

Abstract
Simple, fast, and precise counting of viable bacteria is fundamental to a variety of microbiological applications such as food quality monitoring and clinical diagnosis. To this end, agar plating, microscopy, and emerging microfluidic devices for single bacteria detection have provided useful means for counting viable bacteria, but they also have their limitations ranging from complexity, time, and inaccuracy. We present herein our new method RAPiD (Resazurin-Amplified Picoarray Detection) for addressing this important problem. In RAPiD, we employ vacuum-assisted sample loading and oil-driven sample digitization to stochastically confine single bacteria in Picoarray, a microfluidic device with picoliter-sized isolation chambers (picochambers), in <30 s with only a few minutes of hands-on time. We add AlamarBlue, a resazurin-based fluorescent dye for bacterial growth, in our assay to accelerate the detection of “microcolonies” proliferated from single bacteria within picochambers. Detecting fluorescence in picochambers as an amplified surrogate for bacterial cells allows us to count hundreds of microcolonies with a single image taken via wide-field fluorescence microscopy. We have also expanded our method to practically test multiple titrations from a single bacterial sample in parallel. Using this expanded “multi-RAPiD” strategy, we can quantify viable cells in E. coli and S. aureus samples with precision in ~3 h, illustrating RAPiD as a promising new method for counting viable bacteria for microbiological applications.
Graphical Abstract
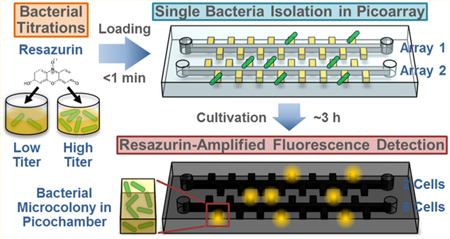
Precise counting of viable bacteria is critical to diverse microbiological applications ranging from ensuring food safety1—3 to diagnosing bacterial infections (e.g., urinary tract infections).4—6 To this end, plating bacteria of interest in agar plates is the most established and widely used method because it uses only a simple setup and ensures that only viable bacterial cells grown into colony forming units (CFU) are enumerated. The drawback to agar plating, however, is the lengthy culturing time that can take up to several days, which delays proper inspection of food quality and timely diagnosis of infections. Microscopy presents an alternative technique that can directly and rapidly count the total number of bacteria. But the high magnification required to observe individual cells restricts the field of view under the microscope and therefore limits its use to samples with relatively high bacterial concentrations. Moreover, the small field of view also necessitates that multiple images are typically taken for counting, which is cumbersome and can still lead to inaccurate and imprecise enumeration.7—12 As such, tools that can marry the simplicity of plating and the speed of microscopy to achieve simple, fast, and precise counting of viable bacteria can be extremely helpful for various microbiological applications.
Recently, “single-cell” microfluidic devices that can isolate individual bacterial cells in nanoliter or subnanoliter volumes and then cultivate the isolated bacteria have emerged as enabling tools for microbiology.13—15 Such single-cell resolution is well suited for fast and precise counting of viable bacteria. Among these devices, those utilizing microfluidic droplets16—24 have been the most prevalent, mainly because they can reliably isolate single bacteria at high throughputs based on stochastic confinement.16,25 In addition, droplet-based devices have been routinely coupled with fluorescent bacteria indicator dyes19,21,26 including resazurin16,22 for rapid detection of single bacterial cells. As the fluorescent molecules can diffuse throughout the droplets, such diffusible fluorescent signals provide an amplified surrogate for detecting bacteria, thus improving the detection speed and sensitivity.22 Despite these advantages, microfluidic droplet devices require skilled personnel and delicate flow control. Moreover, although microscopic imaging of droplets is commonly performed, unwanted merging or destabilization of droplets often occurs when they are transferred between droplet generation devices to incubation tubes and then to imaging chambers.27—29 Microscopic imaging of droplets can also be obscured by the leakage of bacterial staining dye molecules between neighboring droplets and into the surrounding oil phase.21,26,28,30
Alternatively, single bacteria have been isolated and cultivated in microfluidic arrays of spots,31,32 channels,33—39 and chambers.40—48 These array-based devices present practical advantages over droplet-based devices because they allow direct detection via microscopy without the need of transferring droplets or the risk of droplet merging and destabilization. However, because the majority of these array-based devices40—46 are designed for studying the physiology, growth, and replication of individual bacterial cells over time, they rely on submicron trapping structures such as narrow constrictions or shallow chambers to ensure a sufficient number of single bacteria can be captured for the experiments. Such tiny traps can complicate device fabrication and render loading of bacteria into devices tedious and time-consuming (e.g., air-bubble-based cell loading45). Moreover, in these devices, individual traps are often connected to a feeding channel for replenishing culture broth;40—47 such “semi-isolated” designs cannot prevent fluorescent molecules from diffusing to neighboring traps and are therefore incompatible with resazurin-amplified fluorescence detection. Finally, in a recently reported array-based device48 that stochastically encapsulates and fully isolates single bacteria, detection is achieved via bacterial fluorescent proteins and therefore may take up to 24 h for enumerating single bacteria in a sample. Consequently, no array-based devices to date have leveraged resazurin-amplified fluorescence detection as a surrogate for improving the speed and sensitivity of bacteria detection.
In response, we present RAPiD (Resazurin-Amplified Picoarray Detection), a simple, fast, and precise method for counting viable bacteria. In RAPiD, bacteria are first stochastically isolated via a simple process in our Picoarray, a microfluidic device with arrays of picoliter-sized isolation chambers (picochambers). We employ AlamarBlue49,50 (a resazurin-based commercial dye) to achieve resazurin-amplified fluorescence detection of bacteria in RAPiD. In the presence of viable and proliferating bacteria, resazurin molecules in AlamarBlue can be reduced by intracellular electron receptors (e.g., NADH and FADH) into strongly fluorescent resorufin molecules,49,50 thus providing an effective surrogate for detecting the “microcolonies” that have proliferated from individual bacteria within picochambers. This strategy not only accelerates the detection of individual microcolonies but also provides a convenient means for counting hundreds of such microcolonies in the Picoarray device in parallel via wide-field fluorescence microscopy. Moreover, we have designed our Picoarray to harbor multiple identical but independent units for enumerating multiple titrations of a bacterial sample with RAPiD in parallel, thereby enhancing the precision for quantifying the bacteria sample. For initial demonstration, we use our “multi-RAPiD” strategy to quantify the viable cells of E. coli and S. aureus samples with precision in only 3 h.
