

Abstract
Early infantile epileptic encephalopathy (EIEE) is a severe disorder associated with epilepsy, developmental delay and intellectual disability, and in some cases premature mortality. We report the case of a female infant with EIEE and strikingly suppressed respiratory dysfunction that led to death. Postmortem research evaluation revealed hypoplasia of the arcuate nucleus of the medulla, a candidate region for respiratory regulation. Genetic evaluation revealed heterozygous variants in the related genes NRXN1 (c.2686C>T, p.Arg896Trp) and NRXN2 (c.3176G>A, p.Arg1059Gln), one inherited from the mother with family history of sudden infant death syndrome (SIDS) and one from the father with family history of febrile seizures. Although there are no previous reports with the digenic combination of NRXN1 and NRXN2 variants, patients with biallelic loss of NRXN1 in humans and double neurexin 1α/2α knockout mice have severe breathing abnormalities, corresponding to the respiratory phenotype of our patient. These observations and the known interaction between the NRXN1 and NRXN2 proteins lead us to hypothesize that digenic variants in NRXN1 and NRXN2 contributed to the phenotype of EIEE, arcuate nucleus hypoplasia, respiratory failure, and death.
Keywords: aplasia/hypoplasia of the brainstem, frontoparietal cortical dysplasia, infantile encephalopathy, respiratory failure
INTRODUCTION
The developmental and epileptic encephalopathy (DEE) syndromes that present in infancy include Ohtahara syndrome, or early infantile epileptic encephalopathy (EIEE), which is characterized by a burst suppression pattern on electroencephalogram (EEG) and a generally poor prognosis, with high mortality rates in infancy (Beal et al. 2012). Anecdotally, we have observed that EIEE is associated with dysfunctional regulation of respiration, which has also been described in the related epileptic encephalopathy West syndrome (Jansen et al. 2013). The anatomic basis of respiratory dysfunction in such cases has not been studied in detail, and hypotheses include the role of central nervous system depression that may be exacerbated by medications given for seizure control as well as dysregulation of the normal processes of developmental maturation of respiration. Neurexins are cell adhesion proteins that play a role in synaptic transmission through interaction with neuroligins. Deletions within NRXN1 have been associated with a range of neurodevelopmental disorders including intellectual disability, developmental delay, speech and language delay, autism spectrum disorders, schizophrenia, and epilepsy (Curran et al. 2013; Møller et al. 2013; Lal et al. 2015). Heterozygous loss of function of NRXN1 has been associated with susceptibility to generalized epilepsy (Olson et al. 2014; Lal et al. 2015). Interestingly, epilepsy is a more consistent feature in patients with more carboxy-terminal deletions, including the β-isoform (Schaaf et al. 2012; Dabell et al. 2013). Homozygous disruption of NRXN1 in humans causes a severe Pitt–Hopkins-like syndrome including early-onset epilepsy, breathing difficulties, severe intellectual disability, absent speech, stereotypies, and autistic features (Zweier et al. 2009; Harrison et al. 2011; Duong et al. 2012; Béna et al. 2013; Imitola et al. 2014). Variants and truncations in NRXN2, although less frequently described, have been associated with autism spectrum disorder and language delay (Gauthier et al. 2011). Deletion of the Nrxn2α gene in mice can replicate some of the core symptoms of autism (Dachtler et al. 2014). Here we present the case of a female infant with EIEE with striking respiratory dysfunction, particularly during sleep, who was found to have heterozygous variants in both NRXN1 and NRXN2, one inherited from each parent. Although premortem neuroimaging had been assessed as normal, extensive postmortem neuroimaging and neuropathology were performed to evaluate the regions that contribute to regulation of respiratory function and that are thought to contribute to premature death in conditions such as sudden infant death syndrome (SIDS), sudden unexpected death in childhood with febrile seizures, and sudden unexpected death in epilepsy patients (SUDEP).
RESULTS
Clinical Presentation and Family History
The patient was an infant girl who initially presented at 8 d of life for poor feeding, lethargy, and weight loss. She was born to a 25-yr-old G2P2 mother, who was on levothyroxine and insulin for hypothyroidism and gestational diabetes. Labor was induced at 38 weeks of gestation for decreased fetal movement and concerning nonstress tests, but the patient emerged vigorous with Apgar scores of 8 at 1 and 5 min. Her examination was notable for a weak cry, and laryngoscopy revealed supraglottic edema. She was discharged but continued to have difficulties with breastfeeding and became increasingly lethargic. She presented at 8 d of life with significant lethargy and hypertonicity of the extremities. Initial diagnostic evaluation included MRI of the brain and spine, which were considered normal. Video-EEG monitoring was performed to evaluate whether apneic episodes were seizures. Although those episodes were not associated with EEG seizures, the study revealed a markedly abnormal background with a burst suppression pattern, as well as multiple focal seizures arising from the midline and bilateral parietal regions that manifested clinically as bilateral tonic arm extension. Based on her seizures, encephalopathy, and EEG findings, she was diagnosed with EIEE. She was treated with multiple antiepileptic drugs (AEDs), including phenobarbital, levetiracetam, zonisamide, and intermittent diazepam. Her clinical seizure frequency declined, but she continued to have frequent subclinical seizures with continued burst suppression pattern in the background. She had empiric trials of pyridoxine, pyridoxal-5-phosphate, folic acid, and biotin, with no effects on her clinical status or EEG. She also had a comprehensive metabolic evaluation (including serum lactate, pyruvate, amino acids, and cerebrospinal fluid amino acids), which did not identify an etiology for her presentation.
