

Abstract
Acetylation of the microtubule-associated protein tau promotes its polymerization into neurofibrillary tangles that are implicated in the pathology of Alzheimer’s disease (AD). The gaseous neurotransmitter nitric oxide (NO) regulates cell signaling through the nitrosylation of proteins. We found that NO production and tau acetylation at Lys280 occurred in the brain tissue in mice and in cultured mouse cortical neurons in response to exposure to amyloid-β1–42 (Aβ1–42), a peptide that is also implicated in AD. An increased abundance of NO facilitated the S-nitrosylation (SNO) of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). S-nitrosylated GAPDH (GAPDH-SNO) promoted the acetylation and activation of the acetyltransferase p300 and facilitated the nitrosylation and inactivation of the deacetylase sirtuin 1 (SIRT1). The abundance of GAPDH-SNO was increased in postmortem brain samples from AD patients. Preventing the increase in GAPDH-SNO abundance in both cultured neurons and mice, either by overexpression of the nitrosylation mutant of GAPDH (GAPDH C150S) or by treatment with the GAPDH nitrosylation inhibitor CGP3466B (also known as omigapil), abrogated Aβ1–42-induced tau acetylation, memory impairment, and locomotor dysfunction in mice, suggesting that this drug might be repurposed to treat patients with AD.
INTRODUCTION
Alzheimer’s disease (AD) is a common neurodegenerative disorder that impairs memory and higher cognitive functions and, as an aging- associated disease, is increasing in incidence (1–5). Plaque deposits of amyloid, mostly composed of amyloid-β (Aβ1–42), are a key feature of AD and are implicated in its pathology (6–8). Numerous studies have shown that a high or toxic amount of recombinant oligomeric Aβ1–42 elicits neuronal death, synaptic dystrophy, loss, and dysfunction, and learning and memory deficits in AD patients and transgenic mice models of AD (6, 8, 9). Brain regions that are often affected by the deposition of the amyloid plaque include the hippocampus, the cortex, and subcortical structures, such as the basal forebrain and the amygdala (10, 11).
The protein tau is also implicated in AD and regulates neuronal functions by binding and stabilizing microtubules (12–15). However, the acetylation of tau impairs its interaction with microtubules and promotes its aggregation (16–20). Lys280-acetylated tau is detected in postmortem AD patient brain tissue (21) and in insoluble fractions in brain lysates from both transgenic mouse models of AD, in which the abundance of Lys280-acetylated tau increased with age in the cortex (21), suggesting that Lys280 acetylation of tau promotes its aggregation and its role in AD. Notably, acetylation of tau at this epitope is preceded by other modifications, including phosphorylation, and is succeeded by truncation modifications and cell death in AD.
Numerous studies have shown that an increase in nitrosative stress is associated with protein misfolding and aggregation, mitochondrial dysfunction, and endoplasmic reticulum stress, ultimately leading to synapse loss or neuronal cell death, which are common in several neurodegenerative diseases, including AD (22–24). Nitric oxide (NO) mediates cell signaling through the nitrosylation of proteins, in which NO is covalently incorporated into a cysteine residue in the protein, forming an S-nitrosothiol (SNO) (25). We previously reported that p300- mediated acetylation of Lys160-nitrosylated glyceraldehyde-3-phosphate dehydrogenase (GAPDH-SNO), in turn, promotes GAPDH-mediated acetylation of p300 (26), which is critical for its catalytic activity (26). Elsewhere, we have also shown that GAPDH-SNO interacts directly with the deacetylase sirtuin 1 (SIRT1), upon which the NO group is transferred from GAPDH-SNO to SIRT1 at Cys387/390, which impairs its catalytic activity (27). Here, we investigated whether these NO-induced effects on GAPDH, p300, and SIRT1 have a role in AD with regard to tau and amyloid pathology.
RESULTS
Aβ1–42 induces tau acetylation at Lys280 residue in a NO-dependent manner
To investigate whether Aβ1–42 has any influence on tau acetylation, we isolated primary neurons from the cortex of mice, treated them with Aβ1–42 oligomers, and monitored Lys280 acetylation on tau by Western blotting and confocal microscopy. We found that tau acetylation at Lys280 increased significantly in response to Aβ1–42, but not in the presence of the neuronal nitric oxide synthase (nNOS) inhibitor L-NG-nitroarginine methyl ester (L-NAME) (Fig. 1A and B, and fig. S1, A and B). We repeated these analyses with wild-type or Nos1 -null (hereafter nNOS−/−) mice that had been intracortically injected with a control peptide or with Aβ1–42 and again found the acetylation of tau at Lys280 to be nNOS-dependent (Fig. 1, C and D, and fig. S1, C and D). In addition, tau acetylation was increased in primary neurons isolated from nNOS+/+ mice compared to those from nNOS−/− mice treated with Aβ1–42 (Fig. 1E and fig. S1E). Together, these data suggest that Aβ1–42 up-regulates tau acetylation at Lys280 in an NO-dependent manner in neurons.
Fig. 1. Nitric oxide regulates tau acetylation at Lys280.
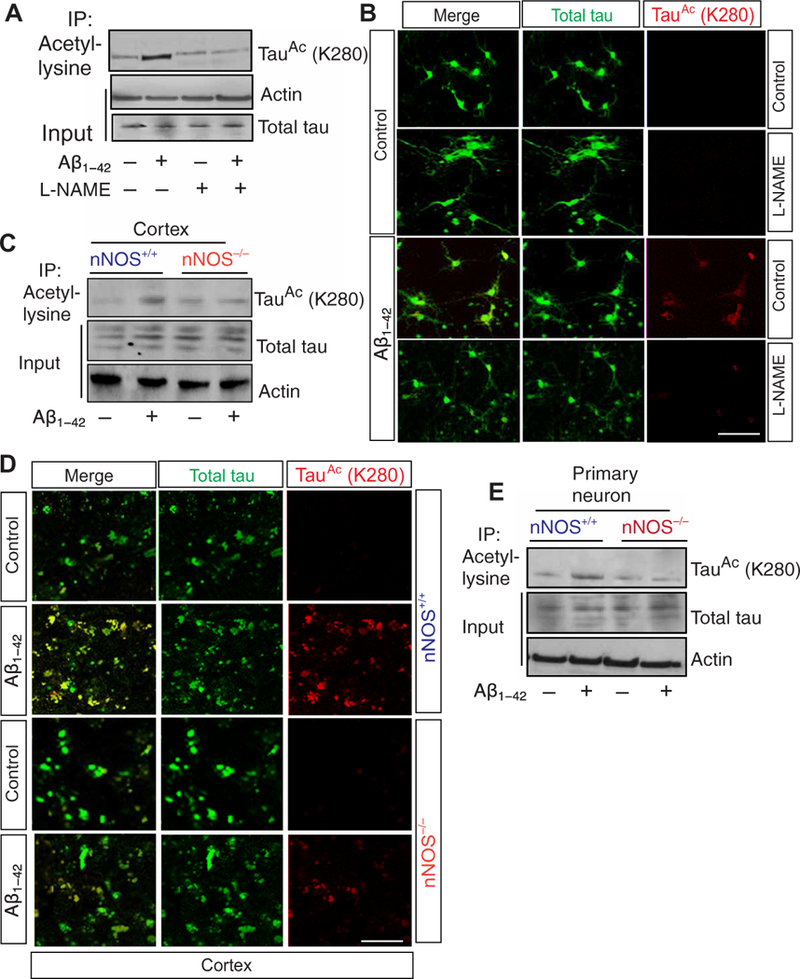
(A) Coimmunoprecipitation (Co-IP) assay to detect tau acetylation at Lys280 (K280) in primary neurons treated with amyloid-p1–42 (Ap1–42) for 24 hours in the presence or absence of L-NG-nitroarginine methyl ester (L-NAME). (B) Confocal microscopic analysis of acetylation of tau at Lys280 in primary neurons treated with Ap1–42 for 24 hours with or without L-NAME. (C) Co-IP assay to detect acetylation of tau in the cortical lysates isolated from neuronal nitric oxide synthase (nNOS)+/+ and nNOS−/− mice after intracortical infusion of Ap1–42. (D) Confocal microscopy analysis of tau acetylation in the cortex of nNOS+/+ and nNOS−/− mice after infusion of Ap1–42 into the cortex. The acety- lated tau was monitored by red fluorescent signal (quantified), and total tau was determined by green fluorescent signal. Scale bar, 100 mm. (E) Co-IP assay to detect acetylation of tau in primary neuron isolated from nNOS+/+ and nNOS−/− mice. Data are quantified in the Supplementary Materials; all blots and microscopy are representative of three independent experiments from five to seven mice each condition.
