

Abstract
Pathogenic variation in MAPT, GRN, and C9ORF72 accounts for at most only half of frontotemporal lobar degeneration (FTLD) cases with a family history of neurological disease. This suggests additional variants and genes remain to be identified as risk factors for FTLD. We conducted a case-control genetic association study comparing pathologically diagnosed FTLD patients (n = 94) to cognitively normal older adults (n = 3,541), and found suggestive evidence that gene-wide aggregate rare variant burden in MFSD8 associates with FTLD risk. Because homozygous mutations in MFSD8 cause neuronal ceroid lipofuscinosis (NCL), similar to homozygous mutations in GRN, we assessed rare variants in MFSD8 for relevance to FTLD through experimental follow-up studies. Using post-mortem tissue from middle frontal gyrus (MFG) of patients with FTLD and controls, we identified increased MFSD8 protein levels in MFSD8 rare variant carriers relative to non-variant carrier patients with sporadic FTLD and healthy controls. We also observed increases in lysosomal and autophagy-related proteins in MFSD8 rare variant carrier and sporadic FTLD patients relative to controls. Immunohistochemical analysis revealed that MFSD8 was expressed in neurons and astrocytes across subjects, without clear evidence of abnormal localization in patients. Finally, in vitro studies identified marked disruption of lysosomal function in cells from MFSD8 rare variant carriers, and identified one rare variant that significantly increased the cell surface levels of MFSD8. Considering the growing evidence for altered autophagy in the pathogenesis of neurodegenerative disorders, our findings support a role of NCL genes in FTLD risk and suggest that MFSD8-associated lysosomal dysfunction may contribute to FTLD pathology.
Keywords: autophagy, frontotemporal dementia, genetics, lysosomes, neurodegeneration, neuronal ceroid lipofuscinosis
INTRODUCTION
Frontotemporal lobar degeneration (FTLD) accounts for 5-10% of all dementia cases and is a leading cause of pre-senile dementias [46, 52]. Pathologically diagnosed FTLD is defined by neurodegeneration in the frontal and temporal lobes, leading to heterogeneous clinical symptomatology classified as frontotemporal dementia (FTD) [34, 50]. This pattern of degeneration slowly alters complex traits that make us most human, including behavior and language. Changes in personality and behavior are a hallmark of behavioral variant FTD (bvFTD) patients [59], while the nonfluent variant primary progressive aphasia (nfvPPA) and semantic variant PPA (svPPA) syndromes are characterized by impaired speech and language expression and comprehension [16].
Approximately 40% of all FTLD patients have a family member with a neurological or psychiatric disorder, and an estimated 10-20% of cases are caused by a single inherited pathogenic variant [60, 63]. The majority of genetic FTLD cases are due to pathogenic variation in one of three genes, MAPT, GRN, and C9ORF72, each resulting in different patterns of protein aggregation (broadly speaking, tau in MAPT and TAR DNA-Binding Protein (TDP)-43 in GRN and C9ORF72) with distinctive neuroanatomical patterns across the cortical and subcortical brain regions. Rare and pathogenic variants in other genes have been established to cause less than 5% of familial FTLD cases (i.e., VCP, CHMP2B, TARDBP, FUS, OPTN, and TBK1 [51, 56, 63, 66]). Nevertheless, nearly half of all patients with a family history suggestive of a genetic etiology do not carry a pathogenic variant in a known FTLD-associated gene, making it likely that additional genetic risk factors exist [51].
The application of next-generation sequencing (NGS) in association studies facilitates the identification of novel genetic risk factors or causes for many complex diseases. In particular, whole genome and whole exome sequencing (WGS and WES, respectively) have increased our understanding of how rare variation, which often has larger biological effects than common variation, contributes to disease [13]. The development of burden-based statistical tests has also accelerated the characterization of how gene-wide rare variation contributes to disease risk, especially for diseases with relatively small patient populations [33]. Importantly, these analyses can assess whether different rare variants occurring in the same gene are enriched in affected versus unaffected individuals. These advances have increased appreciation for the contribution of rare variants to the heritable portion of complex disease phenotypes unexplained by common variants. In the context of FTLD, genetic discoveries have informed potential pathogenic mechanisms for disease, and highlight how dysfunction in the endo-lysosomal system may lead to a loss of neuronal proteostasis, ultimately contributing to disease development [17].
In this study, we analyzed NGS data from pathologically diagnosed, sporadic FTLD patients to identify new genetic risk factors for FTLD. We found that aggregate rare variant burden in MFSD8 was enriched in FTLD patients relative to clinically normal older controls. We further assessed MFSD8 for its relevance to FTLD through biochemical analysis of post-mortem tissue from FTLD patients carrying the same rare variants in MFSD8 associated with disease risk. This revealed disturbances in protein levels of MFSD8, as well as lysosomal and autophagy-related markers compared to clinically normal older controls. We also localized MFSD8 protein to neurons and astrocytes. Finally, we demonstrated marked disruption of lysosomal function in cells derived from MFSD8 rare variant carriers, and at least one rare variant associated with FTLD risk was found to increase the cell surface levels of MFSD8 in transfected cell lines. These findings implicate rare variation in MFSD8 as a novel candidate risk factor for FTLD, and support a role for autophagy and lysosomal dysfunction in FTLD pathobiology.
MATERIALS AND METHODS
Participants and clinical assessment
Patients from across the FTLD spectrum (n = 94) were assessed and clinically diagnosed at the University of California, San Francisco Memory and Aging Center (UCSF MAC). None of the participants in this study carried a known disease causing pathogenic variant. All participants underwent clinical assessment during an in-person visit to the UCSF MAC that included a neurological exam, cognitive assessment, and medical history [47, 58]. Each participant’s study partner (i.e., spouse or close friend) was also interviewed regarding functional abilities. A multidisciplinary team composed of a neurologist, neuropsychologist, and nurse then established clinical diagnoses for cases according to consensus criteria for FTD and its subtypes [14, 16]. All cases underwent an autopsy through the UCSF Neurodegenerative Disease Brain Bank and were diagnosed with an FTLD spectrum disorder. Patients with FTLD-tau (Pick’s disease, corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), or unclassifiable), argyrophilic grain disease, FTLD-TDP (Types A, B, C, or unclassifiable), FTLD-UPS (Ubiquitin positive), or FTLD-FUS (atypical FTLD-U) pathology were included across all stages of analysis (see Table S1 for more details).
Clinically normal older controls were obtained from the Alzheimer’s Disease Sequencing Project (ADSP; n = 3,541). All ADSP controls were originally recruited through the Alzheimer’s Disease Genetics Consortium and the Cohorts for Heart Aging Research in Genomic Epidemiology consortia. The discovery and refined analyses included participants who were clinically assessed for dementia and/or pathological features of a neurodegenerative disorder upon autopsy. All ADSP sample phenotype and demographic data were obtained from dbGAP (study accession phs000572.v7.p4; table accessions pht004306.v4.p4.c1, pht004306.v4.p4.c2, pht004306.v4.p4.c5, pht004306.v4.p4.c6). Detailed demographic information is included in Table 1. This study was approved by the UCSF Institutional Review Board and written informed consent was obtained from all participants and surrogates per UCSF Institutional Review Board protocol.
Table 1.
Study participant characteristics.
| Analysis | Variable | FTLD | Controls | P-Value |
|---|---|---|---|---|
|
Discovery (n = 2,661) |
n | 62 | 2,599 | NA |
| Age at death or last visit (Mean ± SD) | 68.6 ± 7.34 | 85.9 ± 3.55 | < 0.001 | |
| Sex (M / F) | 35 / 27 | 1,163 / 1,436 | NS | |
| Age of Onset (n = 32)* | 60.0 ± 7.35 | NA | NA | |
| Disease Duration (n = 32)* | 9.56 ± 3.58 | NA | NA | |
| Pathological Type (Tau / TDP) | 24 / 38 | NA | NA | |
|
Refined (n = 3,635) |
n | 94 | 3,541 | NA |
| Age at death or last visit (Mean ± SD) | 67.8 ± 8.39 | 86.0 ± 3.63 | < 0.001 | |
| Sex (M / F) | 49 / 45 | 1,419 / 2,122 | < 0.05 | |
| Age of Onset (n = 62)* | 59.2 ± 8.80 | NA | NA | |
| Disease Duration (n = 62)* | 8.98 ± 3.32 | NA | NA | |
| Pathological Type (Tau / TDP / FUS / UPS) | 51 / 37 / 5 / 1 | NA | NA | |
Summary demographic and clinical information for participants included in the discovery and refined analyses. The refined analysis includes 54 FTLD cases and 2,085 controls from the discovery analysis, and an additional 40 FTLD cases and 1,456 controls
Age of onset and disease duration was available for the indicated number of participants included in each analysis. M, male; F, female; SD, standard deviation; NA, not applicable; NS, not significant. TDP, TAR DNA-binding protein 43; FUS, Fused in sarcoma; UPS, ubiquitin positive.
Sequencing data generation
All pathologically diagnosed FTLD patient DNA samples from UCSF underwent WGS at the New York Genome Center (New York City, NY) or HudsonAlpha Institute for Biotechnology (Huntsville, AL) on an Illumina HiSeq-X, with 150 base pair (bp) paired-end reads to obtain 30x sequencing coverage. All rare variants in MFSD8 identified in UCSF patients across all stages of analysis were confirmed with direct Sanger sequencing at the UCLA Neuroscience Genomics Core (Los Angeles, CA). ADSP samples underwent WES (average coverage 30-70x) at one of three NHGRI funded large-scale sequencing centers, either the Human Genome Sequencing Center at Baylor College of Medicine, Broad Institute, or Genome Institute at Washington University. Whole exome capture was performed using either the Illumina Rapid Capture Exome kit at the Broad Institute or Nimblegen VCRome v2.1 kit at Baylor College of Medicine and Washington University (Roche).
Sequencing data alignment and variant calling
Paired-end reads from WGS were aligned to the same version of the GRCh37 human reference genome used by the 1000 Genomes Project with the Burrows-Wheeler Aligner (BWA-MEM v0.7.8) [37], and processed using the Broad Institute’s Genome Analysis Toolkit (GATK) best-practices pipeline that includes marking of duplicate reads by the use of Picard tools (v1.83, http://picard.sourceforge.net), realignment around small insertions and deletions (INDELs), and base recalibration via GATK v3.2.2 [43]. Single nucleotide polymorphisms (SNPs) and INDEL variants were called using GATK HaplotypeCaller. To improve variant call accuracy, multiple single-sample genomic variant call files (GVCFs) were jointly genotyped using GATK GenotypeGVCFs, which generates a multi-sample variant call file (VCF).
The tools and parameters used to process ADSP WES data at the three NHGRI sequencing centers are detailed on the National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site (https://www.niagads.org/adsp/content/sequencing-pipelines). For the current study, ADSP WES data was downloaded as the overall ADSP VCF from combination of two project-level VCFs, and BAM files containing sequences that aligned to the protein coding exons +/− 1 bp surrounding each exon of MFSD8 (Table S2). The overall VCF was used in the discovery analysis (n = 2,661), while the BAM files for MFSD8 were used for joint variant calling across all cases and controls included in the refined analyses (n = 3,635). Sequencing data from the NHGRI sites was aligned to different versions of the GRCh37 human reference genome, and we verified that there was 100% concordance between the sequences for MFSD8 across these versions of the reference genomes. To further ensure data quality, we used GATK’s DepthOfCoverage tool to determine that the average sequencing depth across all target regions in BAM files from all sequencing centers was >30x, which is sufficient for reliable variant calling [36, 78]. Based on these assessments, we determined the WES control data to be of sufficient quality and reliability to combine with our WGS data and performed joint variant calling across BAM files containing reads aligned to the coding exons of MFSD8 +/− 1 bp surrounding each exon from all data sources using the same methods described above.
