

Abstract
Background:
Vascular smooth muscle cells (SMC) synthesize extracellular matrix (ECM) that contributes to tissue remodeling following revascularization interventions. The cytokine transforming growth factor-β (TGF-β) is induced upon tissue injury and regulates tissue remodeling and wound healing, but dysregulated signaling results in excess ECM deposition and fibrosis. The LIM domain protein LMO7 is a TGF-β1 target gene in hepatoma cells, but its role in vascular physiology and fibrosis is unknown.
Methods:
We employ carotid ligation and femoral artery denudation models in mice with global or inducible smooth muscle-specific deletion of LMO7, and knockout, knockdown, overexpression, and mutagenesis approaches in mouse and human SMC, and human arteriovenous fistula (AVF) and cardiac allograft vasculopathy (CAV) samples to assess the role of LMO7 in neointima and fibrosis.
Results:
We demonstrate that LMO7 is induced post-injury and by TGF-β in SMC in vitro. Global or SMC-specific LMO7 deletion enhanced neointimal formation, TGF-β signaling, ECM deposition and proliferation in vascular injury models. LMO7 loss of function in human and mouse SMC enhanced ECM protein expression at baseline and following TGF-β treatment. TGF-β neutralization or receptor antagonism prevented the exacerbated neointimal formation and ECM synthesis conferred by loss of LMO7. Notably, loss of LMO7 coordinately amplified TGF-β signaling by inducing expression of Tgfb1 mRNA, TGF-β protein, αv and β3 integrins that promote activation of latent TGF-β, and downstream effectors pSMAD3 and CTGF. Mechanistically, the LMO7 LIM domain interacts with AP-1 transcription factor subunits c-FOS and c-JUN and promotes their ubiquitination and degradation, disrupting AP-1-dependent TGF-β autoinduction. Importantly, preliminary studies suggest that LMO7 is upregulated in human intimal hyperplastic arteriovenous fistula (AVF) and cardiac allograft vasculopathy (CAV) samples, and inversely correlates with pSMAD3 in CAV.
Conclusions:
LMO7 is induced by TGF-β and serves to limit vascular fibrotic responses through negative feedback regulation of the TGF-β pathway. This mechanism has important implications for intimal hyperplasia, wound healing, and fibrotic diseases.
Keywords: smooth muscle, extracellular matrix, fibrosis, transcription factors, TGF-β, LMO7
Introduction
Cardiovascular disease is the leading cause of death globally, with atherosclerotic lesions leading to myocardial infarction, stroke and peripheral vascular disease. While focal lesions can be treated by percutaneous intervention or bypass graft, these procedures can be complicated by intimal hyperplasia1. Smooth muscle cells (SMC) retain a plasticity that allows for wound healing but also contributes to intimal hyperplasia. In response to vessel injury, quiescent, differentiated medial SMC undergo a phenotypic modulation to a myofibroblast phenotype in which they proliferate and migrate to the vessel lumen where they secrete extracellular matrix (ECM) and form a neointima. SMC proliferation is an early response to injury, but lesion growth continues over time due to SMC deposition of ECM, including collagens and fibronectin2. Since this matrix can comprise up to 80% of the lesion volume3, understanding the mechanisms underlying SMC ECM synthesis remains an important question.
Transforming growth factor-β (TGF-β) is a central regulator of ECM deposition in vascular injury and in injury-induced fibrosis in many tissues4. TGF-β precursor is synthesized intracellularly and secreted into the ECM as an inactive latent complex. Upon binding to activating molecules such as MMPs or αv integrins, this complex undergoes a conformational change or proteolysis to release the active TGF-β dimer that binds to the cell surface receptor TGFβR2 to elicit downstream signaling through canonical and non-canonical pathways5. The canonical SMAD2/3 pathway mainly mediates fibrotic responses, as SMAD3 knockout is sufficient to inhibit fibrosis in fibroblast and inflammatory cells6. TGF-β stimulates SMAD2 and SMAD3 phosphorylation and nuclear translocation to activate gene transcription in association with SMAD45.
TGF-β-mediated ECM synthesis is essential for wound repair, but excess ECM deposition leads to fibrosis. Thus, identifying mechanisms that promote appropriate resolution of TGF-β signaling is critical. We identify the protein LIM domain only 7 (LMO7) as a novel negative feedback regulator of TGF-β signaling and ECM deposition. LMO7 is a 150-kDa scaffolding protein containing protein-protein interaction domains including calponin homology, PDZ, F-Box, and LIM domains7, 8. LMO7 localizes to plasma membrane, cytoskeleton and nucleus and has been shown to play roles in adherens junction assembly7, cell migration9, and gene transcription10. Little is known regarding detailed molecular mechanisms or in vivo phenotypes, as adult mice with germline deletion of Lmo7 appear normal, but aged knockout mice develop spontaneous lung adenocarcinoma11. LMO7 plays roles in skeletal muscle transcription and in cardiac development10, 12, but its role in SMC has not been investigated. Because LMO7 has been shown to be induced by TGF-β1 in hepatoma cells13, we aimed to investigate the function of LMO7 in ECM synthesis and vascular injury response in SMC downstream of TGF-β.
Methods
Additional detailed methods available in data Supplement.
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure from the corresponding author upon reasonable request.
Mice.
Lmo7−/− mice in C57BL/6J background were obtained from Dr. Jun Miyoshi (Osaka Medical Center, Japan)11. Experimental Lmo7−/− and littermate control mice were generated from heterozygous knockout breeding pairs. Lmo7fl/fl mice obtained from Dr. Ju Chen (UCSD)14 were originally in Black Swiss background and backcrossed to BL6 for over six generations. Female Lmo7fl/fl mice were crossed with male Myh11-CreERT2 (on Y-chromosome, Jackson Laboratory) mice to generate female Lmo7fl/+ and male Lmo7fl/+/Myh11-CreERT2, which were intercrossed to generate male Lmo7fl/fl/Myh11-CreERT2 or Lmo7+/+/Myh11-CreERT2. Male Lmo7fl/fl/Myh11-CreERT2 mice were then crossed with littermate female Lmo7fl/fl to generate experimental Lmo7fl/fl/Myh11-CreERT2 mice, and Lmo7+/+/Myh11-CreERT2 were crossed with littermate female Lmo7+/+ to generate Lmo7+/+/Myh11-CreERT2 (control) mice. Experimental and control mice were age- and size-matched. Lmo7 deletion was induced by injecting 6-week-old mice with 50 mg/kg tamoxifen for 5 days, followed by 5 day recovery prior to surgery. Control mice also received tamoxifen. Genotyping was performed by PCR. Mice were housed in pathogen-free conditions in Yale University animal facilities. All experiments were approved by the Yale University Institutional Animal Care and Use Committee.
Animal experiments.
Male mice were used for all intimal hyperplasia experiments. For experimental global Lmo7−/− mice, littermate Lmo7+/+ mice were used as control. For experimental Lmo7fl/fl/Myh11-CreERT2 mice, age- and size-matched Lmo7+/+/Myh11-CreERT2 mice were used as control. For carotid artery ligation, 8–16-week-old mice were anesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine. The left common carotid artery was exposed, separated from the vagus nerve and completely ligated using 6–0 silk suture immediately proximal to the carotid bifurcation15. Mice were sacrificed at between 3 – 28 days after surgery by cardiac perfusion with PBS+sodium nitroprusside (SNP) followed by 4% paraformaldehyde (PFA). The injured and contralateral uninjured arteries were dissected, fixed, dehydrated, and embedded in OCT compound (Tissue Tek, Elkhart, IN) for cryosectioning. The femoral artery wire denudation model was performed and analyzed as described16 on 20g mice using a 0.010” guidewire (Modern Grinding, Port Washington, WI).
Human tissue samples.
Deidentified matured patent human arteriovenous fistula (AVF) samples were provided by Yale Vascular Surgery. Samples were obtained after at least 6 months of hemodialysis during surgical revision of the fistula due to severe anastomotic stenosis. The samples analyzed are of a segment of normally remodeling patent vein from the fistulae. Control veins were obtained from renal disease patients at time of initial AVF creation. Human coronary arteries from cardiac allograft vasculopathy or normal patients were provided post-autopsy by the Yale Research Histology Core Facility in accordance with YSOP#116. Sample procurement with informed consent approved by Human Investigation Committee of Yale University IRB HIC#1005006865.
Statistical analysis.
Values are presented as mean ± standard error of the mean (SEM). Statistical analysis was performed using Prism 7 (Graph-Pad). Comparisons between two samples were performed using a nonparametric Mann-Whitney U test. Data sets with independent groups were analyzed by one-way ANOVA followed by Sidak multiple comparisons test. Experiments comparing two independent variables were analyzed by two-way ANOVA with additive interaction model followed by Holm-Sidak multiple comparisons testing. Comparisons between two curves were performed by nonlinear regression with extra sum-of-squares F test. Tests are two-sided. P-values less than 0.05 were considered significant.
Results
Loss of LMO7 in SMC exacerbates intimal hyperplasia and ECM deposition.