Aβ1–42 induces GAPDH-SNO, which activates p300 to acetylate tau
Previously, it was demonstrated that tau can be acetylated by activation of the acetyltransferase p300 (19), and we have shown that activation of p300 can be activated by the nitrosylation of GAPDH in human embryonic kidney (HEK) 293 cells, macrophages, and dopaminergic neuroblastoma SHSY5Y cells (26). Given our results above that Aβ1-42 induces tau acetylation through a nitrosylating enzyme, we hypothesized that the mechanism may be mediated by the nitrosylation of GAPDH and subsequent activation of p300. To test our hypothesis, we first assessed the amount of nitrosylated GAPDH (GAPDH-SNO) that was present in postmortem cortical samples from AD patients. We found significantly greater amounts of nitrosylated GAPDH in AD samples than in controls (postmortem cortical samples from individuals of similar age range with no known dementia) (Fig. 2A and fig. S2A). A biotin-switch assay in cortical lysates from mice revealed that GAPDH nitrosylation was inducible by administration of Aβ1–42 (Fig. 2B). Previously, we reported that GAPDH can be nitrosylated at the Cys150 residue (26,28). To see whether Aβ1–42 nitrosylates GAPDH at that residue, we overexpressed wild-type GAPDH or expressed mutant (C150S) GAPDH in primary neurons isolated from mice and treated them with Aβ1–42. Aβ1–42-induced nitrosylation of GAPDH was abolished in cells that expressed GAPDH C150S (Fig. 2C).
Fig. 2. Nitrosylated GAPDH activates p300 and acetylates tau.
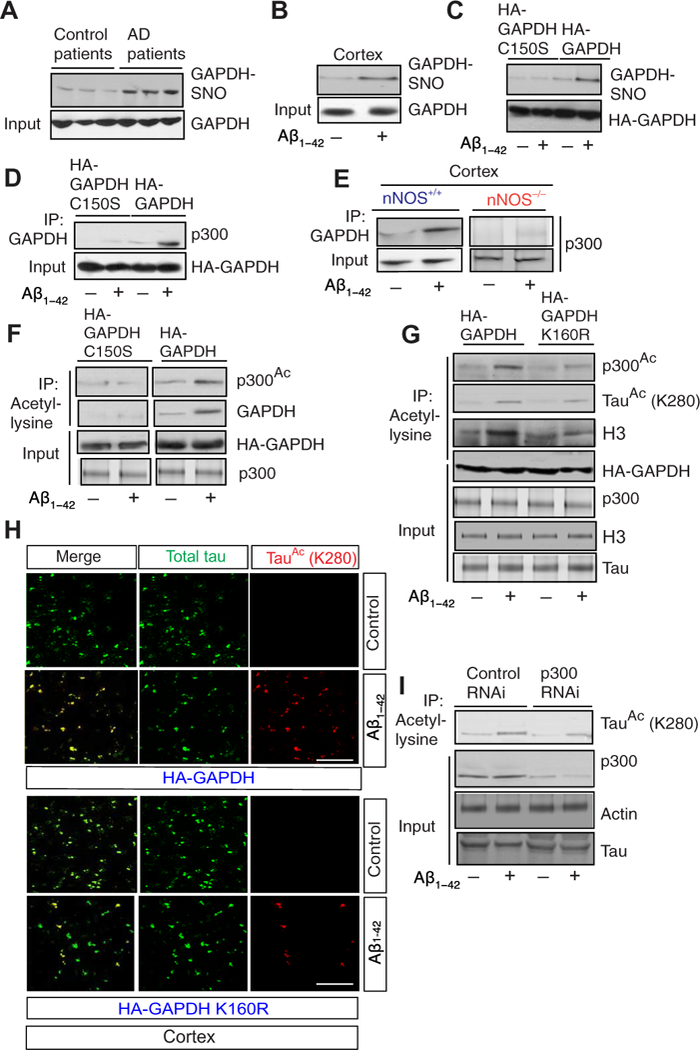
(A) S-nitrosylation of glyceraldehyde-3-phosphate dehydrogenase (GAPDH-SNO) was measured using the biotin-switch assay in lysates from post-mortem cortical samples from human patients [n = 7 Alzheimer’s disease (AD) patient samples; n = 3 control samples]. (B) Biotin-switch assay to detect GAPDH-SNO in cortex isolated from mice administered with Aβ1-42. (C) Nitrosylation of GAPDH assessed by biotin-switch assay in primary neurons that overexpressed GAPDH and GAPDH C150S and was treated with Aβ1-42. (D) GAPDH or GAPDH C150S was overexpressed in primary neurons isolated from nNOS+/+ mice and treated with Aβ1-42. The interaction between GAPDH and p300 was assessed by co-IP. (E) Co-IP between GAPDH and p300 in cortical lysates isolated from nNOS+/+ and nNOS−/− mice after administration of Aβ1-42. (F) The acetylation of p300 in primary neurons that overexpressed GAPDH or GAPDH C150S and was treated with Aβ1–42, as assessed by co-IP with an antibody to acetyl-lysine and Western blotting with an antibody to acetylated p300. (G) The acetylation of p300, H3, and tau in primary neurons that overexpressed GAPDH or GAPDH K160R and were treated with Aβ1–42, assessed by co-IP with an antibody to acetyl-lysine and subsequent Western blotting. (H) Immunofluorescence signals for acetylated tau (red) and total tau (green) in the cortex from mice overexpressing GAPDH or GAPDH K160R and injected with Aβ1–42. Scale bar, 100 μm. (I) Co-IP assay to assess tau acetylation in primary neurons isolated from nNOS+/+ mice and treated with either control or p300 small interfering RNA (siRNA) before administration of Aβ1–42. Data are quantified in the Supplementary Materials; all blots and microscopy are representative of three independent experiments from five to seven mice each condition. HA, hemagglutinin; RNAi, RNA interference.