The patient's clinical course was additionally marked by an irregular breathing pattern, with prolonged periods of apnea associated with desaturation. These episodes did not correlate with EEG changes and were thus not thought to be due to seizure activity; of note, they often occurred during clinical sleep periods. Over the hospital course, her respiratory support requirement escalated from none initially, to supplemental oxygen via nasal cannula, to noninvasive positive pressure support (CPAP and BiPAP), and finally to intubation on day of life 38. Attempts were made to minimize the use of as-needed diazepam for clusters of clinical seizures in the event that this intervention was affecting her respiratory reserve, but even in the absence of additional diazepam respiratory effort was compromised. Given the patient's escalating respiratory support requirements and overall grim prognosis due to her EIEE diagnosis and persistent burst suppression pattern on EEG, a multidisciplinary and family meeting was held on day of life 43, at which point the decision was made to redirect care, and she died on day of life 49 following extubation. The patient's parents requested to donate her brain to research; postmortem neuroimaging and neuropathology studies were performed after consent for autopsy and enrollment into studies approved by the Boston Children's Hospital Institutional Review Board.
The patient's family history was notable for SIDS (mother's half-sister), as well as generalized tonic–clonic seizure and/or febrile seizures in multiple family members across three generations in her paternal grandmother's family. The patient's father and her father's sister both had multiple febrile seizures in childhood and were developmentally normal (Fig. 1).
Figure 1.

Pedigree of the family. (FS) Febrile seizures in childhood, (GTCS) generalized tonic–clonic seizures, (SIDS) sudden infant death syndrome.
Pre- and Postmortem Phenotypic Analyses
Neuroimaging
Conventional premortem neuroimaging performed at 10 d of age had not revealed a significant abnormality apart from mildly increased T2 signal intensity in the bilateral dentate nuclei of the cerebellar hemispheres. Notably, coronal imaging detailing the hippocampus was not performed as it is not part of the routine neonatal imaging protocol.
High-resolution postmortem imaging performed on a research basis revealed asymmetry of the hippocampal formations, with a globular shape and slightly larger size of the left hippocampus (Fig. 2A). Also, note was made of excessive folding and hyperconvolution of the dentate gyrus bilaterally, lending a serrated appearance to the dentate gyrus, more appreciably on the right (Fig. 2B,C). Close examination of the brainstem showed small inferior olivary nuclei and hypoplasia of the arcuate nucleus (Fig. 2E).
Figure 2.
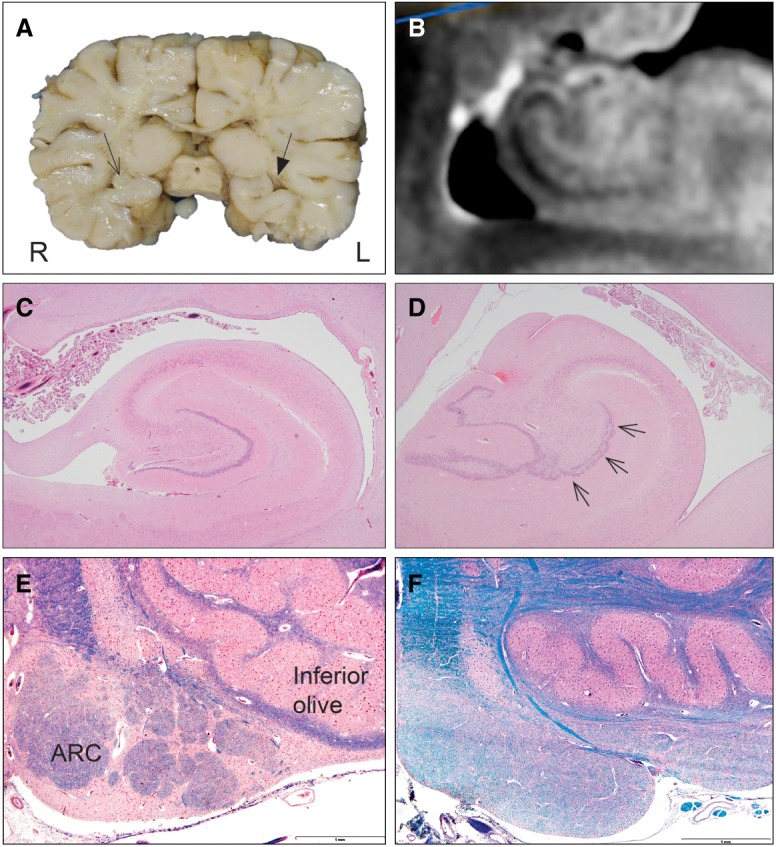
Postmortem neuroanatomy and neuropathology. (A) Macroscopic abnormalities of the brain with a globular shape and slightly larger size of the left hippocampus (filled triangle arrow). (B) High-resolution T2 postmortem magnetic resonance imaging of the right hippocampus. (C) Control hippocampus. H&E staining, 2×. (D) Pathology of the patient's left hippocampus demonstrating focal granule cell bilamination in the dentate gyrus (small arrows). H&E staining, 2×. (E) Control arcuate nucleus (ARC, arrows) in clusters along the pyramid at the ventral surface of the medulla. H&E, 4×. (F) Absence of ARC clusters in the brainstem of the patient. LFB staining, 4×.