SMC contribute to vascular remodeling through phenotypic switching, proliferation, and ECM deposition. Because the function of LMO7 is unknown in SMC, we initially assessed its expression in response to vascular injury. Wild type (WT) mice were subjected to carotid artery ligation to induce intimal hyperplasia15. Immunostaining showed that LMO7 was expressed at modest levels in uninjured vessels and was highly induced from day 7 post-injury, peaking at day 10 but remaining elevated through 28 days (Fig. 1A). Notably, the initial induction of LMO7 followed the initiation of TGF-β signaling as indicated by p-SMAD3 staining (Fig. 1A). Because TGF-β1 is induced over time in response to vascular injury and contributes to intimal hyperplasia through SMC proliferation and ECM synthesis17, we next determined whether LMO7 is a TGF-β1 target gene in SMC. LMO7 mRNA (Suppl Fig. 1A) and protein (Suppl Fig. 1B, C) was induced by TGF-β1 treatment in human coronary artery SMC (CASMC) in a concentration- and time-dependent manner, with peak protein expression at 0.5 ng/ml TGF-β1 up to 48 hours.
Figure 1. Loss of LMO7 in SMC exacerbates intimal hyperplasia and ECM deposition.

(A) WT mice were subjected to carotid artery ligation and injured vessels were harvested at time points ranging from 3 to 28 days. Cryosections were immunostained for LMO7 (green) (left panel). Adjacent sections were immunostained for p-SMAD3 (green) (right panel). DAPI was used to visualize nuclei. Representative images from two mice at each time point are shown. 20 sections were stained for LMO7 or pSMAD3 per individual per time point. (B) EVG staining of tissue sections from control or Lmo7iΔSM mice 28 days after carotid artery ligation. (C) Quantification of intimal size and intimal/medial ratio of injured carotid arteries (n=10). (D) Trichrome staining of cross-sections of contralateral uninjured and injured arteries of control or Lmo7iΔSM mice after carotid ligation. (E) Picro-Sirius Red staining of cross-sections of injured arteries of control or Lmo7iΔSM mice at 28 days post carotid ligation by phase contrast (upper panel) or polarized light (lower panel) microscopy. (F) Quantification of relative integrated intensity of the signal in medial and intimal layers shown in polarized light images in (E) (n=5). (G) Mouse aortic SMCs were treated with 0.5ng/ml TGF-β1 for 24hrs and mRNA was harvested for qPCR analysis of ECM genes (n=4 independent experiments). Two-way ANOVA revealed a significant effect of LMO7 depletion and TGF-β1 treatment on mRNA expression. For Col1a2 mRNA only, LMO7 depletion significantly enhanced the TGF-β1 effect on expression (p=0.0009). Scale bar=50 μm. Data are expressed as mean ± S.E.M. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
To study the function of LMO7 in vascular injury, mice with germline deletion of Lmo7 (Lmo7−/−)11 were subjected to carotid artery ligation or femoral artery denudation18. Elastic Van Gieson (EVG) staining revealed that while the contralateral uninjured vessels showed no significant morphologic difference between Lmo7+/+ and Lmo7−/− mice at baseline, neointimal expansion was more than doubled in Lmo7−/− mice in both injury models (Suppl Fig. 2A-D). Since SMC play a central role in intimal hyperplasia, we generated smooth muscle-specific inducible Lmo7 knockout mice by crossing Lmo7fl/fl 14 with Myh11_creERT2 (Lmo7iΔSM), allowing for efficient tamoxifen-induced deletion (Suppl Fig. 2E). Lmo7+/+/Myh11_creERT2 mice treated with tamoxifen served as controls. Lmo7iΔSM mice exhibited exacerbated neointimal expansion over controls of similar magnitude as in the global knockout in both injury models (Fig 1B-C, Suppl Fig. 2F-G), indicating that the loss of LMO7 in SMC is sufficient to enhance neointima formation. Ki67 immunohistochemistry revealed enhanced intimal and medial proliferation after carotid ligation at 28 days (Suppl Fig. 3A-B) or femoral endothelial denudation injury at 10 days (Suppl Fig. 3C-D), in Lmo7−/− mice. Lmo7−/− mouse aortic SMC (SMC) (Suppl Fig. 3E) or human CASMC with LMO7 knockdown (Suppl Fig. 3F-G) also exhibited increased proliferation compared to controls.
Vascular ECM deposition also differed between Lmo7iΔSM and control injured mice. While there was no obvious difference between the uninjured arteries of the two groups, Masson’s Trichrome staining (Fig. 1D, Suppl Fig. 4A) and Picrosirius Red (PSR) staining with polarized light microscopy (Fig. 1E-F) revealed increased collagen in injured Lmo7iΔSM medial and intimal layers versus control. Increased ECM deposition was also observed in germline knockout mice with both vascular injury models (Suppl Fig. 4B-E). Consistent with these in vivo findings, expression of major ECM genes Col1a1, Col1a2, Col3a1 and Fn1 was higher in aortic SMC cultured from Lmo7−/− mice, with up to 10-fold more than in WT SMC (Fig. 1G). As expected, TGF-β1 treatment induced expression of ECM genes in WT and Lmo7−/− cells; however, the total levels of ECM genes after TGF-β1 treatment were much greater in Lmo7−/− cells, reaching up to 25-fold over the WT baseline levels (Fig. 1G). The medial layer of freshly isolated mouse aorta (uninjured mice) also demonstrated a modest induction of ECM gene mRNAs in Lmo7−/− mice (Suppl Fig. 4F). While both proliferation and ECM deposition contribute to neointimal expansion and are inter-related, we sought to assess the relative contribution of these processes to the LMO7 loss of function phenotype. For carotid ligation injury, there was a nominally greater increase in neointimal size (3.76±3.10 fold) relative to the increase in neointimal cell number (2.16±1.15 fold) in Lmo7iΔSM vs controls. Similarly, in the femoral denudation model there was a nominally greater increase of 1.92±0.97 fold in neointimal size relative to the increase of 1.42±0.41 fold in neointimal cell number (compare Fig. 1B-C and Suppl Fig. 2F-G to Suppl Fig. 5A-B). While this is only a trend and cannot account for potential changes in cell volume, we additionally determined that there is more ECM gene mRNA on a per cell basis in Lmo7−/− SMCs compared to controls (Suppl Fig. 5C). These data collectively suggest that LMO7 is induced by TGF-β1 in SMC and that LMO7 deficiency leads to hyperproliferation and enhanced ECM deposition, which both contribute to exaggerated neointimal formation following vascular injury.
TGF-β signaling and ECM gene expression are augmented in LMO7 deficient SMCs.
Given that injured LMO7 deficient vessels display exaggerated intimal hyperplasia and ECM deposition, and that LMO7 is itself a TGF-β1-target gene, we predicted that LMO7 induction may serve as a negative feedback regulator of TGF-β signaling that functions to limit the injury response. To test this hypothesis, we assessed SMAD3 phosphorylation (p-SMAD3) as an indicator of TGF-β activity post-injury. p-SMAD3 immunostaining was stronger in the neointima of Lmo7iΔSM vessels than in controls at 28 days post-injury, and was mainly nuclear (Fig. 2A), indicating increased TGF-β activity. ACTA2 levels were elevated at 28 days post-injury in the Lmo7iΔSM mouse, also consistent with elevated TGF-β signaling (Fig. 2A). Immunostaining for TGF-β19 or its effector CTGF also revealed enhanced TGF-β activity in Lmo7iΔSM vessels 28 days post-injury (Fig. 2B). Enhanced TGF-β signaling was also observed in injured femoral arteries from Lmo7iΔSM mice and Lmo7−/− mice (Suppl Fig. 6). Importantly, we noted an increase in TGF-β or CTGF at all time points assessed from 3–28 days following carotid ligation injury in Lmo7iΔSM compared to control mice. While CTGF peaked at day 14 and then declined in control mice, there was no such decline in Lmo7iΔSM mice (Fig. 2C), indicating that loss of LMO7 induced an increased and more persistent TGF-β signal. Consistent with these findings, TGF-β1 induced higher levels of p-SMAD3 in Lmo7−/− mouse SMC which was significant at 0.5 ng/ml TGF-β1, the dose used throughout this study (Fig. 2D).
Figure 2. TGF-β signaling and ECM gene expression are augmented in LMO7 deficient SMCs.