Previously, we determined that nitrosylated GAPDH interacts with the acetyltransferase p300 and that this interaction mediates the acetylation of both p300 and GAPDH (26). Coimmunoprecipitation (Co-IP) assays confirmed that the interaction between GAPDH and p300 was substantially induced by Aβ1–42 in primary neurons overexpressing wild-type but not C150S-mutant GAPDH (Fig. 2D), as well as in nNOS+/+ but not nNOS−/− mice (Fig. 2E). Furthermore, the acetylation of both p300 GAPDH was markedly increased in primary neurons that overexpressed wild-type but not C150S- mutant GAPDH (Fig. 2F).
Our previous findings also indicated that GAPDH is acetylated at Lys160 and that preventing acetylation of GAPDH inhibits the acetylation of p300, which is indispensable for its catalytic activity (26). Overexpression of a nonacetylatable mutant (K160R) of GAPDH in primary neurons significantly inhibited Aβ1–42-induced acetylation of p300 and of H3 histone, a marker p300 acetyltransferase activity (Fig. 2G and fig. S2B). Confocal microscopy analysis further confirmed that Lys280-acetylated tau was increased in the cortex of mice injected with Aβ1–42 and which overexpressed wild-type but not K160R-mutant GAPDH (Fig. 2H and fig. S2C). Furthermore, RNA interference [RNAi; specifically small interfering RNA (siRNA)] mediated depletion of p300 in primary neurons before exposure to Aβ1–42-inhibited tau acetylation (Fig. 2I and fig. S2D). However, disruption of p300 decreased Aβ1–42-induced tau acetylation by roughly only half in cells and in vivo (Fig. 2, G and I, and fig. S2, B and D); this suggests that activation of p300 may be only partially responsible for the increase in tau acetylation.
Nitrosylation of SIRT1 by GAPDH-SNO prevents deacetylation of tau after administration of Aβ1–42
Given the partial effects of p300 inhibition and our previous findings that the deacetylase SIRT1 is inactivated through transnitrosylation by GAPDH-SNO (27), we hypothesized that concurrent inactivation of a deacetylase, perhaps SIRT1, may also contribute to tau acetylation. To test this hypothesis, we tested the interaction between tau and SIRT1 in the cortex of nNOS+/+ and nNOS−/− mice after administration of Aβ1–42. We found that the interaction between SIRT1 and tau after administration of Aβ1–42 was decreased significantly in nNOS+/+ mice compared to nNOS−/− mice (Fig. 3A and fig. S3A), indicating that SIRT1 may regulate tau acetylation in an NO-dependent manner. Accordingly, we found that Aβ1–42 induces SIRT1- SNO (Fig. 3B) along with an interaction of GAPDH with SIRT1 (Fig. 3C) in the cortex of nNOS+/+ mice.
Fig. 3. Nitrosylation of GAPDH induces SIRT1 nitrosylation and tau acetylation.
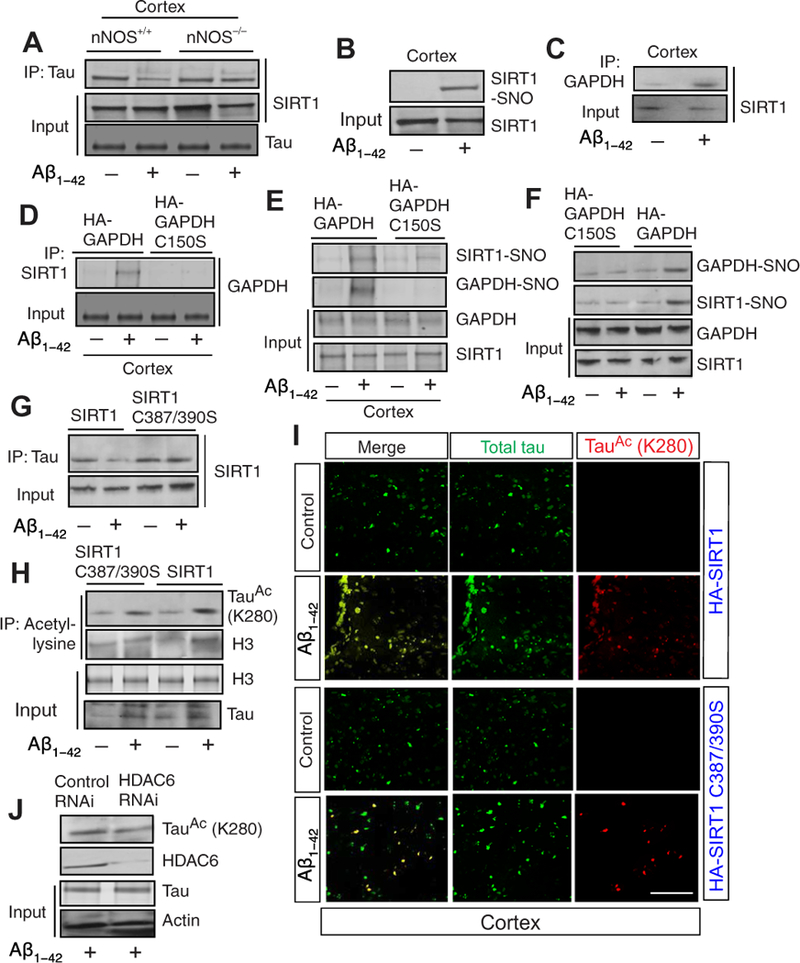
(A) The interaction between deacetylase sirtuin 1 (SIRT1) and tau was monitored by co-IP assay using lysates from cortex isolated from both nNOS+/+ and nNOS−/− mice administered with or without Aβ1-42. (B) Biotinswitch assay was performed to monitor nitrosylation of SIRT1 using the cortical lysates after administration of Aβ1-42. (C) The interaction between GAPDH and SIRT1 was monitored by co-IP assay using cortical lysates after administration of Aβ1-42. (D) Primary neurons isolated from nNOS+/+ mice were overexpressed with either GAPDH or GAPDH C150S construct before administration of Aβ1-42. The interaction between SIRT1 and GAPDH was monitored by co-IP assay. (E) GAPDH or GAPDH C150S was overexpressed in the cortex, and nitrosylation of GAPDH and SIRT1 was monitored by biotin-switch assay. (F) Primary neurons isolated from nNOS+/+ mice were overexpressed with either GAPDH or GAPDH C150S construct before administration of Aβ1-42. The nitrosylation of GAPDH and SIRT1 was then assessed and monitored by biotin-switch assay. (G) Primary neurons isolated from nNOS+/+ mice overexpressed with either SIRT1 or SIRT1 C387/390S were treated with Aβ1-42. Cell lysates were used to do co-IP assay to monitor the interaction between SIRT1 and tau. (H) Primary neurons isolated from nNOS+/+ mice overexpressed with either SIRT1 or SIRT1 C387/390S were treated with Aβ1-42. Acetylation of tau (Lys280) and H3 were assessed in cell lysates using a co-IP assay. The change in the amount of acetylated H3 and tau was quantitated for each sample. (I) Confocal microscopic analysis of total (green) and acetylated (red) tau in the cortex after administration of Aβ1-42. Scale bar, 100 μm. (J) Primary neurons isolated from nNOS+/+ mice were treated with either control or HDAC6 siRNA before administration with Aβ1–42. The tau acetylation at Lys280 was assessed by co-IP assay with antibody to acetyl-lysine and subsequent Western blotting. Data are quantified in the Supplementary Materials; all blots and microscopy are representative of three independent experiments from five to seven mice each condition.