EXPERIMENTAL SECTION
Picoarray Device Fabrication.
Our Picoarray devices were fabricated based on the polydimethylsiloxane (PDMS) soft lithography technique.51 A single photomask with all fluidic features of Picoarray, including inlets, outlets, branch channels, connecting channels, and picochambers was designed in L-Edit v16.0 (Tanner EDA, Monrovia, CA) and printed onto a high-quality transparency (CAD/Art Services, Inc., Bandon, OR). Using the photomask, master molds were microfabricated onto 4-in. silicon wafers via a single-step, alignment-free, standard SU8 photolithography process. PDMS fluidic layers were replicated from the master molds using 10:1 (w/w) SYLGARD 184 (Dow Corning, Midland, MI). Fluidic access holes were punched into PDMS fluidic layers with sharpened needles (McMaster-Carr, Elmhurst, IL). Blank, ~100-μm-thick PDMS cover layers were separately fabricated using 15:1 (w/w) SYLGARD 184. Each Picoarray was then assembled with a PDMS fluidic layer, a PDMS cover layer, and a glass cover layer (43 mm × 50 mm, thickness = ~0.19 to 0.25 mm; Ted Pella, Inc., Redding, CA). The PDMS cover layer was first bonded to the glass layer following O2 plasma treatment (45 s, 30 W, 500 mTorr). Finally, the exposed surface of the PDMS cover layer and the PDMS fluidic layer were treated with O2 plasma and bonded. All completely assembled Picoarray devices were stored at 80 °C for at least 24 h before use. Procedures for fabricating Picoarray devices are provided in more detail in the Supporting Information.
Bacteria Loading, Digitization, Incubation, and Detection in Picoarray.
Prior to all experiments, devices were placed in a vacuum chamber for >2 h. Samples containing cation-adjusted Mueller-Hinton broth (Becton, Dickinson and Company, Franklin Lakes, NJ), AlamarBlue (DAL1025, Thermo Fisher Scientific, Waltham, MA), and either E. coli (ATCC 25922, ATCC, Manassas, VA) or S. aureus (ATCC 29213, ATCC, Manassas, VA) at predetermined concentrations were prepared before each experiment, drawn into blunt-end needles, and immediately loaded into Picoarray devices via vacuum-assisted sample loading52—54 without external pressure. The sample-containing blunt-end needles were kept in the devices for 2—3 min to ensure that all picochambers were filled with samples. Subsequently, 100 cSt silicone oil (378364, Sigma-Aldrich, St. Louis, MO) carried by blunt-end needles and Tygon tubings was injected into the devices at 10 psi. The silicone oil flowed through branch channels and out of device outlets in <1 min and, in the process, completely separated picochambers and digitized bacteria within picochambers in the devices. The pressure was reduced to 1 psi, and device outlets were sealed with sealing plugs. The devices were incubated at 37 °C on a flat-bed PCR machine (ProFlex 2× flat PCR System, Thermo Fisher Scientific, Waltham, MA) to facilitate bacterial replication within picochambers. Fluorescence detection of picochamber arrays in Picoarray devices was performed with a fluorescence microscope (BX51, Olympus, Japan) equipped with a mercury lamp, an AlamarBlue-compatible filter cube (49305; Chroma Technology Corp., Bellows Falls, VT), a 1.25× magnification objective lens (Olympus PlanAPO N 1.25 × /0.04 NA), and a digital CCD camera (Retiga EXi Fast 1394, QImaging, Canada). Detailed experimental procedures for preparing, loading, digitizing, incubating, and detecting bacteria in Picoarray devices are explained extensively in the Supporting Information.
Data Acquisition and Analysis.
Data acquisition from fluorescence images and downstream data analysis were performed using ImageJ55 (1.48v), Microsoft Excel 2010, and Origin 8.0. More specifically, fluorescence intensities in the picochambers and background fluorescence were measured via ImageJ. Background-normalization of fluorescence intensities and histogram analysis were executed in Excel. Fluorescence intensity thresholds for differentiating negative from positive picochambers were calculated via Origin. Finally, all data shown in this work were plotted with Origin. Complete data acquisition and analysis procedures are detailed in the Supporting Information.
RESULTS AND DISCUSSION
Overview of RAPiD.
RAPiD is based on isolating single bacteria in Picoarray and leveraging resazurin-based fluorescence to accelerate and parallelize the detection of bacterial microcolonies in the device. We employ soft lithography to fabricate Picoarray out of air-permeable PDMS; this material allows us to create a vacuum within the device prior to performing our assay (Figure 1, pre-assay step). Consequently, when we begin the assay by loading the bacterial sample containing Mueller-Hinton broth and AlamarBlue, the sample is assisted by the vacuumed device to flow autonomously into the picochambers in ~15 s without external pressure (Figure 1, step 1). Multiple titrations of a given bacterial sample can be loaded into independent array units, where decreasing titrations proportionally decrease the number of picochambers within each array unit to be occupied by single cells, in accordance with Poisson distribution. For separating picochambers and isolating single bacteria, we then inject partitioning oil into the device, which flows through the channels and displaces the bacterial sample in the channel in ~10 s but does not flow into the picochambers occupied by the incompressible sample (Figure 1, step 2). This represents a much simpler technique for achieving stochastic confinement of single bacterial cells than previous microfluidic-based techniques. While the device is incubated at 37 °C, each bacterial cell confined within a picochamber replicates into essentially a microcolony that reduces resazurin into fluorescent resorufin (Figure 1, step 3). As fluorescent resorufin molecules diffuse throughout the entire picochamber, the effect of bacterial growth is amplified to the entire picochamber. As such, by detecting strong fluorescence within picochambers as a surrogate of detecting bacteria, we accelerate the detection of individual microcolonies to 3 h. Moreover, we can reliably count microscale colonies in the Picoarray in parallel via wide-field fluorescence microscopy in a single image, thus significantly simplifying the counting process (Figure 1, step 4).
