

Abstract
Cubic metallacages were arranged into multidimensional (one-, two-, and three-dimensional) suprastructures via multistep assembly. Four new shape-controllable, hybrid metallacages with modified substituents and tunable electronic properties were prepared using dicarboxylate ligands with various substituents (sodium sulfonate, nitro, methoxyl, and amine), tetra-(4-pyridylphenyl) ethylene, and cis-(PEt3)2Pt(OTf)2. The as-prepared metallacages were used as building blocks for further assembly. Diverse suprastructures with tunable emissions (λmax from 451 to 519 nm) and various substituents (−SO3Na, −NO2, −OCH3, and −NH2) were prepared depending on the substituents and solvents used.
Graphical Abstract
INTRODUCTION
In nature, self-assembly enables the building of sophisticated suprastructures,1 from multicellular organisms to plant structures, through the interaction of a vast number of unrelated species. For example, three-dimensional (3D) structures in biology form naturally to provide essential functions in even the most basic forms of life. Inspired by assemblies in the natural world, diverse suprastructures with distinct spatial arrangements can be provided by artificial self-assembly.2–12 The use of molecular species,2–5 macromolecules,6–8 and polymers9 as building blocks combined with self-assembly approaches13–16 involving hydrogen-bonding,13 π−π stacking,14 solvophobicity15 and/or crystallization16 has led to a range of important advances. A wide variety of functional species with diverse dimensions,17–22 including micelles,17 vesicles,18 ribbons,19 films,20 fibers,21 and tubes,22 have been generated. For example, two-dimensional (2D) thin films have potential applications23 in various fields. 3D mesostructures24–27 have potential applications in biomedical devices,24 metamaterials,25 and energy storage.26 However, the realization of efficient life functions mostly depends on their multilevel structure and complexity.28 For example, most proteins in living organisms depend on primary, secondary, tertiary, and quaternary structures to perform intricate functions. Therefore, the introduction of further complexity through multistage processing and the enhancement of noncovalent interactions represent important goals.
Assembly is undergoing a transition from simple-component to multicomponent assembly and single-step to multistep processing.29,30 First, control over the size, shape, and composition of these building blocks has enabled the formation of suprastructures with substantial structuraldiversity.31–36 Furthermore, harnessing noncovalent interactions to create functional materials in a controlled manner will lead to a better understanding of the dynamics and the formation of complex self-organized patterns. The building blocks used for multiscale assembly37–40 include magnetite cubes, which have been used to fabricate fibers;13 micelles37 and amphiphilic cylindrical block comicelles,38 which have been employed to construct plates; and designed proteins, which have been used to prepare extracellular vesicles. However, for the construction of multiscale self-assemblies with controllable shapes and functions, the selection of a suitable protocol remains challenging.41 Coordination-driven self-assembly provides a bottom-up approach to construct various building blocks with controllable shape and size.42–44 For example, in previous reports, tetra-(4-pyridylphenyl)-ethylene (TPPE) with aggregation-induced emission (AIE)45–50 properties was anchored onto discrete metal− organic complexes (with controllable shape and size) through coordination-driven self-assembly.51 Besides that, delicate suprastructures such as molecular knot has been synthesized by David A. Leigh,52,53 complicated metal−organic complexes based assemblies has been reported by Anderson.54,55 Nitschke used metal−organic complexes56–58 for selective anion extraction,59 signal transduction,60 the triggering of multiple structural transformations,61 etc.
In this paper, we establish a facile route for coordination-driven modular assembly that allows the formation of a diverse range of suprastructures with tunable emissions and surface modifications. Coordination-driven metallacages were synthesized using TPPE (used as faces), dicarboxylate moieties (used as pillars), and cis-(PEt3)2Pt(OTf)2 (used as corners). These metallacages are composed of three modules that can be varied. The first module, TPPE, conveys AIE properties to the metallacages. The second module consists of the changeable dicarboxylate moieties, which confer specific characteristics to the metallacages. The third module is cis-(PEt3)2Pt(OTf)2, which is used to form the metallacage. Through the selective combination of these modules, a series of metallacages with adjustable optical properties were prepared, that were used as building blocks to construct suprastructures (one-dimensional (1D) fibers, 2D plates and 3D spheres) with tunable emissions and surface modifications.
RESULTS AND DISCUSSION
As shown in Figure 1a, multicomponent cages 1−4 were formed from TPPE (5) (Figure S1), cis-(PEt3)2Pt(OTf)2 (6) (Figures S2−S3), and dicarboxylate ligands with various substituents [sodium sulfonate (7) (Figure S4), nitro (8), methoxyl (9), and amine (10)] through coordination interactions. Tetragonal cage 1 was prepared by stirring a mixture of TPPE (5), the sodium sulfonate-functionalized dicarboxylate ligand (7) and 90° Pt(II) acceptor (6) in a 1:2:4 ratio in an acetone/water (v/v = 8:1) mixture. The 31P{1H} and 1H NMR spectra of the reaction mixture suggest the formation of a single, discrete assembly with high symmetry. The 31P{1H} spectrum of cage 1 show two doublets of approximately equal intensity at δ = 6.07 and −0.09 ppm with concomitant 195Pt satellite peaks corresponding to two distinct phosphorus environments (Figure 1c, Figure S5), indicating that the Pt(II) centers on cage 1 possess a heteroligated N,O-coordination motif with one pyridyl and one carboxylate moiety per metal center.51 In the 1H NMR spectrum of cage 1 (Figure 1h, Figure S6), the protons of the pyridyl groups are shifted downfield (Δδ[H5a] = 0.06 ppm; Δδ[H5b] = 0.16 ppm) relative to those of ligand 5 (Figure 1g, Figure S1), consistent with coordination of the N atoms to the platinum centers. The protons that correspond to dicarboxylate ligand 7 are observed at δ = 8.36 and 7.97 ppm (Figure 1h, Figure S6). The well-defined signals in both the 31P{1H} and 1H NMR spectra indicate that a discrete structure was the sole assembly product. Similar to those of cage 1, the 31P{1H} spectrum of metallacage cage 2 also show two doublets of approximately equal intensity at δ = 5.66 and 0.44 ppm with concomitant 195Pt satellite peaks corresponding to two distinct phosphorus environments (Figure 1d, Figure S7), demonstrating that a similar structure was obtained.