Quality control and post-processing
All cohort-wide VCFs were filtered according to previously established criteria [10]. Briefly, we kept variants with genotype quality (GQ) scores greater than 30 and read depth (DP) scores greater than 10. We also excluded variants that failed quality control filters in the Genome Aggregation Database (http://gnomad.broadinstitute.org) [35], which includes variants with low inbreeding coefficient, that fall in low complexity and segmental duplication regions, and with no observed high quality non-reference genotype calls. The resulting filtered VCF was annotated with gene names, variant type (i.e., exonic, intronic, etc.), and amino acid change for all exonic variants using Annovar [73]. Once annotated with gene names, we also excluded variants in false positive genes that exhibit a high frequency of rare protein-altering variants in whole exome and genome sequencing studies since these genes likely do not contribute to FTLD risk [15, 65]. For the discovery analysis, separate, filtered VCFs of joint calls from FTLD cases (n = 62) or ADSP controls (n = 2,599) were converted to PLINK format and merged using PLINK [57]. The merged dataset was filtered to remove variants with genotyping rates below 95%, and minor allele frequencies (MAF) greater than 0.5%. For the refined analysis, a separate VCF was generated from joint variant calling across target regions of MFSD8 for all FTLD cases and controls. This VCF was filtered as mentioned above, converted to PLINK format, and filtered again to remove variants with genotyping rates below 99% and MAFs greater than 0.5%, followed by removal of individuals with genotyping rates below 90% in targeted regions of MFSD8. Gene variant sets for the discovery analysis were created from SNPs alone, while the gene variant set for the refined analysis included SNPs and small INDELs (<5 bp to increase reliability of calls). Variants included in the analysis were classified as missense, nonsense, or splice site variants to reflect variation that is most likely to have a strong biological effect.
Antibodies
The HA.11 monoclonal antibody used to detect EGFP-HA-MFSD8 was from BioLegend and the transferrin receptor (TfR) monoclonal antibody was from Life Technologies. The HPA044802 polyclonal antibody against human MFSD8 was from ATLAS Antibodies. We verified the specificity of the HPA044802 antibody in cells with MFSD8 expression knocked down using two different shRNA constructs, and confirmed by immunoblot that the immunoreactive band corresponding to MFSD8 in control cells was almost undetectable in the knock down cells (see Supplementary Materials for more information and Fig. S1).
Post-mortem human brain tissue processing and case selection
Post-mortem human brain tissue was obtained from the USCF Neurodegenerative Disease Brain Bank. Consent for brain donation was obtained from all subjects or their surrogates in accordance with the Declaration of Helsinki and research was approved by the Institutional Review Board of the UCSF Committee on Human Research. At autopsy, brain handling depended on where procurement occurred. Cases procured remotely underwent hemibrain immersion fixation in 10% buffered formalin. The opposite hemibrain was cut into 1 cm coronal slabs and frozen. Locally procured cases were cut freshly into 1 cm thick bihemispheric coronal slabs and alternately fixed for 48-72 hours in 10% buffered formalin or frozen. Neuropathological diagnoses were made following consensus diagnostic criteria [20, 38, 39, 42] using previously described histological and immunohistochemical methods [25, 71].
Subjects for post-mortem tissue studies included cognitively normal healthy controls (n = 3), patients with FTLD carrying MFSD8 rare variants associated with FTLD risk in our genetic analyses (n = 5), and patients with sporadic FTLD (n = 5) selected to represent the same primary neuropathological diagnosis as the rare variant carriers. The MFSD8 rare variant carriers included four of five identified as rare variant carriers in our genetic analyses and one who came to autopsy during the course of this study and was found to carry the G385R variant through targeted sequencing of MFSD8 (see Table S4 for all participant details and demographics). The middle frontal gyrus (MFG) was chosen for all post-mortem tissue analyses because it is affected across FTLD subtypes. Whenever possible, adjacent fixed and frozen tissues were used (local cases); otherwise, fixed and frozen tissues were taken from the same (MFG) region in opposite hemispheres.
Immunoblot and densitometric analysis of tissue and transfected cell lysates
Freshly frozen post-mortem tissue from the subjects described above was analyzed by immunoblot (Table S4). Tissue lysates were prepared in 0.25 M Sucrose with 7 M Urea, incubated 15 min on ice, and followed by centrifugation at 25,000 x g for 15 min. Protein concentration in the supernatant (extracted proteins) was quantified by bicinchoninic acid, and then incubated at 30°C for 10 min after addition of Laemli sample buffer and subjected to SDS-PAGE [72]. Gels were transferred to nitrocellulose membrane, blocked with low-fat milk and incubated overnight with the primary antibody against MFSD8 (HPA044802, ATLAS). The proteins were visualized using peroxidase-conjugated secondary antibodies and chemiluminescent reagent (PerkinElmer) in a LAS-3000 Imaging System (Fujifilm). Densitometric quantification was performed on unsaturated images using ImageJ Software (NIH). Immunoblot membranes were stained with Ponceau S and the density of each band was normalized to the total densitometric value of the Ponceau S staining for each sample. Normalization to Ponceau S staining was preferred to common housekeeping proteins (i.e., Glyceraldehyde-3-phosphate dehydrogenase or Actin), as their protein levels are often affected in conditions with altered intracellular protein degradation.
Immunoblotting of cell lysates was done essentially as in Sirkis et al. [68]. Briefly, cells were harvested on ice by washing with cold PBS followed by lysing in a buffer containing 100 mM NaCl, 10 mM Tris-Cl, pH 7.6, 1% (v/v) Triton X-100 and Complete protease inhibitor cocktail (Roche). Triton-insoluble material was sedimented by centrifugation at 16,000 x g for 10 min at 4°C. Supernatants were mixed with 5X SDS-PAGE sample buffer supplemented with DTT, then heated at 55°C for 10 min prior to running in 4–20% acrylamide gradient gels (Invitrogen). After SDS-PAGE, proteins were transferred onto PVDF membranes (Merck Millipore), blocked in 5% non-fat milk (dissolved in PBS containing 0.1% Tween-20), and probed with HA.11 and TfR antibodies at 1:2,000 and 1:10,000, respectively. Blots were developed using enhanced chemiluminescence and imaged on a ChemiDoc digital imager (Bio-Rad). Protein signals were quantified by densitometry using the Fiji distribution of ImageJ (NIH).
Immunohistochemical analysis of post-mortem tissue
MFSD8 immunochemistry (IHC) was performed on 8 μm-thick sections cut from formalin-fixed, paraffin-embedded tissue blocks of MFG from the same subjects described above and analyzed by immunoblot (Table S4). The sections were deparaffinized in xylene for 30 min, washed in 100% and 95% ethanol, and then the endogenous peroxidase was quenched by 3% hydrogen peroxide in 10% methanol for 30 min. In order to retrieve antigenicity, sections were boiled within 0.01 M citrate-buffer saline (pH 6) for 5 min at 121°C. After cooling for 15 min, the sections were washed PBS-T (0.2% Tween-20 in 0.01 M PBS, pH 7.4) for 15 min, blocked for 30 min in blocking solution (10% goat serum in PBS-T), and incubated in primary antibody (anti-MFSD8, 1:50, HPA044802, ATLAS) at room temperature overnight. After two changes of PBS-T, the sections were incubated with biotinylated secondary antibody (BA-1000, 1:200, Vector Laboratories) for 40 min at room temperature, washed, then incubated with avidin-biotin-peroxidase complexes (ABC, 1:100, PK-6100, Vector Laboratories). After washing, immunostaining was visualized with 5% chromogen 3,3’-diaminobenzidine (DAB) solution, counterstained with hematoxylin (SH26-500D, Fisher Scientific), and dehydrated, then cover slips were applied. Images were captured with a Nikon Eclipse 80i microscope.
Patient-derived cell lines
Primary fibroblasts isolated from skin biopsies of healthy donors and FTD patients harboring MFSD8 rare variants (2 lines: E336Q or G385R) were obtained from UCSF/Gladstone Institute. The MFSD8 rare variant carrier patient cell lines were derived from clinically diagnosed FTD patients and were therefore not included in genetic analyses of pathologically diagnosed FTLD cases. Rare variants in these patients were identified by exome chip analysis (E336Q) or targeted sequencing (G385R) of MFSD8, and both were confirmed by Sanger sequencing as described above. Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Sigma-Aldrich), in the presence of 10% fetal bovine serum (FBS), 50 μg/ml penicillin and 50 μg/ml streptomycin at 37°C with 5% CO2 and tested for mycoplasma contamination every two weeks using DNA staining protocol with Hoechst 33258 dye or MycoSenser PCR Assay Kit (Stratagene).
Immunofluorescence
Primary fibroblasts grown on coverslips were fixed with 4% PFA, blocked and then incubated with the primary and corresponding fluorophore-conjugated secondary antibodies following standard procedures [22, 53]. Mounting medium contained DAPI (4,6-diamidino-2-phenylindole) to highlight the cell nucleus. All images were acquired with an Axiovert 200 fluorescence microscope (Carl Zeiss Microscopy) with a 100x or 63x objective and 1.4 numerical aperture, mounted with an ApoTome.2 slider, and prepared using Adobe Photoshop CS3 (Adobe Systems) and ImageJ Software (NIH). The number of puncta per cell was quantified using the ‘analyze particles' function of ImageJ after adjusting the threshold in non-saturated images. The percentage of co-localization was determined by the ‘JACoP' plugin in ImageJ after adjusting the threshold of individual frames [8].
Intracellular protein degradation assay
Measurement of intracellular protein degradation was performed by metabolic labeling (with [3H] leucine (2 μCi/ml) (NENPerkinElmer Life Sciences) for 48 hrs at 37°C) and pulse-chase experiments as described before [5, 54]. After labeling, cells were extensively washed and maintained in medium with an excess of unlabeled leucine. Aliquots of the medium taken at different times were precipitated in trichloroacetic acid, and proteolysis measured as the amount of acid-precipitable radioactivity (protein) transformed in acid-soluble radioactivity (amino acids and small peptides) at each time. Lysosomal-dependent degradation was inhibited using 20 mM NH4Cl and 100 μM leupeptin (Sigma) added directly to the culture media.
Cell-surface biotinylation
Cell-surface labeling was carried out in a manner similar to that performed in Sirkis et al. [68]. Briefly, cells were washed at RT with PBS and labeled with the EZ-Link Sulfo-NHS-SSBiotin reagent (ThermoFisher) at 1 mg/ml in PBS for 10 min. Cells were then placed on ice, washed with cold Tris-buffered saline to quench the biotin reagent, then washed with cold PBS and finally lysed and clarified as described above. To capture biotinylated proteins, StrepTactin resin (iba) was added to the clarified lysates and the mixtures rotated at 4°C for 1 h. The resin was then pelleted and washed multiple times with lysis buffer. Finally, 2X SDS-PAGE sample buffer supplemented with DTT was added to the washed resin, and the samples were vortexed, heated and prepared for immunoblotting as described above. For the analysis of surface-labeled MFSD8, we normalized the signal derived from all surface-labeled MFSD8 bands in a given lane to the overall MFSD8 signal derived from the corresponding cell-lysate lanes (surface/total), and expressed these ratios relative to reference sequence MFSD8 (WT).
Statistical analyses
All FTLD cases (n = 94) and clinically normal older controls (n = 3,541) analyzed were confirmed to be unrelated by calculating Pi-Hat values for all pairs of individuals in PLINK, and only individuals with Pi-Hat values <0.125 across all comparisons to other individuals were retained for analysis. All cases (n = 62) and controls (n = 2,599) in the discovery cohort self-reported race as ‘white’, except for one case with a self-reported primary race as ‘unknown’, who was retained in the cohort due to limited sample size. Multidimensional scaling (MDS) analysis was performed on FTLD cases and controls to confirm European ancestry using WGS data from CEU, YRI, and CHB samples in the 1000 Genomes Project as reference populations [1]. Non-European individuals were excluded from the refined analysis due to an insufficient number of participants for sub-group analysis and potential for confounding background genetics, and the first two vectors of the MDS analysis were included as covariates in the refined analysis (Fig. S2).