Figure 1.
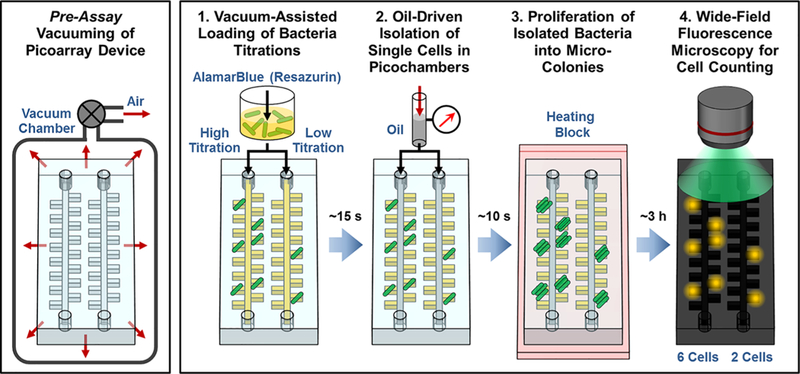
Overview of RAPiD (Resazurin-Amplified Picoarray Detection) for Simple, Fast, and Precise Counting of Viable Bacteria. An air-permeable, PDMS-based Picoarray device is kept under a vacuum prior to the assay (Pre-Assay Step). The vacuum created within the device enables autonomous loading (i.e., without external pressure) of bacterial samples containing Mueller-Hinton broth and AlamarBlue (a resazurin-based dye for bacterial growth detection) into the device in ~15 s. In the device, each picoliter-sized isolation chamber (picochamber) stochastically confines either 0 or 1 cell based on Poisson distribution (step 1). The bacterial sample can be divided into multiple titrations and tested in our multiplexed device, which can improve precision for quantifying the bacterial sample concentration. Partitioning oil is subsequently injected into the device, where it flows through the channels and separates the picochambers in ~10 s, thereby isolating single bacterial cells within picochambers (step 2). As each isolated cell replicates into a “microcolony” (step 3), resazurin molecules are reduced by bacterial metabolites into fluorescent resorufin molecules that diffuse throughout the entire picochamber. As a result, wide-field fluorescence microscopy can be employed to detect and count microscale colonies in the Picoarray in parallel after only ~3 h (step 4).
Picoarray Device and Operation.
The simple sample loading and digitization workflow, coupled with resazurin-amplified detection, allows us to simplify the design of our Picoarray device. As such, our device features a planar architecture of uniform height (~50 μm) and involves neither micrometer or sub-micrometer trapping structures nor micro-fluidic valves. Each Picoarray device in the current iteration houses five identical yet independent units, with each unit having a symmetrical, single-inlet—single-outlet design. The inlets and outlets of the device are designed to interface with blunt-end needles and syringes, which function as de facto sample holders (Figure 2A). Within each unit, there are 14 100-μm-wide branch channels that split at the inlet, travel parallel to each other throughout the unit, and reconnect at the outlet (Figure 2A, inset i). Fifty pairs of 100 μm (L) × 50 μm (W) chambers are connected to each branch channel via 50 μm (L) × 25 μm (W) connecting channels, forming an array of 1400 chambers at the center of each unit, equivalent to 7000 chambers per device. At a height of 50 μm, each chamber is 250 pL in volume and hence referred to as a picochamber; each Picoarray therefore has the capacity to analyze 1.75 μL of samples. A large pitch of 100 μm is spaced between adjacent picochambers to prevent diffusion of fluorescence molecules and artifacts of fluorescence imaging from interfering with counting individual picochambers (Figure 2A, inset ii). This modular design would allow us to easily scale up the number of picochambers and array units for widening the dynamic range and expanding to a number of testing conditions, respectively.
Figure 2.