Figure 1.

(a) [2 + 8 + 4] assembly of 5, 6 and 7 to furnish cage 1; 5, 6 and 8 to furnish cage 2; 5, 6 and 9 to furnish cage 3; and 5, 6 and 10 to furnish cage 4 via heteroligation-directed self-assembly of 90° Pt(II) acceptors and pyridyl and carboxylate ligands. (b−k) Partial 31P{1H} (b−f) and 1H NMR (g−k) spectra (CD2Cl2) of building blocks 5 (g) and 6 (b) and tetragonal cage 1 (c, h), cage 2 (d, i), cage 3 (e, j), and cage 4 (f, k) highlighting the changes associated with the transformation from diverse building blocks into discrete metallacages.
In the 1H NMR spectrum of cage 2 (Figure 1i, Figure S8), the protons of the pyridyl groups are shifted downfield (Δδ[H5a] = 0.05 ppm; Δδ[H5b] = 0.19 ppm) relative to those of ligand 5, consistent with the coordination of the N atoms to the platinum centers. The protons corresponding to dicarboxylate ligand 8 (Figure S9) are observed at δ = 8.65 and 8.39 ppm (Figure S8). A comparison of the 31P{1H} and 1H NMR spectrum of the four assemblies shows that the cage 3 (Figures 1e,j, Figures S10−12) and cage 4 (Figure 1f,k, Figures S13− 15) has a molecular structure similar to that of cages 1 and 2 suggesting that they also contain a tetragonal prismatic core. The stoichiometry of discrete cages is further supported by the electrospray ionization time-of-flight mass spectrometry (ESITOF-MS) results. The ESI-TOF-MS spectrum for cage 1 shows peaks for the assigned [2 + 8 + 4] assembly, including peaks corresponding to an intact entity with charge states resulting from the loss of OTf− counterions (m/z = 1951.16 for [M − 4OTfNa − 3OTf]3+) (Figure 2a, Figure S16). The ESI-TOF-MS results for cages 2−4 also show peaks corresponding to an intact entity with charge states from the loss of OTf− counterions (Figure 2b–d, Figures S17−S19). All peaks were isotopically resolved, and they agreed well with their calculated theoretical distributions.
Figure 2.

Theoretical (top) and experimental (bottom) ESI-TOF-MS spectra of (a) cage 1 [M − 4OTfNa − 3OTf]3+, (b) cage 2 [M − 3OTf]3+, (c) cage 3 [M − 3OTf]3+, and (d) cage 4 [M − 3OTf]3+.
The optical properties of the compounds were closely related to the substituents and solvents, and the properties were first evaluated by UV−vis (ultraviolet−visible) spectroscopy to determine the excitation wavelength for further fluorescence experiments (Figure S20). Fluorescence spectroscopy indicated that the solvent polarity62,63 was found to play a critical role in tuning the emission.63
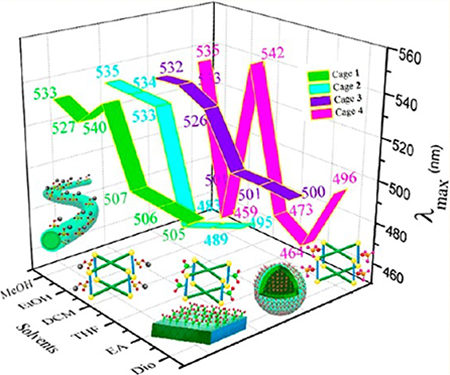
On the basis of the differences in solvent polarity among dioxane, ethyl acetate, tetrahydrofuran, dichloromethane, ethanol, and methanol, the λmax values in the fluorescence spectra (Figure S21) were divided into two wavelength regions, < 507 (464−507 nm) in dioxane, ethyl acetate, and tetrahydrofuran, and >526 (the only exception is cage 4 in ethanol) in dichloromethane, ethanol, and methanol. As shown in the green area in Figure 3a, the peaks of cage 1 were centered at 506 nm in dioxane, tetrahydrofuran, and ethyl acetate, but the peaks ranged from 527 to 540 nm in dichloromethane, ethanol, and methanol. For cage 2, a similar tendency was observed, further demonstrating solvent-dependent emission, as shown in the cyan area.
Figure 3.

(a) λmax values of cages 1−4 in six solvents. (b) Photographs of cage 1, cage 2, cage 3 and cage 4 (from top to bottom) in six solvents. Dio, dioxane; EA, ethyl acetate; THF, tetrahydrofuran; DCM, dichloromethane; EtOH, ethanol; MeOH, methanol.
