

ABSTRACT
Although exact causes of Parkinson disease (PD) remain enigmatic, mitochondrial dysfunction is increasingly appreciated as a key determinant of dopaminergic neuron susceptibility in both familial and sporadic PD. Two genes associated with recessive, early-onset PD encode the ubiquitin (Ub) kinase PINK1 and the E3 Ub ligase PRKN/PARK2/Parkin, which together orchestrate a protective mitochondrial quality control (mitoQC) pathway. Upon stress, both enzymes cooperatively identify and decorate damaged mitochondria with phosphorylated poly-Ub (p-S65-Ub) chains. This specific label is subsequently recognized by autophagy receptors that further facilitate mitochondrial degradation in lysosomes (mitophagy). Here, we analyzed human post-mortem brain specimens and identified distinct pools of p-S65-Ub-positive structures that partially colocalized with markers of mitochondria, autophagy, lysosomes and/or granulovacuolar degeneration bodies. We further quantified levels and distribution of the ‘mitophagy tag’ in 2 large cohorts of brain samples from normal aging and Lewy body disease (LBD) cases using unbiased digital pathology. Somatic p-S65-Ub structures independently increased with age and disease in distinct brain regions and enhanced levels in LBD brain were age- and Braak tangle stage-dependent. Additionally, we observed significant correlations of p-S65-Ub with LBs and neurofibrillary tangle levels in disease. The degree of co-existing p-S65-Ub signals and pathological PD hallmarks increased in the pre-mature stage, but decreased in the late stage of LB or tangle aggregation. Altogether, our study provides further evidence for a potential pathogenic overlap among different forms of PD and suggests that p-S65-Ub can serve as a biomarker for mitochondrial damage in aging and disease.
Abbreviations: BLBD: brainstem predominant Lewy body disease; CCCP: carbonyl cyanide m-chlorophenyl hydrazone; DLB: dementia with Lewy bodies; DLBD: diffuse neocortical Lewy body disease; EOPD: early-onset Parkinson disease; GVB: granulovacuolar degeneration body; LB: Lewy body; LBD: Lewy body disease; mitoQC: mitochondrial quality control; nbM: nucleus basalis of Meynert; PD: Parkinson disease; PDD: Parkinson disease with dementia; p-S65-Ub: PINK1-phosphorylated serine 65 ubiquitin; SN: substantia nigra; TLBD: transitional Lewy body disease; Ub: ubiquitin
KEYWORDS: Aging, alpha-synuclein, autophagy, lewy body disease, MAPT, mitochondria, mitophagy, PARK2, parkin, parkinson disease, phospho-ubiquitin, PINK1, SNCA, tau, ubiquitin
Introduction
Parkinson disease (PD) is the second most common neurodegenerative disease, with aging being the most important risk factor [1]. Incidence rates increase with age from approximately 1% in the 60s to 4–5% in the 80s [2]. Clinically, PD is characterized by motor symptoms [3] arising from the selective loss of dopamine neurons in the substantia nigra (SN) that project to the caudate nucleus and putamen [4]. During the disease course, neuronal degeneration also affects other brain regions (e.g., olfactory bulb, nucleus basalis of Meynert [nbM], hippocampus, amygdala and neocortex), contributing to comorbid non-motor symptoms. Neuropathologically, PD is characterized by the presence of fibrillar SNCA/alpha-synuclein aggregates in neuronal somata (Lewy bodies; LBs) and processes (Lewy neurites) [5]. Yet, there is considerable phenotypic overlap in a clinicopathological continuum of neuropathologically diagnosed LB disease (LBD) cases. The clinical spectrum includes pure PD, PD with dementia (PDD) and dementia with Lewy bodies (DLB), and the clinical manifestations predominantly depend on the anatomical distribution and load of SNCA pathology [6,7]. In addition, hyperphosphorylated MAPT (microtubule associated protein tau; p-MAPT) and resulting neurofibrillary tangles have been observed in several brain areas in PD [8]. Both SNCA and MAPT pathologies may act synergistically [9], and variants in genes encoding these 2 proteins are the most significant genetic PD risk factors known [10].
Familial forms of PD have been identified in 5–10% of all patients and have been attributed to mutations in more than 20 different disease genes and risk factors [11]. Loss-of-function mutations in PINK1 (PTEN induced putative kinase 1) and PRKN/PARK2 (parkin RBR E3 ubiquitin protein ligase) are the most common causes of hereditary early-onset PD (EOPD; defined as an age of onset ≤ 50 yr) [12]. While the encoded enzymes PINK1 and PRKN are broadly cytoprotective, they appear particularly critical for survival of dopamine neurons [13,14]. Though exact disease mechanisms remain unknown, accumulating evidence suggests that neuroprotection may occur through PINK1-PRKN-mediated mitochondrial quality control (mitoQC). This multifaceted pathway is thought to regulate various aspects of mitochondrial health and function including but not limited to antioxidant mechanisms, stress response, morphology, transport, biogenesis, and degradation [15,16]. Upon mitochondrial stress, the ubiquitin (Ub) kinase PINK1 accumulates on the mitochondrial surface and phosphorylates serine 65 of Ub (p-S65-Ub) and of PRKN in order to activate and locally recruit this E3 Ub ligase from the cytosol [17]. Both enzymes then jointly amplify the formation of phosphorylated poly-Ub chains on mitochondria. This specific label is decoded by autophagy receptors that bind members of the MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) family on phagophores, the precursors to autophagosomes, and thereby direct damaged mitochondria to lysosomes for clearance (mitophagy) [18,19].
Mitophagy can be impaired at multiple steps, but the outcomes with respect to p-S65-Ub levels may differ. Not only stress and PINK1-PRKN, but also autophagic and lysosomal capacities play critical roles for the effective turnover of damaged mitochondria. Complete loss of PINK1 or PRKN causes EOPD and prevents the induction or flux of mitophagy through absent/reduced levels of the p-S65-Ub degradation label. In addition to EOPD, reduced activities of either enzyme may also contribute to later onset PD or at least modify rates of progression and clinical phenotypes [20,21]. Yet, in the presence of functional PINK1-PRKN, enhanced mitochondrial dysfunctions as well as defects in autophagic/lysosomal clearance, both well-documented pathogenic features of PD [22,23], may result in detectable accumulation of this transient ‘mitophagy tag’. Regardless of this difference, mitoQC is emerging as a candidate for a potential great pathogenic overlap between different forms of familial and sporadic PD [24].
While mitoQC has been intensely investigated in cell lines, the physiological significance in neurons, in vivo and the specific contribution of its failure to the pathogenesis of PD remain uncertain. Here, we evaluated p-S65-Ub in human post-mortem brain tissue to better understand the association between alterations in mitoQC, age and PD neuropathology. Using novel antibodies [25,26] we characterized the morphology and subcellular localization of p-S65-Ub along with organelle and disease markers. In addition, we quantified p-S65-Ub levels across 5 regions from large cohorts of neurologically normal controls and cases with clinically diagnosed parkinsonian syndrome and pathologically confirmed LBD. Correlations of findings with clinicopathological data, revealed an age- and a disease-dependent increase of the mitophagy label in human brain. Moreover, p-S65-Ub not only co-existed but also correlated with SNCA and MAPT pathology in distinct areas of LBD brains. Our data emphasizes the relevance of PINK1-PRKN-directed mitoQC and suggest that p-S65-Ub may serve as a novel biomarker for mitochondrial damage in aging and disease.
Results
Morphology and distribution of p-S65-Ub immunopositive structures in human brains
To analyze mitoQC in human post-mortem brain, PD vulnerable regions including the SN, hippocampus, amygdala, putamen and nbM were stained with the anti-p-S65-Ub mitophagy marker. We had previously developed and extensively characterized these antibody tools [25,26]. Twenty-eight cases from a neurologically normal cohort (hereafter referred to as controls) and 28 cases with pathologically-confirmed LBD from a spectrum of clinically diagnosed PD, PDD, and DLB patients were included in the study (see Table 1 for an overview of the cohorts as well as Tables S1 and S2 for details of controls and LBD cases, respectively).
Table 1.
Subject characteristics.
| Cohort | N | Median age (25%-75%) | Male (%) | Braak tangle stage (25%-75%) | Thal amyloid phase (25%-75%) | Braak PD stage (25%-75%) |
|---|---|---|---|---|---|---|
| Normal* | 28 | 68 (54–82) | 17 (61%) | I (0-II) | 0 (0–1) | 0 (0–0) |
| Sporadic LBD* | 28 | 78 (70–84) | 21 (75%) | II-III (I-III) | 1 (0–2) | 4 (3–5) |
| Mutation carriers | 9 | 78 (72–94) | 5 (56%) | IV-V (II-V) | 5 (1–5) | 4 (3–5) |
Neuropathological scores are presented as median (25th, 75th percentile). Individual information regarding each individual is provided in Tables S1-S3.
*Subgroups of the cohorts used in different studies are indicated in Tables S1 and S2.
Abbreviations: PD, Parkinson disease; LBD, Lewy body disease.
Close inspection of brain sections revealed 3 major types of p-S65-Ub immunopositive structures in both controls and LBD cases: granules, granulovacuolar degeneration bodies (GVBs) and beaded neurites (Figure 1A). The specificity of the staining was corroborated on adjacent brain sections with another commercially available antibody against p-S65-Ub that revealed exactly the same pattern, though was overall less sensitive in multiple applications (Figure S1). All 3 p-S65-Ub-positive structures had distinct distributions across different brain regions (Figure 1(B)). Punctate p-S65-Ub granules were frequently found in the cytoplasm of neuromelanin-containing cells in the SN and occasionally in neurons in the amygdala and nbM. Meanwhile, strong immunoreactivity for p-S65-Ub was observed in small spherical structures within pyramidal neurons in the hippocampus and amygdala that were morphologically consistent with GVBs seen in aging and Alzheimer disease. Whereas these structures have been reported to stain positive with various phospho-specific antibodies [27], GVBs also contain ubiquitinated proteins and have been described as specific forms of residual autophagic bodies with incomplete degradation [28,29]. p-S65-Ub labeled GVBs were found in the nbM as well, but to a much lesser extent. In general, somatic p-S65-Ub staining, including granules and GVBs, were most abundant in the SN and hippocampus, followed by amygdala and then nbM. In contrast, p-S65-Ub-positive beaded neurites were frequent in the nbM, less in the SN, and even less common in the hippocampus and amygdala. Putamen was almost devoid of any p-S65-Ub-immunopositive structures.