To further substantiate whether SIRT1 was transnitrosylated by GAPDH-SNO upon administration of Ab1–42, we overexpressed either wild-type or C150S-mutant GAPDH in the cortex of mice and administered Aβ1–42. We found that the interaction between GAPDH and SIRT1 (Fig. 3D) and the nitrosylation of SIRT1 and GAPDH (Fig. 3E and fig. S3B) were increased in the cortices of mice overexpressing wild-type GAPDH, but not GAPDH C150S after administration of Aβ1–42 (Fig. 3D). Consistent with this in vivo data, we found that nitrosylation of SIRT1 was decreased in cells that overexpressed GAPDH C150S (Fig. 3F). We infer that a decrease in interaction between GAPDH and SIRT1 results in a loss of nitrosylation of SIRT1, which indicates that SIRT1 can be nitrosylated by transnitrosylation reaction through its interaction with nitrosylated GAPDH. Thus, blocking nitrosylation of GAPDH results in prevention of SIRT1 nitrosylation.
SIRT1 is nitrosylated at residues Cys387/390 and mutation of these residues abolishes its nitrosylation (27). To test whether overexpression of SIRT1 C387/390S has any influence on the interaction between SIRT1 and tau and the acetylation of tau in cells, we isolated primary neurons and transfected them with either SIRT1 C387/390S or wild-type SIRT1 constructs before we administered Aβ1–42 to the cultures. We found that the interaction between tau and SIRT1 was increased substantially in cells that overexpressed SIRT1 C387/390S compared to cells that overexpressed SIRT1 (Fig. 3G and fig. S3C). To see whether an interaction between tau and nitrosylated SIRT1 affects tau acetylation, we monitored tau acetylation at residue Lys280 by Western blot hybridization. We found that acetylation of tau was decreased by not more than 50% (Fig. 3H and fig. S3D), although the interaction between SIRT1 and tau was increased significantly in cells that overexpress SIRT1 C387/390S compared to cells that overexpress SIRT1 (Fig. 3H). As a control, we tested acetylation of H3 after administration of Aβ1–42 in cells that overexpress SIRT1 or SIRT1 C387/390S. We found that H3 acetylation was increased significantly in cells that overexpress SIRT1 C387/390S compared to cells that overexpressed SIRT1 (Fig. 3H). The influence of nitrosylation of SIRT1 on tau acetylation was further assessed by confocal microscopy after overexpression of either SIRT1 or SIRT1 C387/390S in the cortex. We found that tau acetylation was increased substantially in cortical cells that overexpressed SIRT1 but not SIRT1 C387/390S (Fig. 3I and fig. S3E).
These data suggest that nitrosylation of SIRT1 is sufficient to block its deacetyltransferase activity but is not enough to reduce the acetylation of tau. These data can be explained in two ways: (i) that it may be possible that another deacetylase, such as HDAC6, is involved in tau acetylation at Lys280 and/or (ii) that tau acetylation at Lys280 depends on both activation of p300 and inactivation of SIRT1. Thus, preventing only the inactivation of SIRT1 will not rescue the tau acetylation at residue Lys280. To test whether another deacetylase, such as HDAC6 (16), is involved in tau acetylation at Lys280, we depleted HDAC6 in primary neurons before administration of Aβ1–42 and monitored the acetylation of tau by Western blot hybridization. We found that tau acetylation at Lys280 remains unaltered in cells depleted of HDAC6 (Fig. 3J and fig. S3F). Thus, we concluded that HDAC6 may not be responsible for acetylation upon Aβ1–42 and excluded the first possibility.
Blocking GAPDH nitrosylation abolishes tau acetylation in vivo
Given that our data show that an increase in GAPDH-SNO is responsible for activation of p300 and inactivation of SIRT1, we anticipated that reducing the amount of GAPDH-SNO would prevent both events and consequently Aβ1–42-induced acetylation of tau at Lys280. To test this hypothesis, we overexpressed the nitrosylation mutant of GAPDH (GAPDH C150S) into the cortex before administration of Aβ1–42 and measured tau acetylation in that brain region. We found that tau acetylation at Lys280 was increased after administration of Aβ1–42 in mice that overexpressed wild-type GAPDH but not those that overexpressed GAPDH C150S (Fig. 4A and fig. S4A). These data suggest that the ni-trosylation of GAPDH is critical for tau acetylation. This was further confirmed by confocal microscopy analysis wherein acetylation of tau was increased significantly despite no change to total tau abundance (Fig. 4, B and D, and fig. S4B).
Fig. 4. Inhibition of GAPDH nitrosylation results in a reduction in tau acetylation.
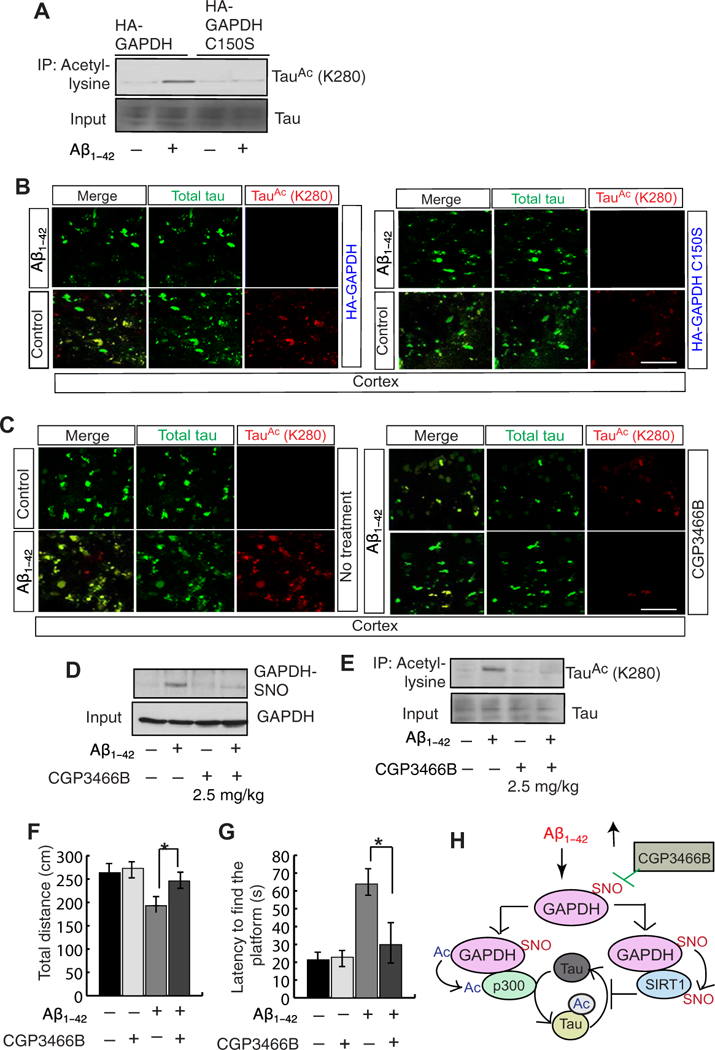
(A) GAPDH or GAPDH C150S was overexpressed in the cortex before administration of Aβ1–42. tau acetylation was assessed by co-IP assay with an antibody to acetyl-lysine and subsequent Western blotting. (B) GAPDH or GAPDH C150S was overexpressed in cortex before administration of Aβ1–42. Confocal microscopic analysis then assessed total (green) and acetylated (red) tau in the cortical tissue. Scale bar, 100 μm. (C) Confocal microscopic analysis then assessed total (green) and acetylated (red) tau in the cortical tissue from mice treated with CGP3466B (top, 0.5 mg/ kg; bottom, 2.5 mg/kg) before administration of Aβ1–42 into the cortex (right) compared with Aβ1–42 alone or controls (left). Scale bar, 100 μm. (D and E) Nitrosylation of GAPDH (D) and the acetylation of tau (E) were assessed in primary neurons cultured with Aβ1–42 with or without CGP3466B. Data above are quantified in the Supplementary Materials; all blots and microscopy are representative of three independent experiments from five to seven mice each condition. (F) Total distance calculated in open-field tests in mice treated with Aβ1–42 and with or without CGP3466B.n = 15 to 20 mice; **P < 0.001, Wilcoxon two-sample test (nonpara-metric version of two sample t test). (G) Latency to find the platform in Morris water maze test was monitored in mice administered with Aβ1–42 with or without CGP3466B treatment.n = 15 to 20 mice; **P < 0.001, Wilcoxon two-sample test. (H) A working model of our findings suggesting that Ap1–42 leads to an induction in the level of nitric oxide (NO), which, in turn, nitrosylates GAPDH. Nitrosylated GAPDH interacts with an acetyltransferase p300 and a deacetylase SIRT1. The interaction between p300 and GAPDH facilitates an activation of p300 and an induction of tau acetylation. The interaction between SIRT1 and GAPDH results in nitrosylation of SIRT1, which is unable to deacetylate tau. Inhibition of GAPDH-SNO reduces tau acetylation and improves behavioral impairments.