Neuropathological examination revealed bilateral maldevelopment of cortical and hippocampal folding with focal cortical dysplasia and granule cell dispersion in the dentate gyrus, with partial bilamination (right hippocampus shown in Fig. 2B and left hippocampus in Fig. 2C, with left dentate gyrus bilamination in Fig. 2C). In addition to the cortical abnormalities, there was anomalous development of the olivo-arcuate system in the brainstem, with significant hypoplasia of the arcuate nucleus along the ventral medullary surface (Fig. 2E). There was mild gliosis of the reticular formation of the medulla, including the paragigantocellularis lateralis, the putative central rhythm generator in the rostral medulla of experimental animals homologous to the pre-Bötzinger complex. Other cardiorespiratory-related nuclei throughout the brainstem (e.g., the nucleus of the solitary tract) were unremarkable.
Genomic Analyses
The patient had been evaluated for a genetic diagnosis with a range of investigations including initially commercial chromosomal microarray and single-gene sequencing of PHOX2B, associated with congenital central hypoventilation syndrome (CCHS), which were unrevealing. A comprehensive epilepsy gene panel, including deletion/duplication analysis, revealed variants of unknown significance in GAMT (NM_00156.4, c.79T>C, p.Tyr27His), MBD5 (NM_018328.4, c.2000T>G, p.Leu667Trp), and NRXN1 (NM_004801.4, c.2686C>T, p.Arg896Trp), all of which were inherited. Because these findings could not fully explain the EIEE and respiratory phenotype, whole-exome sequencing (WES) of the patient and her parents was performed and revealed an additional variant in NRXN2 (NM_138732.2, c.3176G>A, p.Arg1059Gln). We did not find de novo, homozygous recessive, or compound heterozygous variants that could definitively explain the phenotype of our patient (Supplemental data). In addition, we did not find variants in phenotype-related genes or from a large candidate gene list that could explain the phenotype. The variant in NRXN1 was shown to be maternally inherited and located in exon 15, in the carboxyl terminus of the α-neurexin form. The NRXN2 variant was paternally inherited and located in exon 16, in the carboxyl terminus of the α-neurexin form (Supplemental Fig. S1). The information of the variants is shown in Table 1. The allele frequency and pathogenicity prediction of the variants are shown in Table 2. Both variants are rare/absent in the general population and are predicted to be deleterious according to different tools of pathogenicity prediction. In addition, both genes are under functional constraint (Lek et al. 2016). Both variants have been classified using the ACMG guidelines (Richards et al. 2015) as variants of unknown significance.
Table 1.
Variant table
| Gene | Variant | Chr | HGVS DNA Ref. | HGVS protein Ref. | Variant type | Predicted effect | dbSNP ID | Genotype | ClinVar ID |
|---|---|---|---|---|---|---|---|---|---|
| NRXN1 | c.2686C>T (p.Arg896Trp) | 2 | NM_004801 | NP_004792 | Missense | Substitution | rs79605277 | Heterozygous | SCV000241889.13 |
| NRXN2 | c.3176G>A (p.Arg1059Gln) | 11 | NM_138732 | NP_620060 | Missense | Substitution | rs777033569 | Heterozygous | SCV000863537 |
(Chr) Chromosome, (HGVS) Human Genome Variation Society.
Table 2.
Functional prediction and conservation scores of the two variants
| Gene | Variant | AF (gnomAD) | SIFT | PP2-HDIV | MT | CADD | z-score | pLI | GERP-RS | ClinVar |
|---|---|---|---|---|---|---|---|---|---|---|
| NRXN1 | c.2686C>T (p.Arg896Trp) | 0 | D | D | DC | 26.5 | 3.02 | 1 | 4.52 | VUS |
| NRXN2 | c.3176G>A (p.Arg1059Gln) | 1.22 × 105 | D | D | DC | 29.7 | 5.81 | 1 | 4.13 | N/A |
(AF) Allele frequency, (CADD) combined annotation-dependent depletion, (D) deleterious, (DC) disease causing, (GERP) Genomic Evolutionary Rate Profiling, (MT) MutationTaster, (N/A) not available, (pLI) loss-of-function constraint metric, (PP-2) PolyPhen-2, (SIFT) Sorting Intolerant from Tolerant, (VUS) variant of uncertain significance, (z-score) missense constraint metric.
An in silico analysis within Decipher revealed 19 patients with NRXN1 and seizures. One patient with a copy-number variation (CNV) loss also had breathing dysregulation, generalized tonic–clonic seizures, and severe developmental delay (ID: 266445). There were no patients with NRXN2 and seizures in Decipher. About 13/18 patients with variants or CNVs in NRXN2 had intellectual disability or neurodevelopmental delay and one patient presented with asthma.
DISCUSSION
We present the case of a female infant with EIEE and strikingly suppressed respiratory drive during sleep. Although it is not uncommon for infants with severe epilepsy to require intubation when having many seizures and receiving multiple AEDs, respiratory failure occurred in our patient in the setting of only a modest combination of medications, and persistent respiratory failure could not be explained by the number of seizures nor the degree of medication that was administered to treat seizures. The premortem observations of deficient respiratory drive and reserve correlate with our postmortem observations—on neuropathology and neuroimaging—of near-complete absence of the arcuate nucleus. The diagnosis of EIEE itself warrants a thorough evaluation for etiology, including genetic etiology with a focus on epilepsy-related genes (Olson et al. 2017). In addition, the profound respiratory phenotype, in combination with the arcuate hypoplasia and temporal lobe abnormalities noted postmortem, prompted additional research analysis with WES.