Figure 1.
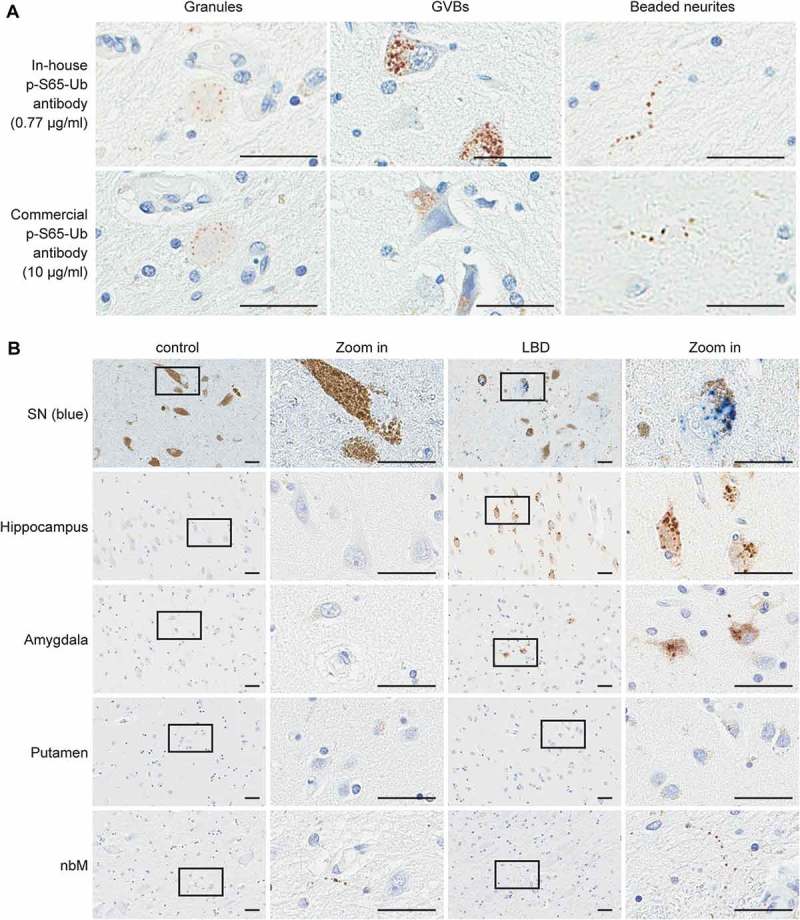
Morphology and distribution of p-S65-Ub-immunopositive structures. (A) Adjacent sections from LBD brains were stained with different p-S65-Ub antibodies at the indicated concentrations. Representative images of p-S65-Ub-immunopositive structures from various brain regions show the same 3 major patterns: granules (left panels), GVBs (middle panels) and beaded neurites (right panels). (B) Representative images of human brain sections from normal (left) and LBD cases (right) stained with p-S65-Ub antibody are shown. Chromogen used for the SN was HighDef Blue (blue) to distinguish the positive signal from dark brown neuromelanin in DA neurons. Conventional brown 3,3ʹ-diaminobenzidine was used as a chromogen for all other brain regions (hippocampus, amygdala, putamen, and nbM). A magnified image of the boxed area is shown to the right. Scale bars: 20 µm.
In order to further validate the specificity of the p-S65-Ub antibodies in human post-mortem brain tissue, we included available samples from PINK1 or PRKN mutation carriers (Table 1 and Table S3). Compared to age-matched sporadic LBD, PINK1 or PRKN mutant cases showed a dramatic reduction of somatic p-S65-Ub levels in the SN (Figure 2; p = 0.0006 and p = 0.006, respectively). This is consistent with roles of both enzymes in joint catalysis and amplification of p-S65-Ub chains and impairments in labeling damaged mitochondria due to loss of either function. Among 2 algorithms we developed for unbiased p-S65-Ub quantification, the positive pixel count algorithm allowed capture of all p-S65-Ub structures, but was very sensitive to nonspecific signals from lipofuscin and other non-cellular structures; the positive cell count algorithm had superior specificity and was equally sensitive for granular and vacuolar somatic p-S65-Ub (Figure S2). Though beaded neurites were undetectable with the latter algorithm, we here chose this more stringent approach and focused on the cell soma where most mitochondria, autophagosomes and lysosomes are located.
Figure 2.
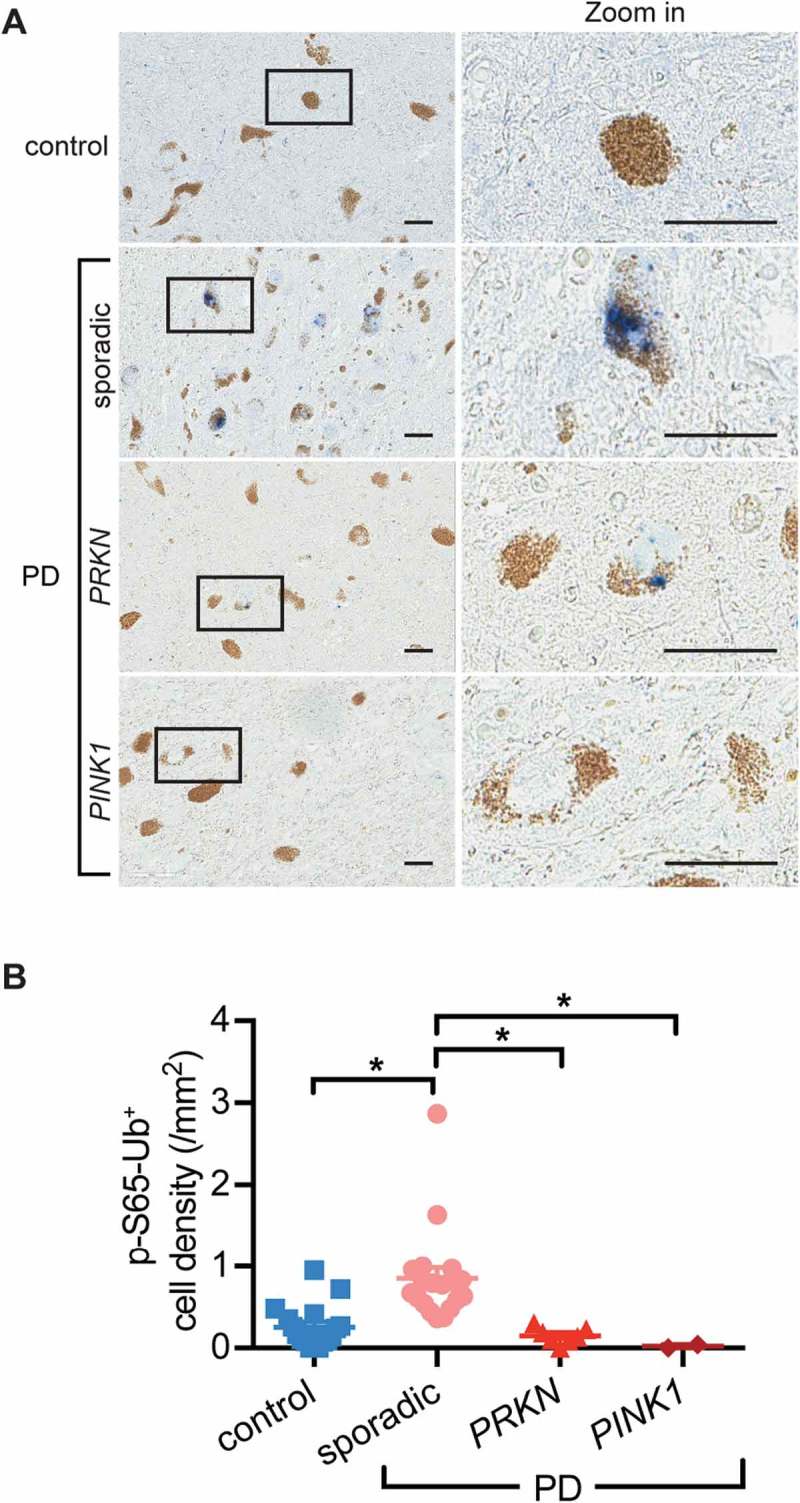
Reduced or absent p-S65-Ub staining in brains from PRKN or PINK1 mutation carriers. (A) SN sections from controls, sporadic LBD and cases with PINK1 or PRKN mutations were stained with p-S65-Ub antibody. Representative images of stained sections show enhanced p-S65-Ub levels in sporadic PD, but reduced or absent signal in SN of PRKN or PINK1 mutant cases. A magnified image of the boxed area is shown to the right. (B) Quantification of p-S65-Ub signal in age-matched controls, sporadic LBD cases and mutation carriers (Kruskal-Wallis test, *p < 0.01). n = 17 for normal, n = 19 for sporadic PD, n = 7 for PD with PRKN mutation, and n = 2 for PD with PINK1 mutation.
High resolution imaging analyses of p-S65-Ub-positive structures
To better characterize the p-S65-Ub-positive structures on a subcellular level, we used super-resolution microscopy of LBD sections double labeled with other cellular markers. In the SN, small somatic p-S65-Ub granules showed partial colocalization with the mitochondrial marker PPIF/cyclophilin F (peptidylprolyl isomerase F), and greater colocalization with the lysosomal marker CTSD (cathepsin D) (Figure 3(A,B)). Interestingly, beaded neurites also colocalized with mitochondrial, but not lysosomal markers. In line with the idea that the autophagic degradation of mitochondria may occur predominantly in the cell body, co-labeling allowed the identification of distinct p-S65-Ub structures during mitoQC that potentially represented the mitochondrial pools in cell processes and lysosomal pools in the soma, respectively. In addition, as remnants of autophagic organelles [30], CSNK1D (casein kinase 1 delta)-positive GVBs in the hippocampus strongly colocalized with both p-S65-Ub and SSBP1 (single stranded DNA binding protein 1), another mitochondrial marker (Figure 3(C)), suggesting an accumulation of p-S65-Ub-labeled mitochondrial components in autophagic bodies.