All cohorts were analyzed using a sequence kernel association test (SKAT), and followed previously published criteria by limiting the discovery analysis to genes with four or more SNPs meeting criteria described earlier, and SNPs and small INDELs in MFSD8 meeting the same criteria for the refined analysis [33]. Gene-based association tests were performed using the optimal unified test (SKAT-O) in the R-based “SKAT” package [33]. Multiple testing correction of P-values was done using the Bonferroni method. A priori, genome-wide statistical significance for the discovery analysis was set at PBonf <0.05 and for the gene-based refined analysis at Praw <0.05. Differences in demographics between cases and controls were assessed by t-test (continuous data) or X2 test (categorical data) in R. The odds ratio (OR) with 95% confidence intervals (CI) and X2 test for association of gene-wide, aggregate rare variant burden with FTLD was calculated in R. All numerical results in the experimental studies are reported as mean and standard error of the mean (s.e.m.). Statistical significance of the differences in protein levels between experimental groups in studies with transfected cells and post-mortem tissue was established by one-way ANOVA with Dunnett’s and Tukey’s test, respectively, used for post-hoc analyses. Demographic and experimental differences were considered statistically significant for P <0.05.
Data sharing
Data generated by the UCSF MAC are available upon request. Data requests can be submitted through the UCSF Memory and Aging Center Resource Request form: http://memory.ucsf.edu/resources/data. For questions related to UCSF data use and access, please contact Rosalie Gearhart at Rosalie.Gearhart@ucsf.edu. Investigators who wish to obtain ADSP data must apply for access. Please see https://www.niagads.org/adsp/ for more details.
RESULTS
Overview of Cohorts
The stage 1 “discovery” analysis consisted of 2,661 participants: 62 pathologically diagnosed, sporadic FTLD cases and 2,599 clinically normal older controls. All participants were self-reported ‘white’, except for one case with a self-reported race of ‘unknown’, which was included in this cohort due to limited sample size. The goal of this stage was to identify novel candidate genes for rigorous follow-up analyses. In the stage 2 “refined” analysis, all 3,635 participants were of European ancestry confirmed by MDS covariates derived from genetic data and using reference populations from the 1000 Genomes Project (Fig. S2), including 94 FTLD cases (54 discovery cohort cases plus an additional 40 sporadic FTLD cases) and 3,541 controls (2,085 discovery cohort controls and an additional 1,456 controls). Demographic characteristics are provided by stage in Table 1. There was a difference in sex across participants in the refined analysis. As expected, we found that cases were younger than controls across all cohorts; we intentionally included controls that were older than our cases to reduce the likelihood that individuals defined as controls at the time of our study might go on to develop FTLD in the future.
Rare variant burden in MFSD8 is enriched in FTLD cases
Stage 1: Discovery Analysis
We analyzed our discovery cohort (62 cases and 2,599 controls) using SKAT-O to identify candidate genes in which gene-wide rare variant burden associated with sporadic FTLD risk. After quality control of variant level data, we analyzed 14,459 genes that each had four or more rare, protein altering SNPs (n = 233,334). This unbiased analysis identified MFSD8 as the most significant gene in which rare variant burden was associated with FTLD risk (PRaw = 4.9×10−6, PBonf = 0.07, Table 2). Even though the strength of this association is slightly above our threshold for statistical significance after multiple testing correction (PBonf < 0.05), we pursued this finding in follow-up studies for several reasons. First, prior studies have found that recessive pathogenic variants in MFSD8 cause late infantile neuronal ceroid lipofuscinosis (NCL) [2, 30, 40, 67], a childhood neurodegenerative lysosomal-storage disorder. Second, the link between MFSD8 and NCL is akin to observations with GRN, for which heterozygous pathogenic variants result in autosomal dominant FTLD and homozygous pathogenic variants cause NCL. Third, variation in another NCL gene, CTSF, has been associated with FTD risk [80]. Taken together, with the near significant association between rare variant burden in MFSD8 and FTLD risk in the discovery analysis, we expanded on this finding.
Table 2.
Results of discovery and refined analyses of MFSD8 rare variant burden in FTLD cases.
| Analysis | Gene | Variants Tested |
FTLD MAC (% carriers) |
Control MAC (% carriers) |
SKAT-O PRaw |
SKAT-O PBonf |
|---|---|---|---|---|---|---|
|
Discovery (n = 2,661) |
MFSD8 | 9 | 4 (6.5%) | 10 (0.4%) | 4.9×10−6 | 0.07 |
| TEDC2 | 9 | 6 (9.7%) | 17 (0.7%) | 2.0×10−5 | 0.29 | |
| HAUS5 | 18 | 7 (11%) | 40 (1.5%) | 2.5×10−5 | 0.36 | |
| HSF5 | 13 | 3 (4.8%) | 15 (0.6%) | 3.4×10−5 | 0.49 | |
| TIAF1 | 5 | 4 (6.5%) | 12 (0.5%) | 5.5×10−5 | 0.79 |
| Analysis | Gene | Variants Tested |
FTLD MAC (% carriers) |
Control MAC (% carriers) |
SKAT-O PRaw |
X2 test PRaw |
|---|---|---|---|---|---|---|
|
Refined (n = 3,635) |
MFSD8 | 19 | 5 (5.3%) | 39 (1.1%) | 6.2×10−3 | 2.3×10−4 |
Top panel, the top five genes associated with FTLD risk identified by SKAT-O analysis of rare, protein-altering SNPs in the discovery cohort (62 FTLD cases and 2,599 controls). Raw P-values (PRaw) for the 14,459 genes tested in the discovery analysis were corrected for multiple testing using the Bonferroni method (PBonf). Bottom panel, SKAT-O and X2 statistical test results for association of aggregate rare variant burden in MFSD8 with FTLD risk in the refined analysis, which includes 54 FTLD cases and 2,085 controls from the discovery analysis, and an additional 40 FTLD cases and 1,456 controls. The X2 test was performed on minor allele counts in all FTLD cases and controls. Percentages represent the proportion of FTLD cases and healthy controls carrying the minor allele of a rare variant. SNP, single nucleotide polymorphism; MAC, minor allele count.
Stage 2: Refined Analysis
Since our discovery analysis had several caveats, including analyzing variants identified by separate variant calling strategies and using self-reported race, we took additional steps to assess and refine our stage 1 results in a more rigorous analysis. First, we performed WGS on an additional 40 sporadic, pathologically diagnosed FTLD cases to fold into this analysis. We also included an additional 1,456 clinically normal older controls with WES data from the ADSP cohort. Second, we performed an MDS analysis to confirm European ancestry of all participants, which resulted in the removal of eight cases, two of which are MFSD8 rare variant carriers, and 514 controls with genetic data suggestive of mixed ancestry. Even though all participants were confirmed to be of European ancestry at this point, we included the first two vectors from the MDS analysis as covariates in the SKAT-O refined analysis to further account for within-population variability. Third, since variants included in the discovery analysis were not identified through joint variant calling across cases and controls, we identified protein-altering variants for the refined analysis by performing joint variant calling across the 12 protein coding exons of MFSD8 +/− 1 bp surrounding each exon for all 3,635 participants included at this stage. Although we did not observe an association between MFSD8 rare variant burden and FTLD risk when the additional FTLD cases and controls were analyzed alone (SKAT-O P = 0.11), we did observe a significant association between MFSD8 rare variant burden and FTLD risk in the refined analysis of 94 FTLD cases and 3,541 controls (P = 6.2×10−3; Table 2). A X2 test comparing minor allele counts of rare MFSD8 variants between all cases and controls corroborated the SKAT analysis (P = 2.3×10−4; odds ratio [OR] = 5.0, 95% CI [1.95, 12.9]). In total, three FTLD-tau and two FTLD-TDP cases were identified as MFSD8 rare variant carriers (5.3% of 94 FTLD cases). This is compared to 39 out of 3,541 (1.1% of all controls) cognitively healthy adults carrying a rare variant in MFSD8. Table S3 provides a summary of the 19 rare variants in MFSD8 included in the refined analysis across all FTLD cases and controls. Finally, all MFSD8 rare variants identified by WGS in FTLD cases (n = 5) were confirmed by Sanger sequencing. These findings suggest that rare variation in MFSD8 may contribute risk to FTLD.
MFSD8 protein levels are elevated in MFSD8 rare variant carriers
To gain insight into the possible contributions of MFSD8 rare variants to the pathogenesis of FTLD, we first characterized MFSD8 protein in freshly frozen post-mortem tissue from cognitively normal healthy controls (n = 3), FTLD patients carrying MFSD8 rare variants (n = 5), and patients with sporadic FTLD (n = 5). All FTLD cases were matched for age, sex, and pathological diagnosis (Table S4). After confirming the specificity of the MFSD8 antibody (Fig. S1), immunoblot analysis of brain tissue from MFG, a region of the brain known to be affected in FTLD, was performed to assess changes in MFSD8 protein across FTLD patients and controls. This revealed increased levels, on average, of MFSD8 across all MFSD8 rare variant carriers, particularly in those with FTLD-tau, relative to healthy controls and sporadic FTLD (Fig. 1A, B). Although this increase was not statistically significant due to high inter-patient variability and a small number of MFSD8 rare variant carriers available for analysis, these findings support a role for MFSD8 contributing to FTLD risk.
Fig. 1. Immunoblot of MFSD8 in post-mortem tissue from FTLD patients carrying rare variants in MFSD8.
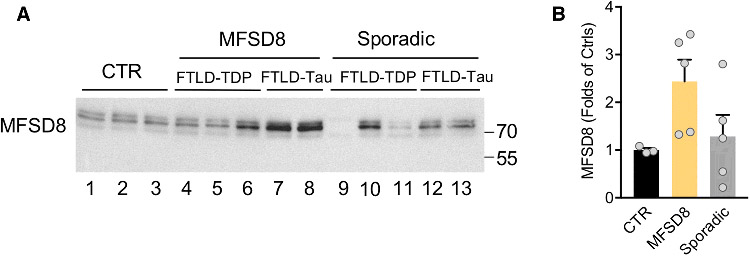
(A) Immunoblot for MFSD8 in tissue from middle frontal gyrus of clinically normal, healthy controls (CTR, n = 3), FTLD patients carrying MFSD8 rare variants (MFSD8, n = 5) and patients with sporadic FTLD (Sporadic, n = 5). (B) Quantification of the levels of MFSD8 normalized to Ponceau S. Values are expressed relative to control subjects as mean +/− s.e.m. See Table S4 for more details on specific pathological diagnoses and demographics of all FTLD patients and controls.
Neurons have the strongest expression of MFSD8
Immunohistochemistry was performed on MFG to assess cell type specificity and subcellular localization of MFSD8. Immunoreactivity was generally strongest in neuronal cytoplasm across all diagnostic groups, but it was also detected in astrocytes (Fig. 2). Although most of the MFSD8 staining was cytoplasmic, nuclear staining in neurons was also observed in FTLD cases and controls with the highest MFSD8 immunoreactivity (Fig. 2B, C, F, and G). We observed no distinguishing features regarding the subcellular localization of MFSD8 in rare variant carrier cases relative to sporadic FTLD cases or controls, and there was no suggestion that MFSD8 protein adopted spatial patterns reminiscent of tau or TDP-43 neuronal cytoplasmic inclusions in patients. Nevertheless, the presence of MFSD8 in neurons of MFG suggests a possible role for MFSD8 in FTLD pathology.
Fig. 2. Immunohistochemical staining of MFSD8 in post-mortem tissue from FTLD patients carrying rare variants in MFSD8.

Representative images of immunostaining for MFSD8 in middle frontal gyrus of clinically normal, healthy controls (Controls, HC, panels A-E), FTLD patients carrying MFSD8 rare variants (Carriers, panels F-J), and patients with sporadic FTLD (Sporadic, panels K-O). All FTLD patients were matched for age, sex, and pathological diagnosis. Panels (D-E) are higher magnification images of MFSD8 immunoreactivity in neurons of the same healthy controls depicted in (A) and (C). MFSD8 Immunoreactivity in neurons (→) and astrocytes (►) are highlighted. See Table S4 for more details on specific pathological diagnoses and demographics of all FTLD patients and controls. All sections were counterstained with hematoxylin. Scale bar: 10 μm in (D-E), 20 μm in (O). TDP-B, TAR DNA-Binding Protein 43 - Type B; CBD, corticobasal degeneration; PSP, progressive supranuclear palsy.