The peaks in dioxane, tetrahydrofuran, and ethyl acetate ranged from 483 to 495 nm, and the peaks in dichloromethane, ethanol, and methanol ranged from 533 to 535 nm. As reported in the previous literature,64 λmax changed with a change in solvent polarity. The present results support a previous study that demonstrated that the nature of the solvent effect is a key parameter for tuning the emission. A solvent-dependent effect was also observed for cage 3, as shown in the purple area. The first region was centered at 501 nm, and the second region ranged from 526 to 533 nm. For cage 4, the peaks ranged from 464 to 496 nm in dioxane, ethyl acetate, and tetrahydrofuran. The peak ranges (Figure 3a) for the cages in dioxane, tetrahydrofuran, and ethyl acetate were 505−507 nm (cage 1), 483−495 nm (cage 2), 500−501 nm (cage 3), and 464−496 nm (cage 4). By contrast, the λmax values in dichloromethane, ethanol, and methanol were in the ranges of 527−540 nm (cage 1), 533−535 nm (cage 2), 526−533 nm (cage 3), and 459−542 nm (cage 4).
As shown in UV−vis spectra (Figure S20), the four cages have similar absorption bands. The fluorescence spectra (Figure S21) exhibit similar emissions of cage 1 (λmax= 540 nm), cage 2 (λmax= 533 nm), cage 3 (λmax= 526 nm), and cage 4 (λmax= 542 nm) in dichloromethane. Both the UV−vis and fluorescence spectra indicate the weak influence of different substituents on the electrical properties of TPPE. Thus, it is deduced that the polarity of the solvents affected the wavelength shifts,65 which might be due to the different solubilities of the metallacages in the six solvents.
In addition to the solvent influence, the effect of substituents also plays an important role in tuning the emission. As the substituent changed from −SO3Na to −NH2, the cage emission showed more colors. The digital photo of cage 1 under a UV lamp at 365 nm shown in Figure 3b (top) revealed several colors in these solvents. Generally, these metallacages emitted two different sets of colors, one set in dioxane, ethyl acetate, and tetrahydrofuran and another set in dichloromethane, ethanol and methanol. Cage 1 showed green emission in dioxane, ethyl acetate, and tetrahydrofuran, which red-shifted to yellowish emission in dichloromethane, ethanol and methanol.
For cage 2, blue emission instead of green emission was observed in dioxane, ethyl acetate, and tetrahydrofuran; light orange emission in dichloromethane, light green emission in ethanol, and pale green emission in methanol were observed. When the substituents on the metallacages were changed to OCH3 (cage 3), pale-blue colors were observed in dioxane, ethyl acetate, and tetrahydrofuran. Light orange emission in dichloromethane, yellow-green emission in ethanol, and weak pale orange emission in methanol. For cage 4, the following emissions were observed: pale blue in dioxane, light blue in ethyl acetate, dark blue in tetrahydrofuran, light orange in dichloromethane, gray in ethanol, and light orange in methanol. The wavelength range (Figure 4a, Figure S21) in the fluorescence spectra ranged from 35 nm (cage 1) to 83 nm (cage 4), consistent with the digital photos of the six solvents in Figure 3b. The CIE diagram shows that the color region changed from narrow to wide as the substituents changed from −SO3Na to −NH2 (Figures 4b,c). Similar to the solvent-dependent effect, this spectral broadening might also be the result of the different solubility and aggregation behaviors caused by the substituents. The metallacages were shown to remain intact after they were dissolved in six solvents, as confirmed by NMR and fluorescence spectra. The well-defined signals in the 31P{1H} NMR spectra demonstrated the stability of the metallacages in dichloromethane, methanol, and ethanol (Figures S22−S24). Figure S25 shows the comparison (Figure S21) of TPPE in dioxane (λmax = 508 nm), ethyl acetate (λmax = 500 nm), and tetrahydrofuran (λmax= 510 nm). All of these cages showed one peak and different emission to TPPE.
Figure 4.

(a) Fluorescence spectra of cage 1, cage 2, cage 3 and cage 4 in dioxane, ethyl acetate, tetrahydrofuran, dichloromethane, ethanol, and methanol (λex = 365 nm, c = 25.0 μM). (b,c) CIE diagram of cage 1 and cage 4 in six solvents.
The further assembly behaviors were then investigated by changing the solvents and substituents. The differences among the four metallacages, i.e., cages 1−4, may be due to the properties of the substituents and the ratio of hydrophobic/hydrophilic moieties. For cage 1, the charged substituent −SO3Na was introduced into the metallacages. As shown in Figure 5a, after the sample (25 μM) was aged at room temperature in water for 1 week, a filamentous suspension was observed with the naked eye. The size of the suspension network increased continuously as the incubation time increased. Green emission was observed under UV-light irradiation at 365 nm (Figure 5a inset, Figure S26), and the morphology of the suspension was examined by scanning electron microscope (SEM). The intensity observed over a large area of the sample revealed a fiber-based network, as shown in Figures 5a–c. Transmission electron microscopy (TEM) and SEM image measurements also indicated a fiber-based network structure, as shown in Figures 5d–f. The suspension had a green emission, which indicated that these assemblies were composed of cage 1. As shown in Figure 5g–l, energy dispersive X-ray analysis (EDX) mapping analysis confirmed the homogeneous distributions of N, Pt, P, F, S and O across the microfibers; and the analysis provided evidence that the microfibers were composed of TPPE, cis-Pt(PEt3)2(OTf)2, and the carboxylate ligand. Taking the fiber in Figure 5f as an example, the length and diameter of the fiber were 4.0 μm and 200 nm. These data indicate that the aggregates were composed of at least hundreds of metallacages. On the basis of this information, we propose a possible self-assembly mechanism for the cage 1-based microfiber (Figure S27). TPPE drives the π−π stacking and then promotes the cage 1-based aggregation, leading to the formation and elongation of aggregate-based microfibers. In addition to one-dimensional microfibers, 2D assemblies could also be formed by cage 2 in tetrahydrofuran, as demonstrated by SEM and shown in Figures 6a–c. Blue emission was observed under UV-light irradiation at 365 nm (Figure 6a inset, Figure S28). The TEM (Figure 6d) results were consistent with the SEM observations. The STEM and magnified images (Figures 6e,f) support the formation of microplates.