Figure 3.
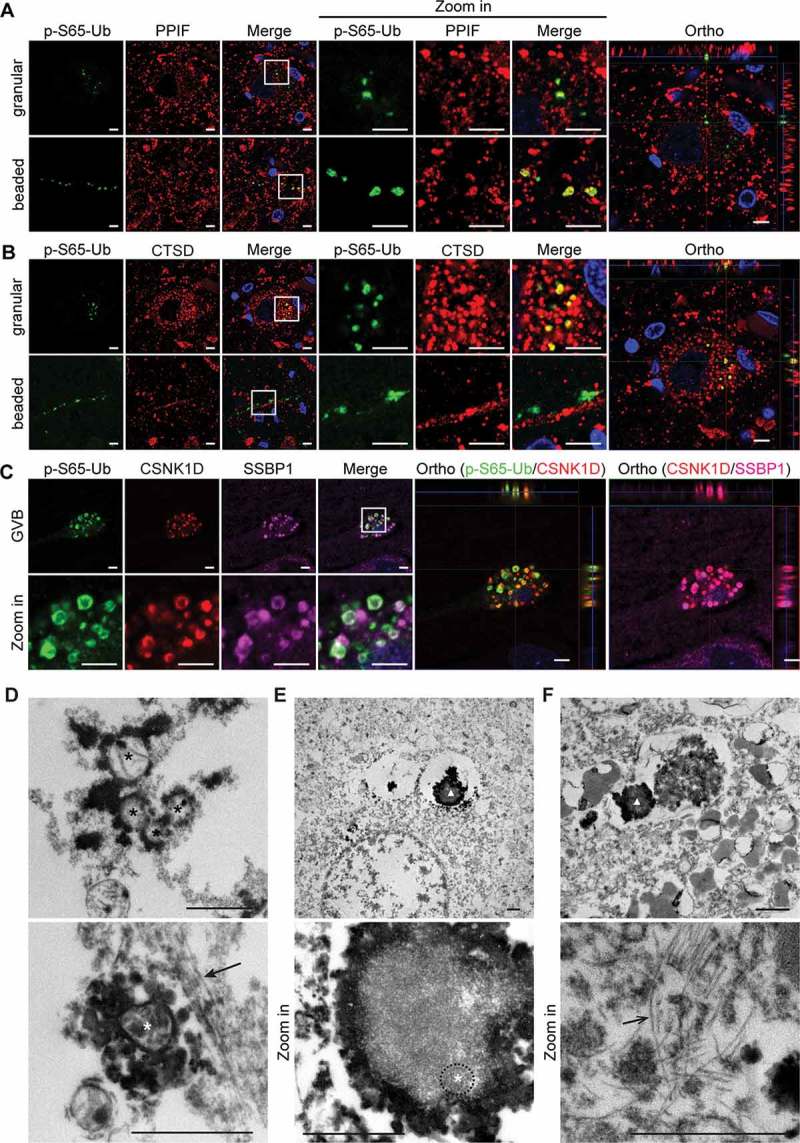
High-resolution imaging and ultrastructural examination of human brain tissue immunolabeled with p-S65-Ub. Human brain sections from LBD cases were multicolor labeled with antibodies against p-S65-Ub and (A) PPIF (mitochondrial marker), (B) CTSD (lysosomal marker) or (C) CSNK1D (GVB marker) and SSBP1 (mitochondrial marker). Representative Airyscan confocal images from SN and hippocampus are shown. p-S65-Ub-immunopositive granules partially colocalized with both mitochondrial and lysosomal markers; beaded neurites were co-labeled only with mitochondrial, but not lysosomal markers. GVBs were partially co-labeled with CSNK1D and mitochondrial markers. The colocalization of p-S65-Ub with the respective organelle markers is also demonstrated in the orthogonal (Ortho) view presented to the right. Scale bars: 5 µm. (D) Top: Mitochondria (black asterisks) are intensely labeled with p-S65-Ub on the outer membrane. Bottom: Some mitochondria (white asterisk) were found next to a neurofilament-like structure (solid arrow). (E, F) Top: At lower magnification, structures labeled (triangle) are consistent with GVBs seen at light microscopy. Bottom: At higher magnification, spherical double-membrane structures, possibly mitochondria, are found in the GVBs (white asterisk and circled by dashed line). Some labeled GVBs are adjacent to paired helical filaments (open arrow). Scale bars: 1 µm.
To further examine the ultrastructure of p-S65-Ub-positive labels, we developed pre-embedding techniques for immuno-electron microscopy. As a control, we first performed p-S65-Ub immunostainings in HeLa cells stably expressing PRKN. Increased p-S65-Ub levels in the cytoplasm were observed specifically upon treatment with the depolarizer carbonyl cyanide m-chlorophenyl hydrazone (CCCP) with both immunofluorescent and immunocytochemical staining methods (Figure S3A, B). Consistent with the colocalization of p-S65-Ub and TOMM20 (translocase of outer mitochondrial membrane 20), examination of cells with immunoelectron microscopy confirmed p-S65-Ub signals at the mitochondrial outer membrane (Figure S3C). Similarly, analyses of human post-mortem tissue stained with p-S65-Ub antibodies showed intense signals on mitochondrial outer membranes. This was seen in both cytoplasm (Figure 3(D), top) and processes of cells (Figure 3(D), bottom) indicated by the nearby neurofilament-like structure. Vacuolar-shaped GVBs were also labeled with p-S65-Ub, some of which contained spherical structures with double membranes that could represent mitochondria undergoing degradation (Figure 3(E)). In some neurons, distinctly labeled GVB structures were adjacent to paired helical filaments (Figure 3(F)). No immunolabeling was found in sections where the primary or secondary antibodies were omitted (data not shown). Taken together, the patterns seen for p-S65-Ub in light microscopy were confirmed by electron microscopy.
p-S65-Ub increases with age and disease, and enhanced levels in LBD brain are age- and Braak tangle stage-dependent
To assess mitoQC during aging, intracellular p-S65-Ub inclusions in the above 5 brain regions vulnerable to Lewy-related pathology were quantified in controls and analyzed based on age at death, concurrent neurofibrillary (Braak tangle stage) and amyloid (Thal amyloid phase) pathology (Figure 4(A–C) and Figure S4A-C). Of note, Braak tangle stages I-III are seen with normal aging and do not necessarily imply dementia [31]. Significant correlations of p-S65-Ub levels with age were observed in the hippocampus, amygdala, and nbM. In the same brain regions, p-S65-Ub levels were also significantly correlated with Braak tangle stage. No age or Braak tangle stage effect was found in the SN or putamen within the control cohort and no effect of Thal amyloid phase was detected in any of the 5 brain regions studied.
Figure 4.
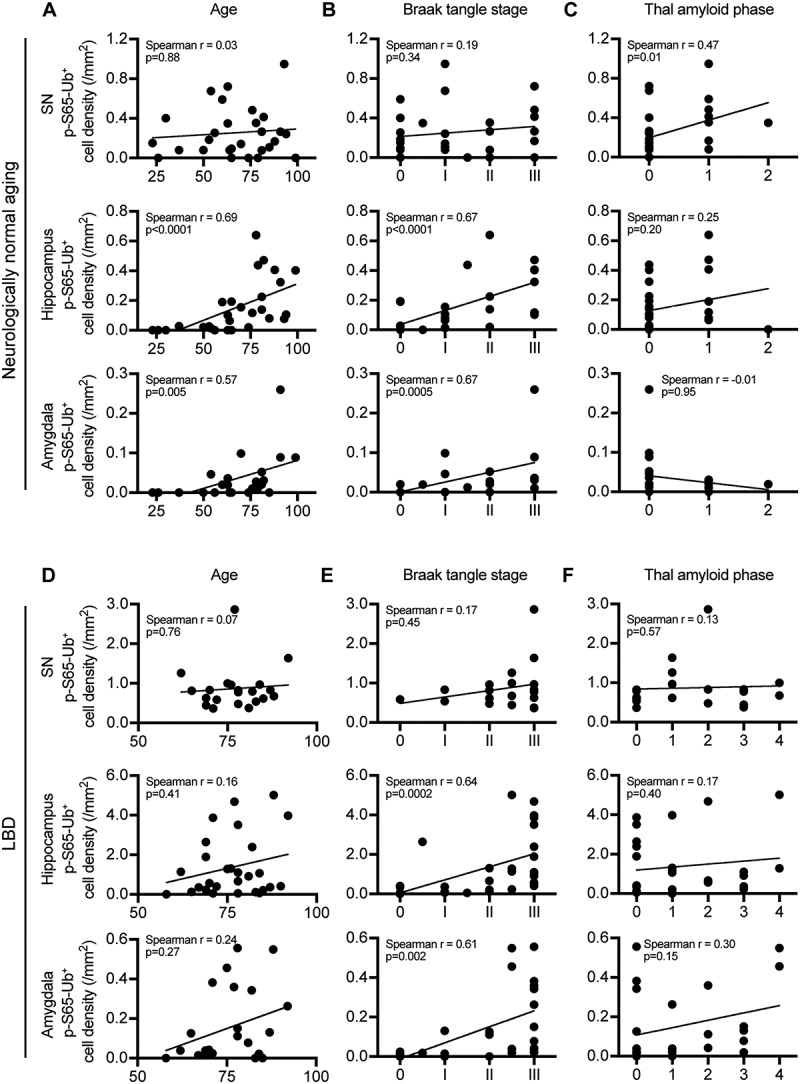
p-S65-Ub cell density increases with advanced aging and Braak tangle stage in select brain regions in neurologically normal controls or in LBD. SN, hippocampus and amygdala from neurologically normal controls and LBD cases were stained with p-S65-Ub antibody. Correlations of p-S65-Ub cell density with age at death, Braak tangle stage and Thal amyloid phase was examined within either normal aging (A–C) or LBD cohort (D–F) (Spearman’s test of correlation, significance level: p < 0.01). In controls, n = 28 for SN, n = 28 for hippocampus, n = 22 for amygdala. Putamen and nbM are shown in Figure S4A-C. In LBD, n = 21 for SN, n = 28 for hippocampus, n = 24 for amygdala. Putamen and nbM are shown in Figure S4D-F.