Lysosomal and autophagy-related proteins are elevated in MFSD8 rare variant carriers
Multiple chaperones and two main proteolytic systems, the ubiquitin proteasome system and the autophagy/lysosomal system, contribute to maintenance of cellular proteostasis [23]. Altered lysosomal degradation by autophagy has also been associated with accelerated neurodegeneration [21, 31, 44, 45, 49]. Immunoblot analysis of freshly frozen post-mortem tissue from controls, FTLD patients carrying MFSD8 rare variants, and patients with sporadic FTLD revealed a trend toward expansion of the autophagic/lysosomal compartments in both MFSD8 rare variant carriers and sporadic FTLD cases relative to controls (Fig. S3A, B). Interestingly, except for levels of LAMP-1 that were significantly higher in the sporadic FTLD cases, the increase of the other two lysosomal markers, LAMP-2 and Cathepsin D (CTSD), was consistently higher in the MFSD8 rare variant carriers (Fig. S3A, B). Similarly, a trend towards higher levels of lipidated LC3 (LC3-II), the most widely used autophagosomal marker [4], was also observed in MFSD8 rare variant carriers relative to sporadic FTLD cases and controls (Fig. S3A, B). Although the increases in lysosomal and autophagosomal proteins did not reach statistical significance due to the limited availability of MFSD8 rare variant carrier cases, these changes resemble the impaired lysosomal function and autophagy described in mice with Mfsd8 ablated [9]. This suggests that MFSD8 in humans may also play a role in lysosomal and autophagic function.
Lysosomal malfunctioning in MFSD8 rare variant carriers
Ablation of Mfsd8 in mice has recently been shown to cause lysosomal alterations such as lysosome enlargement and accumulation of autofluorescent lipofuscin, an indirect measurement of defective lysosomal degradation in vivo [9]. Since our steady-state data from post-mortem tissue also suggested alterations in lysosomal and autophagosomal markers in MFSD8 rare variant carriers, we characterized lysosomal dysfunction in patient-derived cell lines with rare missense variants in MFSD8. To that effect, we measured degradation rates of intracellular long-lived proteins (which preferentially undergo degradation by autophagy) in fibroblasts derived from two FTD patients carrying different MFSD8 rare missense variants (E336Q and G385R) and healthy controls using metabolic labeling [5]. We found a significant decrease in total rates of protein degradation in rare variant carrier cell lines compared to controls (Fig. 3A, B). This decrease in intracellular proteolysis primarily results from impaired lysosomal degradation. The rate of lysosomal degradation in MFSD8 variant carrier cells also remained significantly reduced upon serum removal, a condition known to stimulate autophagy (Fig. 3A, B). Immunofluorescence staining in the same cells revealed an increase in the number of autophagosomes (LC3 positive puncta) in MFSD8 rare variant carrier cells relative to controls (Fig. 3C, D). Interestingly, although there was a trend toward increased number of lysosomes (LAMP-2 positive puncta) in cells from MFSD8 rare variant carriers, the most distinct difference relative to control cells was the lower average size of lysosomes (Fig. 3D). Co-immunostaining revealed that a fraction of cellular MFSD8 was detected in LAMP-2 positive vesicles, consistent with its previously described lysosomal location, and that cells from MFSD8 rare variant carriers displayed significantly higher co-localization of both proteins (Fig. 3E). Furthermore, the observed immunoreactivity of MFSD8 in the nucleus of cells from controls and MFSD8 rare variant carriers was consistent with our IHC findings in post-mortem tissue (Fig. 3E). Taken together, the higher number of autophagosomes without lysosomal expansion in MFSD8 rare variant carrier cell lines suggest that the autophagic failure originates from poor lysosomal clearance of autophagosomes. This phenotype is similar to the one observed in Mfsd8 knock-out mice [9], supporting the hypothesis that rare missense variants in MFSD8 associated with FTLD risk result in reduced MFSD8 protein function.
Fig. 3. Lysosomal function in fibroblasts from FTD patients carrying rare missense variants in MFSD8.
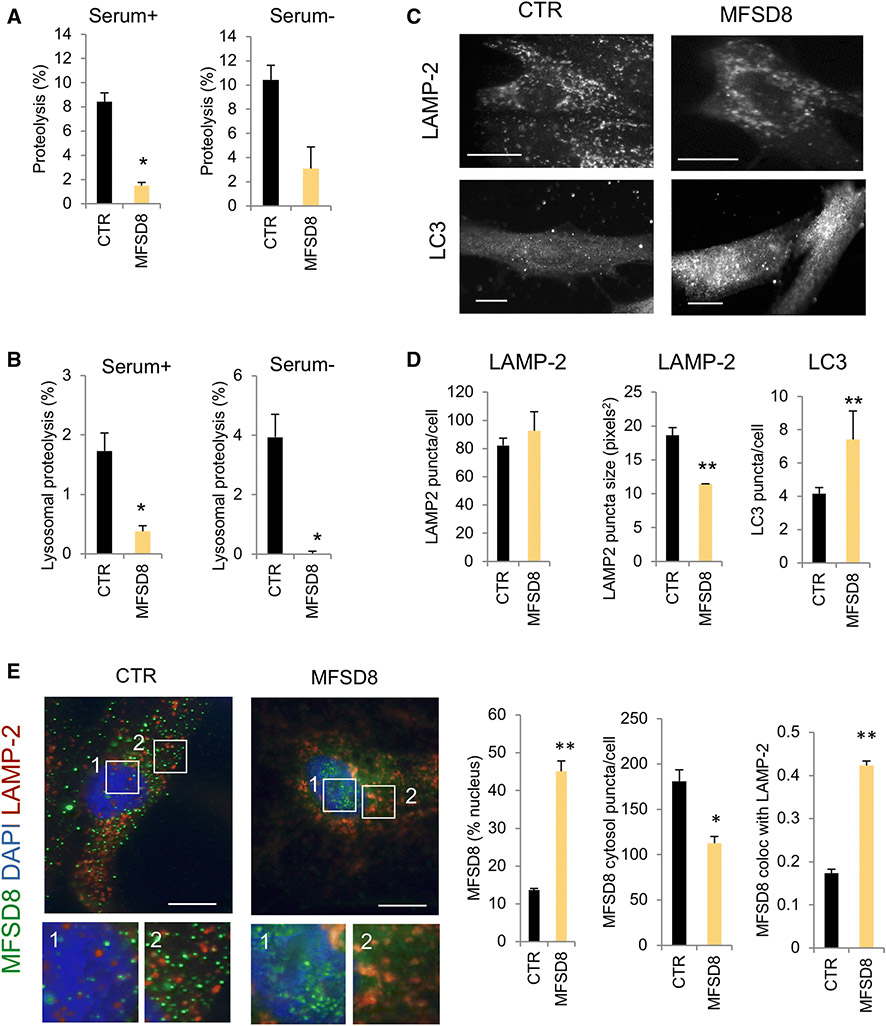
(A) Long half-life intracellular protein degradation in skin fibroblasts from clinically normal, healthy control donors (control, CTR) and MFSD8 rare variant carriers maintained in the presence (left) or absence (right) of serum. Values are expressed as percentage of proteolysis and are mean + s.e.m. of n = 3 experiments. (B) Lysosomal degradation was calculated after addition of ammonium chloride and leupeptin to cells maintained as in (A). Values represent the percentage of protein degradation sensitive to the inhibitors. Differences with control are significant at p < 0.05 (*). (C) Immunostaining for the lysosomal marker LAMP-2 or the autophagosomal marker LC3 of the same cells. Scale bar: 10 μm. (D) Quantification of the number (left) and average size (middle) of puncta positive for LAMP-2 (top) and of the number of puncta positive for LC3 (right). Quantification was done for n = 4 fields. Differences with control are significant at p < 0.05 (*), p < 0.01 (**), or p<0.001 (***). (E) Co-immunostaining for MFSD8 and LAMP-2 in the same cells. Left: representative images of merge fields. Nuclei were stained with Dapi. Inserts show squared regions in the nucleus (1) and cytosol (2) at higher magnification. Right: quantification of MFSD8 in the nucleus (left), cytosol (middle) or associated to LAMP-2 positive compartments (right). Values are mean + s.e.m. of n = 4 fields. Differences with control are significant at p < 0.05 (*) or p < 0.01 (**).
Intriguingly, we found more pronounced changes in the two lysosomal marker proteins, LAMP-2 and CTSD, than LAMP-1 (a broader endo-lysosomal maker) in post-mortem brain tissue from MFSD8 rare variant carriers (Fig. S3). In addition, we observed that not all MFSD8 co-localized with LAMP-2 (Fig. 3E), and we therefore used subcellular fractionation of mouse brain homogenates to further characterize Mfsd8 in different lysosomal subgroups [12]. Mouse Mfsd8 was detected at very low levels in autolysosomes that originate from fusion of autophagosomes and lysosomes [26] (Fig. S4). However, Mfsd8 was very abundant in a population of lysosomes that display high activity for chaperone-mediated autophagy (CMA), a selective form of autophagy where substrate proteins are directly translocated across the lysosomal membrane for degradation [12, 24] (Fig. S4). Increased abundance of Mfsd8 in this group of lysosomes is intriguing since defective CMA has previously been described in several neurodegenerative diseases, including FTLD [53, 74].
The F379S rare missense variant in MFSD8 alters cell surface expression
To further characterize the putative functional consequences of the rare missense variants in MFSD8 associated with FTLD risk, all five variants were cloned and expressed in HEK cells. Immunoblot analysis demonstrated that none of the variants altered total MFSD8 protein abundance (Fig. 4), but changes were observed in the cell surface expression of MFSD8. Specifically, the F379S variant showed a modest but significant increase in cell surface expression as well as accumulation of a lower molecular weight band on the cell surface relative to reference sequence MFSD8 (Fig. 4, *). The G385R variant demonstrated a smaller increase in surface expression that did not reach significance. These findings suggest that the F379S variant may affect MFSD8 function by altering its intracellular trafficking.
Fig. 4. Biochemical characterization of rare variants in MFSD8 associated with FTLD risk.
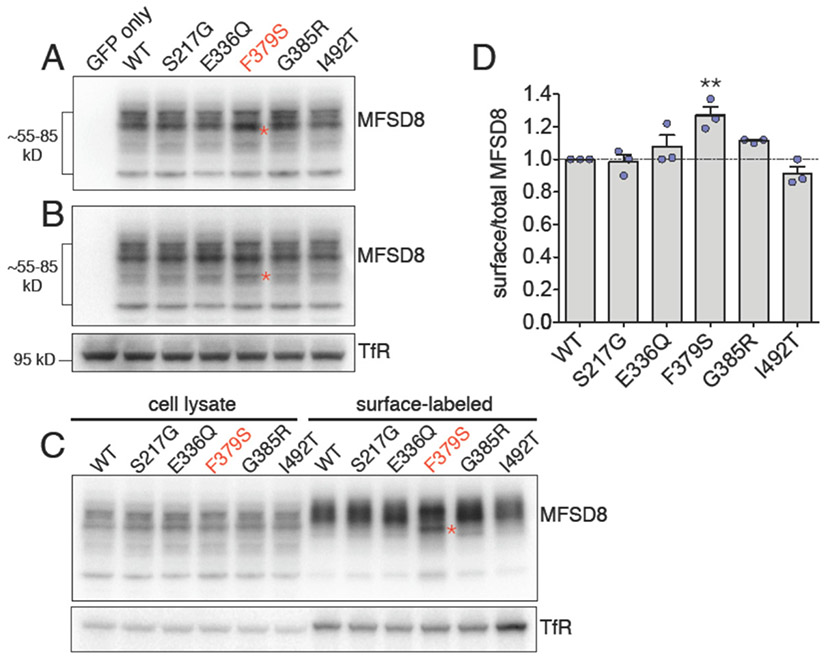
(A-B) Five variants (S217G, E336Q, F379S, G385R, I492T) were generated, dual-tagged with GFP and HA (GFP-HA-MFSD8), transiently expressed in HEK-293T cells and compared to cells expressing human reference sequence (WT) MFSD8 and negative control, GFP-transfected cells. One day after transfection, the cells were lysed and the lysates analyzed by immunoblotting for MFSD8 using an HA antibody. The tagged MFSD8 constructs ran as a collection of specific bands ranging in size from approximately 55-85 kD. All variants displayed grossly normal overall expression, but one variant, F379S, frequently showed subtle increases in several distinct lower-molecular weight (MW) bands, relative to WT MFSD8. Immunoblots from two representative experiments are shown in (A-B), and the accumulated lower-MW bands for variant F379S are indicated by (*). Transferrin receptor (TfR) was used as a loading control. (C-D) Cell-surface biotinylation analysis revealed that variant F379S showed a modest but statistically significant increase in surface expression, relative to WT MFSD8. Cell surface-associated F379S also showed accumulation of a lower-MW band (*, C). TfR was used as a loading control for the cell lysates and as a positive control for the surface labeling. Results were quantified from three independent experiments. Differences with WT are significant at p < 0.01 (**).