Previously, we reported that treatment with CGP3466B reduced the nitrosylation of GAPDH both in vivo and in cultured cells (29–31). To test whether treatment with CGP3466B can reduce tau acetylation, we administered CGP3466B in mice at two concentrations (0.5 and 2.5 mg/kg) before administrating Aβ1–42 into the cortex and monitored tau acetylation by confocal microscopy. The amount of acety- lated tau in Aβ1–42-treated cortex was abolished by treatment with CGP3466B at the higher dose (2.5 mg/kg) (Fig. 4C and fig. S4C). We also measured GAPDH-SNO level and tau acetylation using the lysates prepared from the cortex after administration of Aβ1–42 into the cortex. We found that nitrosylation of GAPDH was prevented (Fig. 4D and fig. S4D), and tau acetylation was reduced significantly (Fig. 4E and fig. S4E) in mice treated with CGP3466B compared to those treated with vehicle. These data further support the conclusion that GAPDH-SNO is a critical mediator of tau acetylation in the brain.
To test whether blocking GAPDH nitrosylation rescues behavioral impairments associated with tau acetylation, we measured locomotor activity by the open-field test and spatial memory by the Morris water maze test in mice pretreated with CGP3466B before being administered with Aβ1–42. Compared to mice administered with Aβ1–42 only, pretreatment with CGP3466B improved the activity of the mice in the open-field test by more than 82% (Fig. 4F) and reduced the latency to find the platform (Fig. 4G). These data suggest that blocking GAPDH nitrosylation improves the locomotor and spatial memory deficits caused by Aβ1–42 exposure.
DISCUSSION
We have shown here that administration of Aβ1–42 nitrosylates GAPDH, which inactivates SIRT1 through augmenting its nitrosylation level via transnitrosylation reaction. At the same time, nitrosylated GAPDH activates p300 by increasing its acetylation level. Both inactivation of SIRT1 and activation of p300 contribute to an augmentation in the level of tau acetylation preferably at Lys280, which was previously shown to contribute to tau aggregation (Fig. 4H).
The direct infusion of oligomeric Aβ1–42 into the wild-type mouse brain provided an in vivo model to acutely increase the amount of Aβ1−−42 in a spatial and temporal manner. The advantage of using wild-type mice for this model avoids the potential compensation or side effects from the mutations introduced in transgenic mouse lines (32–34). Past studies have shown that infusing toxic amount of Aβ1–42 into the brain results in tau phosphorylation (35, 36). Our study provides insight into pathological tau acetylation after infusion of Aβ1–42 into the cortex.
Our data indicate an augmentation in tau acetylation contributed significantly by inactivation of SIRT1 and activation of p300. Nota-bly, either blocking p300 or activating SIRT1 singularly did not reduce tau acetylation at Lys280. The deacetylation of tau is also reportedly regulated by SIRT1 (19, 37) and HDAC6 (38, 39), and blocking HDAC6 activity or genetically reducing HDAC6 expression reportedly alters tau acetylation (40). However, our results suggest that HDAC6 does not regulate the acetylation of tau at Lys280 specifically, but that SIRT1 has a substantial role.
An induction of NO in response to Aβ1–42 has been demonstrated previously (41–43). Their studies have highlighted the importance of nitrosylated proteins in the pathogenesis of AD. They have shown that nitrosylation of mitochondrial fission protein DRP1 (44) and cyclin-dependent kinase 5 (42) facilitates mitochondrial dysfunction and synaptic alteration, which contributes significantly to the pathogenesis of AD. Our study shows that nitrosylation of GAPDH and SIRT1 regulates pathological tau acetylation at Lys280. The nitrosylation of SIRT1 was identified a few years ago (27), however, its role in the pathogenicity of neurological disorders is not established. Our study suggests that preventing the ni-trosylation of SIRT1 by reducing GAPDH-SNO abundance will be instrumental in reducing pathological tau acetylation at Lys280.
The nitrosylation of GAPDH has been implicated in several neurodegenerative diseases, such as Parkinson’s disease (PD) (31), mental disorders (45), and in mouse models of hyperactivity induced by a drug of abuse, such as cocaine (30). However, the role of nitrosylation of GAPDH in tauopathies has not been identified yet. Our study could be the first example where nitrosylation of GAPDH significantly contributes to the acetylation of tau. Moreover, inhibition of nitrosylation of GAPDH by administration of CGP3466B has been shown to provide neuroprotection in 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine-treated mice model of PD (31), ketamine- induced mental disorder (45), and cocaine-induced hyperactivity in mice (30). Here, we have shown that treatment with CG- P3466B reduces tau acetylation and provides neuroprotection against Aβ-induced toxicity. Moreover, treatment with CGP3466B reverses the deficits in memory and lack of locomotor function induced by Aβ1–42. Together, our data provide a framework to develop a novel therapeutic strategy to reduce tau acetylation, considered as one of the major causes for tau aggregation and consequent toxicity in neurons.