Next, we quantified p-S65-Ub in LBD cases (Figure 4(D–F) and Figure S4D-F) with a significant loss of neuromelanin-containing cells in the SN (p < 0.0001) (Figure S5). Significant correlations of p-S65-Ub levels with Braak tangle stage were observed in the hippocampus and amygdala, but not with age at death or Thal amyloid phase in any of the 5 brain regions. We then examined correlation of p-S65-Ub with 2 scoring systems for SNCA pathology [7,32,33]. While no difference was observed among LBD subtypes (brainstem predominant [BLBD], transitional [TLBD], and diffuse neocortical [DLBD]), a trend of correlation with Braak PD stage was detected in the amygdala (p = 0.01) (Figure S6). No correlations were found with any of the other demographic characteristics including primary presenting clinical feature (parkinsonism versus cognitive decline), disease duration or age at onset (Figure S7).
Due to the significant effects of age and Braak tangle stage on p-S65-Ub signal, controls and LBD brains were compared to determine the disease-specific effect (Table 1 and Figure 5(A))). Younger controls were excluded to obtain age-matched cohorts (see Table S1 for details). Overall, LBD brains had significantly higher p-S65-Ub levels in the SN and hippocampus (p < 0.0001 and p = 0.001, respectively), which remained significant (p ≤ 0.001) after adjusting individually for age, sex and Braak tangle stage. Of note, adjustment for amyloid-β plaques was not possible due to lack of higher Thal amyloid phase in controls. While, increased p-S65-Ub levels in the amygdala and nbM were age driven, the effects in the SN were disease driven. Hippocampal p-S65-Ub levels were associated with both, but showed a stronger association with disease status (control vs. LBD) than age as examined by multivariable linear regression models (Age: p = 0.016, β: 0.012; Disease status: p = 0.0001, β: 0.62).
In the SN, LBD cases had significantly increased p-S65-Ub levels compared to controls in both younger (56–75 years old, p = 0.004) and older (76–94 years old, p = 0.0002) age groups (Figure 5(B)). Such an increase was also observed in the hippocampus in the younger group (p = 0.004). At more advanced Braak tangle stage (Figure 5(C)) LBD cases also showed significantly elevated p-S65-Ub levels in the SN and hippocampus (p = 0.0002 and p = 0.0004, respectively). Regardless of age or Braak tangle stage, no significant differences in p-S65-Ub levels were found between controls and LBD cases in the amygdala, putamen or nbM.
Figure 5.
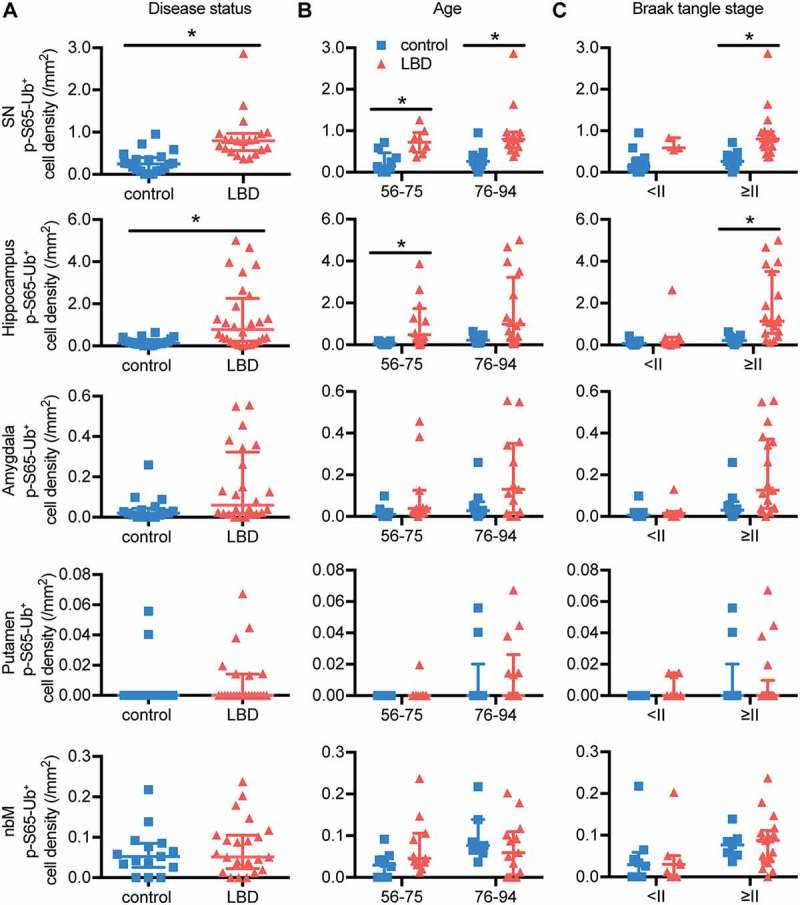
LBD brains have significantly higher p-S65-Ub levels compared to age-matched controls in SN and hippocampus. (A) p-S65-Ub-positive cell density from all 5 brain regions was compared between age-matched controls and LBD cases (Wilcoxon rank sum test, *p < 0.01). p-S65-Ub-positive cell density was also compared within (B) different age and (C) Braak tangle stage groups (Wilcoxon rank sum test followed by adjustment with Bonferroni correction, *p < 0.005). In controls, n = 20 for SN, n = 20 for hippocampus, n = 16 for amygdala, n = 16 for putamen, and n = 15 for nbM. In LBD, n = 21 for SN, n = 28 for hippocampus, n = 24 for amygdala, putamen, and nbM. All data are presented in scatter-plots showing the median and interquartile range.
Interactions between p-S65-Ub, SNCA and MAPT in the LBD cohort
To investigate the interaction of p-S65-Ub with other PD pathological markers, we quantified LB and tangle density by immunostaining for correlations and used super-resolution microscopy for colocalization studies. As expected, LBD cases had significantly more LBs than age-matched controls in all 5 brain regions (p < 0.0001) (Figure 6(A) and Figure S8A). A significant correlation of p-S65-Ub and LB density was identified in the amygdala (Figure 6(B)). Interestingly, in the SN from LBD cases, super-resolution microscopy revealed clear colocalization of SNCA immunoreactivity (LB509) with p-S65-Ub-positive beaded neurites, but only partial colocalization with somatic p-S65-Ub granules (Figure 6(C)). Neither of the p-S65-Ub positive structures colocalized with phosphorylated SNCA (p-S129-SNCA), the pathological form of SNCA (Figure S8C). Moreover, we observed a negative association of p-S65-Ub levels and LB pathology progression. Cells with small SNCA aggregates (pre-LBs) had abundant p-S65-Ub granules, whereas cells with dense-core (more mature) LBs only occasionally had surrounding p-S65-Ub granules that closely decorated the surface of the LB (Figure 6(D)). Strikingly, no p-S65-Ub signals were found in cells with typical brainstem type mature LBs.
Figure 6.
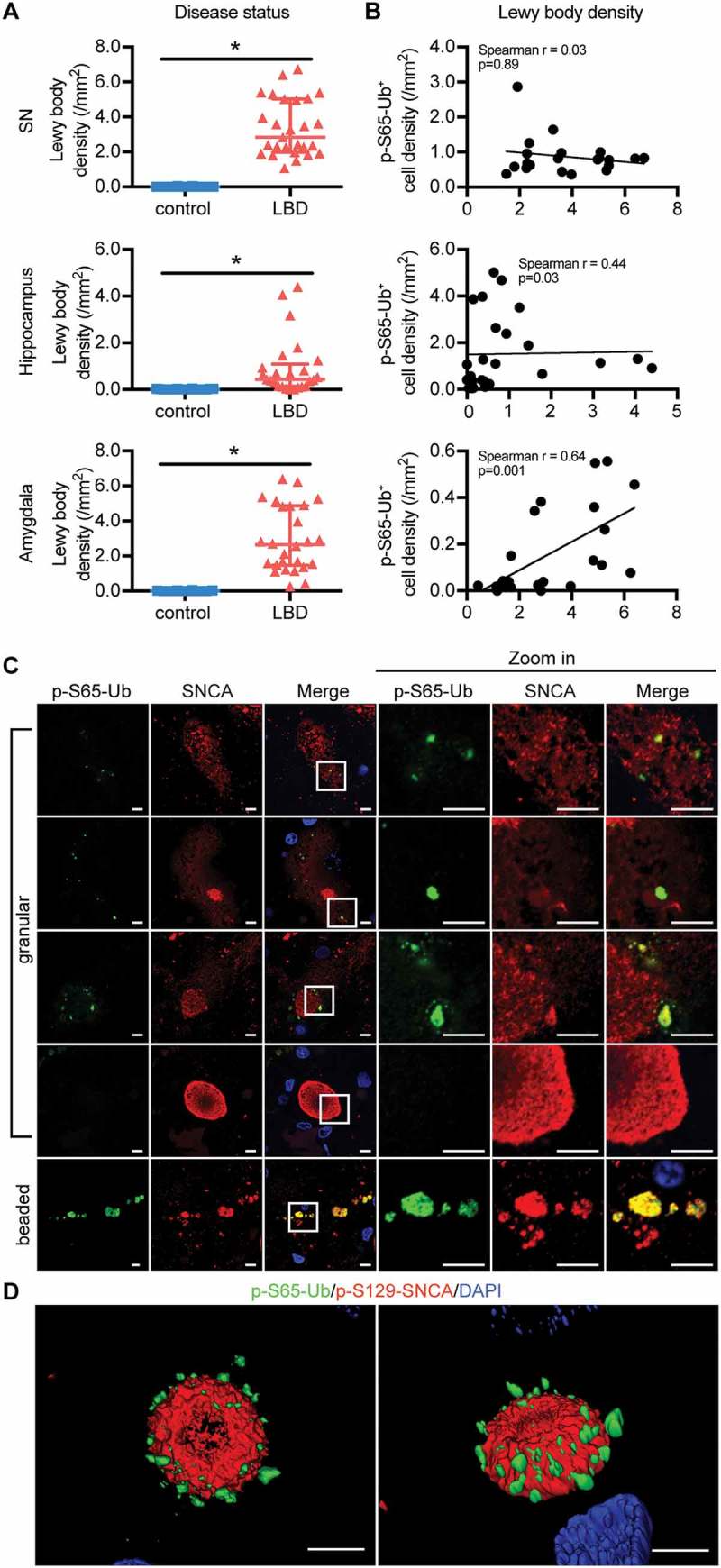
Correlation and interaction of p-S65-Ub and SNCA pathology in LBD brains. (A) Lewy body density in SN, hippocampus and amygdala was compared between age-matched normal controls and LBD cases (Wilcoxon rank sum test, *p < 0.01). Data are presented in scatter-plots showing the median and interquartile range. (B) The correlation between p-S65-Ub and Lewy body density in LBD brains was examined, and significant correlations were observed in the amygdala (Spearman’s test of correlation, significance level: p < 0.01). In controls, n = 20 for SN, n = 20 for hippocampus, n = 16 for amygdala. In LBD, n = 21 for SN, n = 28 for hippocampus, n = 24 for amygdala. Putamen and nbM are shown in Figure S7A, B. (C) SN sections of LBD cases were double immunostained with p-S65-Ub and SNCA antibodies. Cells with different stages of SNCA-positive aggregations are shown. A magnified image of the boxed area is shown to the right. p-S65-Ub (green) colocalizes with SNCA (red) more in beaded neurites than in granules in soma. p-S65-Ub levels appear to decline with maturation of SNCA into LBs. For double labeling with p-S129-SNCA, see Figure S7C. (D) The spatial relationship of p-S65-Ub- and p-S129-SNCA-positive LB is shown in 3D surface rendering. Scale bars: 5 µm.