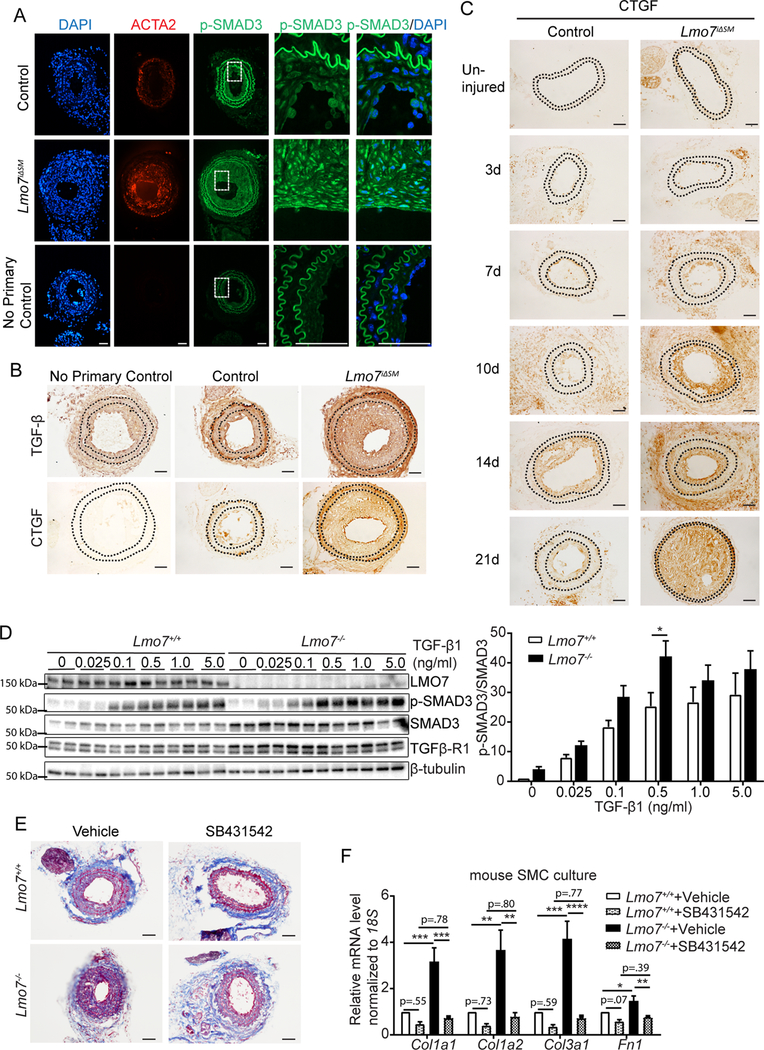
(A, B) Immunostaining of (A) p-SMAD3 (green) or ACTA2 (red) or (B) TGF- β or CTGF in tissue sections from control and Lmo7iΔSM mice after carotid artery ligation. (C) CTGF Immunohistochemistry of arterial sections of control and Lmo7iΔSM mice after carotid artery ligation for durations as indicated. (D) Western analysis of Lmo7+/+ or Lmo7−/− mouse SMCs treated with TGF-β1 at indicated doses for 24 hrs. Quantification of SMAD3 phosphorylation is shown on right (n=6 independent experiments). Two-way ANOVA revealed a significant effect of LMO7 depletion and TGF-β1 dose effect on p-SMAD3 phosphorylation, but there was no significant interaction between Lmo7−/− and TGF-β1 treatment. (E) Lmo7+/+ or Lmo7−/− mice were subjected to carotid ligation and injected intraperitoneally with SB4341542 (10mg/kg/d) or Vehicle (1% DMSO/30% polyethylene/ 1% Tween 80) daily from days 7–28 post-ligation. Trichrome staining of cross-sections of injured arteries is shown. (F) qPCR analysis of ECM genes in Lmo7+/+ or Lmo7−/− mouse SMCs treated with 10 μM SB431542 or Vehicle for 24 hrs (n=5 independent experiments). Two-way ANOVA revealed a significant effect of LMO7 depletion and TGF-β receptor inhibition on mRNA expression. For Col1a1, Col1a2 and Col3a1 mRNA, the magnitude of reduction was greater in Lmo7−/− SMCs (Col1a1 p=0.0057, Col1a2 p=0.019, Col3a1 p=0.0022), indicating LMO7 regulates many ECM genes via TGF-β signaling. Scale bar=50 μm. Data are expressed as mean ± S.E.M. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
To determine whether enhanced TGF-β signaling is responsible for the increased neointima and ECM deposition in Lmo7−/− mice, we blocked this signaling by injecting mice daily post-injury with the TGFβ-R1 inhibitor SB431542. Inhibition of TGF-β signaling was confirmed by reduced p-SMAD3 staining (Suppl Fig. 7A). The administration of TGF-β receptor inhibitor prevented neointima formation in control mice and greatly reduced both neointima and collagen staining in Lmo7−/− mice (Suppl Fig. 7B-C, Fig. 2E). Similarly, treatment with SB431542 reduced ECM gene induction with TGF-β in Lmo7−/− mouse SMC to below baseline control levels (Fig. 2F).
Loss of LMO7 induced elevated expression of TGF-β1 and αvβ3 integrin, a latent TGF-β1 activator.
In order to understand how loss of LMO7 augments TGF-β effects, we examined the expression of Tgfb1 mRNA and activating proteins in control and LMO7 deficient mice. Tgfb1 mRNA levels were higher in Lmo7−/− mice than controls in normal, uninjured aorta (Fig. 3A). Lmo7−/− SMC also expressed higher Tgfb1 mRNA levels than the controls at baseline (Fig. 3B). TGF-β1 is known to transcriptionally induce its own expression in a form of positive feedback regulation20, and this autoinduction resulted in greater maximal level of Tgfb1 in Lmo7−/− SMC compared to control (Fig. 3B). These increases in mRNA were reflected in the secreted protein levels, as a two-fold induction of total TGF-β1 secretion was detected in the conditioned medium from Lmo7−/− SMC versus controls (Fig. 3C). Addition of TGF-β neutralizing antibodies to the media reduced the elevated ECM gene expression in Lmo7−/− cells to the level of the wild type baseline (Fig. 3D).
Figure 3. Loss of LMO7 induced elevated expression of TGF-β1 and αvβ3 integrin, a latent TGF-β1 activator.
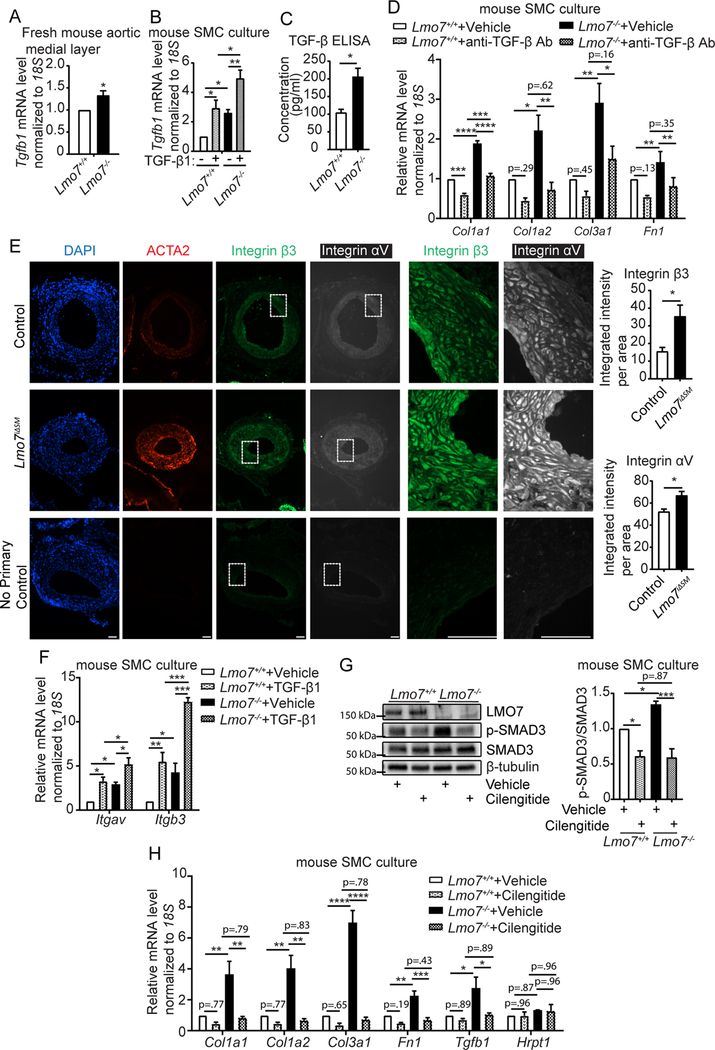
(A) qPCR analysis of Tgfb1 gene expression in medial layers of freshly isolated mouse aortas (n=4 independent experiments). (B) Mouse SMCs were treated with 0.5 ng/ml TGF-β1 for 24hrs and mRNA was harvested for qPCR analysis of Tgfb1 gene (n=4 independent experiments). Two-way ANOVA revealed a significant effect of LMO7 depletion and TGF-β1 treatment on Tgfb1 mRNA expression but there was no significant interaction between Lmo7−/− and TGF-β1 treatment. (C) Lmo7+/+ or Lmo7−/− mouse SMCs with the same confluency were cultured in 0.5% FBS for 24 hrs. Conditioned media was harvested to quantitate total TGF-β secretion by ELISA. (n=4 independent experiments). (D) qPCR analysis of ECM genes from Lmo7+/+ or Lmo7−/− mouse SMCs treated with 40 μg/ml TGF-β neutralizing antibody (Clone #1D11) or Vehicle for 24 hrs (n=3 independent experiments). Two-way ANOVA revealed a significant effect of LMO7 depletion and anti-TGF-β neutralizing antibody treatment on mRNA expression. LMO7 depletion resulted in a greater magnitude of reduction by neutralizing antibody treatment for Col1a1 mRNA (p=0.0022). (E) Immunostaining of ACTA2 (red), β3 Integrin (green) and αv Integrin (white) on tissue sections from control and Lmo7iΔSM mice 21 days after femoral artery injury. Scale bar=50 μm. Cells from each mouse were randomly picked and β3 and αv Integrin staining intensity was measured and normalized to cell area. Quantification is shown on right (n=4). (F) Mouse SMCs were treated with 0.5ng/ml TGF-β1 for 24hrs and mRNA was harvested for qPCR analysis of Itgav and Itgb3 genes (n=3 independent experiments). Two-way ANOVA revealed a significant effect of LMO7 depletion and TGF-β1 treatment on mRNA expression, but there was no significant interaction between Lmo7−/− and TGF-β1 treatment. (G, H) Mouse SMCs were treated with 20 μM Cilengitide or Vehicle for 24hrs and cell lysates were harvested for (G) western analysis of SMAD3 phosphorylation (n=3 independent experiments) or (H) qPCR analysis of ECM and Tgfb1 genes (n=3 independent experiments). Hprt1 was used as a negative control in qPCR analysis. Two-way ANOVA revealed a significant effect of LMO7 depletion and Cilengitide treatment on SMAD3 phosphorylation and mRNA expression, and the magnitude of reduction was greater with Cilengitide treatment in Lmo7−/− SMCs for all the readouts except Tgfb1 mRNA (SMAD3 phosphorylation p=0.013, Col1a1 p=0.030, Col1a2 p=0.0098, Col3a1 p=0.0001, Fn1 p=0.015), indicating LMO7 regulates TGF-β activity as well as Tgfb1 and ECM gene expression by modulating αvβ3 Integrin activity. Data are expressed as mean ± S.E.M. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
A recent study by Henderson et al. revealed αv integrins as a key regulator of activation of latent TGF-β and tissue fibrosis in multiple organs21. In addition to promoting its own transcription, TGF-β can promote its own activation through transcriptional induction of integrin subunits22. Expression of αvβ3 integrin, the most abundant αv integrin in blood vessels, is induced by vascular injury23 and its inhibition using peptidomimetics dramatically attenuates restenosis24. Notably, immunostaining for both αv and β3 subunits in injured vessels revealed enhanced expression in Lmo7iΔSM mice versus controls (Fig. 3E, Suppl Fig. 8A). The medial layer of uninjured Lmo7−/− mouse aorta also contained increased αv and β3 mRNA (Suppl Fig. 8B), and Lmo7−/− mouse SMCs expressed higher baseline levels of αv and β3 mRNA than controls (Fig. 3F). TGF-β1 treatment induced αv and β3 mRNA in control cells, and levels were greatly increased in Lmo7−/− ASMC (Fig. 3F). To assess the role of αvβ3 integrin in the Lmo7−/− phenotype, we treated the mouse ASMCs with cilengitide, a specific αvβ3 integrin inhibitor25. Western and qPCR analysis showed that cilengitide inhibited the increased SMAD3 phosphorylation (Fig. 3G) and the enhanced expression of ECM genes (Fig. 3H) in Lmo7−/− SMCs. Cilengitide also inhibited the increased expression of Tgfb1 mRNA in Lmo7−/− SMCs (Fig. 3H). These data indicate that enhanced αvβ3 expression in Lmo7−/− SMC, likely through activation of latent TGF-β, contributes to upregulation of TGF-β itself, downstream SMAD3 signaling and increased ECM target gene expression.