DISCUSSION
In the present study, we analyzed sequencing data from pathologically diagnosed sporadic FTLD patients and clinically normal older controls to identify genetic risk factors for FTLD. Gene-level association tests implicated aggregate rare variant burden in the NCL-linked gene, MFSD8, in FTLD risk. Follow-up experimental studies were used to investigate the possible role of MFSD8 in FTLD pathobiology and lysosomal function. Our studies revealed modest increases in MFSD8 protein in MFG of FTLD patients harboring rare variants in MFSD8 relative to patients with sporadic FTLD and controls. This effect was most marked in rare variant carriers diagnosed with FTLD-Tau. We also localized MFSD8 protein to neurons and astrocytes. Lysosome marker proteins were consistently increased in all FTLD cases relative to controls, while MFSD8 rare variant carriers also had a trend towards an increase in an autophagosome marker protein. Finally, biochemical studies revealed marked disruption of lysosomal function in cells from FTD patients carrying rare missense variants in MFSD8, and demonstrated that at least one rare FTLD-associated variant in MFSD8 resulted in greater cell surface expression. Together, our results suggest that rare variation in MFSD8 may contribute to FTLD risk by altering autophagy and lysosomal function. These key findings are discussed in turn.
Gene-level statistical analyses implicate rare variation in MFSD8 as a risk factor for FTLD
We sought to identify novel genetic risk factors for FTLD and found a suggestive association between FTLD risk and aggregate rare variant burden in MFSD8, a gene encoding a lysosomal membrane protein of unknown function. Although this finding did not reach exome-wide significance and requires replication in an independent cohort of FTLD cases, we pursued this finding since recessive homozygous pathogenic variants in MFSD8 cause variant late infantile NCL [2, 30, 40, 67], a rare inherited lysosomal storage disorder that primarily affects children and young adults [29]. Interestingly, even though FTLD is a neurodegenerative disorder that affects older adults, previous studies have identified shared genetic risk factors between NCL and FTLD. Heterozygous GRN pathogenic variants cause about 10% of all FTD cases, while homozygous recessive pathogenic variants in GRN have been linked to recessive adult-onset NCL in two separate families [3, 6, 7, 11, 69, 79]. More recently, Ward and colleagues provided further evidence for phenotypic overlap between FTLD and NCL due to pathogenic GRN variants by identifying NCL-like biological hallmarks in haploinsufficient GRN patients [75]. Given the relationship of GRN to NCL, rare variation in other NCL genes, including CTSF, has also been suggested to contribute risk to FTD [80]. We identified five FTLD cases heterozygous for MFSD8 rare variants in the present study, suggesting our findings are akin to GRN but with several differences. First, homozygous pathogenic variants in GRN and MFSD8 lead to different types of NCL that occur during different stages of life; GRN homozygous variants lead to adult NCL whereas MFSD8 homozygous variants lead to late-infantile NCL [48]. Also unlike GRN, our results support a “two-hit” hypothesis in which one rare variant in MFSD8 increases the risk of developing FTLD, but requires an additional “hit” before FTLD may develop. This is further supported by the identification of two rare variants in clinically normal controls that cause early termination codons in MFSD8, both of which are predicted to be loss of function variants in the Genome Aggregation Database. Thus, our findings support a role for rare variants in MFSD8 increasing FTLD risk, but suggest these variants are unable to cause disease in the absence of another, as yet undetermined risk factor.
Beyond being linked to the neurologic defects observed in FTD and NCL, pathogenic variants in both MFSD8 and GRN have also been linked to vision defects and neuronal loss in the retina [61, 76]. A family study previously linked both the E381X and E336Q variants in MFSD8 to individuals with nonsyndromic autosomal recessive macular dystrophy [61]. The same study also identified the E336Q variant as a risk factor for maculopathies and cone disorders, and suggested that this hypomorphic variant may cause cone dysfunction in combination with a more severe variant in MFSD8, such as the E381X variant. Although this is similar to the “two-hit” model we propose for how rare variants in MFSD8 may contribute to FTLD risk, further studies of larger cohorts are required to elucidate if the missense variants that we identified in FTLD cases, particularly E336Q, may lead to disease in combination with other risk factors.
To the best of our knowledge, the only rare variant identified in this study that has previously been linked to NCL is the stop-gain variant, E381X [2] (Table S3). In the current study, a clinically normal control from the ADSP cohort was found to be heterozygous for this variant. We were unable to verify this variant with Sanger sequencing since the data for this participant was derived from a public source, and we did not have access to clinical data for this participant that might indicate any abnormal vision or neurological symptoms. Future studies will be critical to establishing the clinical significance of the five rare variants in MFSD8 that we identified in FTLD cases.
MFSD8 rare variant carriers display elevated levels of MFSD8 protein
We observed increased MFSD8 protein levels in MFG of MFSD8 rare variant carrier patients relative to sporadic FTLD patients and controls, and MFSD8 immunoreactivity was generally strongest in the cytoplasm of neurons across all diagnostic groups. Since MFG is affected by neurodegeneration in FTLD, it is possible that disrupting MFSD8 function may contribute to some of the pathological changes observed in FTLD. Similar observations have been made in studies of post-mortem brain tissue from NCL patients, where individuals with homozygous loss of function variants in MFSD8 show the earliest signs of neuronal loss and prominent neuronal accumulation of autofluorescent ceroid material in regions with the highest expression levels of MFSD8 mRNA, as identified in rat brains [64]. Thus, the elevated expression levels of MFSD8 protein in MFG of rare variant carrier cases relative to controls and sporadic FTLD cases suggests that MFSD8 may be playing a direct role in the pathobiology of FTLD in these cases.
MFSD8 rare variant carriers display changes in autophagic/lysosomal markers
Our experimental results from immunoblots of autophagosomal and lysosomal proteins in post-mortem tissue from MFSD8 rare variant carriers and mouse brain homogenates and the marked decrease in lysosomal-dependent degradation in cells from MFSD8 rare variant carriers support a role for MFSD8 in the autophagic/lysosomal system. Malfunctioning autophagy has been extensively demonstrated in a large number of neurodegenerative conditions, including FTLD [21, 44, 45, 49], and experimental disruption of autophagy in neurons leads to rapid neurodegeneration [19, 28]. Autophagic failure in the context of neurodegeneration may occur at different steps of this process, including defects at the level of cargo recognition, delivery of cargo due to impaired autophagosome formation or defective autophagosome/lysosomal fusion, translocation of cargo across the lysosomal membrane, or degradation of cargo in lysosomes [45, 49, 77]. The increased levels of the autophagosome marker LC3-II in post-mortem tissue together with the higher number of LC3 positive vesicles and marked reduction in lysosomal-dependent degradation rates observed in cells from MFSD8 rare variant carriers suggest that autophagosome clearance, an important step in autophagy, may be affected in these patients. Finally, we found a higher abundance of Mfsd8 in the group of lysosomes usually engaged in CMA. Together, these findings suggest that MFSD8 plays a role in autophagy and, probably more specifically, in CMA. However, detailed analyses of different types of autophagy in Mfsd8 knock-out mice and human cells derived from MFSD8 rare variant carriers are required to confirm involvement of MFSD8 in these degradative pathways.
Rare missense variants associated with FTLD risk alter intracellular trafficking of MFSD8
MFSD8 is a protein of unknown structure and function, making it difficult to assess the functional consequences of the five FTLD-associated rare missense variants identified in our study. However, previous studies suggest it belongs to the Major Facilitator Superfamily (MFS) of proteins and may function as a lysosomal membrane transporter [64, 67]. We used biochemical techniques to begin characterizing how rare missense variants in MFSD8 may alter protein function. These studies revealed a significant increase in cell surface accumulation for the F379S variant of MFSD8, and similar changes for the G385R variant of MFSD8. Multiple lysosomal trafficking pathways exist for delivering proteins to lysosomes, including a direct route from the trans-Golgi network, and an indirect route via the plasma membrane with subsequent endocytosis [62]. MFSD8 has previously been shown to predominantly reach the lysosome via the indirect route with clathrin-mediated endocytosis [70]. The F379S variant may lead to reduced MFSD8 function in lysosomes by disrupting its intracellular trafficking. This variant may also modify glycosylation of MFSD8 at an immediately upstream N-linked glycosylation site (amino acids 376NTT378) in a proposed lysosomal luminal loop [70]. This is supported by the observed increase in abundance of a lower molecular weight band on the immunoblot of cell surface MFSD8 (* in Fig. 4). Although the role of these glycosylation sites in normal MFSD8 function is unknown, disrupting glycosylation may alter MFSD8’s putative function as a lysosomal transporter. Future studies will be important for determining whether there are additional consequences of the F379S variant on MFSD8 function beyond increasing cell surface accumulation and possibly altering glycosylation of MFSD8 and, more importantly, how these changes contribute to FTLD pathobiology.
Beyond disrupting intracellular trafficking of MFSD8, rare missense variants may affect MFSD8 function by disrupting protein structure. Since there is no crystal structure of MFSD8, we used a previously reported computational prediction of MFSD8’s membrane topology to determine in which domains our FTLD-associated variants are located [67] (Fig. S5). Two variants fall in putative transmembrane domain alpha helices (S217G and I492T), one near the membrane-cytosol interface (E336Q), and two in a large loop facing the lysosomal lumen (F379S and G385R). Since alpha helical transmembrane domains are critical structural components that facilitate the transport function of MFS proteins [32, 55], amino acid substitutions in or near these domains may distort the structure of MFSD8 and affect its putative transport function. Previous studies have also suggested that loop regions in transporters may be important for stabilizing different conformational states of the protein [18], and in MFS transporters specifically, for transmitting conformational changes throughout the protein during substrate transport [41]. Thus, amino acid substitutions in loop domains may also disrupt MFSD8’s ability to transport substrates. Future identification of a substrate for MFSD8 will be necessary not only to elucidate any functional consequences of the rare variants we identified associated with FTLD risk, but also to determine the functional role of MFSD8 in lysosomes. These roles include a range of putative functions from maintenance of lysosomal pH to lysosomal calcium storage, or even efflux of small macromolecules or peptides resulting from protein breakdown. It is also interesting to keep in mind that MFSD8 may function outside the lysosomal compartment. In this respect, the Human Protein Atlas describes a fraction of MFSD8 that is detected in the nucleoplasm. We observed nuclear MFSD8 immunoreactivity in neurons and fibroblasts of controls and FTD patients carrying MFSD8 rare variants. The pathogenic relevance of the nuclear accumulation of MFSD8 requires further investigation.
Rare variation in MFSD8 may result in TDP-43 or tau pathology through differing mechanisms linked to lysosomal dysfunction
Given the link between NCL and FTD through GRN, we expected that variation in MFSD8 might contribute more risk to FTLD-TDP since GRN is associated with TDP-43 pathology in the brain; nevertheless, MFSD8 rare variants were observed in both FTLD-TDP and FTLD-tau cases. This raises the possibility that MFSD8 variants may contribute to either TDP-43 or tau pathology through distinct mechanisms. For example, MFSD8 may play a more direct role in the cellular response to aggregated TDP-43 protein in neurons, whereas its function may be indirectly involved with the response to tau aggregates in neurons and glia. Both scenarios are consistent with a role for MFSD8 in overall protein clearance as is suggested by our protein degradation studies in cultured cells. Whether additional genetic risk factors modify a propensity to develop tau versus TDP-43 pathology remains an open question, and future studies will be required to elucidate putative differences in the role of MFSD8 in the different pathological subtypes of FTLD.