The combination of premortem diagnosis and a coordinated postmortem neuroradiological and neuropathological assessment led to a more detailed phenotype that incorporated previously unrecognized abnormalities of not only the cerebral cortex and hippocampi but also the brainstem in the form of almost complete absence of the arcuate nucleus. Our group has shown previously that the arcuate nucleus is cytologically and spatially homologous to the ventral respiratory chemosensitive zones in cats on the ventral medullary surface (Filiano et al. 1990). We also reported severe arcuate hypoplasia in ∼5% of SIDS cases (Filiano and Kinney 1992; Kinney et al. 2016), as well as binding defects in the arcuate nucleus in SIDS cases to neurotransmitters implicated in ventral chemosensory control—for example, muscarinic cholinergic receptors (Kinney et al. 1995) and serotonergic receptors (Panigrahy et al. 2000). Interestingly, the arcuate nucleus has been previously implicated in the respiratory function and chemoreception in a few case reports of CCHS (Folgering et al. 1979; Tomycz et al. 2010). Hypoplasia of the arcuate nucleus and neurochemical abnormalities in SIDS (Filiano and Kinney 1992; Kinney et al. 1995; Panigrahy et al. 1997, 2000) raise the possibility that at least a subset of SIDS cases have impaired central chemosensitivity that may contribute to respiratory dysfunction and death. Recent studies in adults have underscored the possible role of the arcuate nucleus at the ventral medullary surface as critical for central chemosensitivity (Benarroch et al. 2007) in human adult disorders. Besides arcuate nucleus hypoplasia, the neuropathological assessment of our case also showed hippocampal asymmetry and dentate gyrus anomalies. We have reported these findings previously in toddlers with sudden death and febrile seizures (Kinney et al. 2016), raising the question of whether these anatomical defects, thought to be developmental, contribute to sudden death through an interaction with respiratory control mechanisms, although the precise mechanisms have yet to be elucidated.
Detailed postmortem neuropathological assessments of patients with EIEE are limited to a few case reports, including one from our group describing a patient with a pathogenic variant in SCN2A (Touma et al. 2013). We had hypothesized that the profound respiratory dysfunction in our patient was due to an independent defect in the gene PHOX2B, but this proved not to be the case. We did not identify a genetic cause of the patient's epilepsy, suspected genetic, with the first tier of clinical testing with CMA and epilepsy panel. However, the nature of the epileptic encephalopathy and the positive family history on both sides of the family suggested that a combination of genetic factors may have together contributed to the patient's phenotypes. Having identified two disease-relevant variants in two related genes, NRXN1 and NRXN2, we hypothesize a digenic process in which neither variant is sufficient to produce severe disease on its own but in combination the two together could have led to our patient's phenotype. Digenic inheritance is a hypothesized form of complex genetic disease, only a small number of conditions have been reported with digenic inheritance—for example, some forms of deafness, long QT syndrome, and Bardet–Biedl syndrome (Schäffer 2013). An operational definition by Schäffer says: “inheritance is digenic when the variant genotypes at two loci explain the phenotypes of a patient more clearly than the genotype at one locus alone” (Schäffer 2013). The family history of SIDS on the maternal side and febrile seizures on the paternal side suggest that there may be some contribution of the neurexin-related variants in other phenotypes. Unfortunately, DNA is not available from additional family members, including the child who died of SIDS, to further explore this hypothesis within the pedigree.
Neurexins are neuronal adhesion molecules located in the presynaptic terminal, where they interact with postsynaptic neuroligins to form a trans-synaptic complex required for efficient neurotransmission in the brain. In mammals, three neurexin genes (NRXN1, NRXN2, and NRXN3) have been described. They are mainly localized at the presynaptic terminal and form heterophilic interactions with neuroligins, which are localized to the postsynaptic compartment. Thus, the neurexin–neuroligin complex forms a trans-synaptic adhesion complex. Each neurexin gene encodes a long α-neurexin isoform and a short β-neurexin isoform. In addition to the two major isoforms, there are five conserved alternative splice sites in α-neurexins (splice site [SS]#1 to SS#5) and two in β-neurexins (SS#4 and SS#5), whereby they can be transcribed in thousands of alternatively spliced isoforms, contributing to distinct structural domains and variability (Reissner et al. 2013; Südhof 2017). α-Neurexins contain six LNS (laminin-neurexin-sex hormone binding globulin) domains with three epidermal growth factor (EGF)-like domains interspersed. β-Neurexins are identical to the carboxyl terminus of α-neurexins starting from the last LNS domain but have a unique amino-terminal stretch of 37 histidine-rich residues. The LNS domains are thought to behave like glycan-binding lectins. The variants of our patient in NRXN1 and NRXN2 are located in LNS4 and LNS5, respectively. A general function of the LNS domains has not been demonstrated so far, but, for example, dystroglycan requires a specific glycosylation to bind to LNS4-5 (Wizemann et al. 2003). In addition, the variants are located nearby SS#3. Although we do not have functional studies, these findings might support the possibility that the variants influence splicing and the transcription alternative splice isoforms.