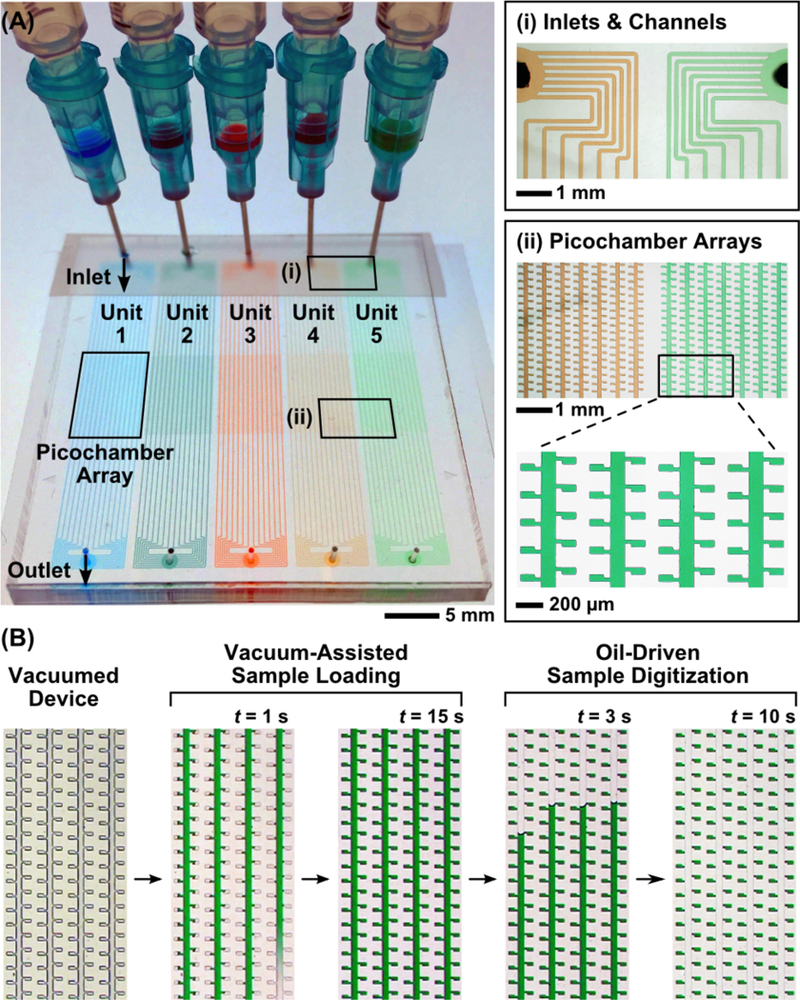
Simple design and rapid operation of Picoarray device. (A) Picoarray features five independent units with identically symmetrical, single-inlet−single-outlet design and conveniently interfaces with blunt-end needles and syringes, which function as sample holders. Within each unit, the inlet splits into 14 parallel branch channels (inset i) that reconnect at the outlet. Fifty pairs of chambers are connected to each branch channel via connecting channels (inset ii), forming an array of 1400 250-pL “picochambers” at the center of each unit. (B) Rapid, simple, and robust isolation of single bacteria in picochambers of our device is enabled by vacuum-assisted sample loading and oil-driven sample digitization. The device is vacuumed prior to sample loading. The vacuum created within the device allows the sample to autonomously flow through the branch channels and fill 100% of all picochambers in ∼15 s. Silicone oil is subsequently injected into the device. The oil flows through the branch channels but not into sample-filled picochambers, allowing each picochamber to be separated from its neighbors and the sample within to be digitized at 100% efficiency in ∼10 s.
Vacuum-assisted sample loading and oil-driven sample digitization within Picoarray can be completed in <30 s (Figure 2B). Moreover, the entire process requires only a few minutes of hands-on time. Prior to sample loading, we cover the inlets and outlets of the device with tape, which allows the negative pressure to build up within the device during vacuum. Once the device is removed from the vacuum, we immediately insert blunt-end needles and syringes holding food dyes as mock samples through the tape and into the inlets, whereupon the samples autonomously flow through the branch channels within seconds and fill 100% of all picochambers in ~15 s (Figure 2B and Video S-1). Importantly, all picochambers in all five units can be filled via vacuum-assisted sample loading in a sequential and timely manner. After removing the tape covering the outlets, we pressurize (10 psi) silicone oil into the device, where it flows through the branch channels to the outlet but not into sample-filled picochambers, thus partitioning each picochamber from its neighbors and achieving 100% sample digitization in ~10 s (Figure 2B and Video S-2). Notably, our planar design supports 100% sample digitization even though the branch channels, the connecting channels, and the picochambers all have the same height, unlike complex, multiheight designs reported in the literature.56 Finally, we decrease the pressure applied to the partitioning oil to 1 psi and seal the outlets with epoxy-filled needle plugs, which prevents bacteria from escaping the device during cultivation and detection.
The user-friendliness of Picoarray and its operation allows untrained users to adopt our method with ease. As a demonstration, we shipped several Picoarray devices to Stanford University School of Medicine and asked a collaborator who has no microfluidic expertise or prior experience with our device to replicate our method. After only walking through the experimental procedures via video conferencing, our collaborator successfully achieved vacuum-assisted sample loading and oil-driven sample digitization (Figure S-1). These results clearly demonstrate the robustness, reproducibility, and accessibility of our method to an inexperienced user.
Stochastic Confinement and Fluorescent Detection of Bacteria in Picochambers.
Single bacterial cells are reliably stochastically confined, cultivated, and detected via fluorescence in our Picoarray. To demonstrate, we loaded 8 × 105 CFU/mL of a previously quantified E. coli stock (ATCC 25922) with Mueller-Hinton broth and AlamarBlue into our device. This concentration results in a mean occupancy (λ) of 0.2 in our picochamber array. That is, ~20% of the picochambers were expected to be occupied by a single bacterial cell and then detected with strong fluorescence. We incubated the device at 37 °C for 3 h before we qualitatively estimated the number of strongly fluorescent picochambers. We indeed observed ~20% of picochambers with strong fluorescence dispersed stochastically within our observation area, which matches our expectation and strongly supports that single bacteria are stochastically confined in accordance to Poisson distribution (Figure 3A). We only detected fluorescence in picochambers and dark background in branch channels, which indicates that resazurin and resorufin molecules are retained in picochambers by our silicone oil and that E. coli are indeed trapped in oil-partitioned picochambers. When observing the array under a bright field and without resazurin fluorescence, we could not differentiate any picochambers with or without E. coli (Figure S-2), which validates the importance of using resazurin-amplified fluorescence for detecting bacteria in picochambers. We next used bright-field microscopy at 400× magnification to detect bacteria within picochambers; we verified that picochambers with weak fluorescence were indeed empty, whereas picochambers with strong fluorescence indeed enclosed microcolonies of viable E. coli cells proliferated from a single isolated cell (Figure 3A). Finally, we confirm that, for those picochambers with strong fluorescence, the intensities indeed increased hourly from 0 to 3 h (Figure S-3), suggesting that E. coli cells isolated in those picochambers have been replicating and yielding fluorescence.
Figure 3.