Figure 5.

(a) SEM image of the cage 1-based microfibers (inset: optical image showing the white-green emission of the microfibers in water). (b,c) SEM images of microfibers at different magnifications. (d) TEM image of the microfibers. (e) An enlarged SEM image of a microfiber. (f) A STEM image of the self-assembly of the cage 1-based microfibers. EDX mapping images of the cage 1-based microfibers: the distributions of (g) N, (h) Pt, (i) P, (j) F, (k) S, and (l) O.
Figure 6.

(a) SEM image of cage 2-based microplates (inset: optical image showing the blue emission of the microplates). (b,c) SEM images of microplates at different magnifications. (d) TEM image of microplates. (e,f) STEM image of microplates at different magnifications. (g) EDX mapping images of the distributions of N, O, S, Pt, F, and P (scale bar, 500 nm).
Elemental mapping analysis (Figure 6g) revealed homogeneous distributions of the characteristic elements across the microplates (Figure S29). In ethanol, cage 2 formed a 2D structure, and as shown in Figure S30, microfilms were obtained. These microfilms ranged from a few hundred nanometers to a few micrometers in size, and they exhibited yellow-green emission under UV-light irradiation (Figure S30a inset, Figure S31). The elemental mapping analysis demonstrated that the microfilms were composed of TPPE, cis-Pt(PEt3)2(OTf)2, and the nitro-modified carboxylate ligand (Figure S30h−m). Similar to the cage 2, when cage 4 assembled in tetrahydrofuran, the leaf-like structure was the dominant morphology, and blue emission was observed under UV-light irradiation (Figures S32−S33). Elemental mapping analysis demonstrated homogeneous distributions of N, O, Pt, F, S, and P across the microleaf structures (Figure S32). Three-dimensional (3D) microspheres were obtained in THF when the substituents were changed to −OCH3. As shown in Figure 7a–c, spherical objects with diameters ranging from a few hundred nanometers to a few micrometers were observed, and these spheres exhibited blue emission (Figure 7a inset, Figure S34) under UV-light irradiation. Magnification of an individual sphere revealed a hollow structure (Figure 7d). Elemental mapping analysis (Figure 7e–j) indicated homogeneous distributions of N, O, Pt, F, S, and P across the microspheres, providing evidence that the microspheres were composed of TPPE, cis-Pt(PEt3)2(OTf)2, and the carboxylate ligand. Based on these observations, a possible self-assembly mechanism of cage 3 is shown in Figure S35. Microspheres developed to minimize the free energy of the system. In addition, spherical aggregates with diameters of tens of nanometers and strong green emission were obtained by cage 1 assembly in ethanol and tetrahydrofuran (Figures S36−S39). The intact structures of the metallacages after further assembly were characterized by 31P{1H} NMR and fluorescence spectra. As shown in Figures S40−S41, the 31P{1H} NMR spectra confirmed the stability of cage 1 and cage 2 in ethanol after 7 days. Furthermore, aggregates formed at water and THF were centrifuged and redispersed in DCM to confirm the integrity of cage M. Then, 31P{1H} and 1H NMR spectra were used to determine the integrity of cage 1−4. Both the 31P{1H} NMR spectra (Figures S42−46) confirmed that the metallacages were intact after assembly.
Figure 7.

(a) SEM image of cage 3-based hollow microspheres (inset: optical image showing the blue emission of the microspheres). (b−d) SEM images of hollow microspheres at different magnifications. EDX mapping images of the distributions of (e) N, (f) O, (g) Pt, (h) F, (i) S, and (j) P in hollow microspheres.
As mentioned above, platinum metallacages are tetragonal prismatic structures in which the TPPE units, dicarboxylate moieties, and 90° Pt (II) ions construct the faces, pillars, and corners, respectively. This type of structure possesses two key features: the hydrophobic cavity consisting of two TPPE units, four dicarboxylate derivatives, and eight 90° Pt(II) corners, and the changeable substituents in the pillar, which convey a specific character to cages 1−4. The construction of metallacage-based suprastructures is summarized in Scheme 1. Dicarboxylate ligands with various substituents (−SO3Na, −NO2, −OCH3, −NH2), TPPE, and cis-(PEt3)2Pt(OTf)2 were used as building blocks to fabricate multicomponent metallacages through coordination interactions. Their optical properties were closely related to the substituents and solvents. Cage 1 emitted orange light in dichloromethane, ethanol, and methanol but green light in dioxane, tetrahydrofuran, and ethyl acetate. As the substituents changed from −SO3Na to −NH2, the emission spectra became broader (the spectral broadening might be the result of the different solubility and aggregation behaviors caused by the substituents).