To date, only six case reports of biallelic loss of NRXN1 in humans have been described (Table 3; Zweier et al. 2009; Harrison et al. 2011; Duong et al. 2012). Patients with biallelic loss of NRXN1 have a variable neurodevelopmental phenotype, presenting with epilepsy, breathing abnormalities, speech delay, and abnormal sleep–wake cycle. Some of the reported patients with biallelic loss of NRXN1 variants have severe breathing abnormalities. Although highly similar, our patient had a more severe phenotype than the patients with biallelic loss of NRXN1. She presented with arcuate nucleus hypoplasia, hippocampal asymmetry, and dentate gyrus anomalies, raising the question, “Did these anomalies contribute to the death of our patient?” Thus, it seems plausible that biallelic loss of NRXN1 in humans could lead to a fully penetrant, severe neurodevelopmental phenotype, whereas the heterozygous deletion likely represents a susceptibility factor for variable cognitive, neurological, and psychiatric disorders (Béna et al. 2013). Functional studies in mice revealed that deletion of α-neurexins did not impair prenatal viability but markedly compromised postnatal survival. Triple-knockout mice died on the first day and most double-knockout mice died within the first week. Remarkably, combined Nrxn1a−/− and Nrxn2a−/− and triple-knockout mice had the same level of breathing difficulties. In addition, they showed that the irregular respiratory rhythm of these mice was an intrinsic property of the brainstem (Missler et al. 2003; Etherton et al. 2009).
Table 3.
Clinical characteristics of patients with biallelic NRXN1 and NRXN1–NRXN2 variants
| Patients | Our proband | Zweier et al. (2009) | Harrison et al. (2011)— sibling 1 | Harrison et al. (2011)— sibling 2 | Duong et al. (2012) | Béna et al. (2013) | Imitola et al. (2014)— sibling 1 | Imitola et al. (2014)— sibling 2 |
|---|---|---|---|---|---|---|---|---|
| NRXN variants | NRXN1: c.2686C>T; p.Arg896Trp | NRXN1: 113 kb del (2:51001003–51113677) | NRXN1: 79 kb del (2:50214717–50293739) | NRXN1: 79 kb del (2:50214717–50293739) | NRXN1: 451 kb del (2:50812891–51263512) | NRXN1: 181 kb del (2:50963194–51144527) | NRXN1: 130 kb del (2:51047025–51176742) | NRXN1: 107 kb del (2:51047060–51154995) |
| NRXN2: c.3176G>A; p.Arg1059Gln | NRXN1: p.Ser979Ter | NRXN1: 287 kb del (2:51008023–51294599) | NRXN1: 287 kb del (2:51008023–51294599) | NRXN1: c.2880-1G>A; p.Tyr961ThrfsX31 | NRXN1: 401 kb del (2:50963194–51364465) | NRXN1: 135 kb del (2:51184729–51319450) | NRXN1: 174 kb del (2:51154996–51328796) | |
| Age | 49 da | 18 yr | 16 yr | 11 yr | 33 yr | 10 yr | 7 yr | 7 yr |
| Gender | F | F | F | F | M | F | M | F |
| Developmental delay | N/A | Severe | Severe | Severe | Severe | Severe | Severe | Severe |
| Seizure onset | 2 d | None | 6 mo | 4 mo | 7 mo | 6.5 yr | 5 yr | None |
| Seizure type | EIEE | None | EIEE | EIEE | IS, TC | AA, TC, HT | AA | None |
| Age of walking | N/A | 2 yr | 5 yr | None | 3 yr | 26 mo | 2 yr | 2 yr |
| Speech | N/A | None | 20–25 words | None | None | Few words | 2–10 words | 2–5 words |
| Breathing abnormality | Irregular breathing with periods of severe apnea | Hyperventilation | Hyperventilation | Breath holding | N/A | Recurrent airway infections | No | Hyperventilation |
| Stereotypies | N/A | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Constipation | N/A | Yes | Yes | Yes | N/A | Yes | Yes | Yes |
| Abnormal sleep–wake cycle | N/A | Yes | Yes | Yes | N/A | Yes | Yes | Yes |
| Brain MRI | Normal | N/A | Normal | N/A | Moderate atrophy and cyst deflecting right hippocampus | Normal | Normal | – |
| Other | Extreme lethargy and hypertonicity of the extremities | Strabismus, protruding tongue, excessive drooling, broad mouth | GORD, scoliosis, pulmonary stenosis, hypotonia, early-onset, puberty, self-harm behavior | GORD, scoliosis, pulmonary stenosis, hypotonia, early-onset, puberty | Strabismus, perceptive hearing impairment, autism, dysmorphic features | Recurrent FS during her first 2 yr, feeding problems and GORD, autism, no obvious dysmorphic features | Dysphagia, aggressive and self-harm behavior, and ventricular septal defect | Aggressive behavior, strabismus, hypotonia and ataxia |
(AA) atypical absence, (EIEE) early infantile epileptic encephalopathy, (F) female, (GORD) gastroesophageal reflux disease, (HT) hypotonic seizures, (IS) infantile spasms, (M) male, (MRI) magnetic resonance imaging, (N/A) not available, (TC) tonic–clonic seizures.
aDeceased.