MATERIALS AND METHODS
Aβ injections and in vivo transfection in mice
Before injection, the Aβ1–42 peptide was dissolved in a physiological saline solution at a concentration of 5 mg/ml and incubated at 37°C for 72 hours to induce aggregation (46,47). All the animal studies conducted were according to the Committee on Animal Use for Research and Education at Georgia Regents University and the University of Pittsburgh in compliance with National Institutes of Health guidelines, as described previously by others and our laboratory (48–51). For the Aβ1–42 injections, male C57BL/6 mice (8 to 10 weeks) were divided into four groups (n = 5) per treatment. Both nNOS+/+ and nNOS−/− mice were anesthetized with xylazine (8 mg/kg) and ketamine (60 mg/kg) by intraperitoneal injection, mounted in a stereotaxic frame, and injected with 100 μM Aβ1–42, as described before (34, 50). Motor function of the mice was assessed with the open-field test, and memory function was assessed with the Morris water maze test after 7 days after infusion of Aβ1–42 into the cortex (both behavior experiments are described below). Mice were treated with CGP3466B (2.5 mg/kg) through intraperitoneal injection after 20-min infusion of Aβ1–42 into the cortex. Either hemagglutinin (HA)-GAPDH, HA-GAPDH C150S, HA-GAPDH K160R, SIRT1, or SIRT1 C387/390S was injected into the brain using the in vivo DNA transfection reagent jetPEI (Polyplus- transfection) according to the manufacturer’s protocol with little modification (52–54). Briefly, each construct and jetPEI were separately diluted in a 5% sterile glucose solution. Our formulation corresponds to a nitrogen and phosphate (N/P) ratio of 7. A 5-μl volume of the polyethyleneimine-plasmid complexes was stereotaxically injected bilaterally into the cortex. The control and HDAC6 RNAi (Santa Cruz Biotechnology, catalog no. sc-35545) were injected into the mouse brain using jetSI reagent (Polyplus-transfection) according to the manufacturer’s protocol 4 days before administration of Aβ1–42 into the cortex with little modification (55). All the experiments and analysis were performed in a blinded manner.
Biotin-switch assay
The assay was performed as described (29,49, 56), with minor modifications. Briefly, cells/tissues were first lysed for 10 min on ice in NP-40/ Hepes-EDTA-Neocuproine (HEN buffer adjusted to contain 0.4% NP-40) and then centrifuged at 1000g for 10 min. at 4°C. The pellet was then resuspended in radioimmunoprecipitation assay (RIPA)/ HEN (HEN buffer adjusted to contain 1% Triton X-100, 1% sodium deoxycholate, and 0.1% SDS) and cleared by centrifugation at 16,000g at 4°C. The supernatant was then used for the assay. In addition, labeling was performed in the dark for 2 to 3 hours at room temperature with 5 mM sodium ascorbate and 0.4 mM biotin-HPDP (Pierce), and pull down was performed overnight with high-capacity neutravidin agarose (Pierce).
Morris water maze test
In the Morris water maze test, the hidden platform procedure was performed in a circular tank filled with opaque water as described already with minor modifications (48,49). For training, the mice (8 to 12 weeks old) received Aβ1–42 in the cortex, were placed in the tank at four random points, and were allowed to search for and find the platform. In the event that the mouse did not find the platform within the 60-s trial period, the mouse was manually put on the platform for an extra 30 s. Two trials were given every day, and the latency to find the platform in each trial was recorded. A probe trial was performed on day 6 or 7, in which mice were allowed to swim in the tank for 60 s without the platform, and performance was assessed as the time spent in the quadrant in which the hidden platform was originally located.
Open-field test
Locomotor activity was assessed in mice treated intraperitoneally with Aβ1–42 and with or without CGP3466B (2.5 mg/kg) using open- field tests, performed as described previously (46). Briefly, the animals were placed at the center of the arena and allowed to explore the apparatus freely for 60 min with the experimenter out of the animal’s sight. A black square arena (100 cm × 100 cm × 60 cm) was used for the test. The total distance travelled was analyzed using a video-tracking software.
Cortical primary neuronal cultures, treatment, and Western blotting
The preparation of the primary cortical neurons from the nNOS+/+ and the nNOS−/− mice was performed as described previously (29, 48, 49). Pregnant mice at embryonic day 18.5 gestational age based on the vaginal plug determination method were anesthetized with xylazine (8 mg/kg)/ ketamine (60 mg/kg) and cleaned using 70% ethanol. The uterus was opened, and pups were placed in sterile prechilled Hanks’ balanced salt solution. Each pup was mounted on the stereomicroscope, and cortices were dissected out. After enzyme-based digestion, cortical neurons were counted and plated on the sterilized cover glass or six-well plates in serum-free neurobasal medium supplemented with appropriate growth supplements (B27 and N2, Life Technologies). The medium was changed every 3 to 4 days. For the biochemical experiments, neurons were washed, scraped off, and collected in lysis buffer with protease inhibitors. The protein content was estimated by the Bradford method. Primary neurons were treated with Aβ1–42 (100 μg/ml) for 24 hours in subsequent experiments. All experiments were performed with 10- to 12-day-old cultures, at which time the cultures contain ~95 to 98% neurons and 2 to 5% astrocytes. We overexpressed GAPDH, GAPDH C150S, and GAPDH K160R in primary neurons using jetPRIME reagent (Polyplus- transfection) according to the manufacturer’s protocol. Briefly, the complexes were added to the media of 6- to 8-day-old primary neuronal cultures. After 3 to 4 days, Aβ1–42 was added and cells were incubated for 24 hours then either homogenized for biochemical assays or processed for immunohistochemistry as described below. Neurons were collected into a sample buffer [20 mM tris-HCl (pH 7.6), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, and 1% SDS containing protease inhibitors: 1 mM phenylmethylsulfonyl fluoride, aprotinin (10 μg/ml), leu-peptins (10 μg/ml), and pepstatin A (1 μg/ml)] (57). Then, equal amounts of protein for each sample were run in a SDS-polyacrylamide gel electrophoresis (SDS-PAGE) gel and transferred using polyvinylidene difluoride membrane. The membrane was incubated overnight with the primary antibody for acetylated tau-K280 (AnaSpec; 1:200). After washing, the membrane was incubated with secondary antibody and imaged using Odyssey CLx infrared imaging system as described previously (49,58,59).
Brain tissue lysates, co-IP, and Western blotting
Mouse or human brain tissue samples were lysed in RIPA containing protease inhibitors and phosphatase inhibitors. Samples were spun to remove the insoluble fraction, and the concentration of protein was quantified using a Bradford assay. Brain tissues were suspended and homogenized in 5 vol/g of RIPA buffer [50 mM tris (pH 8.0), 150 mM NaCl, 1% NP-40, 5 mM EDTA, 0.5% sodium deoxycholate, and 0.1% SDS]. Finally, resultant insoluble pellets were extracted in 1 vol/gram of tissue in urea buffer [7 M urea, 2 M thiourea, 4% CHAPS, and 30 mM tris (pH 8.5)]. Soluble and insoluble fractions were analyzed by SDS-PAGE electrophoresis and Western blotting using the indicated antibodies to detect total and acetylated tau proteins, as described previously (16,47, 60). For the co-IP assay, each lysate isolated from cells/tissues was incubated overnight with either antibodies to acetyl-lysine (Cell Signaling Technology; 1:200), tau (AnaSpec; 1:200), or GAPDH (Santa Cruz Biotechnology; 1:1000). Then, samples were pulled down with immunoglobulin G beads and run into SDS-PAGE gel. The membrane was used for Western blotting using antibodies against acetylated tau-K280 (AnaSpec; 1:100), SIRT1 (Cell Signaling Technology; 1:500), p300 (Santa Cruz Biotechnology; 1:500), and GAPDH (Cell Signaling Technology; 1:2000).