Scheme 1.

Multidimensional Suprastructures (1D, 2D, and 3D) with Tunable Surface Modifications and Emissions Were Prepared by Multilevel Assemblya
aIn the first level, dicarboxylate ligands with various substituents (-sodium sulfonate, -nitro, -methoxyl, and -amine), tetra-(4-pyridylphenyl) ethylene, and cis-(PEt3)2Pt(OTf)2 were used as building blocks to fabricate four shape-controllable metallacages. In the next stage, the as-prepared metallacages were used as building blocks for the second-level assembly. Diverse structures with different surface substituents and emissions were prepared on the basis of the nature of the substituents and solvents.
Cages 1−4 were used as a building block for the second stage of assembly. In water, 1D nanofibers with green emission were obtained. In organic solvents, 2D and 3D structures with diverse emissions were obtained through the assembly of cages 1−4 in ethanol and tetrahydrofuran. Among them, cages 2, 4 tended to form 2D structures. For example, cage 2-based nitrofunctionalized microfilms (510 nm) and microplates (461 nm) were obtained in ethanol and tetrahydrofuran, respectively. Amine-functionalized microleaf structures (459 nm) were obtained in tetrahydrofuran. However, cage 1, 3 tended to form 3D structures in organic solvents. For example, 3D methoxyl-functionalized hollow microspheres (451 nm) and sodium sulfonate-functionalized microspheres (506 nm) were fabricated in tetrahydrofuran. The above experiments indicate that diverse suprastructures with tunable substituents −X = −SO3Na, −NO2, −OCH3, −NH2) and broad emissions (λmax from 451 to 519 nm) can be obtained by adjusting the substituents and solvents.
CONCLUSION
In this report, TPPE-based metallacages were arranged into complex architectures with multicolor emissions. First, dicarboxylate ligands with various substituents (-sodium sulfonate, -nitro, -methoxyl, and -amine substituents) were synthesized. Then, metallacages were prepared via the combination of dicarboxylate ligand derivatives, TPPE, and cis-(PEt3)2Pt(OTf)2. Finally, hybrid metallacages were used as building blocks and further assembled, to form 1D, 2D, and 3D suprastructures with broad emissions (λmax from 451 to 519 nm) and tunable substituents (−SO3Na, −NO2, −OCH3, and −NH2). This investigation not only bridges the functional gap between small molecules and suprastructures but will also help researchers explore the connection between spatial order and function on multiple scales, thus paving the way for interesting applications in optoelectronic material science.
EXPERIMENTAL SECTION
Materials and Methods.
All reagents were commercially available and used as supplied without further purification. Deuterated solvents were purchased from Cambridge Isotope Laboratory (Andover, MA). Compounds 5,51 6,51 7−1051,66 were prepared according to modified literature procedures. NMR spectra were recorded at room temperature. 1H chemical shifts are reported relative to the residual solvent signals, and 31P{1H} NMR chemical shifts are referenced to an external unlocked sample of 85% H3PO4 (δ = 0.0). Mass spectra were recorded on a Waters synapt G2 tandem mass spectrometer using electrospray ionization with a MassLynx operating system. Ultraviolet−visible experiments were conducted on a Hitachi U-4100 absorption spectrophotometer. Fluorescence experiments were conducted on a Hitachi F-7000 fluorescence spectrophotometer. Transmission electron microscopy investigations were performed on a JEM-2100EX instrument. For TEM, dispersions of the assemblies were dried onto carbon-coated copper support grids. A Zeiss Supra55 field-emission scanning electron microscope was used to investigate the assemblies. For scanning electron microscopy, dispersions of the assemblies were dried onto silicon wafers. High-resolution TEM and elemental mapping images were obtained using a Tecnai G2 F30 S-TWIN instrument.
Synthesis of Cage 1.
Tetra(4-pyridylphenyl)ethylene compound 5 (3.20 mg, 5.00 μmol), cis-Pt(PEt3)2(OTf)2 6 (14.60 mg, 20.00 μmol), and sodium sulfate-functionalized carboxylate ligand 7 (3.12 mg, 10.00 μmol) were placed in a vial, followed by the addition of H2O (0.40 mL) and acetone (1.20 mL). After heating at 70 °C for 24 h, all the solvent was removed by a N2 flow, and the solid was dried under vacuum. Acetone (1.00 mL) was then added to the resultant mixture, and the solution was stirred for 30 min at room temperature. Then, the mixture was filtered to remove insoluble materials. The resulting tetragonal cage 1 was precipitated with diethyl ether, isolated and dried under reduced pressure and dissolved in CD2Cl2 for characterization. 31P {1H} NMR (CD2Cl2, room temperature, 121.4 MHz) δ (ppm): 6.07 ppm (d, 2JP−P = 19.4 Hz), −0.09 ppm (d, 2JP−P = 19.4 Hz). 1H NMR (CD2Cl2, room temperature) δ (ppm) 8.66 (d, 16H), 8.36 (s, 8H), 7.97 (s, 4H), 7.68 (d, 16H), 7.17 (d, 16H), 7.15 (d, 16H). ESI-TOF-MS (m/z) calcd for [M − 4OTfNa − 3OTf]3+, (C221H316F3N8O31P16Pt8S53+, 1951.16 (100%); found, 1951.16, Fragment, calcd for [M/2 − 2NaOTf − 2OTf−]2 + (C110H158N4O14P8Pt4S22+, 1425.89 (100%), found 1425.79.