We have assessed the findings in our patient in the context of patients with biallelic loss of NRXN1 and studies in mice involving Nrxn1 and Nrxn2. The reported studies provide some evidence to support the assertion that, in principle, digenic pathogenic variants in NRXN1 and NRXN2 such as those in our patient act together to produce a severe phenotype. However, we did not perform functional studies of the effect of the specific variants in our patient. There are no other reported human cases with digenic variants in NRXN1/NRXN2 to which we can compare our patient. Although the NRXN2 variant is present in three individuals in gnomAD, overall NRXN2 is very intolerant for missense variants (constraint missense z-score of 5.81; http://exac.broadinstitute.org/). In the current study, we performed WES analysis, which is associated with a high sequencing depth of the protein-coding regions. Because WES targets only the protein-coding regions (1%–2% of the genome), we may have missed variants in regulatory regions or small intragenic CNVs, which might account instead for the phenotype. We did evaluate for larger CNVs with a commercial chromosomal microarray.
CONCLUSION
We present a case with EIEE, strikingly suppressed respiratory effort, and arcuate nucleus hypoplasia identified on postmortem research evaluation in the setting of two variants in NRXN1 and NRXN2, one inherited from the patient's mother who had a family history of SIDS and one from her father who had a history of febrile seizures and family history of epilepsy. Patients with biallelic loss of NRXN1 in humans and double α-neurexin knockout mice have severe breathing abnormalities, corresponding to our patient's respiratory phenotype. Heterozygous NRXN1 variants can be disease-associated, and the NRXN1 and NRXN2 proteins have a known interaction. Taken together, these findings lead us to hypothesize that our patient's digenic variants in NRXN1 and NRXN2 contributed to the combined phenotype of EIEE and arcuate nucleus hypoplasia, which in turn contributed to her respiratory failure and ultimately death. We hypothesize that the neuroanatomical abnormalities identified played a role in brainstem-mediated respiratory dysfunction, through mechanisms similar to those hypothesized for SIDS and also associated with loss of function of neurexin.
METHODS
Neuroimaging Technique
Ex vivo postmortem imaging was performed after fixation of the brain on a 3-Tesla scanner using a 64-channel coil. High-resolution T1- and T2-weighted imaging of the whole brain was performed at 300-micron resolution. Additional 200-micron resolution small field of view imaging of the hippocampi was performed as well. Finally, multishell multi B-value 1 mm isotropic resolution diffusion-weighted imaging was performed.
Genomic Analyses
The microarray was performed by Claritas Genomics using a 4 × 180k Agilent array with a resolution of 5–20 kb for known disease regions and around OMIM genes and with a backbone resolution of 100 kb.
DNA extracted from whole blood underwent capture for exome sequencing using the Agilent SureSelect XTHuman All Exon v4 (Broad Institute). Sequencing of 100-bp paired-end reads was obtained using Illumina HiSeq (Illumina). Coverage was >90% meeting 20× coverage. Our data analysis and variant calling methods have been described previously (Olson et al. 2017). We utilized the BCH Connect Genomics Gateway integrated with the WuXi NextCODE analysis platform (Gudbjartsson et al. 2016) for variant interrogation and analysis. We performed WES for the proband and his parents and evaluated for de novo, homozygous recessive and/or compound heterozygous variants. In addition, we filtered the WES data for variants in a large list of candidate genes (n = 512) and for disease-associated genes. Functional prediction scores were obtained from the dbNSFP database version 3.5 (August 2017; http://varianttools.sourceforge.net/). In total we used five prediction scores (SIFT, PolyPhen-2-HVAR, PolyPhen-2-HDIV, MutationTaster, CADD) and one conservation score (GERP++). We also looked for the functional constraint gene scores (Lek et al. 2016). We classified the pathogenicity of the variants using the ACMG guidelines (Richards et al. 2015). We checked Decipher (https://decipher.sanger.ac.uk) for additional CNVs in NRXN1/2.
ADDITIONAL INFORMATION
Data Deposition and Access
Raw data were not deposited but may be available by contacting the authors. The variants were deposited to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and can be found under accession numbers SCV000241889.13 and SCV000863537.
Ethics Statement
The study was approved by the Boston Children's Hospital Institutional Review Board, with full written consent obtained from the parents, providing consent for the proband.
Acknowledgments
We are grateful to the family of the patient for their participation in research, and we thank the staff of the Intensive Care Unit of Boston Children's Hospital who cared for the patient and family and conveyed and honored the family's requests for research participation. We also thank the Aaron's Ohtahara foundation, whose early support for our group allowed our Epilepsy Genetics Program to pursue genetic studies for Ohtahara syndrome. We are grateful to the thoughtful reviewers of the paper who provided valuable feedback.
Author Contributions
All authors were involved in the conception or design of the study, analysis and interpretation of the data, and/or drafting the article or revising it critically for important intellectual content.
Funding
A.M.R. was supported by a Fellowship of the Belgian American Educational Foundation and by a Fulbright Program grant sponsored by the Bureau of Educational and Cultural Affairs of the United States Department of State and administered by the Institute of International Education. A.H.P. was supported by the Boston Children's Hospital Translational Research Program. A.M.R., R.D.G., and A.H.P. were supported by the Isaac Stone Award from Citizens United for Research in Epilepsy.