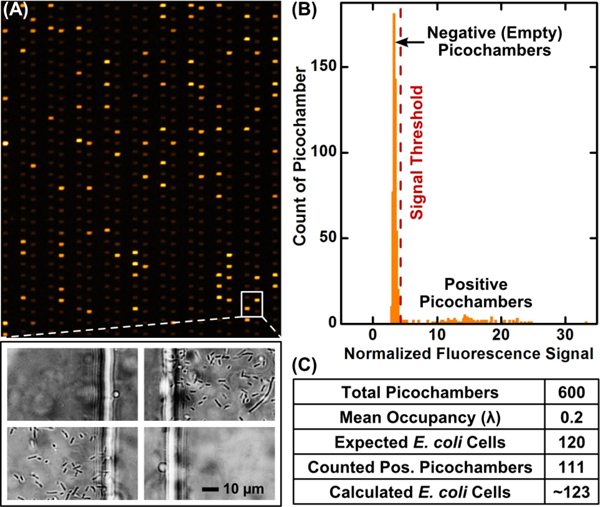
Stochastic confinement and fluorescent detection of bacterial microcolonies in picochambers. (A) An 8 × 105 CFU/mL E. coli sample with Mueller-Hinton broth and AlamarBlue is loaded into Picoarray such that ~20% of picochambers are expected to be occupied by a single bacterial cell (i.e., mean occupancy (λ) of 0.2). After incubation, ~20% of stochastically dispersed picochambers in an observation area of 600 picochambers yield strong fluorescence, suggesting that E. coli cells are indeed stochastically confined in the picochambers in accordance to Poisson distribution. The correlation between strong fluorescence and the presence of E. coli “micro-colonies” proliferated from a single cell within picochambers is verified via bright-field microscopy (400× magnification). (B) Background-normalized fluorescence signals of the 600 picochambers are plotted into a histogram, which shows a subpopulation of strongly fluorescent picochambers that have confined E. coli (i.e., positive) and a large subpopulation of weakly fluorescent picochambers that are empty. The two subpopulations are separated by a signal threshold (red dash line) determined via histogram analysis. (C) The number of E. coli cells in the sample is calculated from the positive picochambers, which may trap multiple cells. Thus, although 111 positive picochambers are counted, the number of E. coli cells in the sample is 123 (see main text for explanation).
For reliably counting fluorescent microcolonies in the Picoarray, we have established a strategy for consistently discarding picochambers that experience sample evaporation at 37 °C and may distort the fluorescence detection and counting process. In our device, picochambers located in the periphery of each array experience more significant sample evaporation but essentially protect and reduce sample evaporation in picochambers located near the center of the array (Figure S-4). We thus designate these peripheral picochambers as the “evaporation buffer” and exclude them from analysis. As a result, we only measure the fluorescence of the same 600 centrally located picochambers across all array units, devices, and experiments. Of note, we elect to analyze 600 picochambers because this number is on par with the upper limit of the number of colonies that can be counted on typical agar plates.
We have subsequently developed a quantitative analysis protocol for counting the number of E. coli in this sample and other bacteria in subsequent samples. In our analysis, we first use ImageJ to measure the fluorescence intensities of the 600 picochambers in our observation area. We then calculate the background-normalized fluorescence signals of these picochambers and plot the signals in a histogram (Figure 3B). The histogram shows a smaller subpopulation of strongly fluorescent picochambers that confined E. coli (i.e., positive) and a larger subpopulation of weakly fluorescent empty picochambers, with a ~ 5-fold difference in the fluorescence signals between the two populations. Histogram analysis via Gaussian peak fitting allows us to set the fluorescence signal threshold for counting the positive picochambers (red dash line, Figure 3B; see Supporting Information for detailed explanation). Subsequently, using a methodology analogous to digital PCR,57 we calculate the number of bacteria and account for positive picochambers occupied by multiple cells by correcting the counts based on the total number of picochambers (i.e., 600 in this example) and counted positive picochambers with
| (1) |
For example, based on our quantitative analysis, we counted 111 positive picochambers in our sample, which is equivalent to ~123 E. coli cells after correction (Figure 3C). The number of calculated E. coli cells using our device is thus close to the expected number of 120 cells. Finally, we quantified three E. coli samples at 8 × 105 CFU/mL with three different devices and counted 134.6 ± 21.5 E. coli cells after correction (Figure S-5); these results demonstrate the reproducibility of counting single bacterial cells for samples with the same concentration in our Picoarray devices.
Enumerating Bacteria via Multi-RAPiD.
Our strategy for improving the precision in quantifying bacteria in a sample is to enumerate multiple titrations of the bacterial sample with parallelized RAPiD in our Picoarray. This “multi-RAPiD” strategy is practical and analogous to plating multiple titrations of the bacteria sample in multiple plates, a standard technique in microbiology laboratories. To demonstrate the feasibility of multi-RAPiD, we first qualitatively verified that reducing the bacterial input concentrations resulted in fewer positive picochambers. For example, when we loaded and incubated 10-fold serial-dilutions of a new E. coli stock (ATCC 25922; 4.0 × 109 CFU/mL; estimated via triplicated plating; Figure S-6) with Mueller-Hinton broth and AlamarBlue in separate units of a Picoarray (at expected concentrations of 8.0 × 105, 8.0 × 104, and 8.0 × 105 CFU/mL), we indeed detected fewer picochambers with strong fluorescence that decreased by ~10-fold with each dilution of E. coli (Figure 4A).
Figure 4.