Synthesis of Cage 2.
According to the same procedure for the synthesis of cage 1, cage 2 was prepared from ligand 8 (2.60 mg, 10.00 μmol). 31P {1H} NMR (CD2Cl2, room temperature, 121.4 MHz) δ (ppm): 5.66 ppm (d, 2JP−P = 27.9 Hz), 0.44 ppm (d, 2JP−P = 27.9 Hz). 1H NMR (CD2Cl2, room temperature) δ (ppm) 8.65 (d, 24H), 8.39 (s, 4H), 7.71 (d, 16H), 7.50 (d, 16H), 7.19 (d, 16H). ESIT O F - M S (m/z) calcd for [M − 3OTf]3 +, (C225H316F15N12O39P16Pt8S53+, 2104.48 (100%); found, 2014.48, Fragment, calcd for [M/2 − 2OTf−]2+ (C112H158F6N6O18P8Pt4S22+, 1540.87 (100%), found 1540.77.
Synthesis of Cage 3.
According to the same procedure for the synthesis of cage 1, cage 3 was prepared from ligand 9 (2.40 mg, 10.00 μmol). 31P {1H} NMR (CD2Cl2, room temperature, 121.4 MHz) δ (ppm): 6.14 ppm (d, 2JP−P = 21.8 Hz), 0.52 ppm (d, 2JP−P = 21.8 Hz). 1H NMR (CD2Cl2, room temperature) δ (ppm) 8.67 (d, 16H), 7.65 (d, 16H), 7.51 (s, 4H), 7.45 (d, 16H), 7.20 (s, 8H), 7.18 (d, 16H), 3.82 (s, 12H). ESI-TOF-MS (m/z) calcd for [M − 3OTf]3+, (C229H328F15N8O35P16Pt8S53+, 2084.52 (100%); found, 2084.52, Fragment, calcd for [M/2 − 2OTf−]2+ (C114H164F6N4O16P8Pt4S22+,1525.90 (100%), found 1525.81.
Synthesis of Cage 4.
According to the same procedure for the synthesis of cage 1, cage 4 was prepared from ligand 10 (2.25 mg, 10.00 μmol). 31P {1H} NMR (CD2Cl2, room temperature, 121.4 MHz) δ (ppm): 6.38 ppm (d, 2JP−P = 20.6 Hz), 0.30 ppm (d, 2JP−P = 20.6 Hz). 1H NMR (CD2Cl2, room temperature) δ (ppm) 8.67 (d, 16H), 7.71 (d, 16H), 7.50 (d, 16H), 7.37 (s, 4H), 7.24 (s, 8H), 7.19 (d, 16H). ESI-TOF-MS (m/z) calcd for [M − 3OTf]3+, (C225H324F15N12O31P16Pt8S53+, 2064.52 (100%); found, 2064.54, Fragment, calcd for [M/2 − 2OTf−]2+ (C112H162F6N6O14P8Pt4S24+, 1510.90 (100%), found 1510.80.
Supplementary Material
ACKNOWLEDGMENTS
Y.S. thanks the National Natural Science Foundation of China (21503185, 51573194). P.J.S. thanks the NIH (Grant R01-CA215157) for financial support. X.L. thanks NSF (CHE-1506722) and NIH (1R01GM128037–01) for support. Y.S. also acknowledges the Priority Academic Program Development of Jiangsu Higher Education Institutions and the testing center of Yangzhou University. Y.S. thanks L. Cao, X. Chang, and Y. Sun (Yue Sun) for discussions and checking the data.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b05809.
Synthesis, optical properties, and self-assembly of metallacages (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Laurent J; Blin G; Chatelain F; Vanneaux V; Fuchs A; Larghero J; Thery M Nat. Bio. Engineering 2017, 1, 939. [DOI] [PubMed] [Google Scholar]
- (2).Bell OA; Wu G; Haataja JS; Brommel F; Fey N; Seddon AM; Harniman RL; Richardson RM; Ikkala O; Zhang X; Faul CF J. J. Am. Chem. Soc 2015, 137, 14288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Burgess NC; Sharp TH; Thomas F; Wood CW; Thomson AR; Zaccai NR; Brady RL; Serpell LC; Woolfson DN J. Am. Chem. Soc 2015, 137, 10554. [DOI] [PubMed] [Google Scholar]
- (4).Xiao Y; Sun H; Du JJ Am. Chem. Soc 2017, 139, 7640. [DOI] [PubMed] [Google Scholar]
- (5).Dora Tang T-Y; Hak RC; Thompson AJ; Kuimova MK; Williams DS; Perriman AW; Mann S Nat. Chem 2014, 6, 527. [DOI] [PubMed] [Google Scholar]
- (6).Sun Y; Guo F; Zuo T; Hua J; Diao G Nat. Commun 2016, 7, 12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Sun Y; Yao Y; Yan C-G; Han Y; Shen M ACS Nano 2010, 4, 2129. [DOI] [PubMed] [Google Scholar]
- (8).Sun Y; Yao Y; Yan C-G; Han Y; Shen M Adv. Funct. Mater 2008, 18, 3981. [Google Scholar]
- (9).Avakyan N; Greschner AA; Aldaye F; Serpell CJ; Toader V; Petitjean A; Sleiman HF Nat. Chem 2016, 8, 368. [DOI] [PubMed] [Google Scholar]
- (10).Karimi M; Zangabad PS; Baghaee-Ravari S; Ghazadeh M; Mirshekari H; Hamblin MR J. Am. Chem. Soc 2017, 139, 4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zou Q; Abbas M; Zhao L; Li S; Shen G; Yan XJ Am. Chem. Soc 2017, 139, 1921. [DOI] [PubMed] [Google Scholar]