LMO7 inhibits AP-1 activity and TGF-β1 autoinduction.
Our data suggest that LMO7 represses TGF-β signaling, by reducing expression and integrin-mediated activation of TGF-β. We next sought to investigate the molecular mechanism by which LMO7 regulates TGF-β1 and αvβ3 integrin expression. TGF-β1 autoinduction is mediated by the transcription factor AP-126, a heterodimer comprised of subunits of the JUN, FOS and ATF families. TGF-β1 can induce the expression of AP-127. AP-1 contributes to tumorigenesis28, SMC proliferation29 and, interestingly, to TGF-β126 and β3 integrin30 gene transcription. Since TGF-β1 and AP-1 form a positive feedback loop to enhance TGF-β1 signaling, we determined whether LMO7 regulates AP-1 activity. qPCR analysis showed a significantly higher mRNA level of the AP-1 subunits Jun and Fos in medial layer of Lmo7−/− mouse aorta relative to controls (Fig. 4A). Expression of Jun and Fos was also elevated in Lmo7−/− mouse SMCs compared to WT, and treatment with TGF-β1 further induced their expression in both control cells and Lmo7−/− SMCs (Fig. 4B). Human CASMCs with LMO7 siRNA knockdown exhibited an increased expression of c-JUN and c-FOS protein, as well as elevated p-SMAD3 and β3 integrin expression (Fig. 4C). Both the TGFB1 and ITGB3 genes contain AP-1 binding sites (TRE elements) in their proximal promoter regions implicated in enhanced expression of these genes26, 30. Increased AP-1 binding to these elements was detected in hCASMC after LMO7 knockdown in chromatin immunoprecipitation (ChIP)-qPCR assays using antibodies to endogenous c-JUN or c-FOS (Suppl Fig. 9, Fig. 4D).
Figure 4. LMO7 inhibits AP-1 activity and TGF-β1 autoinduction.
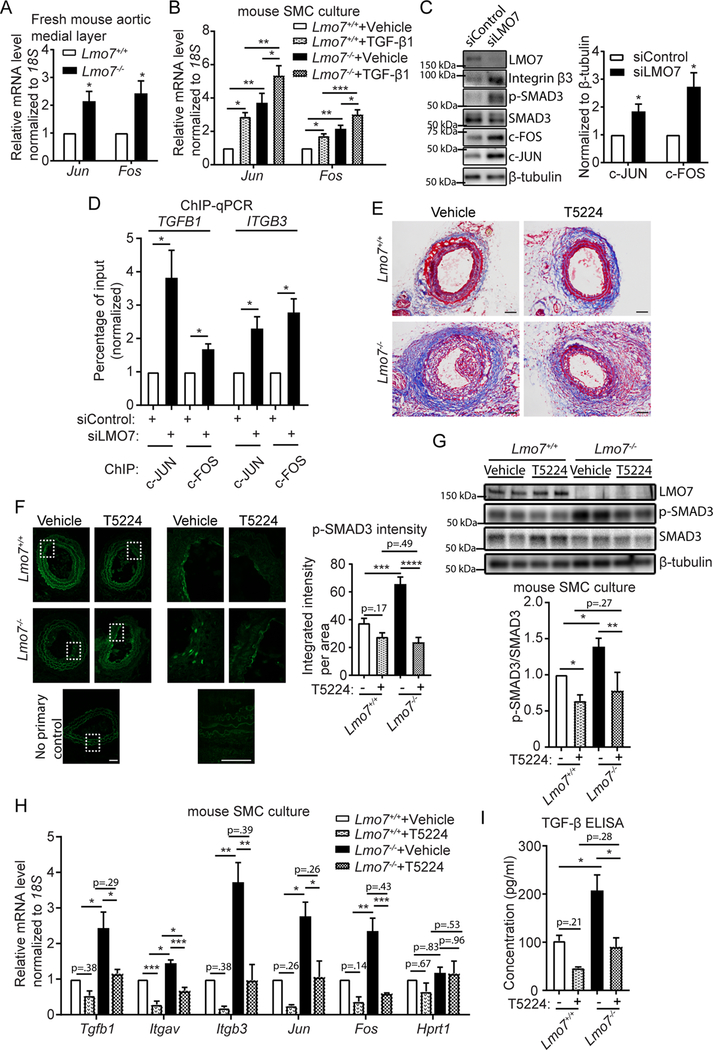
(A) qPCR analysis of Jun and Fos genes in medial layers of freshly isolated mouse aortas (n=4 independent experiments). (B) Mouse SMCs were treated with 0.5ng/ml TGF-β1 for 24hrs and mRNA was harvested for qPCR analysis of Jun and Fos genes (n=5 independent experiments). Two-way ANOVA revealed a significant effect of LMO7 depletion and TGF-β1 treatment on mRNA expression but there was no significant interaction between Lmo7−/− and TGF-β1 treatment. (C) Western analysis of human CASMCs transfected with control or LMO7 siRNA for 72 hrs. Quantification of c-JUN and c-FOS is shown on right (n=4 independent experiments). (D) Chromatin immunoprecipitation (ChIP) with c-JUN or c-FOS antibodies from human CASMCs transfected with control or LMO7 siRNA for 72 hrs, followed by qPCR analysis using primers flanking the AP-1 binding sites in TGFB1 or ITGB3 promoters (n=4 independent experiments). (E, F) Lmo7+/+ or Lmo7−/− mice were subjected to carotid ligation and injected intratracheally with T5224 (30mg/kg/d) or Vehicle (polyvinylpyrrolidone solution) daily from day 4 post-ligation. Mice were sacrificed at day 14 and carotid arteries were harvested. Trichrome (E) or p-SMAD3 (F) staining of cross-sections of injured arteries is shown. p-SMAD3 staining intensity per cell area were measured and shown on right (n=4). Two-way ANOVA revealed a significant effect of LMO7 depletion and T5224 treatment on p-SMAD3 staining intensity and the magnitude of reduction by T5224 treatment was greater in Lmo7−/− mice (p=0.0013), indicating LMO7 regulates TGF-β activity by modulating AP-1 activity. (G, H) Mouse SMCs were treated with 40 μM T5224 for 24hrs and cell lysates were harvested for (G) western analysis of SMAD3 phosphorylation (n=3 independent experiments) or (H) qPCR analysis of Tgfb1, Itgav, Itgb3, Jun and Fos genes (n=3 independent experiments) Hprt1 was used as a negative control in qPCR analysis. Two-way ANOVA revealed a significant effect of LMO7 depletion and T5224 treatment on p-SMAD3 phosphorylation and mRNA expression and the magnitude of reduction by T5224 treatment was greater in Lmo7−/− SMCs for Itgb3 (p=0.026) and Fos (p=0.019) mRNA only. (I) Lmo7+/+ or Lmo7−/− mouse SMCs with the same confluency were treated with 40 μM T5224 or Vehicle for 24 hrs. Conditioned media was harvested to quantitate total TGF-β secretion by ELISA (n=3 independent experiments). Two-way ANOVA revealed a significant effect of LMO7 depletion and T5224 treatment on TGF-β1 production, but there was no significant interaction between Lmo7−/− and T5224 treatment. Data are expressed as mean ± S.E.M. *P < 0.05, **P < 0.01, ***P < 0.001
To determine whether AP-1 plays a causal role in the enhanced TGF-β signaling in Lmo7−/− SMC, we employed the small molecule T5224, a specific inhibitor of c-FOS/AP-1 that targets the basic region/leucine zipper domain31. Notably, T5224 was reported to effectively inhibit TGF-β-induced collagen expression and tissue fibrosis in scleroderma32. Similarly, in vivo administration of T5224 significantly inhibited the enhanced ECM deposition (Fig. 4E) and neointimal formation (Suppl Fig. 10A, B) in Lmo7−/− mice compared to vehicle-treated controls following carotid ligation surgery. This was accompanied by reduced p-SMAD3 staining in the injured Lmo7−/− carotid artery (Fig. 4F). Cell culture experiments confirmed that the enhanced SMAD3 phosphorylation in Lmo7−/− SMCs was also inhibited by T5224 treatment (Fig. 4G). Furthermore, the upregulation of genes in Lmo7−/− relative to control SMC, including Tgfb1, Itgav, and Itgb3, was reduced to the wild type baseline or lower by T5224 treatment (Fig. 4H). As AP-1 factors also undergo transcriptional auto-induction33, we similarly noted AP-1-dependent regulation of Fos and Jun in Lmo7−/− cells (Fig. 4H). TGF-β1 secretion by Lmo7−/− SMC was also inhibited by T5224 (Fig. 4I). As AP-1 can positively regulate SMC proliferation29, the enhanced proliferation of Lmo7−/− cells may also be attributable to the induced AP-1 expression. Indeed, T5224 reduced Ki67 staining in injured Lmo7−/− artery (Suppl Fig. 10C, D) as well as proliferation in cultured Lmo7−/− SMCs (Suppl Fig. 10E). Cyclin D1 (Ccnd1), another AP-1 target gene34, was also induced by LMO7 deletion (Suppl Fig. 10F), suggesting another potential mechanism for enhanced cell proliferation.