Competing Interest Statement
The authors have declared no competing interest.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Beal JC, Cherian K, Moshe SL. 2012. Early-onset epileptic encephalopathies: Ohtahara syndrome and early myoclonic encephalopathy. Pediatr Neurol 47: 317–323. 10.1016/j.pediatrneurol.2012.06.002 [DOI] [PubMed] [Google Scholar]
- Béna F, Bruno DL, Eriksson M, van Ravenswaaij-Arts C, Stark Z, Dijkhuizen T, Gerkes E, Gimelli S, Ganesamoorthy D, Thuresson AC, et al. 2013. Molecular and clinical characterization of 25 individuals with exonic deletions of NRXN1 and comprehensive review of the literature. Am J Med Genet B Neuropsychiatr Genet 162B: 388–403. 10.1002/ajmg.b.32148 [DOI] [PubMed] [Google Scholar]
- Benarroch EE, Schmeichel AM, Low PA, Parisi JE. 2007. Depletion of putative chemosensitive respiratory neurons in the ventral medullary surface in multiple system atrophy. Brain 130: 469–475. 10.1093/brain/awl357 [DOI] [PubMed] [Google Scholar]
- Curran S, Ahn JW, Grayton H, Collier DA, Ogilvie CM. 2013. NRXN1 deletions identified by array comparative genome hybridisation in a clinical case series—further understanding of the relevance of NRXN1 to neurodevelopmental disorders. J Mol Psychiatry 1: 4 10.1186/2049-9256-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabell MP, Rosenfeld JA, Bader P, Escobar LF, El-Khechen D, Vallee SE, Dinulos MBP, Curry C, Fisher J, Tervo R, et al. 2013. Investigation of NRXN1 deletions: clinical and molecular characterization. Am J Med Genet A 161: 717–731. 10.1002/ajmg.a.35780 [DOI] [PubMed] [Google Scholar]
- Dachtler J, Glasper J, Cohen RN, Ivorra JL, Swiffen DJ, Jackson AJ, Harte MK, Rodgers RJ, Clapcote SJ. 2014. Deletion of α-neurexin II results in autism-related behaviors in mice. Transl Psychiatry 4: e484–e488. 10.1038/tp.2014.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong L, Klitten LL, Møller RS, Ingason A, Jakobsen KD, Skjødt C, Didriksen M, Hjalgrim H, Werge T, Tommerup N. 2012. Mutations in NRXN1 in a family multiply affected with brain disorders: NRXN1 mutations and brain disorders. Am J Med Genet B Neuropsychiatr Genet 159B: 354–358. 10.1002/ajmg.b.32036 [DOI] [PubMed] [Google Scholar]
- Etherton MR, Blaiss CA, Powell CM, Südhof TC. 2009. Mouse neurexin-1α deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc Natl Acad Sci 106: 17998–18003. 10.1073/pnas.0910297106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filiano JJ, Kinney HC. 1992. Arcuate nucleus hypoplasia in the sudden infant death syndrome. J Neuropathol Exp Neurol 51: 394–403. 10.1097/00005072-199207000-00002 [DOI] [PubMed] [Google Scholar]
- Filiano JJ, Choi JC, Kinney HC. 1990. Candidate cell populations for respiratory chemosensitive fields in the human infant medulla. J Comp Neurol 293: 448–465. 10.1002/cne.902930308 [DOI] [PubMed] [Google Scholar]
- Folgering H, Kuyper F, Kille JF. 1979. Primary alveolar hypoventilation (Ondine's curse syndrome) in an infant without external arcuate nucleus. Case report. Bull Eur Physiopathol Respir 15: 659–665. [PubMed] [Google Scholar]
- Gauthier J, Siddiqui TJ, Huashan P, Yokomaku D, Hamdan FF, Champagne N, Lapointe M, Spiegelman D, Noreau A, Lafrenière RG, et al. 2011. Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Hum Genet 130: 563–573. 10.1007/s00439-011-0975-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudbjartsson H, Georgsson GF, Gudjónsson SA, Valdimarsson RP, Sigurdsson JH, Stefánsson SK, Másson G, Magnússon G, Pálmason V, Stefánsson K. 2016. GORpipe: a query tool for working with sequence data based on a Genomic Ordered Relational (GOR) architecture. Bioinformatics 32: 3081–3088. 10.1093/bioinformatics/btw199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison V, Connell L, Hayesmoore J, McParland J, Pike MG, Blair E. 2011. Compound heterozygous deletion of NRXN1 causing severe developmental delay with early onset epilepsy in two sisters. Am J Med Genet A 155: 2826–2831. 10.1002/ajmg.a.34255 [DOI] [PubMed] [Google Scholar]
- Imitola J, Walleigh D, Anderson CE, Jethva R, Carvalho KS, Legido A, Khurana DS. 2014. Fraternal twins with autism, severe cognitive deficit, and epilepsy: diagnostic role of chromosomal microarray analysis. Semin Pediatr Neurol 21: 167–171. 10.1016/j.spen.2014.04.027 [DOI] [PubMed] [Google Scholar]
- Jansen K, Varon C, Huffel S, Lagae L. 2013. Early respiratory dysfunction as a biomarker for epileptic encephalopathy. Acta Neurol Scand 128: 381–385. 10.1111/ane.12133 [DOI] [PubMed] [Google Scholar]
- Kinney HC, Filiano JJ, Sleeper LA, Mandell F, Valdes-Dapena M, White WF. 1995. Decreased muscarinic receptor binding in the arcuate nucleus in sudden infant death syndrome. Science 269: 1446–1450. 10.1126/science.7660131 [DOI] [PubMed] [Google Scholar]