Overall considerations
While our findings provide compelling evidence for the contribution of MFSD8 to FTLD risk and pathobiology, several limitations exist when interpreting our results. First, although rare protein-altering variants in MFSD8 occurred at a higher frequency in our FTLD patients relative to controls, we also identified clinically normal older controls carrying MFSD8 rare variants. This finding is consistent with rare variation in MFSD8 increasing the risk of developing FTLD, but likely not being the sole disease-causing pathogenic variant (e.g., in an autosomal dominant mode of inheritance). Second, even though our study design included patients of European descent, we cannot exclude the possibility of ascertainment bias since participants in both genetic analyses were recruited and diagnosed at the same research center. Our study also included samples sequenced by different methods at five different sequencing centers. Although we observed sufficient depth of coverage for reliable variant calling across samples analyzed by whole genome and whole exome sequencing (>30x), we cannot eliminate the possibility of technical variability leading to differences in variant detection across samples or false-negative findings in the unbiased discovery study. Confirming that rare variation in MFSD8 is associated with increased FTD risk will require replication in large, independent FTD cohorts that include patients from non-European populations, and that are sequenced by the same method at a single facility. The former may be particularly relevant since recent studies found recessive pathogenic variants in MFSD8 linked to NCL cases in South America and China [27]. Finally, post-mortem tissue was only available from five MFSD8 rare variant carriers. Further work is required with additional samples to assess whether there are biological differences across different genetic variants that may contribute to heterogeneity in risk penetrance and protein pathology.
In summary, we provide evidence suggesting that rare variants in MFSD8 are associated with FTLD risk. This statistical enrichment was corroborated by experimental studies showing elevated protein levels for MFSD8 and consistent increases in lysosome and autophagosome marker proteins in MFG of MFSD8 rare variant carriers. We also found marked disruption of lysosomal function in cells from FTD patients carrying rare missense variants in MFSD8. Overall, these findings support a role for autophagy and lysosomal dysfunction in the pathobiology of FTLD. MFSD8 may generally be altered in the context of FTLD, either directly or indirectly through its yet-to-be-determined function. Future studies elucidating the specific function of MFSD8 in lysosomes will help determine if modulating lysosomal function, possibly through MFSD8, may be a viable therapeutic strategy for modifying FTLD trajectories.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Jason Chen and the New York Genome Center for technical support of whole genome sequencing. Primary support for this study was provided by the Rainwater Charitable Foundation (JSY, AMC, SEL, GC, WWS, YH). Additional support was provided by the Bluefield Project to Cure FTD (JSY, WWS), Association for Frontotemporal Degeneration Susan Marcus Memorial Fund Clinical Research Grant (JSY), Larry L. Hillblom Foundation 2016-A-005-SUP (JSY), National Institute on Aging K01 AG049152 (JSY), John Douglas French Alzheimer’s Foundation (JSY, GC), National Institute on Aging P01 AG1972403 (BLM), National Institute on Aging P50 AG023501 (BLM), National Institute on Aging R01 AG023501, AG048030, NS079725 (YH), and R01 AG054108 (AMC). National Institutes of Health F32 AG050404 (DWS), RC1 AG035610 (GC), and R01 AG26938 (GC). Takeda Pharmaceutical Company Limited (GC). We acknowledge the support of the National Institute of Neurological Disorders and Stroke Informatics Center for Neurogenetics and Neurogenomics, P30 NS062691 (GC). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
COMPETING INTERESTS
YH is a co-founder and SAB member for E-Scape Bio, Inc. AMC is a co-founder and SAB member for Selphagy Inc.
REFERENCES
- 1.1000 Genomes Project Consortium RA, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR, Chang Y, Feng Q, Fang X, Guo X, Jian M, Jiang H, Jin X, Lan T, Li G, Li J, Li Y, Liu S, Liu X, Lu Y, Ma X, Tang M, Wang B, Wang G, Wu H, Wu R, Xu X, Yin Y, Zhang D, Zhang W, Zhao J, Zhao M, Zheng X, Lander ES, Altshuler DM, Gabriel SB, Gupta N, Gharani N, Toji LH, Gerry NP, Resch AM, Flicek P, Barker J, Clarke L, Gil L, Hunt SE, Kelman G, Kulesha E, Leinonen R, McLaren WM, Radhakrishnan R, Roa A, Smirnov D, Smith RE, Streeter I, Thormann A, Toneva I, Vaughan B, Zheng-Bradley X, Bentley DR, Grocock R, Humphray S, James T, Kingsbury Z, Lehrach H, Sudbrak R, Albrecht MW, Amstislavskiy VS, Borodina TA, Lienhard M, Mertes F, Sultan M, Timmermann B, Yaspo M-L, Mardis ER, Wilson RK, Fulton L, Fulton R, Sherry ST, Ananiev V, Belaia Z, Beloslyudtsev D, Bouk N, Chen C, Church D, Cohen R, Cook C, Garner J, Hefferon T, Kimelman M, Liu C, Lopez J, Meric P, O’Sullivan C, Ostapchuk Y, Phan L, Ponomarov S, Schneider V, Shekhtman E, Sirotkin K, Slotta D, Zhang H, McVean GA, Durbin RM, Balasubramaniam S, Burton J, Danecek P, Keane TM, Kolb-Kokocinski A, McCarthy S, Stalker J, Quail M, Schmidt JP, Davies CJ, Gollub J, Webster T, Wong B, Zhan Y, Auton A, Campbell CL, Kong Y, Marcketta A, Gibbs RA, Yu F, Antunes L, Bainbridge M, Muzny D, Sabo A, Huang Z, Wang J, Coin LJM, Fang L, Guo X, Jin X, Li G, Li Q, Li Y, Li Z, Lin H, Liu B, Luo R, Shao H, Xie Y, Ye C, Yu C, Zhang F, Zheng H, Zhu H, Alkan C, Dal E, Kahveci F, Marth GT, Garrison EP, Kural D, Lee W-P, Fung Leong W, Stromberg M, Ward AN, Wu J, Zhang M, Daly MJ, DePristo MA, Handsaker RE, Altshuler DM, Banks E, Bhatia G, del Angel G, Gabriel SB, Genovese G, Gupta N, Li H, Kashin S, Lander ES, McCarroll SA, Nemesh JC, Poplin RE, Yoon SC, Lihm J, Makarov V, Clark AG, Gottipati S, Keinan A, Rodriguez-Flores JL, Korbel JO, Rausch T, Fritz MH, Stütz AM, Flicek P, Beal K, Clarke L, Datta A, Herrero J, McLaren WM, Ritchie GRS, Smith RE, Zerbino D, Zheng-Bradley X, Sabeti PC, Shlyakhter I, Schaffner SF, Vitti J, Cooper DN, Ball E V, Stenson PD, Bentley DR, Barnes B, Bauer M, Keira Cheetham R, Cox A, Eberle M, Humphray S, Kahn S, Murray L, Peden J, Shaw R, Kenny EE, Batzer MA, Konkel MK, Walker JA, MacArthur DG, Lek M, Sudbrak R, Amstislavskiy VS, Herwig R, Mardis ER, Ding L, Koboldt DC, Larson D, Ye K, Gravel S, Swaroop A, Chew E, Lappalainen T, Erlich Y, Gymrek M, Frederick Willems T, Simpson JT, Shriver MD, Rosenfeld JA, Bustamante CD, Montgomery SB, De La Vega FM, Byrnes JK, Carroll AW, DeGorter MK, Lacroute P, Maples BK, Martin AR, Moreno-Estrada A, Shringarpure SS, Zakharia F, Halperin E, Baran Y, Lee C, Cerveira E, Hwang J, Malhotra A, Plewczynski D, Radew K, Romanovitch M, Zhang C, Hyland FCL, Craig DW, Christoforides A, Homer N, Izatt T, Kurdoglu AA, Sinari SA, Squire K, Sherry ST, Xiao C, Sebat J, Antaki D, Gujral M, Noor A, Ye K, Burchard EG, Hernandez RD, Gignoux CR, Haussler D, Katzman SJ, James Kent W, Howie B, Ruiz-Linares A, Dermitzakis ET, Devine SE, Abecasis GR, Min Kang H, Kidd JM, Blackwell T, Caron S, Chen W, Emery S, Fritsche L, Fuchsberger C, Jun G, Li B, Lyons R, Scheller C, Sidore C, Song S, Sliwerska E, Taliun D, Tan A, Welch R, Kate Wing M, Zhan X, Awadalla P, Hodgkinson A, Li Y, Shi X, Quitadamo A, Lunter G, McVean GA, Marchini JL, Myers S, Churchhouse C, Delaneau O, Gupta-Hinch A, Kretzschmar W, Iqbal Z, Mathieson I, Menelaou A, Rimmer A, Xifara DK, Oleksyk TK, Fu Y, Liu X, Xiong M, Jorde L, Witherspoon D, Xing J, Eichler EE, Browning BL, Browning SR, Hormozdiari F, Sudmant PH, Khurana E, Durbin RM, Hurles ME, Tyler-Smith C, Albers CA, Ayub Q, Balasubramaniam S, Chen Y, Colonna V, Danecek P, Jostins L, Keane TM, McCarthy S, Walter K, Xue Y, Gerstein MB, Abyzov A, Balasubramanian S, Chen J, Clarke D, Fu Y, Harmanci AO, Jin M, Lee D, Liu J, Jasmine Mu X, Zhang J, Zhang Y, Li Y, Luo R, Zhu H, Alkan C, Dal E, Kahveci F, Marth GT, Garrison EP, Kural D, Lee W-P, Ward AN, Wu J, Zhang M, McCarroll SA, Handsaker RE, Altshuler DM, Banks E, del Angel G, Genovese G, Hartl C, Li H, Kashin S, Nemesh JC, Shakir K, Yoon SC, Lihm J, Makarov V, Degenhardt J, Korbel JO, Fritz MH, Meiers S, Raeder B, Rausch T, Stütz AM, Flicek P, Paolo Casale F, Clarke L, Smith RE, Stegle O, Zheng-Bradley X, Bentley DR, Barnes B, Keira Cheetham R, Eberle M, Humphray S, Kahn S, Murray L, Shaw R, Lameijer E-W, Batzer MA, Konkel MK, Walker JA, Ding L, Hall I, Ye K, Lacroute P, Lee C, Cerveira E, Malhotra A, Hwang J, Plewczynski D, Radew K, Romanovitch M, Zhang C, Craig DW, Homer N, Church D, Xiao C, Sebat J, Antaki D, Bafna V, Michaelson J, Ye K, Devine SE, Gardner EJ, Abecasis GR, Kidd JM, Mills RE, Dayama G, Emery S, Jun G, Shi X, Quitadamo A, Lunter G, McVean GA, Chen K, Fan X, Chong Z, Chen T, Witherspoon D, Xing J, Eichler EE, Chaisson MJ, Hormozdiari F, Huddleston J, Malig M, Nelson BJ, Sudmant PH, Parrish NF, Khurana E, Hurles ME, Blackburne B, Lindsay SJ, Ning Z, Walter K, Zhang Y, Gerstein MB, Abyzov A, Chen J, Clarke D, Lam H, Jasmine Mu X, Sisu C, Zhang J, Zhang Y, Gibbs RA, Yu F, Bainbridge M, Challis D, Evani US, Kovar C, Lu J, Muzny D, Nagaswamy U, Reid JG, Sabo A, Yu J, Guo X, Li W, Li Y, Wu R, Marth GT, Garrison EP, Fung Leong W, Ward AN, del Angel G, DePristo MA, Gabriel SB, Gupta N, Hartl C, Poplin RE, Clark AG, Rodriguez-Flores JL, Flicek P, Clarke L, Smith RE, Zheng-Bradley X, MacArthur DG, Mardis ER, Fulton R, Koboldt DC, Gravel S, Bustamante CD, Craig DW, Christoforides A, Homer N, Izatt T, Sherry ST, Xiao C, Dermitzakis ET, Abecasis GR, Min Kang H, McVean GA, Gerstein MB, Balasubramanian S, Habegger L, Yu H, Flicek P, Clarke L, Cunningham F, Dunham I, Zerbino D, Zheng-Bradley X, Lage K, Berg Jespersen J, Horn H, Montgomery SB, DeGorter MK, Khurana E, Tyler-Smith C, Chen Y, Colonna V, Xue Y, Gerstein MB, Balasubramanian S, Fu Y, Kim D, Auton A, Marcketta A, Desalle R, Narechania A, Wilson Sayres MA, Garrison EP, Handsaker RE, Kashin S, McCarroll SA, Rodriguez-Flores JL, Flicek P, Clarke L, Zheng-Bradley X, Erlich Y, Gymrek M, Frederick Willems T, Bustamante CD, Mendez FL, David Poznik G, Underhill PA, Lee C, Cerveira E, Malhotra A, Romanovitch M, Zhang C, Abecasis GR, Coin L, Shao H, Mittelman D, Tyler-Smith C, Ayub Q, Banerjee R, Cerezo M, Chen Y, Fitzgerald TW, Louzada S, Massaia A, McCarthy S, Ritchie GR, Xue Y, Yang F, Gibbs RA, Kovar C, Kalra D, Hale W, Muzny D, Reid JG, Wang J, Dan X, Guo X, Li G, Li Y, Ye C, Zheng X, Altshuler DM, Flicek P, Clarke L, Zheng-Bradley X, Bentley DR, Cox A, Humphray S, Kahn S, Sudbrak R, Albrecht MW, Lienhard M, Larson D, Craig DW, Izatt T, Kurdoglu AA, Sherry ST, Xiao C, Haussler D, Abecasis GR, McVean GA, Durbin RM, Balasubramaniam S, Keane TM, McCarthy S, Stalker J, Bodmer W, Bedoya G, Ruiz-Linares A, Cai Z, Gao Y, Chu J, Peltonen L, Garcia-Montero A, Orfao A, Dutil J, Martinez-Cruzado JC, Oleksyk TK, Barnes KC, Mathias RA, Hennis A, Watson H, McKenzie C, Qadri F, LaRocque R, Sabeti PC, Zhu J, Deng X, Sabeti PC, Asogun D, Folarin O, Happi C, Omoniwa O, Stremlau M, Tariyal R, Jallow M, Sisay Joof F, Corrah T, Rockett K, Kwiatkowski D, Kooner J, Tịnh Hiê’n T, Dunstan SJ, Thuy Hang N, Fonnie R, Garry R, Kanneh L, Moses L, Sabeti PC, Schieffelin J, Grant DS, Gallo C, Poletti G, Saleheen D, Rasheed A (2015) A global reference for human genetic variation. Nature 526:68–74. doi: 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aiello C, Terracciano A, Simonati A, Discepoli G, Cannelli N, Claps D, Crow YJ, Bianchi M, Kitzmuller C, Longo D, Tavoni A, Franzoni E, Tessa A, Veneselli E, Boldrini R, Filocamo M, Williams RE, Bertini ES, Biancheri R, Carrozzo R, Mole SE, Santorelli FM (2009) Mutations in MFSD8/CLN7 are a frequent cause of variant-late infantile neuronal ceroid lipofuscinosis. Hum Mutat 30:E530–E540. doi: 10.1002/humu.20975 [DOI] [PubMed] [Google Scholar]