Immunochemical staining
Neurons were fixed with 3 to 4% paraformaldehyde containing sucrose for 15 min at room temperature, as described previously (26, 48,49). Briefly, cells were permeabilized using 0.1% Triton X-100 in phosphate- buffered saline (PBS) (0.1 M, pH 7.4), then incubated in blocking serum, to which antibodies were added [antibodies to acetylated tau (1:200 dilution; K280, AnaSpec), total tau (1:200 dilution; AnaSpec), and actin (1:3500 dilution; Cell Signaling Technology)]. Cells were then incubated with secondary antibodies conjugated to Alexa Fluor 594 and fluorescein isothiocyanate, and coverslips were mounted on a poly-L-lysine-coated glass slide in a mounting medium containing 4’,6-diamidino-2-phenylindole (DAPI). For the brain samples, 8- to 10-μm frozen sections were rehydrated and permeabilized by 0.1% Triton X-100 in PBS. Sections were then blocked using a serum and incubated with primary antibodies. Antigen retrieval was performed by incubating sections with 0.5 N HCl containing trypsin, as described previously. Tagged secondary antibodies were applied, and sections were washed again and mounted in medium containing DAPI. Sections were analyzed under confocal Zeiss 710 Meta microscope. Images were obtained using integrated image acquisition software, ZEN 2012, and analyzed in a blinded manner.
Supplementary Material
Acknowledgments:
We thank S. Snyder (Johns Hopkins University School of Medicine) for sharing the constructs related to GAPDH and SIRT1. We also thank W. Lariviere (University of Pittsburgh) for editing the manuscript.
Funding: This research was supported by the NIH grants R01NS094516 and R01EY025622.
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Parihar MS, Hemnani T, Alzheimer’s disease pathogenesis and therapeutic interventions. J. Clin. Neurosci. 11, 456–467 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Jahn H, Memory loss in Alzheimer’s disease. Dialogues Clin. Neurosci. 15, 445–454 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hebert LE, Scherr PA, Beckett LA, Albert MS, Pilgrim DM, Chown MJ, Funkenstein HH, Evans DA, Age-specific incidence of Alzheimer’s disease in a community population. JAMA 273, 1354–1359 (1995). [PubMed] [Google Scholar]
- 4.Wimo A, Jonsson L, Winblad B, An estimate of the worldwide prevalence and direct costs of dementia in 2003. Dement. Geriatr. Cogn. Disord. 21, 175–181 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Blennow K, de Leon MJ, Zetterberg H, Alzheimer’s disease. Lancet 368, 387–403 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Murphy MP, LeVine III H, Alzheimer’s disease and the amyloid-β peptide. J.Alzheimers Dis. 19, 311–323 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goure WF, Krafft GA, Jerecic J, Hefti F, Targeting the proper amyloid-beta neuronal toxins: A path forward for Alzheimer’s disease immunotherapeutics. Alzheimer’s Res. Ther. 6, 42 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mucke L, Selkoe DJ, Neurotoxicity of amyloid β-protein: Synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2, a006338 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mokhtar SH, Bakhuraysah MM, Cram DS, Petratos S, The beta-amyloid protein of Alzheimer’s disease: Communication breakdown by modifying the neuronal cytoskeleton. Int. J. Alzheimer’s Dis. 2013, 910502 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT, Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1, a006189 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kar S, Slowikowski SPM, Westaway D, Mount HTJ, Interactions between β-amyloid and central cholinergic neurons: Implications for Alzheimer’s disease. J. Psychiatry Neurosci. 29, 427–441 (2004). [PMC free article] [PubMed] [Google Scholar]
- 12.Ando K, Maruko-Otake A, Ohtake Y, Hayashishita M, Sekiya M, Iijima KM, Stabilization of microtubule-unbound tau via tau phosphorylation at Ser262/356 by Par-1/MARK contributes to augmentation of AD-related phosphorylation and Aβ42-induced tau toxicity. PLOS Genet. 12, e1005917 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kadavath H, Hofele RV, Biernat J, Kumar S, Tepper K, Urlaub H, Mandelkow E, Zweckstetter M, Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl. Acad. Sci. U.S.A. 112, 7501–7506 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Medina M, Hernández F, Avila J, New features about tau function and dysfunction. Biomolecules 6, E21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stoothoff WH, Johnson GVW, Tau phosphorylation: Physiological and pathological consequences. Biochim. Biophys. Acta 1739, 280–297 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, Lee VMY, The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2, 252 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Irwin DJ, Cohen TJ, Grossman M, Arnold SE, Xie SX, Lee VM-Y, Trojanowski JQ, Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain 135, 807–818 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tracy TE, Sohn PD, Minami SS, Wang C, Min SW, Li Y, Zhou Y, Le D, Lo I, Ponnusamy R, Cong X, Schilling B, Ellerby LM, Huganir RL, Gan L, Acetylated tau obstructs KIBRA-mediated signaling in synaptic plasticity and promotes tauopathy- related memory loss. Neuron 90, 245–260 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Min S-W, Cho S-H, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, Meyers D, Cole PA, Ott M, Gan L, Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67, 953–966 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cook C, Stankowski JN, Carlomagno Y, Stetler C, Petrucelli L, Acetylation: A new key to unlock tau’s role in neurodegeneration. Alzheimer’s Res. Ther. 6, 29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gorsky MK, Burnouf S, Dols J, Mandelkow E, Partridge L, Acetylation mimic of lysine 280 exacerbates human Tau neurotoxicity in vivo. Sci. Rep. 6, 22685 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gu Z, Nakamura T, Lipton SA, Redox reactions induced by nitrosative stress mediate protein misfolding and mitochondrial dysfunction in neurodegenerative diseases. Mol. Neurobiol. 41, 55–72 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakamura T, Lipton SA, Emerging roles of S-nitrosylation in protein misfolding and neurodegenerative diseases. Antioxid. Redox Signal. 10, 87–101 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Nakamura T, Lipton SA, Cell death: Protein misfolding and neurodegenerative diseases. Apoptosis 14, 455–468 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Sen N, Snyder SH, Protein modifications involved in neurotransmitter and gasotransmitter signaling. Trends Neurosci. 