- (12).Jiang W; Pacella M; Athanasiadou D; Nelea V; Vali H; Hazen RM; Gray JJ; McKee MD Nat. Commun 2017, 8, 15066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Singh G; Chan H; Baskin A; Gelman E; Repnin N; Kral P; Klajn R Science 2014, 345, 1149. [DOI] [PubMed] [Google Scholar]
- (14).Wong CK; Mason AF; Stenzel MH; Thordarson P Nat. Commun 2017, 8, 1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhu J; Zhang S; Zhang K; Wang X; Mays JW; Wooley KL; Pochan DJ Nat. Commun 2013, 4, 2297. [DOI] [PubMed] [Google Scholar]
- (16).Rupar PA; Chabanne L; Winnik MA; Manners I Science 2012, 337, 559. [DOI] [PubMed] [Google Scholar]
- (17).Mozhdehi D; Luginbuhl KM; Simon JR; Dzuricky M; Berger R; Varol HS; Huang FC; Buehne KL; Mayne NR; Weitzhandler I; Bonn M; Parekh SH; Chilkoti A Nat. Chem 2018, 10, 496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Matosevic S; Paegel BM Nat. Chem 2013, 5, 958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Siavashpouri M; Wachauf CH; Zakhary MJ; Praetorius F; Dietz H; Dogic Z Nat. Mater 2017, 16, 849. [DOI] [PubMed] [Google Scholar]
- (20).Mohammadi E; Zhao C; Meng Y; Qu G; Zhang F; Zhao X; Mei J; Zuo J-M; Shukla D; Diao Y Nat. Commun 2017, 8, 16070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Avakyan N; Greschner AA; Aldaye F; Serpell CJ; Toader V; Petitjean A; Sleiman HF Nat. Chem 2016, 8, 368. [DOI] [PubMed] [Google Scholar]
- (22).Wagenbauer KF; Sigl C; Dietz H Nature 2017, 552, 78. [DOI] [PubMed] [Google Scholar]
- (23).Richardson JJ; Bjornmalm M; Caruso F Science 2015, 348, aaa2491. [DOI] [PubMed] [Google Scholar]
- (24).Tian B; Liu J; Dvir T; Jin L; Tsui JH; Qing Q; Suo Z; Langer R; Kohane D; Lieber CM Nat. Mater 2012, 11, 986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zheng X; Lee H; Weisgraber T; Shusteff M; DeOtte J; Duoss EB; Kuntz JD; Biener MM; Ge Q; Jackson JA; Kucheyev SO; Fang NX; Spadaccini CM Science 2014, 344, 1373. [DOI] [PubMed] [Google Scholar]
- (26).Zhang H; Yu X; Braun PV Nat. Nanotechnol 2011, 6, 277. [DOI] [PubMed] [Google Scholar]
- (27).Noorduin W; Grinthal A; Mahadevan L; Aizenberg J Science 2013, 340, 832. [DOI] [PubMed] [Google Scholar]
- (28).Typas A; Sourjik V Nat. Rev. Microbiol 2015, 13, 559. [DOI] [PubMed] [Google Scholar]
- (29).Chen RP; Blackstock D; Sun Q; Chen W Nat. Chem 2018, 10, 474. [DOI] [PubMed] [Google Scholar]
- (30).Yuan S; Qin J-S; Li J; Huang L; Feng L; Fang Y; Lollar C; Pang J; Zhang L; Sun D; Alsalme A; Cagin T; Zhou H-C Nat. Commun 2018, 9, 808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hu S; Dasbiswas K; Guo Z; Tee Y-H; Thiagarajan V; Hersen P; Chew T-L; Safran SA; Zaidel-Bar R; Bershadsky AD Nat. Cell Biol 2017, 19, 133. [DOI] [PubMed] [Google Scholar]
- (32).Yang M; Chan H; Zhao G; Bahng JH; Zhang P; Kral P; Kotov NA Nat. Chem 2017, 9, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Praetorius F; Dietz H Science 2017, 355, 1283. [DOI] [PubMed] [Google Scholar]
- (34).Xu S; Yan Z; Jang K-I; Huang W; Fu H; Kim J; Wei Z; Flavin M; McCracken J; Wang R; Badea A; Liu Y; Xiao D; Zhou G; Lee J; Chung HU; Cheng H; Ren W; Banks A; Li X; Paik U; Nuzzo RG; Huang Y; Zhang Y; Rogers JA Science 2015, 347, 154. [DOI] [PubMed] [Google Scholar]
- (35).Guo J; Tardy BL; Christofferson AJ; Dai Y; Richardson JJ; Zhu W; Hu M; Ju Y; Cui J; Dagastine RR; Yarovsky I; Caruso F Nat. Nanotechnol 2016, 11, 1105. [DOI] [PubMed] [Google Scholar]
- (36).Chaikeeratisak V; Nguyen K; Khanna K; Brilot AF; Erb ML; Coker JKCC; Vavilina A; Newton GL; Buschauer R; Pogliano K; Villa E; Agard DA; Poliano J Science 2017, 355, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hudson ZM; Boott CE; Robinson ME; Rupar PA; Winnik MA; Manners I Nat. Chem 2014, 6, 893. [DOI] [PubMed] [Google Scholar]