In addition to SNCA pathology, LBD cases also had more neurofibrillary tangles in the nbM and a trend in the hippocampus (p = 0.0006 and p = 0.01, respectively) compared to controls (Figure 7(A) and Figure S9A). Significant correlations of p-S65-Ub and tangle levels were found in both hippocampus and amygdala of LBD cases (Figure 7(B)). Super-resolution microscopy on hippocampal sections from LBD cases showed that p-S65-Ub-positive GVBs partially overlapped with phosphorylated MAPT staining for both MAPT antibodies marking early (CP13; p-S202-MAPT) and late (PHF-1; p-S396- and p-S404-MAPT) MAPT pathology (Figure 7(C) and Figure S9C) [29]. Of note, some p-S65-Ub-positive granules appeared trapped in large pretangle filamentous networks (Figure 7(D) and Figure S9D). In general, p-S65-Ub-positive GVBs were abundant in cells with pre-tangles, but were less frequent in neurons with mature tangles. In summary, the degree of colocalization of p-S65-Ub, a marker of mitoQC, with SNCA and MAPT varied depending on progression of LB and tangle formation.
Figure 7.
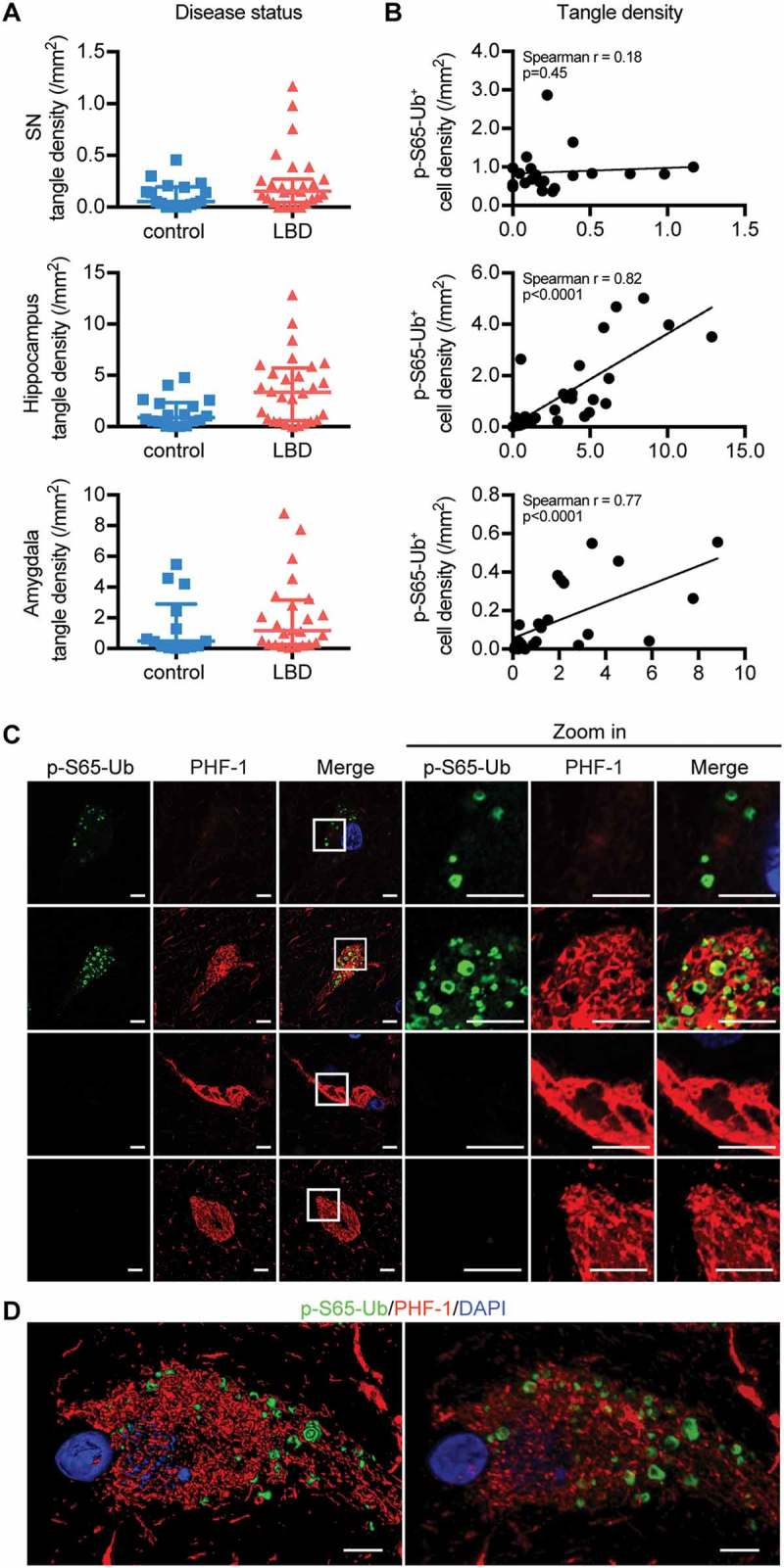
Correlation and interaction of p-S65-Ub and MAPT pathology in LBD brains. (A) tangle density in SN, hippocampus and amygdala is compared between age-matched normal controls and LBD cases (Wilcoxon rank sum test, *p < 0.01). Data are presented in scatter-plots with the median and interquartile range. (B) The correlation of p-S65-Ub with tangle density was examined in LBD, and significant correlations were observed in the hippocampus and amygdala (Spearman’s test of correlation, significance level: p < 0.01). In controls, n = 20 for SN, n = 20 for hippocampus, n = 16 for amygdala. In LBD, n = 21 for SN, n = 28 for hippocampus, n = 24 for amygdala. Putamen and nbM are shown in Figure S8A, B. (C) Hippocampal sections of LBD cases were double stained with p-S65-Ub and p-MAPT antibodies. A magnified image of the boxed area is shown to the right. Intracellular p-S65-Ub GVBs (green) are embedded in the p-MAPT network (red) labeled by late stage p-MAPT marker (PHF-1). For early stage p-MAPT marker CP13, see Figure S8C, D. p-S65-Ub levels appear to increase with maturation of tangles, but disappear in late-stage tangles. (D) The spatial relationship of p-S65-Ub and PHF-1 is shown in 3D surface (left) and maximum (right) rendering. Scale bars: 5 µm.
Discussion
Despite enormous insights gained into the molecular mechanisms of PINK1-PRKN-directed mitoQC over the past few years [17], truly disease-relevant data remain scarce. Studies in primary neurons or in model organisms have been hampered by the lack of sensitive tools and the dynamics of mitophagy, where flux through the degradation arm and counterbalancing mitochondrial biogenesis challenge unambiguous interpretation. Of note, an earlier study had already described altered mitophagy in post-mortem LBD brains [34], long before the discovery of the PINK1-PRKN pathway [35–38]. Here, we confirm the disease relevance and fill an important gap between the initial pathological observation and an extensive body of predominantly cellular/biochemical studies. We characterized p-S65-Ub morphology by high-resolution microscopy, and objectively quantified levels of this novel mitophagy tag in neurologically normal controls and LBD cases across 5 different brain regions. Our findings indicate that alterations of mitoQC occur during normal aging and with progression of PD. Moreover, LBD brains accumulate p-S65-Ub already at a relatively early age in both the SN and hippocampus. Significant correlations of p-S65-Ub with SNCA and MAPT pathology and their co-existence at pre-mature stages were discovered, suggesting a potential early interaction. The disease-dependent increase of p-S65-Ub in the SN and its distinct staining pattern with SNCA provide further evidence linking mitoQC dysfunction with PD pathology. Interestingly, we found close interaction between p-S65-Ub, GVBs and p-MAPT in the hippocampus, revealing a region-specific crosstalk between mitochondrial damage/mitophagy and tauopathy in PD and possibly also other neurodegenerative disorders [39].