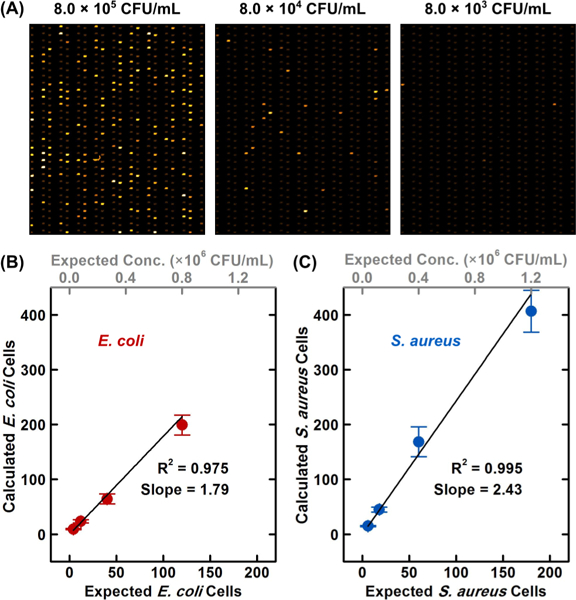
Quantification of bacteria via multi-RAPiD. Precision for quantifying bacterial samples can be improved by measuring multiple titrations of the same sample in parallel via RAPiD in the multiplexed Picoarray. (A) As a qualitative verification of this “multi-RAPiD” strategy, 10-fold decreasing titrations of an E. coli sample (e.g., 8.0 × 105, 8.0 × 104, and 8.0 × 103 CFU/mL) loaded into separate units of a Picoarray indeed yield ~ 10-fold decreasing numbers of positive picochambers. Multi-RAPiD is performed in triplicate (i.e., in three separate devices) to count viable cells from four titrations of three samples of (B) E. coli and (C) S. aureus (error bars represent standard deviations from triplicate experiments). Strong linearity between the calculated number of cells and the expected number of cells for both E. coli and S. aureus indicate that RAPiD can precisely enumerate bacteria from the four titrations, is applicable for both bacterial species, and is highly reproducible across three devices. The greater-than-1 slopes for both E. coli and S. aureus suggest that RAPiD enumerates more viable cells than plating. The factors for such overenumeration are subjects to ongoing investigation.
We subsequently validated multi-RAPiD by quantifying the new E. coli stock in three separate Picoarray devices. Here, we diluted each E. coli aliquot to expected concentrations of 8.0 × 105, 2.7 × 105, 8.0 × 104, and 2.7 × 104 CFU/mL with Mueller-Hinton broth and AlamarBlue. From these four input concentrations, we expected to detect approximately 120, 40, 12, and 4 cells (from 600 picochambers)—in a linearly decreasing trend—across four array units of each device. Following quantitative analysis (Figure S-7), we indeed observed a strongly linear trend from the calculated number of E. coli cells at these four expected concentrations (Figure 4B; R2 = 0.975). Notably, the average coefficient of variation (CV) of multi-RAPiD compared favorably to that of plating (12.2% to 33.5%; Figure S-8). These results indicate that RAPiD can precisely measure the four E. coli titrations and is highly reproducible across three devices. Finally, we note that the slope of the linear fit line between the calculated number of E. coli cells and the expected number of E. coli cells (Figure 4B; slope = 1.79) suggests that the concentration of viable E. coli measured via RAPiD is ~7.2 × 109 CFU/mL. The quantitative difference between RAPiD and plating may be that some E. coli cells are “viable” enough to form microcolonies in Picoarray but not “culturable” enough to form visible colonies in plates (i.e., viable but nonculturable58,59), though the exact causes are a subject of our ongoing investigation.
We have also used multi-RAPiD to enumerate an S. aureus stock in triplicate, showing the applicability of our method to Gram-positive bacteria. Here, we diluted each S. aureus aliquot (ATCC 29213; 1.8 × 1010 CFU/mL; estimated via triplicated plating; Figure S-9) to expected concentrations of 1.2 × 106, 4.0 × 105, 1.2 × 105, and 4.0 × 104 CFU/mL with Mueller-Hinton broth and AlamarBlue such that we expected to detect 180, 60, 18, and 6 cells (from 600 picochambers) in each Picoarray. Following quantitative analysis (Figure S-10), we observed a similarly strong linear trend from the calculated number of S. aureus cells at the four expected concentrations (Figure 4C; R2 = 0.995). The average CV from multi-RAPiD again outpaced that of plating (10.9% to 24.7%; Figure S-11). These results provide additional evidence for the reproducibility and the precision of our method. Finally, we note that the slope of the linear fit line between the calculated number of S. aureus cells and the expected number of S. aureus cells (Figure 4C; slope = 2.43) suggests not only that the concentration of viable S. aureus measured via RAPiD is ~4.4 × 1010 CFU/mL but also that RAPiD generally enumerates more cells than plating. In addition to viable but nonculturable cells, other causes for the overenumeration by RAPiD compared to plating are subjects of a systematic investigation.
CONCLUSIONS
We have developed Resazurin-Amplified Picoarray Detection, or RAPiD, a new method for simple, fast, and precise counting of viable bacteria. RAPiD is based on isolating single bacteria in our Picoarray devices with a simple and fast workflow and leveraging resazurin/AlamarBlue for accelerated and parallelized detection of bacterial microcolonies grown in the devices via wide-field fluorescence microscopy. Indeed, we have stochastically confined single E. coli and S. aureus cells in the picochambers of our Picoarray devices in <30 s via vacuum-assisted sample loading and oil-driven sample digitization, with only a few minutes of hands-on time. By incorporating AlamarBlue in our assay and detecting fluorescence in picochambers as an amplified surrogate for detecting bacteria, we have accelerated the detection of microcolonies that have replicated from single bacteria in 3 h, and we have also parallelized the detection of up to 600 microcolonies with a single, wide-field fluorescence image. Finally, using our multi-RAPiD quantification strategy, we have measured the concentrations of viable E. coli and S. aureus across four titrations of each bacterial sample in our Picoarray devices. These results illustrate that RAPiD presents a simple, fast, and precise method for counting viable bacteria.