- 3.Almeida MR, Macário MC, Ramos L, Baldeiras I, Ribeiro MH, Santana I (2016) Portuguese family with the co-occurrence of frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis phenotypes due to progranulin gene mutation. Neurobiol Aging 41:200.e1-200.e5. doi: 10.1016/j.neurobiolaging.2016.02.019 [DOI] [PubMed] [Google Scholar]
- 4.Asanuma K, Tanida I, Shirato I, Ueno T, Takahara H, Nishitani T, Kominami E, Tomino Y (2003) MAP-LC3, a promising autophagosomal marker, is processed during the differentiation and recovery of podocytes from PAN nephrosis. FASEB J 17:1165–7. doi: 10.1096/fj.02-0580fje [DOI] [PubMed] [Google Scholar]
- 5.Auteri JS, Okada A, Bochaki V, Fred Dice J (1983) Regulation of intracellular protein degradation in IMR-90 human diploid fibroblasts. J Cell Physiol 115:167–174. doi: 10.1002/jcp.1041150210 [DOI] [PubMed] [Google Scholar]
- 6.Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442:916–9. doi: 10.1038/nature05016 [DOI] [PubMed] [Google Scholar]
- 7.Bird TD (2009) Progranulin plasma levels in the diagnosis of frontotemporal dementia. Brain 132:568–9. doi: 10.1093/brain/awp009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolte S, Cordelières FP (2006) A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224:213–32. doi: 10.1111/j.1365-2818.2006.01706.x [DOI] [PubMed] [Google Scholar]
- 9.Brandenstein L, Schweizer M, Sedlacik J, Fiehler J, Storch S (2016) Lysosomal dysfunction and impaired autophagy in a novel mouse model deficient for the lysosomal membrane protein Cln7. Hum Mol Genet 25:777–91. doi: 10.1093/hmg/ddv615 [DOI] [PubMed] [Google Scholar]
- 10.Carson AR, Smith EN, Matsui H, Brækkan SK, Jepsen K, Hansen J-B, Frazer KA (2014) Effective filtering strategies to improve data quality from population-based whole exome sequencing studies. BMC Bioinformatics 15:125. doi: 10.1186/1471-2105-15-125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin J-J, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C (2006) Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442:920–4. doi: 10.1038/nature05017 [DOI] [PubMed] [Google Scholar]
- 12.Cuervo AM, Dice JF, Knecht E (1997) A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J Biol Chem 272:5606–15 [DOI] [PubMed] [Google Scholar]
- 13.Do R, Kathiresan S, Abecasis GR (2012) Exome sequencing and complex disease: practical aspects of rare variant association studies. Hum Mol Genet 21:R1–9. doi: 10.1093/hmg/dds387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dubois B, Feldman H, Jacova C (2014) Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol 13:614–29. doi: 10.1016/S1474-4422(14)70090-0 [DOI] [PubMed] [Google Scholar]
- 15.Fuentes Fajardo K V, Adams D, NISC Comparative Sequencing Program CE, Mason CE, Sincan M, Tifft C, Toro C, Boerkoel CF, Gahl W, Markello T (2012) Detecting false-positive signals in exome sequencing. Hum Mutat 33:609–13. doi: 10.1002/humu.22033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, Ogar JM, Rohrer JD, Black S, Boeve BF, Manes F, Dronkers NF, Vandenberghe R, Rascovsky K, Patterson K, Miller BL, Knopman DS, Hodges JR, Mesulam MM, Grossman M (2011) Classification of primary progressive aphasia and its variants. Neurology 76:1006–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Götzl JK, Lang CM, Haass C, Capell A (2016) Impaired protein degradation in FTLD and related disorders. Ageing Res Rev 32:122–139. doi: 10.1016/j.arr.2016.04.008 [DOI] [PubMed] [Google Scholar]
- 18.Han L, Zhu Y, Liu M, Zhou Y, Lu G, Lan L, Wang X, Zhao Y, Zhang XC (2017) Molecular mechanism of substrate recognition and transport by the AtSWEET13 sugar transporter. Proc Natl Acad Sci U S A 114:10089–10094. doi: 10.1073/pnas.1709241114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441:885–9. doi: 10.1038/nature04724 [DOI] [PubMed] [Google Scholar]
- 20.Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters H V, Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Trojanowski JQ, Vinters H V, Hyman BT (2013) National Insitute on Aging- Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement 8:1–13. doi: 10.1016/j.jalz.2011.10.007.National [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson CW, Melia TJ, Yamamoto A (2012) Modulating macroautophagy: a neuronal perspective. Future Med Chem 4:1715–31. doi: 10.4155/fmc.12.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaushik S, Cuervo AM (2009) Methods to monitor chaperone-mediated autophagy. Methods Enzymol 452:297–324. doi: 10.1016/S0076-6879(08)03619-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaushik S, Cuervo AM (2015) Proteostasis and aging. Nat Med 21:1406–15. doi: 10.1038/nm.4001 [DOI] [PubMed] [Google Scholar]
- 24.Kaushik S, Cuervo AM (2018) The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol 19:365–381. doi: 10.1038/s41580-018-0001-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim E-J, Sidhu M, Gaus SE, Huang EJ, Hof PR, Miller BL, DeArmond SJ, Seeley WW (2012) Selective frontoinsular von Economo neuron and fork cell loss in early behavioral variant frontotemporal dementia. Cereb Cortex 22:251–9. doi: 10.1093/cercor/bhr004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koga H, Kaushik S, Cuervo AM (2010) Inhibitory effect of intracellular lipid load on macroautophagy. Autophagy 6:825–7. doi: 10.1096/fj.09-144519 [DOI] [PubMed] [Google Scholar]
- 27.Kohan R, Pesaola F, Guelbert N, Pons P, Oller-Ramirez AM, Rautenberg G, Becerra A, Sims K, Xin W, Cismondi IA, Noher de Halac I (2015) The neuronal ceroid lipofuscinoses program: A translational research experience in Argentina. Biochim Biophys Acta 1852:2301–2311. doi: 10.1016/j.bbadis.2015.05.003 [DOI] [PubMed] [Google Scholar]
- 28.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441:880–884. doi: 10.1038/nature04723 [DOI] [PubMed] [Google Scholar]
- 29.Kousi M, Lehesjoki A-E, Mole SE (2012) Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum Mutat 33:42–63. doi: 10.1002/humu.21624 [DOI] [PubMed] [Google Scholar]
- 30.Kousi M, Siintola E, Dvorakova L, Vlaskova H, Turnbull J, Topcu M, Yuksel D, Gokben S, Minassian BA, Elleder M, Mole SE, Lehesjoki A-E (2009) Mutations in CLN7/MFSD8 are a common cause of variant late-infantile neuronal ceroid lipofuscinosis. Brain 132:810–819. doi: 10.1093/brain/awn366 [DOI] [PubMed] [Google Scholar]
- 31.Lee J-A, Gao F-B (2008) Roles of ESCRT in autophagy-associated neurodegeneration. Autophagy 4:230–2 [DOI] [PubMed] [Google Scholar]
- 32.Lee J, Sands ZA, Biggin PC (2016) A Numbering System for MFS Transporter Proteins. Front Mol Biosci 3:21. doi: 10.3389/fmolb.2016.00021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, Nickerson DA, Christiani DC, Wurfel MM, Lin X (2012) Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet 91:224–237. doi: 10.1016/j.ajhg.2012.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lehner T, Thomas A, Miller BL, State MW (2016). Genomics, circuits, and pathways in clinical neuropsychiatry
- 35.Lek M, Karczewski KJ, Minikel E V, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won H-H, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, Consortium EA (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536:285–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lelieveld SH, Spielmann M, Mundlos S, Veltman JA, Gilissen C (2015) Comparison of Exome and Genome Sequencing Technologies for the Complete Capture of Protein-Coding Regions. Hum Mutat 36:815–22. doi: 10.1002/humu.22813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mackenzie IR, Neumann M, Baborie A, Sampathu D, Du Plessis D, Jaros E, Perry R, Trojanowski JQ, Mann DM, Lee VM (2011) A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122:111–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mackenzie IRA, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, Ince PG, Kamphorst W, Revesz T, Rozemuller AJM, Kumar-Singh S, Akiyama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ, Mann DMA (2010) Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 119:1–4. doi: 10.1007/s00401-009-0612-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mandel H, Cohen Katsanelson K, Khayat M, Chervinsky I, Vladovski E, Iancu TC, Indelman M, Horovitz Y, Sprecher E, Shalev SA, Spiegel R (2014) Clinico-pathological manifestations of variant late infantile neuronal ceroid lipofuscinosis (vLINCL) caused by a novel mutation in MFSD8 gene. Eur J Med Genet 57:607–12. doi: 10.1016/j.ejmg.2014.09.004 [DOI] [PubMed] [Google Scholar]