While it remains uncertain what exactly triggers activation of PINK1-PRKN-dependent mitoQC in vivo, organelle damage caused by mitochondrial DNA mutations, misfolded mitochondrial proteins and proteotoxic stress may be highly relevant factors [40–42]. In addition, it has become clear that the process is likely very localized and restricted to a few mitochondria or even submitochondrial regions at a time, as opposed to wholesale mitophagy observed in cell lines upon mitochondrial depolarization. p-S65-Ub is a dynamic degradation signal that is rapidly turned over under physiological conditions and therefore detectable only at low levels. Here we identified distinct p-S65-Ub-immunopositive structures that differed in terms of their distributions across brain regions and their colocalization with mitochondrial, autophagic, and/or lysosomal markers. PINK1-PRKN-dependent mitoQC was reported to occur locally in distal axons as well as in the neuronal cell bodies upon retrograde transport in different neuronal cultures [43,44]. However, studies with recently developed fluorescent mitophagy reporter mice indicate that general turnover occurs mostly in neuronal somata in vivo [45]. Observed co-labeling of p-S65-Ub-positive beaded structures with mitochondrial, but not lysosomal markers, may indicate labeled mitochondria en route to lysosomal degradation in the soma. In this context it is important to point out that several other mitophagy pathways have been identified that may operate in parallel to or may cross-talk with PINK1-PRKN-p-S65-Ub signaling. These include different receptor- [46] and lipid-mediated [47,48] pathways that recruit phagophores independent of ubiquitin via direct binding to MAP1LC3A family members.
Unbiased quantification in neurologically normal brains revealed significantly increased p-S65-Ub-positive cells in aged individuals compared to young controls. This is likely indicative of a pathological situation, possibly resulting from greater mitochondrial damage, a general decrease of autophagic/lysosomal clearance, or a combination of both. This is further consistent with a progressive decline in mitochondrial quality and function seen in aging and age-related diseases [49] and a reduced flux of general mitophagy in reporter mice with increased age [50]. The PINK1-PRKN pathway appears to protect organisms from aging-induced mitochondrial dysfunction, mitigate mitochondria DNA mutation accumulation, and foster longevity in animal models [51,52]. Our data provides additional evidence for mitoQC dysfunction in aging human brain, and suggest the hippocampus, amygdala and nbM as particularly vulnerable regions. When comparing age-matched controls and LBD cases, p-S65-Ub granules were prominently increased in the SN across all age groups. During the preparation of this manuscript, a similar punctate p-S65-Ub signal was reported in 1 PD case using the commercial p-S65-Ub antibody though with less resolution of specific colocalization [53]. While PINK1 and PRKN are broadly neuroprotective [54], dopamine neurons appear particularly vulnerable to their dysfunction, possibly due to their high firing rate, Ca2+ load, extreme arborization, and the chemical properties of dopamine itself [14,55]. Our data provide further evidence for considerable mitoQC alterations in dopamine neurons, which could contribute to their susceptibility towards neuronal death in PD. The increased p-S65-Ub level in the hippocampus of LBD cases may contribute to non-motor symptoms, such as cognitive impairment, in LBD.
Both SNCA and MAPT cause impairments of several steps of the mitoQC pathway, from induction of mitochondrial dysfunction to inhibition of autophagy/lysosomal clearance [24,39,56–58], thus possibly resulting in enhanced p-S65-Ub levels as shown in the current study. Impaired mitoQC also has reciprocal effects on SNCA aggregation and MAPT phosphorylation [57,58]. Several of the above mechanisms could work simultaneously to reduce overall neuroprotection of PINK1-PRKN-directed mitoQC, which might be overcome by PINK1 or PRKN overexpression [59,60]. In addition, the observation of more p-S65-Ub in cells with pre-mature SNCA or MAPT pathology in the SN or hippocampus, respectively, suggests that the brain region-specific crosstalk between synucleinopathy or tauopathy and mitoQC might happen early on. The exact mechanisms, distinct SNCA and p-MAPT species and their particular functions or dysfunctions that could have an impact upon mitoQC at different steps need to be studied in the future.
Taken together, increased p-S65-Ub levels demonstrate age- and disease-specific alterations of mitoQC and point to the potential convergence of multiple pathways. Going forward p-S65-Ub may be used as a biomarker and quantitative trait to identify modifiers and determine the relative contributions of mitochondrial dysfunctions, impaired neuronal transport and/or defective proteostasis to the pathogenesis of disease.
Material and methods
Study subjects
A total of 28 neurologically normal controls, 28 sporadic LBD and 9 PINK1 and PRKN mutation carriers were obtained from the Mayo Clinic Florida Brain Bank. All brains were examined in a systematic and standardized manner and obtained between 1998 and 2015. Detailed characteristics of these 3 cohorts are summarized in Tables S1-S3, respectively. All subjects are non-Hispanic Caucasians of European descent. Available clinical information included besides basic demographics, age at onset, disease duration and initial symptoms. The clinical diagnosis of the majority of the LBD cases was PD and PDD, with one case of DLB. The LBD cohort was systematically screened with no mutations in known familial PD genes identified and was further subtyped with SNCA staining into BLBD (n = 7), TLBD (n = 17) and DLBD (n = 4) [7,32,61]. Additional neuropathological information was collected, including age at death, Braak PD stage for SNCA (0–6) and Braak tangle stage for MAPT (0-VI) as well as Thal amyloid phase for amyloid-β plaques (0–5) [33,62]. Cases with higher Braak tangle stages (> III) had been excluded to focus on PD- and not Alzheimer disease-related pathology, and intermediate categories were created for borderline cases. For analysis of low and medium Braak tangle stage groups, we collapsed cases with Braak tangle stage 0, 0-I, I and I-II as ‘< II’ group and cases with Braak tangle stage II, II-III and III as ‘≥ II’ group. For comparisons between controls and LBD cases and between controls, LBD and mutation carriers, we utilized different subgroups of controls and LBD cases that were similar with regard to age at death and Braak tangle stage (compare Tables S1 and S2). The Mayo Clinic brain bank for neurodegenerative disorders operates with approval of the Mayo Clinic Institutional Review Board. All brain samples are from autopsies performed after approval by the legal next-of-kin. Research on de-identified postmortem brain tissue is considered exempt from human subjects regulations by the Mayo Clinic Institutional Review Board.
Immunohistochemistry and image analysis
Sections from paraffin embedded post-mortem brain tissue were cut at a thickness of 5 microns and allowed to dry overnight in a 60°C oven. Following deparaffinization and rehydration, target retrieval was performed by steaming the sections for 30 min in deionized water. Immunostaining was performed with a Dako Autostainer using Envision Plus kit (Agilent, K4011 and K4007). Endogenous peroxidase was blocked for 5 min with 0.03% hydrogen peroxide (Agilent, K4011 and K4007). Sections were then treated with 5% normal goat serum (Invitrogen, 16210072) for 20 min. Subsequently, sections were incubated for 45 min at room temperature using primary antibodies against p-S65-Ub (in-house [25]; 1:1000; Millipore Sigma, ABS1513; 1:50), SNCA (Invitrogen, 180215; 1:3000), or p-S202-MAPT (monoclonal antibody from Peter Davies, Feinstein Institute, CP13; 1:1000). After incubation with primary antibody, sections were incubated in Envision-Plus rabbit- or mouse-labeled polymer HRP (Agilent, K4011 and K4007) for 30 min at room temperature. Peroxidase labeling was visualized with the chromogen solution 3,3ʹ-diaminobenzidine (Agilent, K4011 and K4007). The sections were then counterstained with Lerner 1 hematoxylin (Fisher Scientific, CS400-1D) and coverslipped with Cytoseal mounting medium (Thermo Scientific, 8310). For midbrain sections that contain neuromelanin-positive cells in the SN, peroxidase labeling was visualized with HighDef Blue (Enzo Life Sciences, ADI-950–151). Sections were coverslipped without counterstain using aqueous mounting medium (Thermo Scientific, TA-030-FM). After drying, all sections were scanned with an Aperio AT2 digital pathology scanner (Leica Biosystems, Wetzlar, Germany). Sections stained with p-S65-Ub, SNCA and p-S202-MAPT were traced and quantified using optimized Aperio Immunohistochemistry Image Analysis algorithms to deteermine the p-S65-Ub-positive cell count, LB count and tangle count. p-S65-Ub cell, LB and tangle density was calculated by dividing the positive cell count by the traced area.
Meso scale discovery electrochemiluminescence assays
To detect synthetic p-S65-Ub (custom-made at Thinkpeptides) [25] via Meso Scale Discovery assay, 96-well plates were coated with in-house or commercial (Millipore Sigma, ABS1513) p-S65-Ub antibodies at a final concentration of 3.08 µg/ml in carbonate-bicarbonate coating buffer (15 mM Na2CO3, 35 mM NaHCO3, pH 9.4) and incubated overnight at 4°C. Plates were washed 3 times with TBST (50 mM tris base, pH 7.4, 150 mM NaCl, 0.05% Tween-20 [Sigma-Aldrich, P1379]) blocked with 5% BSA (Boston BioProducts, P-753) in TBST for 2 h, shaking (350 rpm) at room temperature, and then washed again. To compare sensitivity of different p-S65-Ub antibodies, synthetic p-S65-Ub was serially diluted in 1% BSA in TBST to a final concentration of 0.78 ng to 50 ng. Unmodified recombinant Ub (Boston Biochem, U-100H) served as a negative control. Samples were added to the plate and incubated for 1 h at room temperature, shaking at 350 rpm. Plates were washed and detection antibody (Millipore Sigma, MAB1510) diluted 1:500 in 1% BSA TBST was added for 1 h. After 3 washes with TBST goat anti-mouse SULFO TAG detection antibody (Meso Scale Discovery, R32AC; 1: 500 in 1% BSA TBST) was added to each well and incubated with shaking for 1 h at room temperature. Plates were washed and 1x read buffer T with surfactant (Meso Scale Discovery, R92TC) was added to each well, followed by plate reading on the SECTOR Imager 2400A (Meso Scale Discovery, Rockville, MD, USA).
Cell culture, treatment and western blot
HeLa cells stably expressing PRKN [63] were grown in Dulbecco’s modified Eagle medium (Invitrogen, 11965118) supplemented with 10% fetal bovine serum (Biowest, S1620) at 37°C under humidified conditions in 5% CO2:air. Cells were seeded on 6-well plates and incubated with or without 10 µM CCCP (Sigma-Aldrich, C2759) in medium for 4 h.