33, 493–502 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sen N, Hara MR, Kornberg MD, Cascio MB, Bae B-I, Shahani N, Thomas B, Dawson TM, Dawson VL, Snyder SH, Sawa A, Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat. Cell Biol. 10, 866–873 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kornberg MD, Sen N, Hara MR, Juluri KR, Nguyen JVK, Snowman AM, Law L, Hester LD, Snyder SH, GAPDH mediates nitrosylation of nuclear proteins. Nat. Cell Biol. 12,1094–1100 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, Ferris CD, Hayward SD, Snyder SH, Sawa A, S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 7, 665–674 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Sen N, Snyder SH, Neurotrophin-mediated degradation of histone methyltransferase by S-nitrosylation cascade regulates neuronal differentiation. Proc. Natl. Acad. Sci. U.S.A. 108, 20178–20183 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu R, Serritella AV, Sen T, Farook JM, Sedlak TW, Baraban J, Snyder SH, Sen N, Behavioral effects of cocaine mediated by nitric oxide-GAPDH transcriptional signaling. Neuron 78, 623–630 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hara MR, Thomas B, Cascio MB, Bae B-I, Hester LD, Dawson VL, Dawson TM, Sawa A, Snyder SH, Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proc. Natl. Acad. Sci. U.S.A. 103, 3887–3889 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baleriola J, Walker CA, Jean YY, Crary JF, Troy CM, Nagy PL, Hengst U, Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell 158, 1159–1172 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jean YY, Ribe EM, Pero ME, Moskalenko M, Iqbal Z, Marks LJ, Greene LA, Troy CM, Caspase-2 is essential for c-Jun transcriptional activation and Bim induction in neuron death. Biochem. J. 455, 15–25 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sotthibundhu A, Sykes AM, Fox B, Underwood CK, Thangnipon W, Coulson EJ, β-amyloid1–42 induces neuronal death through the p75 neurotrophin receptor. J. Neurosci. 28, 3941–3946 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bolmont T, Clavaguera F, Meyer-Luehmann M, Herzig MC, Radde R, Staufenbiel M, Lewis J, Hutton M, Tolnay M, Jucker M, Induction of tau pathology by intracerebral infusion of amyloid-β-containing brain extract and by amyloid-β deposition in APP x Tau transgenic mice. Am. J. Pathol. 171, 2012–2020 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nisbet RM, Polanco J-C, Ittner LM, Götz J, Tau aggregation and its interplay with amyloid-β. Acta Neuropathol. 129, 207–220 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Min S-W, Chen X, Tracy TE, Li Y, Zhou Y, Wang C, Shirakawa K, Minami SS, Defensor E, Mok SA, Sohn PD, Schilling B, Cong X, Ellerby L, Gibson BW, Johnson J, Krogan N, Shamloo M, Gestwicki J, Masliah E, Verdin E, Gan L, Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat. Med. 21, 1154–1162 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Noack M, Leyk J, Richter-Landsberg C, HDAC6 inhibition results in tau acetylation and modulates tau phosphorylation and degradation in oligodendrocytes. Glia 62, 535–547 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Leyk J, Goldbaum O, Noack M, Richter-Landsberg C, Inhibition of HDAC6 modifies tau inclusion body formation and impairs autophagic clearance. J. Mol. Neurosci. 55, 1031–1046 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Cook C, Carlomagno Y, Gendron TF, Dunmore J, Scheffel K, Stetler C, Davis M, Dickson D, Jarpe M, DeTure M, Petrucelli L, Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum. Mol. Genet. 23, 104–116 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tu S, Okamoto S.-i., Lipton SA, Xu H, Oligomeric Aβ-induced synaptic dysfunction in Alzheimer’s disease. Mol. Neurodegener. 9, 48 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qu J, Nakamura T, Cao G, Holland EA, McKercher SR, Lipton SA, S-Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by β-amyloid peptide. Proc. Natl. Acad. Sci. U.S.A. 108, 14330–14335 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Akhtar MW, Sunico CR, Nakamura T, Lipton SA, Redox regulation of protein function via cysteine S-nitrosylation and its relevance to neurodegenerative diseases. Int. J. Cell Biol. 2012, 463756 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakamura T, Cieplak P, Cho D-H, Godzik A, Lipton SA, S-nitrosylation of Drp1 links excessive mitochondrial fission to neuronal injury in neurodegeneration. Mitochondrion 10,573–578 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harraz MM, Tyagi R, Cortés P, Snyder SH, Antidepressant action of ketamine via mTOR is mediated by inhibition of nitrergic Rheb degradation. Mol. Psychiatry 21, 313–319 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang L, Fang Y, Xu Y, Lian Y, Xie N, Wu T, Zhang H, Sun L, Zhang R, Wang Z, Curcumin improves amyloid β-peptide (1–42) induced spatial memory deficits through BDNF-ERK signaling pathway. PLOS ONE 10, e0131525 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sell GL, Schaffer TB, Margolis SS, Reducing expression of synapse-restricting protein Ephexin5 ameliorates Alzheimer’s-like impairment in mice. J. Clin. Invest. 127, 1646–1650 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sen T, Gupta R, Kaiser H, Sen N, Activation of PERK elicits memory impairment through inactivation of CREB and downregulation of PSD95 following Traumatic Brain Injury. J. Neurosci. 37, 5900–5911 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sen N, Paul BD, Gadalla MM, Mustafa AK, Sen T, Xu R, Kim S, Snyder SH, Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol. Cell 45, 13–24 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jean YY, Baleriola J, Fà M, Hengst U, Troy CM, Stereotaxic infusion of oligomeric amyloid-beta into the mouse hippocampus. J. Vis. Exp. 2015, e52805 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, Chiesa R, Gobbi M, Salmona M, Forloni G, Synthetic amyloid-βoligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. U.S.A. 107, 2295–2300 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abdallah B, Hassan A, Benoist C, Goula D, Behr JP, Demeneix BA, A powerful nonviral vector for in vivo gene transfer into the adult mammalian brain: Polyethylenimine. Hum. Gene Ther. 7, 1947–1954 (1996). [DOI] [PubMed] [Google Scholar]
- 53.Zuckermann M, Hovestadt V, Knobbe-Thomsen CB, Zapatka M, Northcott PA, Schramm K, Belic J, Jones DTW, Tschida B, Moriarity B, Largaespada D, Roussel MF, Korshunov A, Reifenberger G, Pfister SM, Lichter P, Kawauchi D, Gronych J, Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat. Commun. 6, 7391 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamashita T, Ninomiya M, Hernández Acosta P, García-Verdugo JM, Sunabori T, Sakaguchi M, Adachi K, Kojima T, Hirota Y, Kawase T, Araki N, Abe K, Okano H, Sawamoto K, Subventricular zone-derived neuroblasts migrate and differentiate into mature neurons in the post-stroke adult striatum. J. Neurosci. 26, 6627–6636 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Froidevaux M-SC, Berg P, Seugnet I, Decherf S, Becker N, Sachs LM, Bilesimo P, Nygárd M, Pongratz I, Demeneix BA, The co-chaperone XAP2 is required for activation of hypothalamic thyrotropin-releasing hormone transcription in vivo. EMBO Rep. 7, 1035–1039 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sen N, Hara MR, Ahmad AS, Cascio MB, Kamiya A, Ehmsen JT, Aggrawal N, Hester L, Doré S, Snyder SH, Sawa A, GOSPEL: A neuroprotective protein that binds to GAPDH upon S-nitrosylation. Neuron 63, 81–91 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu T, Perry G, Chan HW, Verdile G, Martins RN, Smith MA, Atwood CS, Amyloid-β-induced toxicity of primary neurons is dependent upon differentiation- associated increases in tau and cyclin-dependent kinase 5 expression. J. Neurochem. 88, 554–563 (2004). [DOI] [PubMed] [Google Scholar]
- 58.Sen T, Sen N, Isoflurane-induced inactivation of CREB through histone deacetylase 4 is responsible for cognitive impairment in developing brain. Neurobiol. Dis. 96, 12–21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sen T, Sen N, Treatment with an activator of hypoxia-inducible factor 1, DMOG provides neuroprotection after traumatic brain injury. Neuropharmacology 107, 79–88 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ericsson C, Peredo I, Nistér M, Optimized protein extraction from cryopreserved brain tissue samples. Acta Oncol. 46, 10–20 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.