- 41.Masureel M, Martens C, Stein RA, Mishra S, Ruysschaert J-M, Mchaourab HS, Govaerts C (2014) Protonation drives the conformational switch in the multidrug transporter LmrP. Nat Chem Biol 10:149–55. doi: 10.1038/nchembio.1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor J-P, Weintraub D, Aarsland D, Galvin J, Attems J, Ballard CG, Bayston A, Beach TG, Blanc F, Bohnen N, Bonanni L, Bras J, Brundin P, Burn D, Chen-Plotkin A, Duda JE, El-Agnaf O, Feldman H, Ferman TJ, ffytche D, Fujishiro H, Galasko D, Goldman JG, Gomperts SN, Graff-Radford NR, Honig LS, Iranzo A, Kantarci K, Kaufer D, Kukull W, Lee VMY, Leverenz JB, Lewis S, Lippa C, Lunde A, Masellis M, Masliah E, McLean P, Mollenhauer B, Montine TJ, Moreno E, Mori E, Murray M, O’Brien JT, Orimo S, Postuma RB, Ramaswamy S, Ross OA, Salmon DP, Singleton A, Taylor A, Thomas A, Tiraboschi P, Toledo JB, Trojanowski JQ, Tsuang D, Walker Z, Yamada M, Kosaka K (2017) Diagnosis and management of dementia with Lewy bodies. Neurology 89:88–100. doi: 10.1212/WNL.0000000000004058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. doi: 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, Füllgrabe J, Jackson A, Jimenez Sanchez M, Karabiyik C, Licitra F, Lopez Ramirez A, Pavel M, Puri C, Renna M, Ricketts T, Schlotawa L, Vicinanza M, Won H, Zhu Y, Skidmore J, Rubinsztein DC (2017) Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 93:1015–1034. doi: 10.1016/j.neuron.2017.01.022 [DOI] [PubMed] [Google Scholar]
- 45.Menzies FM, Fleming A, Rubinsztein DC (2015) Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci 16:345–57. doi: 10.1038/nrn3961 [DOI] [PubMed] [Google Scholar]
- 46.Mercy L, Hodges JR, Dawson K, Barker RA, Brayne C (2008) Incidence of early-onset dementias in Cambridgeshire, United Kingdom. Neurology 71:1496–9. doi: 10.1212/01.wnl.0000334277.16896.fa [DOI] [PubMed] [Google Scholar]
- 47.Miller ZA, Mandelli ML, Rankin KP, Henry ML, Babiak MC, Frazier DT, Lobach I V., Bettcher BM, Wu TQ, Rabinovici GD, Graff-Radford NR, Miller BL, Gorno-Tempini ML (2013) Handedness and language learning disability differentially distribute in progressive aphasia variants. Brain 136:3461–3473. doi: 10.1093/brain/awt242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mole SE, Cotman SL (2015) Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim Biophys Acta 1852:2237–41. doi: 10.1016/j.bbadis.2015.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nixon RA (2013) The role of autophagy in neurodegenerative disease. Nat Med 19:983–97. doi: 10.1038/nm.3232 [DOI] [PubMed] [Google Scholar]
- 50.Olszewska DA, Lonergan R, Fallon EM, Lynch T (2016) Genetics of Frontotemporal Dementia. Curr Neurol Neurosci Rep 16:107. doi: 10.1007/s11910-016-0707-9 [DOI] [PubMed] [Google Scholar]
- 51.Olszewska DA, Lonergan R, Fallon EM, Lynch T (2016) Genetics of Frontotemporal Dementia. Curr Neurol Neurosci Rep 16:107. doi: 10.1007/s11910-016-0707-9 [DOI] [PubMed] [Google Scholar]
- 52.Onyike CU, Diehl-Schmid J (2013) The epidemiology of frontotemporal dementia. Int Rev Psychiatry 25:130–7. doi: 10.3109/09540261.2013.776523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Orenstein SJ, Kuo S-H, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, Raya A, Sulzer D, Cuervo AM (2013) Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 16:394–406. doi: 10.1038/nn.3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patel B, Cuervo AM (2015) Methods to study chaperone-mediated autophagy. Methods 75:133–140. doi: 10.1016/j.ymeth.2015.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pedersen BP, Kumar H, Waight AB, Risenmay AJ, Roe-Zurz Z, Chau BH, Schlessinger A, Bonomi M, Harries W, Sali A, Johri AK, Stroud RM (2013) Crystal structure of a eukaryotic phosphate transporter. Nature 496:533–6. doi: 10.1038/nature12042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pottier C, Bieniek KF, Finch N, van de Vorst M, Baker M, Perkersen R, Brown P, Ravenscroft T, van Blitterswijk M, Nicholson AM, DeTure M, Knopman DS, Josephs KA, Parisi JE, Petersen RC, Boylan KB, Boeve BF, Graff-Radford NR, Veltman JA, Gilissen C, Murray ME, Dickson DW, Rademakers R (2015) Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol 130:77–92. doi: 10.1007/s00401-015-1436-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rankin KP, Kramer J, Miller BL (2005) Patterns of cognitive and emotional empathy in frontotemporal lobar degeneration. Cogn Behav Neurol 18:28–36 [DOI] [PubMed] [Google Scholar]
- 59.Rascovsky K, Hodges JR, Kipps CM, Johnson JK, Seeley WW, Mendez MF, Knopman D, Kertesz A, Mesulam M, Salmon DP, Galasko D, Chow TW, Decarli C, Hillis A, Josephs K, Kramer JH, Weintraub S, Grossman M, Gorno-Tempini M-LL, Miller BM (2007) Diagnostic criteria for the behavioral variant of frontotemporal dementia (bvFTD): current limitations and future directions. Alzheimer Dis Assoc Disord 21:S14–8. doi: 10.1097/WAD.0b013e31815c3445 [DOI] [PubMed] [Google Scholar]
- 60.Rohrer JD, Guerreiro R, Vandrovcova J, Uphill J, Reiman D, Beck J, Isaacs AM, Authier A, Ferrari R, Fox NC, Mackenzie IR, Warren JD, de Silva R, Holton J, Revesz T, Hardy J, Mead S, Rossor MN (2009) The heritability and genetics of frontotemporal lobar degeneration. Neurology 73:1451–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roosing S, van den Born LI, Sangermano R, Banfi S, Koenekoop RK, Zonneveld-Vrieling MN, Klaver CCW, van Lith-Verhoeven JJC, Cremers FPM, den Hollander AI, Hoyng CB (2015) Mutations in MFSD8, Encoding a Lysosomal Membrane Protein, Are Associated with Nonsyndromic Autosomal Recessive Macular Dystrophy. Ophthalmology 122:170–179. doi: 10.1016/j.ophtha.2014.07.040 [DOI] [PubMed] [Google Scholar]
- 62.Saftig P, Klumperman J (2009) Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol 10:623–35. doi: 10.1038/nrm2745 [DOI] [PubMed] [Google Scholar]
- 63.See TM, LaMarre AK, Lee SE, Miller BL (2010) Genetic causes of frontotemporal degeneration. J Geriatr Psychiatry Neurol 23:260–268 [DOI] [PubMed] [Google Scholar]
- 64.Sharifi A, Kousi M, Sagné C, Bellenchi GC, Morel L, Darmon M, Hulková H, Ruivo R, Debacker C, El Mestikawy S, Elleder M, Lehesjoki A-E, Jalanko A, Gasnier B, Kyttälä A (2010) Expression and lysosomal targeting of CLN7, a major facilitator superfamily transporter associated with variant late-infantile neuronal ceroid lipofuscinosis. Hum Mol Genet 19:4497–514. doi: 10.1093/hmg/ddq381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shyr C, Tarailo-Graovac M, Gottlieb M, Lee JJ, van Karnebeek C, Wasserman WW (2014) FLAGS, frequently mutated genes in public exomes. BMC Med Genomics 7:64. doi: 10.1186/s12920-014-0064-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sieben A, Van Langenhove T, Engelborghs S, Martin J-J, Boon P, Cras P, De Deyn P-P, Santens P, Van Broeckhoven C, Cruts M (2012) The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol 124:353–72. doi: 10.1007/s00401-012-1029-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Siintola E, Topcu M, Aula N, Lohi H, Minassian BA, Paterson AD, Liu X-Q, Wilson C, Lahtinen U, Anttonen A-K, Lehesjoki A-E (2007) The Novel Neuronal Ceroid Lipofuscinosis Gene MFSD8 Encodes a Putative Lysosomal Transporter. Am J Hum Genet 81:136–146. doi: 10.1086/518902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sirkis DW, Bonham LW, Aparicio RE, Geier EG, Ramos ME, Wang Q, Karydas A, Miller ZA, Miller BL, Coppola G, Yokoyama JS (2016) Rare TREM2 variants associated with Alzheimer’s disease display reduced cell surface expression. Acta Neuropathol Commun 4:1–11. doi: 10.1186/s40478-016-0367-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, Rossi G, Pareyson D, Mole SE, Staropoli JF, Sims KB, Lewis J, Lin W-L, Dickson DW, Dahl H-H, Bahlo M, Berkovic SF (2012) Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet 90:1102–7. doi: 10.1016/j.ajhg.2012.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Steenhuis P, Herder S, Gelis S, Braulke T, Storch S (2010) Lysosomal targeting of the CLN7 membrane glycoprotein and transport via the plasma membrane require a dileucine motif. Traffic 11:987–1000. doi: 10.1111/j.1600-0854.2010.01073.x [DOI] [PubMed] [Google Scholar]
- 71.Tartaglia MC, Sidhu M, Laluz V, Racine C, Rabinovici GD, Creighton K, Karydas A, Rademakers R, Huang EJ, Miller BL, DeArmond SJ, Seeley WW (2010) Sporadic corticobasal syndrome due to FTLD-TDP. Acta Neuropathol 119:365–74. doi: 10.1007/s00401-009-0605-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Towbin H, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A 76:4350–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164. doi: 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow E-M, Cuervo AM, Mandelkow E (2009) Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet 18:4153–70. doi: 10.1093/hmg/ddp367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ward ME, Chen R, Huang H-Y, Ludwig C, Telpoukhovskaia M, Taubes A, Boudin H, Minami SS, Reichert M, Albrecht P, Gelfand JM, Cruz-Herranz A, Cordano C, Alavi MV., Leslie S, Seeley WW, Miller BL, Bigio E, Mesulam M-M, Bogyo MS, Mackenzie IR, Staropoli JF, Cotman SL, Huang EJ, Gan L, Green AJ (2017) Individuals with progranulin haploinsufficiency exhibit features of neuronal ceroid lipofuscinosis. Sci Transl Med 9:eaah5642. doi: 10.1126/scitranslmed.aah5642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ward ME, Taubes A, Chen R, Miller BL, Sephton CF, Gelfand JM, Minami S, Boscardin J, Martens LH, Seeley WW, Yu G, Herz J, Filiano AJ, Arrant AE, Roberson ED, Kraft TW, Farese R V, Green A, Gan L (2014) Early retinal neurodegeneration and impaired Ran-mediated nuclear import of TDP-43 in progranulin-deficient FTLD. J Exp Med 211:1937–45. doi: 10.1084/jem.20140214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wong E, Cuervo AM (2010) Autophagy gone awry in neurodegenerative diseases. Nat Neurosci 13:805–11. doi: 10.1038/nn.2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu L, Yavas G, Hong H, Tong W, Xiao W (2017) Direct comparison of performance of single nucleotide variant calling in human genome with alignment-based and assembly-based approaches. Sci Rep 7:10963. doi: 10.1038/s41598-017-10826-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yu CE, Bird TD, Bekris LM, Montine TJ, Leverenz JB, Steinbart E, Galloway NM, Feldman H, Woltjer R, Miller CA, Wood EM, Grossman M, McCluskey L, Clark CM, Neumann M, Danek A, Galasko DR, Arnold SE, Chen-Plotkin A, Karydas A, Miller BL, Trojanowski JQ, Lee VM, Schellenberg GD, Van Deerlin VM (2010) The spectrum of mutations in progranulin: a collaborative study screening 545 cases of neurodegeneration. Arch Neurol 67:161–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.van der Zee J, Mariën P, Crols R, Van Mossevelde S, Dillen L, Perrone F, Engelborghs S, Verhoeven J, D’aes T, Ceuterick-De Groote C, Sieben A, Versijpt J, Cras P, Martin J-J, Van Broeckhoven C (2016) Mutated CTSF in adult-onset neuronal ceroid lipofuscinosis and FTD. Neurol Genet 2:e102. doi: 10.1212/NXG.0000000000000102 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.