After the treatment, cells were lysed in RIPA lysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 0.1% SDS, 0.5% deoxycholate [Sigma-Aldrich, D6750], 1% NP-40/Igepal [Sigma-Aldrich, I3021] containing protease inhibitor cocktail and phosphatase inhibitors [Sigma-Aldrich, 11697498001 and 04906837001]). Cell lysates were cleared for 15 min at 20817 rcf. Protein content was determined by BCA assay (Thermo Fisher, 23225). Cell lysates were diluted in Laemmli buffer (62.5 mM Tris, pH 6.8, 1.5% SDS, 8.33% glycerol, 1.5% β-Mercaptoethanol, 0.005% bromophenol blue) and boiled at 95°C for 5 min. Protein was subjected to SDS-PAGE using 8–16% Tris-Glycine gels (Invitrogen, EC60485BOX). Proteins were transferred onto polyvinylidene fluoride membranes (Millipore Sigma, IPVH00010). Post transfer, membranes were washed in TBST, blocked in 5% skim milk (Thermo Scientific, OXLP0031B) and incubated with primary antibodies against p-S65-Ub (in-house or Millipore Sigma, ABS1513) at 0.05, 0.25, 0.5 or 1 µg/ml using a multiscreen apparatus overnight at 4°C, followed by washing in TBST. Secondary HRP-conjugated antibodies diluted in 5% milk in TBST (Jackson ImmunoResearch, 111–035-003; 1:10,000) were added for 1 h at room temperature. Bands were visualized after washing in TBST using Immobilon Western Chemiluminescent HRP Substrate (Millipore Sigma, WBKLS0500) on a Fuji LAS-3000 (Fujifilm Life Science USA, Stamford, CT, USA) system using a program with pre-set exposure times. For comparing different antibodies in the detection of peptide, N-terminal biotinylated p-S65-Ub (custom-made at Thinkpeptides) [25] was run on the gel at serial dilution from 10–500 ng and detected with 0.75 µg/ml p-S65-Ub antibodies (in-house [25] or Millipore Sigma, ABS1513). The N-terminal biotinylated non-modified ubiquitin (Boston Biochem, UB-560) was run on the same gel as negative control and HRP-coupled streptavidin (Pierce, 21,130) was used as a loading control.
High-content imaging
To quantify the p-S65-Ub, automated high-content imaging was employed as recently described [25]. In brief, cells were seeded in 96-well imaging plates (Fisher Scientific, 08772225) and allowed to attach overnight. Cells were then treated for 0, 2, 4, or 8 h with 10 µM CCCP. Cells were washed once in PBS (Boston BioProducts, BM-220), fixed for 10 min in 4% paraformaldehyde (Sigma-Aldrich, 441244) and permeabilized with 1% Triton X-100 (Fisher Scientific, BP151) in PBS. Cells were stained using different concentrations of p-S65-Ub antibodies (0.1 and 0.5 µg/ml) (in-house [25] or Millipore Sigma, ABS1513) and with Hoechst 33342 (Invitrogen, H21492l; 1:5000). Plates were imaged on a BD Pathway 855 (BD Biosciences, San Jose, CA, USA) with a 20x objective using a 2 × 2 montage (no gaps) with laser autofocus every second frame. Raw images were processed using the built-in AttoVision V1.6 software. Regions of interest were defined as nucleus and cytoplasm using the built-in ‘RING-2 outputs’ segmentation for the Hoechst channel after applying a shading algorithm. Background signal at 0 h was subtracted for each antibody and values were normalized to the highest value (8 h CCCP in-house antibody).
Immunostaining of cells
Cells were seeded on glass coverslips coated with poly-D-lysine hydrobromide (Sigma-Aldrich, P6407) and incubated with or without 10 µM CCCP in medium for 4 h. After fixation with 4% paraformaldehyde, cells were permeabilized with 0.05% Triton X-100 in PBS and blocked with 10% normal goat serum. For immunofluorescence, cells were incubated with primary antibodies against p-S65-Ub (in-house; 1:260) [25] and TOMM20 (Santa Cruz Biotechnology, sc-17764; 1:50), followed by incubation with secondary antibodies (Invitrogen, A-11034 and A-11004; 1:1000). Nuclei were stained with Hoechst 33342 diluted 1:5,000. Coverslips were mounted onto microscope slides using fluorescence mounting medium (Agilent, S302380). For immunocytochemistry, cells were incubated with primary antibody against p-S65-Ub (in-house; 1:500) [25] and then in Envision-Plus rabbit-labeled polymer HRP (Agilent, K4011). Peroxidase labeling was visualized with 3,3ʹ-diaminobenzidine (Agilent, K4011). Cells on coverslips were then counterstained with Lerner 1 hematoxylin mounted with Cytoseal mounting medium after dehydration and clearing.
Immunofluorescence and imaging of human brain tissue
Following target retrieval, human brain sections were blocked with serum-free protein block (Agilent, X0909) for 1 h at room temperature and incubated in primary antibodies against p-S65-Ub (in-house; 1:500) [25], PPIF (Abcam, ab110324; 1:500), CTSD (Millipore Sigma, IM03; 1:1000), CSNK1D (Santa Cruz Biotechnology, sc-55553; 1:50), SNCA (Invitrogen, 180215; 1:100), p-S129-SNCA (Wako Chemicals USA, 015–25191; 1:3000), p-S202-MAPT (monoclonal antibody from Peter Davies, Feinstein Institute, CP13; 1:250) or p-S396- and p-S404-MAPT (monoclonal antibody from Peter Davies, PHF-1; 1:500) diluted with antibody diluent with background reducing components (Agilent, S302283) at 4°C overnight in a humidity chamber. The next day sections were incubated with secondary antibodies (Invitrogen, A-11034 and A-11004; 1:1000) and DAPI (Sigma-Aldrich, D9542; 1:1000) at room temperature for 1.5 h. Autofluorescence signal was quenched using 3% Sudan black (SPI Supplies, 02560-AB) in 70% MeOH (Pharmco-Aaper, AAP-1031) for 2 min before coverslipping slides using fluorescence mounting medium. Super-resolution confocal (Airyscan) fluorescence images were taken with a Zeiss observer LSM 880 microscope equipped with 405-, 488-, 543-, and 633-nm lasers (Zeiss, Oberkochen, Germany).
Pre-embedding immunoelectron microscopy
Vibratome sections (40 μm) of formalin-fixed human brains were processed for immunohistochemistry using 3,3ʹ-diaminobenzidine as chromogen. The areas with immunolabeled structures were dissected, fixed in 2.5% glutaraldehyde (Electron Microscopy Science, 16020)-0.1 M cacodylate buffer (Electron Microscopy Science, 11652), post-fixed in 2% aqueous osmium tetroxide (Electron Microscopy Science, 19150), en block stained in 2% uranyl acetate (Electron Microscopy Science, 22400) in 50% EtOH (Pharmco-Aaper, 241ACS200CSGL), dehydrated in EtOH and propylene oxide (Electron Microscopy Science, 20401), then infiltrated and embedded in Epon 812 (Polysciences, Inc., 08791). Thin sections were collected on formvar-coated grids (Electron Microscopy Science, FF100-Cu) and examined without counter staining in a Philips 208S electron microscope, fitted with a Gatan 831 Orius CCD digital camera (Gatan, Pleasanton, CA, USA). Digital micrographs were processed with Adobe Photoshop CS5 software.
Statistical analysis
Given that most p-S65-Ub measures were not normally distributed, we used non-parametric tests for all analyses. Specifically, a Wilcoxon rank sum test was used for comparisons of p-S65-Ub measures between 2 groups, whereas a Kruskal-Wallis test was used for comparisons involving more than 2 groups. Additionally, for comparisons of p-S65-Ub measures between normal and LBD groups, in order to adjust for any potential confounding influences of age at death, sex, and Braak tangle stage, we used a van Elteren stratified Wilcoxon rank sum test adjusting individually for these variables [64]. Spearman’s test of correlation was used to assess the association between p-S65-Ub and demographic information or other biomarkers in each brain region. In order to account for the fact that all statistical tests involving p-S65-Ub were performed using measures in 5 different brain regions, we considered p-values of 0.01 or lower as statistically significant after applying a Bonferroni correction for multiple testing. Statistical analyses were performed using SAS (SAS Institute, Inc; version 9.2) and GraphPad Prism (GraphPad Software; version 7).
Funding Statement
This work was supported by multiple funding resources. W.S. is partially supported by the National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (NINDS) [R01 NS085070], the Michael J. Fox Foundation for Parkinson Research and the Foundation for Mitochondrial Medicine, Mayo Clinic Neuroscience Focused Research Team (NFRT) Award, Mayo Clinic Center for Individualized Medicine (CIM), Center for Regenerative Medicine (CRM) and the Center for Biomedical Discovery (CBD), the Marriott Family Foundation, and a Gerstner Family Foundation Career Development Award. Mayo Clinic Florida is a Morris K. Udall Parkinson Disease Research Center of Excellence [NIH/NINDS P50 NS072187 to Z.K.W., O.A.R. and D.W.D.]. X. H. is supported by a pilot grant from the Mayo Clinic Alzheimer Disease Research Center (ADRC) and a fellowship awarded by the American Parkinson Disease Association (APDA). F.C.F. is the recipient of a fellowship from the Younkin Scholar Program and has received support by the APDA. P.T. is supported by an Allergan Medical Educational Grant, a Jaye F. and Betty F. Dyer Foundation Fellowship in progressive supranuclear palsy research, and a Max Kade Foundation postdoctoral fellowship. O.A.R. is supported by NIH/NINDS [R01 NS078086]. Z.K.W. and O.A.R are supported by the Mayo Clinic Center for Individualized Medicine (CIM) and by the Mayo Clinic Center for Regenerative Medicine (CRM). D.W.D. is supported by the Mangurian Foundation for Lewy body research. O.A.R. and D.W.D. are supported by NIH/NINDS [U54 NS100693].