LMO7 suppresses c-FOS and c-JUN by mediating their ubiquitination and proteasomal degradation
Since c-FOS and c-JUN proteins are tightly regulated by the proteasomal degradation pathway35, 36, we determined whether LMO7 regulates AP-1 protein stability. The short half-lives of AP-1 proteins were greatly extended from ~15 min for c-FOS and ~45 min for c-JUN to 60 and 90 min, respectively, in Lmo7−/− SMC (Fig. 5A). LMO7 contains multiple protein-protein interaction domains, including an F-Box motif8. As F-Box motifs can recruit substrates to a Skp-Cullin-F-Box (SCF) E3 ligase complex for degradation, it is possible that LMO7 may regulate AP-1 subunit stability via this motif. We determined that LMO7 co-immunoprecipitated with c-JUN and c-FOS in human CASMC (Fig. 5B). While a modest difference in immunoprecipitated c-FOS or c-JUN was observed between control and siLMO7 hCASMC at baseline, treatment with the 26S proteasome inhibitor MG132 revealed a pronounced ubiquitin laddering pattern in these immunoprecipitates in control but not siLMO7 cells (Fig. 5C), suggesting that LMO7 promotes ubiquitination and degradation of c-FOS and c-JUN.
Figure 5. LMO7 suppresses c-FOS and c-JUN levels by promoting their ubiquitination and proteasomal degradation.
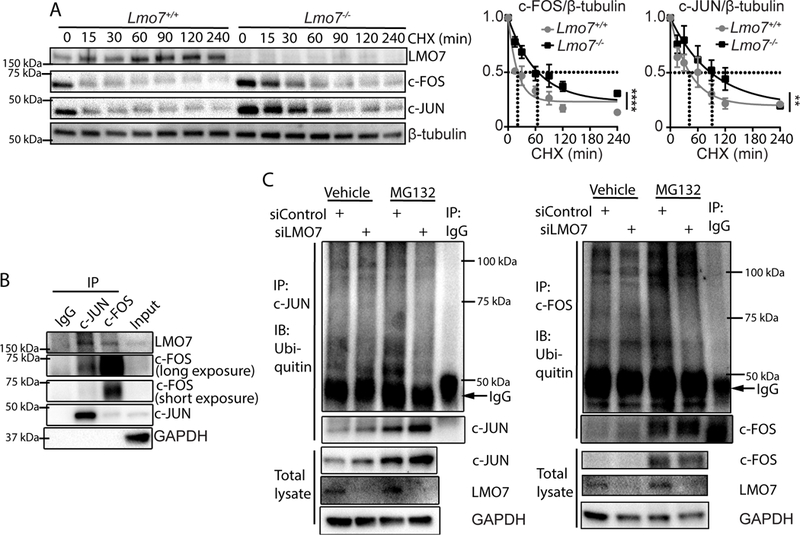
(A) Lmo7+/+ or Lmo7−/− mouse SMCs were treated with 200 μg/ml Cycloheximide and harvested at various time points as indicated for western analysis. Quantification of c-JUN or c-FOS is shown on right (n=3 independent experiments). Stars indicate that the curves are significantly different. (B) Western analysis of immunoprecipitates using antibodies against c-JUN or c-FOS from human CASMCs. (C) Human CASMCs were transfected with control or LMO7 siRNA for 72 hrs and treated with 10 μM MG132 (proteasome inhibitor) or Vehicle for another 6 hrs as indicated. Cell lysates were then immunoprecipitated with antibodies against c-JUN (left panel) or c-FOS (right panel), followed by immunoblotting for ubiquitin. The dark bands just below 50 kDa in the ubiquitin blots are heavy chain of the antibodies used to immunoprecipitate c-JUN or c-FOS. Poly-ubiquitinated bands of modified c-JUN or c-FOS can be noted along the entire length of the blot and are intensified upon inhibition of proteasomal degradation. These bands are absent in the negative control IgG IPs. Immunoblots of these same IP samples blotted for c-JUN or c-FOS are shown immediately below the ubiquitin blots and reveal enhanced total levels of these proteins when degradation is blocked. Controls westerns showing total protein levels are shown at the bottom of the figure.
Because LIM domains are known for scaffolding activity, we tested the hypothesis that the LIM domain mediates interaction between LMO7 and AP-1 components. We mutated the mouse Lmo7 cDNA, deleting the C-terminus which encodes the LIM domain (LMO7ΔC). Intriguingly, full length (FL) LMO7, but not LMO7ΔC, co-immunoprecipitated with c-FOS or c-JUN in HEK293A cells (Fig. 6A). Functionally, FL LMO7 but not the ΔC mutant inhibited the expression of Jun, Fos, Tgfb1 as well as integrin and ECM gene mRNAs (Fig. 6B). Similarly, overexpression of FL LMO7 in Lmo7−/− SMC inhibited c-FOS, c-JUN, and integrin β3 protein expression and SMAD3 phosphorylation, while LMO7ΔC failed to do so (Fig. 6C).These data collectively suggest that the C-terminal LIM domain region of LMO7 is required for interaction with and degradation of c-FOS and c-JUN. LMO7 loss of function leads to an upregulation of AP-1 expression and DNA binding, resulting in upregulation of the TGF-β pathway and its targets (See model, Fig. 7).
Figure 6. The LMO7 LIM domain is required for interaction with c-FOS and c-JUN and regulation of ECM and TGF-β pathway genes.
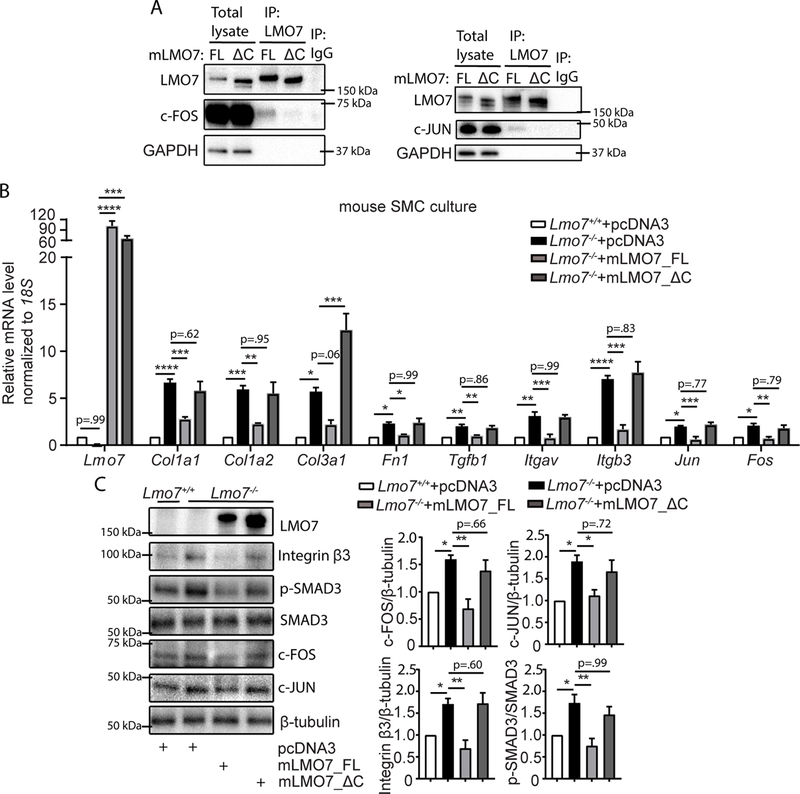
(A) HEK293A cells were transfected with pcDNA3-mLmo7-FL or ΔC and c-FOS (left panel) or c-JUN (right panel) plasmids. After 24 hrs, cell lysates were harvested for immunoprecipitation with LMO7 antibody and western analysis. The image shown is representative of two independent experiments. (B, C) (B) qPCR (n=4 independent experiments) or (C) Western analysis of Lmo7+/+ or Lmo7−/− mouse SMCs transfected with pcDNA3 vector, pcDNA3-mLmo7-FL or ΔC for 24 hrs as indicated. Quantification of c-FOS, c-JUN and β3 integrin expression and SMAD3 phosphorylation in western is shown on right (n=3 independent experiments). One-way ANOVA and multiple comparison with Sidak correction analysis revealed significant differences between conditions as indicated by asterisks in panels (B-C). Data are expressed as mean ± S.E.M. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Figure 7. Model of LMO7 feedback regulation of TGF-β.