We see several readily achievable improvements for RAPiD. For example, we can shorten the detection time and increase the dynamic range of Picoarray by reducing the size of the picochambers while maintaining a detectable resolution under wide-field fluorescence microscopy. Such volume reduction can enhance the signal-to-background ratio within the picochamber22 and increase the number of picochambers without enlarging the footprint of future iterations of our device. Consequently, we can widen the range of testing titrations beyond the 2 orders of magnitude demonstrated in this work, allowing us to tackle applications that require quantification of unknown bacterial sample concentrations. We also envision integrating RAPiD with automated counting methods60,61 to enumerate bacterial cells after exposure to antibacterials, which may accelerate antibiotic susceptibility testing and new compound screening. Finally, if necessary, Picoarray devices can also be disassembled so that viable bacteria can be collected after counting. With these improvements and further development, we believe RAPiD can become a useful alternative to agar plating and microscopy for a wide array of microbiological applications.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dong Jin Shin, Christine O’Keefe, Sarah Friedrich, and Pornpat Athamanolap for assay development, device operation, and helpful discussions during this project. This work is financially supported by the National Institutes of Health (R01AI117032 and R01AI137272), the National Science Foundation (1159771 and 1033744), and Burroughs Wellcome Fund (1017497). K.H. is financially supported through a Hartwell Postdoctoral Fellowship.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.8b02096.
Experimental Section including Bacteria Stock and Storage, Photomask Design and Printing, Master Mold Microfabrication, Picoarray Device Fabrication, Bacterial Sample Preparation, Measurement of Bacterial Sample Concentrations via Plating, Sample Loading and Digitization, Stochastic Confinement of Single Bacteria in Picochambers, Incubation and Fluorescence Detection, Image and Data Analysis, and Derivation of Equation 1 to Calculate Bacteria Counts; Supporting Figures including Picoarray Operation by Untrained OffSite User, Validation of Resazurin-Amplified Fluorescence Detection, Confirmation of Increasing Fluorescence in Picochambers, Sample Evaporation within Picochambers from Incubation, Triplicated Quantification of E. coli Samples at the Same Concentration, Measurement of E. coli Stock Concentration via Plating, Quantitative Analysis in Multi-RAPiD for E. coli, Coefficients of Variation from Plating and RAPiD for E. coli, Measurement of S. aureus Stock Concentration via Plating, Quantitative Analysis in Multi-RAPiD for S. aureus, and Coefficients of Variation from Plating and RAPiD for S. aureus; Supporting Videos including Sample Loading in Picoarray and Sample Digitization in Picoarray (ZIP)
REFERENCES
- (1).Rodriguez-Lazaro D; Lombard B; Smith H; Rzezutka A; D’Agostino M; Helmuth R; Schroeter A; Malorny B; Miko A; Guerra B; Davison J; Kobilinsky A; Hernandez M; Bertheau Y; Cook N Trends Food Sci. Technol. 2007, 18, 306—319. [Google Scholar]
- (2).Jasson V; Jacxsens L; Luning P; Rajkovic A; Uyttendaele M Food Microbiol. 2010, 27, 710—730. [DOI] [PubMed] [Google Scholar]
- (3).Sohier D; Pavan S; Riou A; Combrisson J; Postollec F Front. Microbiol. 2014, 5, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Wilson ML; Gaido L Clin. Infect. Dis. 2004, 38, 1150—1158. [DOI] [PubMed] [Google Scholar]
- (5).Schmiemann G; Kniehl E; Gebhardt K; Matejczyk MM; Hummers-Pradier E Dtsch. Arztebl. Int. 2010, 107, 361—376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Davenport M; Mach KE; Shortliffe LMD; Banaei N; Wang TH; Liao JC Nat. Rev. Urol. 2017, 14, 296—310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kirchman D; Sigda J; Kapuscinski R; Mitchell R Appl. Environ. Microbiol. 1982, 44, 376—382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Roszak DB; Colwell RR Appl. Environ. Microbiol. 1987, 53, 2889—2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Seo EY; Ahn TS; Zo YG Appl. Environ. Microbiol. 2010, 76, 1981—1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Johnson MB; Criss AK J. Visualized Exp. 2013, e50729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Davis CJ Microbiol. Methods 2014, 103, 9—17. [DOI] [PubMed] [Google Scholar]
- (12).Muthukrishnan T; Govender A; Dobretsov S; Abed RMM. J. Mar. Sci. Eng. 2017, 5, 4. [Google Scholar]
- (13).Weibel DB; DiLuzio WR; Whitesides GM Nat. Rev. Microbiol. 2007, 5, 209—218. [DOI] [PubMed] [Google Scholar]
- (14).Hol FJH; Dekker C Science 2014, 346, 1251821. [DOI] [PubMed] [Google Scholar]
- (15).Kaminski TS; Scheler O; Garstecki P Lab Chip 2016, 16, 2168—2187. [DOI] [PubMed] [Google Scholar]
- (16).Boedicker JQ; Li L; Kline TR; Ismagilov RF. Lab Chip 2008, 8, 1265—1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Boedicker JQ; Vincent ME; Ismagilov RF. Angew. Chem., Int. Ed. 2009, 48, 5908—5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Liu WS; Kim HJ; Lucchetta EM; Du WB; Ismagilov RF. Lab Chip 2009, 9, 2153—2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Najah M; Mayot E; Mahendra-Wijaya IP; Griffiths AD; Ladame S; Drevelle A Anal. Chem. 2013, 85, 9807—9814. [DOI] [PubMed] [Google Scholar]