Acknowledgments
We thank the patients and their families who made this study possible. We thank Anneliese Hill for assistance with antibody testing.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental data
Supplemental data can be accessed here.
References
- [1].Lees AJ, Hardy J, Revesz T.. Parkinson’s disease. Lancet. 2009;373(9680):2055–2066. [DOI] [PubMed] [Google Scholar]
- [2].Van Den Eeden SK, Tanner CM, Bernstein AL, et al. Incidence of Parkinson’s disease: variation by age, gender, and race/ethnicity. Am J Epidemiol. 2003;157(11):1015–1022. [DOI] [PubMed] [Google Scholar]
- [3].Langston JW. The Parkinson’s complex: parkinsonism is just the tip of the iceberg. Ann Neurol. 2006;59(4):591–596. [DOI] [PubMed] [Google Scholar]
- [4].Gibb WR, Lees AJ. Anatomy, pigmentation, ventral and dorsal subpopulations of the substantia nigra, and differential cell death in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1991;54(5):388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dickson DW, Braak H, Duda JE, et al. Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol. 2009;8(12):1150–1157. [DOI] [PubMed] [Google Scholar]
- [6].Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord. 2007;22(12):1689–1707. [DOI] [PubMed] [Google Scholar]
- [7].McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology. 2017;89(1):88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wills J, Jones J, Haggerty T, et al. Elevated tauopathy and alpha-synuclein pathology in postmortem Parkinson’s disease brains with and without dementia. Exp Neurol. 2010;225(1):210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Giasson BI, Forman MS, Higuchi M, et al. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300(5619):636–640. [DOI] [PubMed] [Google Scholar]
- [10].Simon-Sanchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41(12):1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bekris LM, Mata IF, Zabetian CP. The genetics of Parkinson disease. J Geriatr Psychiatry Neurol. 2010;23(4):228–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Brooks J, Ding J, Simon-Sanchez J, et al. Parkin and PINK1 mutations in early-onset Parkinson’s disease: comprehensive screening in publicly available cases and control. J Med Genet. 2009;46(6):375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bolam JP, Pissadaki EK. Living on the edge with too many mouths to feed: why dopamine neurons die. Mov Disord. 2012;27(12):1478–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Haddad D, Nakamura K. Understanding the susceptibility of dopamine neurons to mitochondrial stressors in Parkinson’s disease. FEBS Lett. 2015;589(24 Pt A):3702–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Scarffe LA, Stevens DA, Dawson VL, et al. Parkin and PINK1: much more than mitophagy. Trends Neurosci. 2014;37(6):315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mouton-Liger F, Jacoupy M, Corvol JC, et al. PINK1/Parkin-dependent mitochondrial surveillance: from pleiotropy to Parkinson’s disease. Front Mol Neurosci. 2017;10:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Truban D, Hou X, Caulfield TR, et al. PINK1, Parkin, and mitochondrial quality control: what can we learn about Parkinson’s disease pathobiology? J Parkinsons Dis. 2017;7(1):13–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Heo JM, Ordureau A, Paulo JA, et al. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell. 2015;60(1):7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Puschmann A, Fiesel FC, Caulfield TR, et al. Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain. 2017;140(Pt1):98–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dawson TM, Dawson VL. Parkin plays a role in sporadic Parkinson’s disease. Neurodegener Dis. 2014;13(2–3):69–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bose A, Beal MF. Mitochondrial dysfunction in Parkinson’s disease. J Neurochem. 2016;139(Suppl 1):216–231. [DOI] [PubMed] [Google Scholar]
- [23].Dehay B, Martinez-Vicente M, Caldwell GA, et al. Lysosomal impairment in Parkinson’s disease. Mov Disord. 2013;28(6):725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ryan BJ, Hoek S, Fon EA, et al. Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci. 2015;40(4):200–210. [DOI] [PubMed] [Google Scholar]
- [25].Fiesel FC, Ando M, Hudec R, et al. (Patho-)physiological relevance of PINK1-dependent ubiquitin phosphorylation. EMBO Rep. 2015;16(9):1114–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fiesel FC, Springer W. Disease relevance of phosphorylated ubiquitin (p-S65-Ub). Autophagy. 2015;11(11):2125–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dickson DW, Ksiezak-Reding H, Davies P, et al. A monoclonal antibody that recognizes a phosphorylated epitope in Alzheimer neurofibrillary tangles, neurofilaments and tau proteins immunostains granulovacuolar degeneration. Acta Neuropathol. 1987;73(3):254–258. [DOI] [PubMed] [Google Scholar]
- [28].Kohler C. Granulovacuolar degeneration: a neurodegenerative change that accompanies tau pathology. Acta Neuropathologica. 2016;132(3):339–359. [DOI] [PubMed] [Google Scholar]
- [29].Dickson DW, Liu WK, Kress Y, et al. Phosphorylated tau immunoreactivity of granulovacuolar bodies (GVB) of Alzheimer’s disease: localization of two amino terminal tau epitopes in GVB. Acta Neuropathol. 1993;85(5):463–470. [DOI] [PubMed] [Google Scholar]
- [30].Funk KE, Mrak RE, Kuret J. Granulovacuolar degeneration (GVD) bodies of Alzheimer’s disease (AD) resemble late-stage autophagic organelles. Neuropathol Appl Neurobiol. 2011;37(3):295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Braak H, Thal DR, Ghebremedhin E, et al. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70(11):960–969. [DOI] [PubMed] [Google Scholar]
- [32].Kosaka K, Yoshimura M, Ikeda K, et al. Diffuse type of Lewy body disease: progressive dementia with abundant cortical Lewy bodies and senile changes of varying degree–a new disease? Clin Neuropathol. 1984;3(5):185–192. [PubMed] [Google Scholar]
- [33].Braak H, Del Tredici K, Rub U, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211. [DOI] [PubMed] [Google Scholar]
- [34].Zhu JH, Guo F, Shelburne J, et al. Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol. 2003;13(4):473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Geisler S, Holmstrom KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12(2):119–131. [DOI] [PubMed] [Google Scholar]
- [36].Narendra DP, Jin SM, Tanaka A, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8(1):e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Matsuda N, Sato S, Shiba K, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189(2):211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Vives-Bauza C, Zhou C, Huang Y, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA. 2010;107(1):378–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Cai Q, Tammineni P. Alterations in mitochondrial quality control in Alzheimer’s disease. Front Cell Neurosci. 2016;10:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jensen MB, Jasper H. Mitochondrial proteostasis in the control of aging and longevity. Cell Metab. 2014;20(2):214–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jin SM, Youle RJ. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy. 2013;9(11):1750–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Fiesel FC, James ED, Hudec R, et al. Mitochondrial targeted HSP90 inhibitor Gamitrinib-TPP (G-TPP) induces PINK1/Parkin-dependent mitophagy. Oncotarget. 2017;8(63):106233–106248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Cai Q, Zakaria HM, Simone A, et al. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol. 2012;22(6):545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ashrafi G, Schlehe JS, LaVoie MJ, et al. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol. 2014;206(5):655–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].McWilliams TG, Prescott AR, Allen GF, et al. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J Cell Biol. 2016;214(3):333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zimmermann M, Reichert AS. How to get rid of mitochondria: crosstalk and regulation of multiple mitophagy pathways. Biol Chem. 2017;399(1):29–45. [DOI] [PubMed] [Google Scholar]
- [47].Chu CT, Ji J, Dagda RK, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15(10):1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sentelle RD, Senkal CE, Jiang W, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol. 2012;8(10):831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sun N, Youle RJ, Finkel T. The mitochondrial basis of aging. Mol Cell. 2016;61(5):654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sun N, Yun J, Liu J, et al. Measuring in vivo mitophagy. Mol Cell. 2015;60(4):685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Rana A, Rera M, Walker DW. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc Natl Acad Sci USA. 2013;110(21):8638–8643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature. 2015;521(7553):525–528. [DOI] [PubMed] [Google Scholar]
- [53].Shiba-Fukushima K, Ishikawa KI, Inoshita T, et al. Evidence that phosphorylated ubiquitin signaling is involved in the etiology of Parkinson’s disease. Hum Mol Genet. 2017. [DOI] [PubMed] [Google Scholar]
- [54].Feany MB, Pallanck LJ. Parkin: a multipurpose neuroprotective agent? Neuron. 2003;38(1):13–16. [DOI] [PubMed] [Google Scholar]
- [55].Sulzer D, Surmeier DJ. Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov Disord. 2013;28(6):715–724. [DOI] [PubMed] [Google Scholar]
- [56].Xilouri M, Brekk OR, Stefanis L. Autophagy and alpha-synuclein: relevance to Parkinson’s disease and related synucleopathies. Mov Disord. 2016;31(2):178–192. [DOI] [PubMed] [Google Scholar]
- [57].Moors T, Paciotti S, Chiasserini D, et al. Lysosomal dysfunction and alpha-synuclein aggregation in Parkinson’s disease: diagnostic links. Mov Disord. 2016;31(6):791–801. [DOI] [PubMed] [Google Scholar]
- [58].Kerr JS, Adriaanse BA, Greig NH, et al. Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci. 2017;40(3):151–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kamp F, Exner N, Lutz AK, et al. Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010;29(20):3571–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hu Y, Li XC, Wang ZH, et al. Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget. 2016;7(14):17356–17368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Fujishiro H, Ferman TJ, Boeve BF, et al. Validation of the neuropathologic criteria of the third consortium for dementia with Lewy bodies for prospectively diagnosed cases. J Neuropathol Exp Neurol. 2008;67(7):649–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Murray ME, Lowe VJ, Graff-Radford NR, et al. Clinicopathologic and 11C-Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer’s disease spectrum. Brain. 2015;138(Pt 5):1370–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Fiesel FC, Moussaud-Lamodiere EL, Ando M, et al. A specific subset of E2 ubiquitin-conjugating enzymes regulate Parkin activation and mitophagy differently. J Cell Sci. 2014;127(Pt 16):3488–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].van Elteren PH. On the combination of independent two sample tests of Wilcoxon. Bull Inst Int Stat. 1960;37:351–361. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.