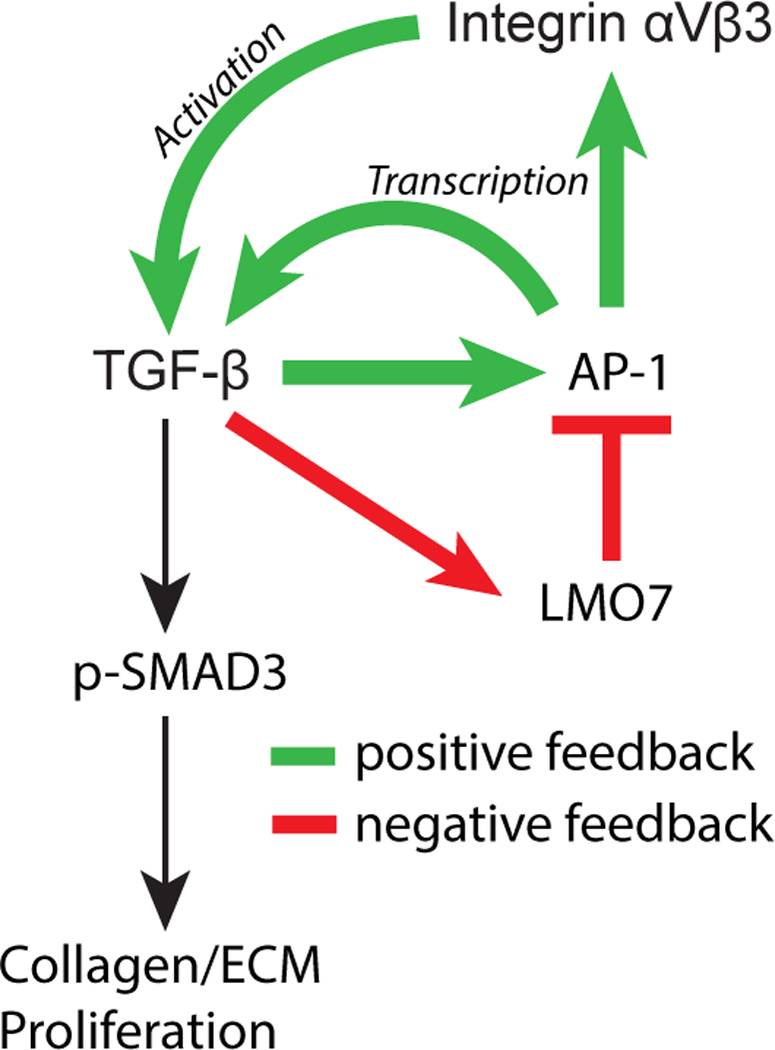
TGF-β signaling autoamplifies through AP-1-dependent transcriptional induction of TGFB1 and its activators, αv (ITGAV) and β3 (ITGB3) integrins. At high doses and later time points, TGF-β signaling induces expression of LMO7, which binds to and promotes the ubiquitination and degradation of AP-1 transcription factors, c-FOS and c-JUN. The resulting reduction in TGF-β activity decreases canonical SMAD3 signaling to ECM target genes including collagen, as well as cell proliferation. Positive feedback processes are indicated by green arrows, negative feedback is indicated by red arrows and lines.
LMO7 expression is inversely correlated with p-SMAD3 in human intimal hyperplasia
To determine whether the role for LMO7 observed in murine vascular injury models translates to human vascular disease, we assessed levels of LMO7 and p-SMAD3 staining in human samples of coronary arteries from patients with cardiac allograft vasculopathy (CAV) (Fig. 8A) and veins from patients with arteriovenous fistula (AVF) (Fig. 8B), both of which are characterized by neointimal hyperplasia associated with elevated TGF-β37, 38. Preliminary analysis of small numbers of rare human samples suggested that LMO7 levels were low in normal control vessels but increased in the disease samples. Furthermore, when quantitated on a per cell basis, an inverse correlation between expression of LMO7 and p-SMAD3 was detected in the neointimal regions in the CAV patient samples (Fig. 8), with a trend toward this relationship in the AVF samples, consistent with our findings in murine injury-induced intimal hyperplasia.
Figure 8. LMO7 expression is inversely correlated with pSMAD3 in human intimal hyperplasia in CAV and patent AVF.
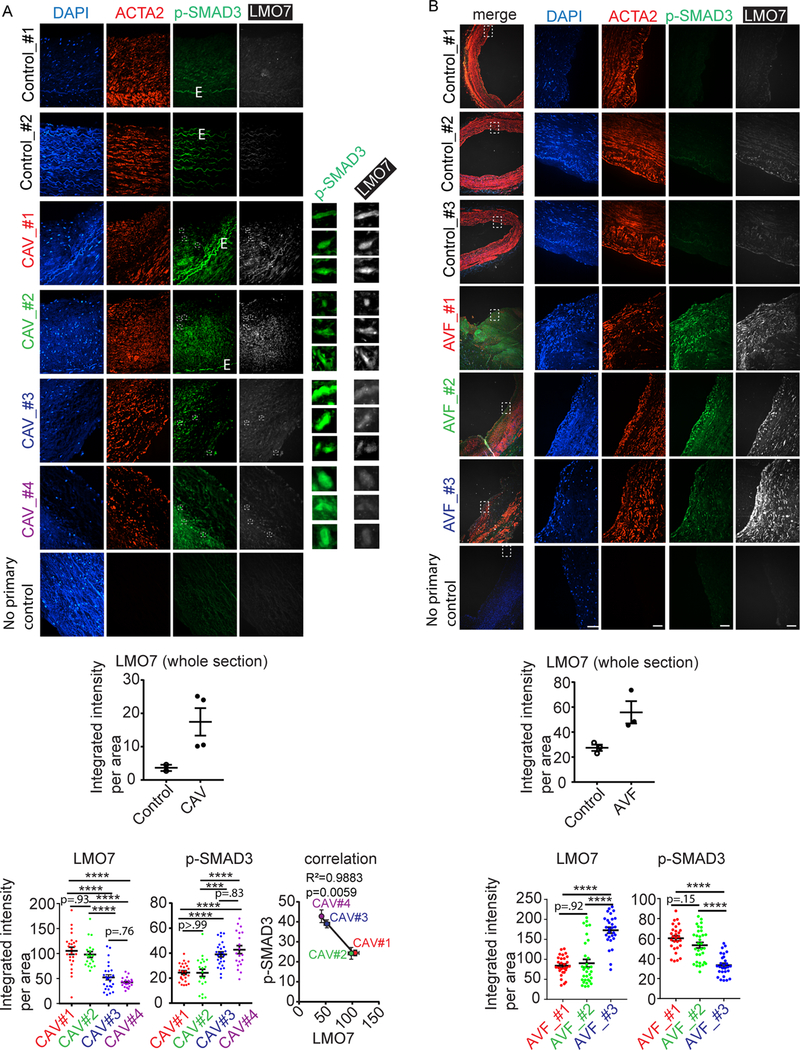
(A, B) Immunofluorescence of p-SMAD3 (green), ACTA2 (red) and LMO7 (white) and DAPI nuclear staining in tissue sections of biopsies from patients with (A) cardiac allograft vasculopathy (CAV, n=4) or normal control coronary arteries (n=2) or (B) patent normally remodeling vein from arteriovenous fistula (AVF, n=3) or normal control veins (n=3). Preliminary analysis of a small number of samples suggests that LMO7 expression level was elevated in the diseased arteries or veins. Quantification of staining in entire section is shown in the upper graph in each panel. White boxes in the CAV images indicate areas shown in high power insets to the right of each CAV images in order to illustrate inverse correlation between p-SMAD3 and LMO7 in representative individual cells. 20–32 cells in total from each of several sections from each patient sample were randomly selected for quantitation of the staining intensity of LMO7 and p-SMAD3 which are plotted in the lower graphs. In CAV samples, the average expression of LMO7 and p-SMAD3 from each patient sample was presented as a single data point in the correlation graphs to confirm the inverse correlation between LMO7 and p-SMAD3 in the diseased patient samples (each individual patient color coded in graphs for clarity). R2 and p value for correlation curve is shown. Data are expressed as mean ± S.E.M. ***P < 0.001, ****P < 0.0001.
Discussion
The molecular mechanisms that distinguish between normal wound healing and progression to fibrosis are poorly understood, but of major importance for many diseases. We identify a key role for the LIM domain protein LMO7 in limiting fibrosis. We report that LMO7 is induced by TGF-β1 and serves as a negative feedback regulator of the TGF-β signaling pathway. By diminishing TGF-β expression, activation, signaling, and target gene expression, our kinetic data suggest that LMO7 contributes to the resolution phase of intravascular wound healing. LMO7 exerts this broad influence on the pathway by regulating the stability of the AP-1 transcription factor subunits c-FOS and c-JUN (Fig. 7).