- Kinney HC, Poduri AH, Cryan JB, Haynes RL, Teot L, Sleeper LA, Holm IA, Berry GT, Prabhu SP, Warfield SK, et al. 2016. Hippocampal formation maldevelopment and sudden unexpected death across the pediatric age spectrum. J Neuropathol Exp Neurol 75: 981–997. 10.1093/jnen/nlw075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal D, Ruppert AK, Trucks H, Schulz H, de Kovel C, Kasteleijn-Nolst Trenité D, Sonsma ACM, Koeleman BPC, Lindhout D, Weber YG, et al. 2015. Burden analysis of rare microdeletions suggests a strong impact of neurodevelopmental genes in genetic generalised epilepsies. PLoS Genet 11: e1005226 10.1371/journal.pgen.1005226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536: 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missler M, Zhang W, Rohlmann A, Kattenstroth G, Hammer RE, Gottmann K, Südhof TC. 2003. α-Neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature 423: 939–948. 10.1038/nature01755 [DOI] [PubMed] [Google Scholar]
- Møller RS, Weber YG, Klitten LL, Trucks H, Muhle H, Kunz WS, Mefford HC, Franke A, Kautza M, Wolf P, et al. 2013. Exon-disrupting deletions of NRXN1 in idiopathic generalized epilepsy. Epilepsia 54: 256–264. 10.1111/epi.12078 [DOI] [PubMed] [Google Scholar]
- Olson H, Shen Y, Avallone J, Sheidley BR, Pinsky R, Bergin AM, Berry GT, Duffy FH, Eksioglu Y, Harris DJ, et al. 2014. Copy number variation plays an important role in clinical epilepsy. Ann Neurol 75: 943–958. 10.1002/ana.24178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson HE, Kelly M, LaCoursiere CM, Pinsky R, Tambunan D, Shain C, Ramgopal S, Takeoka M, Libenson MH, Julich K, et al. 2017. Genetics and genotype–phenotype correlations in early onset epileptic encephalopathy with burst suppression. Ann Neurol 81: 419–429. 10.1002/ana.24883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahy A, Filiano JJ, Sleeper LA, Mandell F, Valdes-Dapena M, Krous HF, Rava LA, White WF, Kinney HC. 1997. Decreased kainate receptor binding in the arcuate nucleus of the sudden infant death syndrome. J Neuropathol Exp Neurol 56: 1253–1261. 10.1097/00005072-199711000-00010 [DOI] [PubMed] [Google Scholar]
- Panigrahy A, Filiano J, Sleeper LA, Mandell F, Valdes-Dapena M, Krous HF, Rava LA, Foley E, White WF, Kinney HC. 2000. Decreased serotonergic receptor binding in rhombic lip-derived regions of the medulla oblongata in the sudden infant death syndrome. J Neuropathol Exp Neurol 59: 377–384. 10.1093/jnen/59.5.377 [DOI] [PubMed] [Google Scholar]
- Reissner C, Runkel F, Missler M. 2013. Neurexins. Genome Biol 14: 213 10.1186/gb-2013-14-9-213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf CP, Boone PM, Sampath S, Williams C, Bader PI, Mueller JM, Shchelochkov OA, Brown CW, Crawford HP, Phalen JA, et al. 2012. Phenotypic spectrum and genotype–phenotype correlations of NRXN1 exon deletions. Eur J Hum Genet 20: 1240–1247. 10.1038/ejhg.2012.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäffer AA. 2013. Digenic inheritance in medical genetics. J Med Genet 50: 641–652. 10.1136/jmedgenet-2013-101713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. 2017. Synaptic neurexin complexes: a molecular code for the logic of neural circuits. Cell 171: 745–769. 10.1016/j.cell.2017.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomycz ND, Haynes RL, Schmidt EF, Ackerson K, Kinney HC. 2010. Novel neuropathologic findings in the Haddad syndrome. Acta Neuropathol 119: 261–269. 10.1007/s00401-009-0599-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touma M, Joshi M, Connolly MC, Grant PE, Hansen AR, Khwaja O, Berry GT, Kinney HC, Poduri A, Agrawal PB. 2013. Whole genome sequencing identifies SCN2A mutation in monozygotic twins with Ohtahara Syndrome and unique neuropathological findings. Epilepsia 54: e81–e85. 10.1111/epi.12137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wizemann H, Garbe JH, Friedrich MV, Timpl R, Sasaki T, Hohenester E. 2003. Distinct requirements for heparin and α-dystroglycan binding revealed by structure-based mutagenesis of the laminin α2 LG4-LG5 domain pair. J Mol Biol 332: 635–642. 10.1016/S0022-2836(03)00848-9 [DOI] [PubMed] [Google Scholar]
- Zweier C, de Jong EK, Zweier M, Orrico A, Ousager LB, Collins AL, Bijlsma EK, Oortveld MAW, Ekici AB, Reis A, et al. 2009. CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt–Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am J Hum Genet 85: 655–666. 10.1016/j.ajhg.2009.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data were not deposited but may be available by contacting the authors. The variants were deposited to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and can be found under accession numbers SCV000241889.13 and SCV000863537.