- (38).Qiu H; Hudson ZM; Winnik MA; Manners I Science 2015, 347, 1329. [DOI] [PubMed] [Google Scholar]
- (39).Votteler J; Ogohara C; Yi S; Hsia Y; Nattermann U; Belnap DM; King NP; Sundquist WI Nature 2016, 540, 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Lai Y-T; Reading E; Hura GL; Tsai K-L; Laganowsky A; Asturias FJ; Tainer JA; Robinson CV; Yeates TO Nat. Chem 2014, 6, 1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Service RF Science 2005, 309, 95. [DOI] [PubMed] [Google Scholar]
- (42).Saha ML; Yan X; Stang P Acc. Chem. Res 2016, 49, 2527. [DOI] [PubMed] [Google Scholar]
- (43).Shanmugaraju S; Mukherjee PS Chem. - Eur. J 2015, 21, 6656. [DOI] [PubMed] [Google Scholar]
- (44).Howlader P; Mondal B; Purba PC; Zangrando E; Mukherjee PS J. Am. Chem. Soc 2018, 140, 7952. [DOI] [PubMed] [Google Scholar]
- (45).Peng H-Q; Zheng X; Han T; Kwok RTK; Lam JWY; Huang X; Tang B-ZJ Am. Chem. Soc 2017, 139, 10150. [DOI] [PubMed] [Google Scholar]
- (46).Liu Y; Deng C; Tang L; Qin A; Hu R; Sun J-Z; Tang B-Z J. Am. Chem. Soc 2011, 133, 660. [DOI] [PubMed] [Google Scholar]
- (47).Wei P; Zhang J-X; Zhao Z; Chen Y; He X; Chen M; Gong J; Sung HH-Y; Williams ID; Lam JWY; Tang BZ J. Am. Chem. Soc 2018, 140, 1966. [DOI] [PubMed] [Google Scholar]
- (48).Xie S; Wong AYH; Kwok RTK; Li Y; Su H; Lam JWY; Chen S; Tang B-Z Angew. Chem., Int. Ed 2018, 57, 5750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Wang D; Su H; Kwok RTK; Hu X; Zou H; Luo Q; Lee MMS; Xu W; Lam JWY; Tang B-Z Chem. Sci 2018, 9, 3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Mei J; Leung NLC; Kwok RTK; Lam JWYL; Tang B-Z Chem. Rev 2015, 115, 11718. [DOI] [PubMed] [Google Scholar]
- (51).Yan X; Cook TR; Wang P; Huang F; Stang P Nat. Chem 2015, 7, 342. [DOI] [PubMed] [Google Scholar]
- (52).Danon JJ; Krüger A; Leigh DA; Lemonnier J; Stephens AJ; Vitorica-Yrezabal IJ; Woltering SL Science 2017, 355, 159. [DOI] [PubMed] [Google Scholar]
- (53).Fielden SDP; Leigh DA; Woltering SL Angew. Chem., Int. Ed 2017, 56, 11166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Rousseaux SAL; Gong JQ; Haver R; Odell B; Claridge TDW; Herz LM; Anderson HL J. Am. Chem. Soc 2015, 137, 12713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Movsisyan LD; Franz M; Hampel F; Thompson AL; Tykwinski RR; Anderson HL J. Am. Chem. Soc 2016, 138, 1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Pilgrim BS; Roberts DA; Lohr TG; Ronson TK; Nitschke JR Nat. Chem 2017, 9, 1276. [Google Scholar]
- (57).Roberts DA; Pilgrim BS; Nitschke JR Chem. Soc. Rev 2017, 47, 626–644. [DOI] [PubMed] [Google Scholar]
- (58).Zhang D; Ronson TK; Mosquera J; Martinez A; Nitschke JR Angew. Chem., Int. Ed 2018, 57, 3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Zhang D; Ronson TK; Mosquera J; Martinez A; Nitschke JR Angew. Chem., Int. Ed 2018, 57, 3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Pilgrim BS; Roberts DA; Lohr TG; Ronson TK; Nitschke JR Nat. Chem 2017, 9, 1276. [Google Scholar]
- (61).Roberts DA; Pilgrim BS; Sirvinskaite G; Ronson TK; Nitschke JR J. Am. Chem. Soc 2018, 140, 9616. [DOI] [PubMed] [Google Scholar]
- (62).Reichardt C Solvents and Solvent Effects in Organic Chemistry, 3rd ed.; Wiley-VCH Publishers, 2003. [Google Scholar]
- (63).Lide DR Handbook of Organic Solvent; CRC Press: Boca Raton, FL, 1994. [Google Scholar]
- (64).Kwak G; Kim H; Kang I; Kim S Macromolecules 2009, 42, 1733. [Google Scholar]
- (65).Klymchenko AS Acc. Chem. Res 2017, 50, 366–375. [DOI] [PubMed] [Google Scholar]
- (66).Yan X; Li S; Cook TR; Ji X; Yao Y; Pollock JB; Shi Y; Yu G; Li J; Huang F; Stang PJ J. Am. Chem. Soc 2013, 135, 14036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.