ECM remodeling is a major component of the tissue repair process. Excessive ECM deposition, however, leads to fibrosis or scarring, altering stiffness and impairing tissue function. Thus, appropriate resolution of wound healing is critical. TGF-β plays a central role in stimulating ECM production4, and TGF-β propagates wound healing through its own AP-1-dependent autoinduction26. Because LMO7 itself is induced by TGF-β, but only after a delay and at higher concentrations, it serves as a sensor of the level of TGF-β signaling. When TGF-β signaling is of sufficient strength and duration to induce LMO7, LMO7 mediates negative feedback that limits ECM production. By regulating TGF-β signaling at multiple levels within the pathway, including transcription of TGF-β as well as its activators αvβ3, LMO7 can coordinately limit TGF-β bioavailability, downregulating expression of TGF-β target genes. While we have noted regulation of canonical TGF-β signaling mediators including p-SMAD3 and CTGF, it remains to be determined whether LMO7-dependent feedback also influences non-canonical TGF-β signaling.
Mechanistically, we find that LMO7 suppresses TGF-β autoinduction and integrin transcription by regulating AP-1 protein stability. AP-1 regulates many key genes and processes involved in ECM remodeling and wound healing, including proliferation29, TGF-β26, collagen39 and MMPs40. Previous studies demonstrated an induction of c-JUN and c-FOS expression in fibrosis in mice and in scleroderma patient dermal fibroblasts, and AP-1 inhibition abrogated TGF-β signaling, myofibroblast phenotype, and ECM deposition in fibroblasts32. The short-lived AP-1 subunits c-FOS and c-JUN are tightly regulated35, 36 by E3 ligases41, 42 as well as a proteasome-independent pathway43. We find that LMO7, through its LIM domain, is able to associate with c-FOS and c-JUN and facilitate their ubiquitination and proteasomal degradation, likely via the LMO7 F-Box motif8. Through attenuation of AP-1 activity, LMO7 coordinately reduces TGF-β signaling. Importantly, we report that AP-1 inhibition rescues the Lmo7−/− phenotype in vitro and in vivo.
LMO7 has previously been implicated in skeletal muscle transcriptional regulation, and in heart development10, 12. While its functions in smooth muscle had not been previously studied, LMO7 has been linked to cancer cell migration downstream of MKL-1 (MRTF-A)9, suggesting that it may potentially also affect SMC phenotype through such pathways. Interestingly, our findings demonstrate an unusual case of dissociation of several key hallmarks of SMC phenotypic modulation. Generally, differentiated SMC are quiescent, non-migratory, and express contractile proteins, while dedifferentiated cells are proliferative, migratory, and downregulate contractile proteins but upregulate ECM synthesis. We report that in the absence of LMO7, SMC demonstrate increased proliferation and ECM, but also increased expression of the contractile protein ACTA2 (See Figs 2A, 3E, Suppl Figs 3C, 6, and 8A). This is consistent with the known role of TGF-β in inducing SMC contractile protein expression, as well as the robust ACTA2 expression characteristic of myofibroblasts. How LMO7 may also modulate fibrotic response through additional pathways such as MRTFs9, 44 may be an interesting topic for future studies.
TGF-β is a key driver of intimal hyperplasia following vascular injury17. While SMC proliferation is limited to the first 2–3 weeks post-injury, human lesions continue to grow due to ongoing ECM deposition, resulting in luminal scar formation that can limit flow and require revascularization2. We identify LMO7 as a factor that is induced by TGF-β and in turn limits TGF-β signaling, ECM deposition and proliferation in murine neointimal lesions. Importantly, preliminary studies also support elevated LMO7 expression relative to control vein or artery in the human neointimal pathologies AVF and CAV, in which TGF-β is also associated with exacerbated neointima formation37, 38. In our control mouse and human specimens, LMO7 levels are very low in normal, mature blood vessels. Our data suggest that the increase in LMO7 that occurs in the context of active vascular remodeling serves to restrain the progression of intimal hyperplasia, as the complete absence of LMO7 in our mouse models results in very severe neointimal formation. The CAV hyperplastic vessels reveal an inverse correlation between LMO7 and p-SMAD3 expression, consistent with our mouse vascular injury findings. There was a similar trend in the relationship between LMO7 and p-SMAD3 expression in the AVF samples, but the small sample size precludes definitive conclusions. Future studies with larger numbers of patients would allow us to elucidate the relationship between LMO7 expression and disease severity in human lesions.
TGF-β contributes to other vascular pathologies as well. In atherosclerosis, TGF-β exerts a protective role by inducing SMC ECM deposition and suppressing leukocyte inflammation and endothelial adhesion molecule expression45. TGF-β signaling exerts protective effects in thoracic aortic aneurysm development46. It will therefore be of interest to evaluate LMO7 function in these diseases. Moreover, feedback regulation of TGF-β signaling may also explain the previous observation of spontaneous lung carcinoma in aged Lmo7−/− mice11, given the known function of TGF-β in tumor metastasis47.
LMO7 has a broad tissue distribution, with high expression in tissues prone to fibrosis11. Even though distinct cell types contribute to fibrosis in different organs (fibroblasts, hepatic stellate cells, and SMCs), these cells share the characteristic of transdifferentiation into myofibroblast-like cells48, the main source of ECM. As progressive fibrosis leads to loss of normal organ function, often with transplantation as the only solution for the end-stage fibrotic diseases49, further elucidating the role of LMO7 in wound healing resolution may have clinical benefit to alleviate adverse fibrogenesis.
Supplementary Material
Clinical Perspective.
What is new?
We identify LMO7 as a TGFβ target gene that provides negative feedback signaling to TGFβ.
By regulating transcription factors that control expression of TGFβ and its activators, LMO7 contributes to the resolution of the wound healing phase in vascular lesions.
What are the clinical implications?
LMO7 expression is low in normal vessels but is induced following vascular injury.
LMO7 negative feedback mechanisms limit the proliferative and fibrotic responses that drive intimal hyperplasia following vascular injury.
LMO7 levels are elevated during hyperplastic vascular remodeling, including in human arteries with cardiac allograft vasculopathy and in veins in maturing arteriovenous fistulae.
LMO7 may be a new mechanistic target for modulation of fibrotic responses.
Acknowledgements:
We thank Dr. Xinbo Zhang for technical assistance. We thank Dr. Jun Miyoshi for Lmo7−/− mice and Dr. Ju Chen for Lmo7f/f mice.
Sources of Funding: This study was supported by grants from the National Institutes of Health to K.A.M. (HL091013, HL118430, and RHL119529), J.H. (HL074190, HL115247, and HL117798), E.L.H. (HLR0125250), A.D. (R01HL128406) and J.Y. (HL126933, HL117064) and by a grant from the American Heart Association to J.Y (SDG).
Footnotes
Disclosures: None.