- (20).Liu X; Painter RE; Enesa K; Holmes D; Whyte G; Garlisi CG; Monsma FJ; Rehak M; Craig FF; Smith CA. Lab Chip 2016, 16, 1636—1643. [DOI] [PubMed] [Google Scholar]
- (21).Scheler O; Kaminski TS; Ruszczak A; Garstecki P ACS Appl. Mater. Interfaces 2016, 8, 11318—11325. [DOI] [PubMed] [Google Scholar]
- (22).Kaushik AM; Hsieh K; Chen LB; Shin DJ; Liao JC; Wang TH Biosens. Bioelectron. 2017, 97, 260—266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lu H; Caen O; Vrignon J; Zonta E; El Harrak Z; Nizard P; Baret JC; Taly V Sci. Rep. 2017, 7, 1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kaushik AM; Hsieh K; Wang TH Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol. 2018, e1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Vincent ME; Liu WS; Haney EB; Ismagilov RF Chem. Soc. Rev. 2010, 39, 974—984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Courtois F; Olguin LF; Whyte G; Theberge AB; Huck WTS; Hollfelder F; Abell C Anal. Chem. 2009, 81, 3008—3016. [DOI] [PubMed] [Google Scholar]
- (27).Chen DLL; Li L; Reyes S; Adamson DN; Ismagilov RF Langmuir 2007, 23, 2255—2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Pompano RR; Liu WS; Du WB; Ismagilov RF. Annu. Rev. Anal. Chem. 2011, 4, 59—81. [DOI] [PubMed] [Google Scholar]
- (29).Debon AP; Wootton RCR; Elvira KS Biomicrofluidics 2015, 9, 024119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Chen YH; Wijaya Gani A; Tang SKY. Lab Chip 2012, 12, 5093—5103. [DOI] [PubMed] [Google Scholar]
- (31).Iino R; Hayama K; Amezawa H; Sakakihara S; Kim SH; Matsumono Y; Nishino K; Yamaguchi A; Noji H Lab Chip 2012, 12, 3923—3929. [DOI] [PubMed] [Google Scholar]
- (32).Iino R; Matsumoto Y; Nishino K; Yamaguchi A; Noji H Front. Microbiol. 2013, 4, 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Balaban NQ; Merrin J; Chait R; Kowalik L; Leibler S Science 2004, 305, 1622—1625. [DOI] [PubMed] [Google Scholar]
- (34).Mannik J; Driessen R; Galajda P; Keymer JE; Dekker C Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 14861—14866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Taniguchi Y; Choi PJ; Li GW; Chen HY; Babu M; Hearn J; Emili A; Xie XS Science 2010, 329, 533—538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wang P; Robert L; Pelletier J; Dang WL; Taddei F; Wright A; Jun S Curr. Biol. 2010, 20, 1099—1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Moffitt JR; Lee JB; Cluzel P Lab Chip 2012, 12, 1487—1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Lu Y; Gao J; Zhang DD; Gau V; Liao JC; Wong PK Anal. Chem. 2013, 85, 3971—3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Long ZC; Olliver A; Brambilla E; Sclavi B; Lagomarsino MC; Dorfman KD Analyst 2014, 139, 5254—5262. [DOI] [PubMed] [Google Scholar]
- (40).Grunberger A; Paczia N; Probst C; Schendzielorz G; Eggeling L; Noack S; Wiechert W; Kohlheyer D Lab Chip 2012, 12, 2060—2068. [DOI] [PubMed] [Google Scholar]
- (41).Gruenberger A; Probst C; Heyer A; Wiechert W; Frunzke J; Kohlheyer D J. Visualized Exp. 2013, e50560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Probst C; Grunberger A; Wiechert W; Kohlheyer D Micromachines 2013, 4, 357—369. [Google Scholar]
- (43).Probst C; Grunberger A; Wiechert W; Kohlheyer DJ Microbiol. Methods 2013, 95, 470—476. [DOI] [PubMed] [Google Scholar]
- (44).Dusny C; Grunberger A; Probst C; Wiechert W; Kohlheyer D; Schmid A Lab Chip 2015, 15, 1822—1834. [DOI] [PubMed] [Google Scholar]
- (45).Grunberger A; Probst C; Helfrich S; Nanda A; Stute B; Wiechert W; von Lieres E; Noh K; Frunzke J; Kohlheyer D Cytometry, Part A 2015, 87, 1101—1115. [DOI] [PubMed] [Google Scholar]
- (46).Probst C; Grunberger A; Braun N; Helfrich S; Noh K; Wiechert W; Kohlheyer D Anal. Methods 2015, 7, 91—98. [Google Scholar]
- (47).Amselem G; Guermonprez C; Drogue B; Michelin S; Baroud CN Lab Chip 2016, 16, 4200—4211. [DOI] [PubMed] [Google Scholar]
- (48).Baltekin Ö; Boucharin A; Tano E; Andersson DI; Elf J Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 9170—9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Page B; Page M; Noel C Int. J. Oncol. 1993, 3, 473—476. [PubMed] [Google Scholar]
- (50).Rampersad SN Sensors 2012, 12, 12347—12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Xia Y; Whitesides GM Annu. Rev. Mater. Sci. 1998, 28, 153—184. [Google Scholar]
- (52).Zhu QY; Gao YB; Yu BW; Ren H; Qiu L; Han SH; Jin W; Jin QH; Mu Y Lab Chip 2012, 12, 4755—4763. [DOI] [PubMed] [Google Scholar]
- (53).Ho JY; Cira NJ; Crooks JA; Baeza J; Weibel DB PLoS One 2012, 7, e41245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Xu LF; Lee H; Jetta D; Oh KW Lab Chip 2015, 15, 3962—3979. [DOI] [PubMed] [Google Scholar]
- (55).Schneider CA; Rasband WS; Eliceiri KW Nat. Methods 2012, 9, 671—675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Ottesen EA; Hong JW; Quake SR; Leadbetter JR Science 2006, 314, 1464—1467. [DOI] [PubMed] [Google Scholar]
- (57).Shen F; Du WB; Kreutz JE; Fok A; Ismagilov RF Lab Chip 2010, 10, 2666—2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Roszak DB; Colwell RR Appl. Environ. Microbiol. 1987, 53, 2889—2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Ding T; Suo Y; Xiang Q; Zhao X; Chen S; Ye X; Liu DJ Microbiol. Biotechnol. 2017, 27, 417—428. [DOI] [PubMed] [Google Scholar]
- (60).Brugger SD; Baumberger C; Jost M; Jenni W; Brugger U; Muhlemann K PLoS One 2012, 7, e33695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Chiang P-J; Tseng M-J; He Z-S; Li C-HJ Microbiol. Methods 2015, 108, 74—82. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.