;48:806–808. doi: 10.1002/1531-8249(200011)48:5<806::AID-ANA18>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 7.Ikeuchi T, Kaneko H, Miyashita A, Nozaki H, Kasuga K, Tsukie T, et al. Mutational analysis in early-onset familial dementia in the Japanese population. The role of PSEN1 and MAPT R406W mutations. Dement Geriatr Cogn Disord. 2008;26:43–49. doi: 10.1159/000141483. [DOI] [PubMed] [Google Scholar]
- 8.Ishikawa A, Piao YS, Miyashita A, Kuwano R, Onodera O, Ohtake H, et al. A mutant PSEN1 causes dementia with Lewy bodies and variant Alzheimer’s disease. Ann Neurol. 2005;57:429–434. doi: 10.1002/ana.20393. [DOI] [PubMed] [Google Scholar]
- 9.Josephs KA, Murray ME, Whitwell JL, Tosakulwong N, Weigand SD, Petrucelli L, et al. Updated TDP-43 in Alzheimer’s disease staging scheme. Acta Neuropathol. 2016;131:571–585. doi: 10.1007/s00401-016-1537-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwok JB, Halliday GM, Brooks WS, Dolios G, Laudon H, Murayama O, et al. Presenilin-1 mutation L271V results in altered exon 8 splicing and Alzheimer’s disease with non-cored plaques and no neuritic dystrophy. J Biol Chem. 2003;278:6748–6754. doi: 10.1074/jbc.M211827200. [DOI] [PubMed] [Google Scholar]
- 11.Le TV, Crook R, Hardy J, Dickson DW. Cotton wool plaques in non-familial late-onset Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:1051–1061. doi: 10.1093/jnen/60.11.1051. [DOI] [PubMed] [Google Scholar]
- 12.Martikainen P, Pikkarainen M, Pöntynen K, Hiltunen M, Lehtovirta M, Tuisku S, et al. Brain pathology in three subjects from the same pedigree with presenilin-1 (PSEN1) P264L mutation. Neuropathol Appl Neurobiol. 2010;36:41–54. doi: 10.1111/j.1365-2990.2009.01046.x. [DOI] [PubMed] [Google Scholar]
- 13.McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology. 2005;65:1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 14.Niwa A, Matsuo K, Shindo A, Yata K, Shiraishi T, Tomimoto H. Clinical and neuropathological findings in a patient with familial Alzheimer disease showing a mutation in the PSEN1 gene. Neuropathology. 2013;33:199–203. doi: 10.1111/j.1440-1789.2012.01340.x. [DOI] [PubMed] [Google Scholar]
- 15.Norton JB, Cairns NJ, Chakraverty S, Wang J, Levitch D, Galvin JE, et al. Presenilin1 G217R mutation linked to Alzheimer disease with cotton wool plaques. Neurology. 2009;73:480–482. doi: 10.1212/WNL.0b013e3181b163ba. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oi Y, Itani M, Hasegawa H, Maki T, Kuzuya A, Yamashita H, et al. A pedigree of familial Alzheimer disease with spastic paraplegia carrying a novel presenilin-1 mutation. J Neurol Sci. 2017;381:1134–1135. doi: 10.1016/j.jns.2017.08.3195. [DOI] [Google Scholar]
- 17.O'Riordan S, McMonagle P, Janssen JC, Fox NC, Farrell M, Collinge J, et al. Presenilin-1 mutation (E280G), spastic paraparesis, and cranial MRI white-matter abnormalities. Neurology. 2002;59:1108–1110. doi: 10.1212/WNL.59.7.1108. [DOI] [PubMed] [Google Scholar]
- 18.Rogaeva E, Bergeron C, Sato C, Moliaka I, Kawarai T, Toulina A, et al. PS1 Alzheimer’s disease family with spastic paraplegia: the search for a gene modifier. Neurology. 2003;61:1005–1007. doi: 10.1212/WNL.61.7.1005. [DOI] [PubMed] [Google Scholar]
- 19.Shea YF, Chu LW, Chan AO, Ha J, Li Y, Song YQ. A systematic review of familial Alzheimer’s disease: differences in presentation of clinical features among three mutated genes and potential ethnic differences. J Formos Med Assoc. 2016;115:67–75. doi: 10.1016/j.jfma.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 20.Shrimpton AE, Schelper RL, Linke RP, Hardy J, Crook R, Dickson DW, et al. A presenilin 1 mutation (L420R) in a family with early onset Alzheimer disease, seizures and cotton wool plaques, but not spastic paraparesis. Neuropathology. 2007;27:228–232. doi: 10.1111/j.1440-1789.2007.00766.x. [DOI] [PubMed] [Google Scholar]
- 21.Sinha N, Grimes D, Tokuhiro S, Sato C, Rogaeva E, Woulfe J. Variant Alzheimer’s disease with spastic paraparesis and supranuclear gaze palsy. Can J Neurol Sci. 2013;40:249–251. doi: 10.1017/S0317167100013822. [DOI] [PubMed] [Google Scholar]
- 22.Smith MJ, Kwok JB, McLean CA, Kril JJ, Broe GA, Nicholson GA, et al. Variable phenotype of Alzheimer’s disease with spastic paraparesis. Ann Neurol. 2001;49:125–129. doi: 10.1002/1531-8249(200101)49:1<125::AID-ANA21>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 23.Takao M, Ghetti B, Hayakawa I, Ikeda E, Fukuuchi Y, Miravalle L, et al. A novel mutation (G217D) in the Presenilin 1 gene ( PSEN1) in a Japanese family: presenile dementia and parkinsonism are associated with cotton wool plaques in the cortex and striatum. Acta Neuropathol. 2002;104:155–170. doi: 10.1007/s00401-002-0536-6. [DOI] [PubMed] [Google Scholar]
- 24.Thal DR, Rüb U, Orantes M, Braak H. Phases of a beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–1800. doi: 10.1212/WNL.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 25.Yokota O, Terada S, Ishizu H, Ishihara T, Ujike H, Nakashima H, et al. Cyclooxygenase-2 in the hippocampus is up-regulated in Alzheimer’s disease but not in variant Alzheimer’s disease with cotton wool plaques in humans. Neurosci Lett. 2003;343:175–179. doi: 10.1016/S0304-3940(03)00339-2. [DOI] [PubMed] [Google Scholar]
- 26.Yokota O, Terada S, Ishizu H, Ujike H, Ishihara T, Namba M, et al. Variability and heterogeneity in Alzheimer’s disease with cotton wool plaques: a clinicopathological study of four autopsy cases. Acta Neuropathol. 2003;106:348–356. doi: 10.1007/s00401-003-0737-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Details of methods. (DOCX 20 kb)
Antibodies used in this study. (DOCX 24 kb)
Data Availability Statement
Not applicable.