References
- 1.Dangas GD, Claessen BE, Caixeta A, Sanidas EA, Mintz GS and Mehran R. In-stent restenosis in the drug-eluting stent era. J Am Coll Cardiol 2010;56:1897–1907. [DOI] [PubMed] [Google Scholar]
- 2.Owens GK, Kumar MS and Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 2004;84:767–801. [DOI] [PubMed] [Google Scholar]
- 3.Fattori R and Piva T. Drug-eluting stents in vascular intervention. Lancet 2003;361:247–249. [DOI] [PubMed] [Google Scholar]
- 4.Leask A and Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J 2004;18:816–827. [DOI] [PubMed] [Google Scholar]
- 5.Massagué J TGFβ signalling in context. Nat Rev Mol Cell Biol 2012;13:616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flanders KC. Smad3 as a mediator of the fibrotic response. Int J Exp Pathol 2004;85:47–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ooshio T, Irie K, Morimoto K, Fukuhara A, Imai T and Takai Y. Involvement of LMO7 in the association of two cell-cell adhesion molecules, nectin and E-cadherin, through afadin and alpha-actinin in epithelial cells. J Biol Chem 2004;279:31365–31373. [DOI] [PubMed] [Google Scholar]
- 8.Cenciarelli C, Chiaur DS, Guardavaccaro D, Parks W, Vidal M and Pagano M. Identification of a family of human F-box proteins. Curr Biol 1999;9:1177–1179. [DOI] [PubMed] [Google Scholar]
- 9.Hu Q, Guo C, Li Y, Aronow BJ and Zhang J. LMO7 mediates cell-specific activation of the Rho-myocardin-related transcription factor-serum response factor pathway and plays an important role in breast cancer cell migration. Mol Cell Biol 2011;31:3223–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holaska JM, Rais-Bahrami S and Wilson KL. Lmo7 is an emerin-binding protein that regulates the transcription of emerin and many other muscle-relevant genes. Hum Mol Genet 2006;15:3459–3472. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka-Okamoto M, Hori K, Ishizaki H, Hosoi A, Itoh Y, Wei M, Wanibuchi H, Mizoguchi A, Nakamura H and Miyoshi J. Increased susceptibility to spontaneous lung cancer in mice lacking LIM-domain only 7. Cancer Sci 2009;100:608–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ott EB, van den Akker NM, Sakalis PA, Gittenberger-de Groot AC, Te Velthuis AJ and Bagowski CP. The lim domain only protein 7 is important in zebrafish heart development. Dev Dyn 2008;237:3940–3952. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura H, Mukai M, Komatsu K, Tanaka-Okamoto M, Itoh Y, Ishizaki H, Tatsuta M, Inoue M and Miyoshi J. Transforming growth factor-beta1 induces LMO7 while enhancing the invasiveness of rat ascites hepatoma cells. Cancer Lett 2005;220:95–99. [DOI] [PubMed] [Google Scholar]
- 14.Lao DH, Esparza MC, Bremner SN, Banerjee I, Zhang J, Veevers J, Bradford WH, Gu Y, Dalton ND, Knowlton KU, Peterson KL, Lieber RL and Chen J. Lmo7 is dispensable for skeletal muscle and cardiac function. Am J Physiol Cell Physiol 2015;309:C470–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar A and Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol 1997;17:2238–2244. [DOI] [PubMed] [Google Scholar]
- 16.Jin Y, Xie Y, Ostriker AC, Zhang X, Liu R, Lee MY, Leslie KL, Tang W, Du J, Lee SH, Wang Y, Sessa WC, Hwa J, Yu J and Martin KA. Opposing Actions of AKT (Protein Kinase B) Isoforms in Vascular Smooth Muscle Injury and Therapeutic Response. Arterioscler Thromb Vasc Biol 2017;37:2311–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Majesky MW, Lindner V, Twardzik DR, Schwartz SM and Reidy MA. Production of transforming growth factor beta 1 during repair of arterial injury. J Clin Invest 1991;88:904–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie Y, Jin Y, Merenick BL, Ding M, Fetalvero KM, Wagner RJ, Mai A, Gleim S, Tucker DF, Birnbaum MJ, Ballif BA, Luciano AK, Sessa WC, Rzucidlo EM, Powell RJ, Hou L, Zhao H, Hwa J, Yu J and Martin KA. Phosphorylation of GATA-6 is required for vascular smooth muscle cell differentiation after mTORC1 inhibition. Sci Signal 2015;8:ra44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dasch JR, Pace DR, Waegell W, Inenaga D and Ellingsworth L. Monoclonal antibodies recognizing transforming growth factor-beta. Bioactivity neutralization and transforming growth factor beta 2 affinity purification. J Immunol 1989;142:1536–1541. [PubMed] [Google Scholar]
- 20.Van Obberghen-Schilling E, Roche NS, Flanders KC, Sporn MB and Roberts AB. Transforming growth factor beta 1 positively regulates its own expression in normal and transformed cells. J Biol Chem 1988;263:7741–7746. [PubMed] [Google Scholar]
- 21.Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, Pellicoro A, Raschperger E, Betsholtz C, Ruminski PG, Griggs DW, Prinsen MJ, Maher JJ, Iredale JP, Lacy-Hulbert A, Adams RH and Sheppard D. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med 2013;19:1617–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mamuya FA and Duncan MK. aV integrins and TGF-beta-induced EMT: a circle of regulation. J Cell Mol Med 2012;16:445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stouffer GA, Hu Z, Sajid M, Li H, Jin G, Nakada MT, Hanson SR and Runge MS. Beta3 integrins are upregulated after vascular injury and modulate thrombospondin- and thrombin-induced proliferation of cultured smooth muscle cells. Circulation 1998;97:907–915. [DOI] [PubMed] [Google Scholar]
- 24.Srivatsa SS, Fitzpatrick LA, Tsao PW, Reilly TM, Holmes DR, Schwartz RS and Mousa SA. Selective alpha v beta 3 integrin blockade potently limits neointimal hyperplasia and lumen stenosis following deep coronary arterial stent injury: evidence for the functional importance of integrin alpha v beta 3 and osteopontin expression during neointima formation. Cardiovasc Res 1997;36:408–428. [DOI] [PubMed] [Google Scholar]
- 25.Dechantsreiter MA, Planker E, Mathä B, Lohof E, Hölzemann G, Jonczyk A, Goodman SL and Kessler H. N-Methylated cyclic RGD peptides as highly active and selective alpha(V)beta(3) integrin antagonists. J Med Chem 1999;42:3033–3040. [DOI] [PubMed] [Google Scholar]
- 26.Kim SJ, Angel P, Lafyatis R, Hattori K, Kim KY, Sporn MB, Karin M and Roberts AB. Autoinduction of transforming growth factor beta 1 is mediated by the AP-1 complex. Mol Cell Biol 1990;10:1492–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pertovaara L, Sistonen L, Bos TJ, Vogt PK, Keski-Oja J and Alitalo K. Enhanced jun gene expression is an early genomic response to transforming growth factor beta stimulation. Mol Cell Biol 1989;9:1255–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eferl R and Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer 2003;3:859–868. [DOI] [PubMed] [Google Scholar]
- 29.Zhan Y, Kim S, Yasumoto H, Namba M, Miyazaki H and Iwao H. Effects of dominant-negative c-Jun on platelet-derived growth factor-induced vascular smooth muscle cell proliferation. Arterioscler Thromb Vasc Biol 2002;22:82–88. [DOI] [PubMed] [Google Scholar]
- 30.Cao X, Ross FP, Zhang L, MacDonald PN, Chappel J and Teitelbaum SL. Cloning of the promoter for the avian integrin beta 3 subunit gene and its regulation by 1,25-dihydroxyvitamin D3. J Biol Chem 1993;268:27371–27380. [PubMed] [Google Scholar]
- 31.Aikawa Y, Morimoto K, Yamamoto T, Chaki H, Hashiramoto A, Narita H, Hirono S and Shiozawa S. Treatment of arthritis with a selective inhibitor of c-Fos/activator protein-1. Nat Biotechnol 2008;26:817–823. [DOI] [PubMed] [Google Scholar]
- 32.Avouac J, Palumbo K, Tomcik M, Zerr P, Dees C, Horn A, Maurer B, Akhmetshina A, Beyer C, Sadowski A, Schneider H, Shiozawa S, Distler O, Schett G, Allanore Y and Distler JH. Inhibition of activator protein 1 signaling abrogates transforming growth factor β-mediated activation of fibroblasts and prevents experimental fibrosis. Arthritis Rheum 2012;64:1642–1652. [DOI] [PubMed] [Google Scholar]
- 33.Angel P, Hattori K, Smeal T and Karin M. The jun proto-oncogene is positively autoregulated by its product, Jun/AP-1. Cell 1988;55:875–885. [DOI] [PubMed] [Google Scholar]
- 34.Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A and Pestell RG. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem 1995;270:23589–23597. [DOI] [PubMed] [Google Scholar]
- 35.Stancovski I, Gonen H, Orian A, Schwartz AL and Ciechanover A. Degradation of the proto-oncogene product c-Fos by the ubiquitin proteolytic system in vivo and in vitro: identification and characterization of the conjugating enzymes. Mol Cell Biol 1995;15:7106–7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Treier M, Staszewski LM and Bohmann D. Ubiquitin-dependent c-Jun degradation in vivo is mediated by the delta domain. Cell 1994;78:787–798. [DOI] [PubMed] [Google Scholar]
- 37.Stracke S, Konner K, Kostlin I, Friedl R, Jehle PM, Hombach V, Keller F and Waltenberger J. Increased expression of TGF-beta1 and IGF-I in inflammatory stenotic lesions of hemodialysis fistulas. Kidney Int 2002;61:1011–1019. [DOI] [PubMed] [Google Scholar]
- 38.Aziz T, Hasleton P, Hann AW, Yonan N, Deiraniya A and Hutchinson IV. Transforming growth factor beta in relation to cardiac allograft vasculopathy after heart transplantation. J Thorac Cardiovasc Surg 2000;119:700–708. [DOI] [PubMed] [Google Scholar]
- 39.Chung KY, Agarwal A, Uitto J and Mauviel A. An AP-1 binding sequence is essential for regulation of the human alpha2(I) collagen (COL1A2) promoter activity by transforming growth factor-beta. J Biol Chem 1996;271:3272–3278. [DOI] [PubMed] [Google Scholar]
- 40.Benbow U and Brinckerhoff CE. The AP-1 site and MMP gene regulation: what is all the fuss about? Matrix Biol 1997;15:519–526. [DOI] [PubMed] [Google Scholar]
- 41.Sasaki T, Kojima H, Kishimoto R, Ikeda A, Kunimoto H and Nakajima K. Spatiotemporal regulation of c-Fos by ERK5 and the E3 ubiquitin ligase UBR1, and its biological role. Mol Cell 2006;24:63–75. [DOI] [PubMed] [Google Scholar]
- 42.Nateri AS, Riera-Sans L, Da Costa C and Behrens A. The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science 2004;303:1374–1378. [DOI] [PubMed] [Google Scholar]
- 43.Hirai S, Kawasaki H, Yaniv M and Suzuki K. Degradation of transcription factors, c-Jun and c-Fos, by calpain. FEBS Lett 1991;287:57–61. [DOI] [PubMed] [Google Scholar]
- 44.Small EM, Thatcher JE, Sutherland LB, Kinoshita H, Gerard RD, Richardson JA, Dimaio JM, Sadek H, Kuwahara K and Olson EN. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res 2010;107:294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grainger DJ. Transforming growth factor beta and atherosclerosis: so far, so good for the protective cytokine hypothesis. Arterioscler Thromb Vasc Biol 2004;24:399–404. [DOI] [PubMed] [Google Scholar]
- 46.Milewicz DM, Prakash SK and Ramirez F. Therapeutics Targeting Drivers of Thoracic Aortic Aneurysms and Acute Aortic Dissections: Insights from Predisposing Genes and Mouse Models. Annu Rev Med 2017;68:51–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Massagué J TGFbeta in Cancer. Cell 2008;134:215–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML and Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol 2007;170:1807–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wynn TA and Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 2012;18:1028–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.