

Abstract
As potent and selective therapeutic agents, peptides and proteins are an important class of drugs, but they typically have sub-optimal pharmacokinetic profiles. One approach to solve this problem is by their conjugation with “stealth” polymers. Conventional methods for conjugation of this class of polymers to peptides and proteins are typically carried out by reactions that have poor yield and provide limited control over the site of conjugation and the stoichiometry of the conjugate. To address these limitations, new chemical and biological approaches have been developed that provide new molecular tools in the bioconjugation toolbox to create stealth polymer conjugates of peptides and proteins with exquisite control over their properties. This review article highlights these recent advances in the synthesis of therapeutic peptide/protein-stealth polymer conjugates.
Graphical Abstract
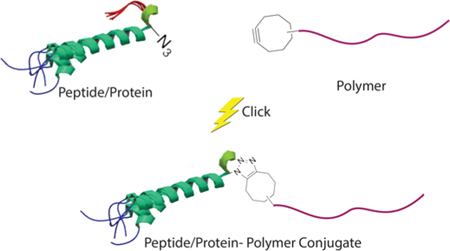
1. INTRODUCTION
Many peptides and proteins have high selectivity and potency, are well tolerated and have a safe in vivo profile, and have therefore become an important class of drugs.1–3 Currently, there are more than 60 peptide therapeutics approved by the U.S. Food and Drug Administration (FDA) on the market, another approximately 140 in clinical trials, and many more in the preclinical pipeline. The annual global sales of peptide therapeutics is estimated to reach US $25.4 billion by 2018, which attests to the importance of this class of drugs.1 With the exception of antibodies and their fusion proteins, which are typically not modified by polymer conjugation, there are 59 FDA approved protein drugs on the market4–6 with combined annual global sales of US $222.7 billion projected by 20197, and many others under clinical development.
Despite their therapeutic use, peptides and most proteins have some significant limitations as drugs; they typically have a very short plasma half-life, and some peptides and proteins have poor stability in circulation. These attributes lead to their rapid in vivo elimination, which necessitates frequent injections that result in high treatment costs and suboptimal patient compliance, and thus limits their utility as therapeutics.3,8 Antibodies —the source of the largest class of protein therapeutics— in contrast are typically highly soluble, and have an extremely long half-life in systemic circulation of up to 3 weeks in humans9, and they hence do not need to be modified by polymer conjugation. Consequently, we do not discuss antibodies in this review.
One approach to overcome the limitations of therapeutic proteins and peptides involves attaching “stealth” polymers —defined as polymers that show minimal interaction with proteins and cells. Polyethylene glycol (PEG) is the gold standard stealth polymer, and its stealth effect stems from its preferential interaction with water molecules through hydrogen bonding, thereby creating a hydration layer surrounding the biomolecule that minimizes its interactions with proteins.10 PEGylated conjugates of peptides and proteins exhibit improved solubility because of the hydrophilicity of the polymer, a much longer plasma half-life than the unmodified peptide/protein due to an increase in hydrodynamic size conferred by the PEG and because they escape opsonization, and they also have lower immunogenicity than the parent molecule because of their protein resistance.11,12 It is important to note, however, that while the attached PEG minimizes the interactions with blood components, and hence enables the conjugate to escape opsonization, it can also reduce the biological activity of the conjugate, though the improved pharmacokinetics and pharmacodynamics of PEG conjugates generally compensate for their somewhat lower bioactivity in terms of in vivo efficacy.
Although PEG conjugates have been on the market for several decades, many of the current methods for synthesis of therapeutic protein/peptide-stealth polymer conjugates result in heterogeneous conjugates where neither the site of conjugation nor the stoichiometry —the number of polymer chains attached to a protein or peptide— are tightly controlled, resulting in a heterogeneous mixture of conjugates that exhibit a range of pharmacokinetics and pharmacodynamics.8,11,13,14 There is hence a great need to develop methods that enable the synthesis of structurally uniform therapeutic protein/peptide-stealth polymer conjugates in terms of their conjugation site and stoichiometry, using polymers with a highly defined structure as defined by their sequence, architecture, chain length and polydispersity. The development of new molecules and methodologies from bioorganic chemistry, chemical biology and polymer science now provide a set of molecular tools that make the goal of synthesis of molecularly precise protein/peptide-polymer conjugates a reality. Here, we provide a brief overview of these tools, and highlight their utility for the synthesis of site-specific and stoichiometric therapeutic protein/peptide-stealth polymer conjugates.
2. SYNTHESIS OF POLYMER-REACTIVE THERAPEUTIC PEPTIDES AND PROTEINS
Of the chemically reactive subset of native amino acids present in peptides and proteins, Lys and Cys are most commonly used for conjugation to polymers, but these residues offer little to no control over the site of conjugation and the stoichiometry of the conjugate, as most proteins and peptides have more than one copy of these residues located at a solvent-accessible site on the surface of the protein. Furthermore, each site can have a different reactivity towards a polymer because of varying degrees of solvent accessibility that is determined by its location on the protein or peptide scaffold. Hence, exploiting these amino acids for polymer conjugation typically results in heterogeneous protein/peptide-stealth polymer conjugates in terms of the conjugation site and stoichiometry, and hence therapeutic activity. In addition, chemically reactive amino acids are often implicated in the biological activity of many protein drugs, especially enzymes, so that the modification of these residues may further decrease the therapeutic activity of the conjugate.15
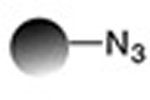
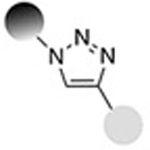
Solving this problem requires reactions that are site-specific, have no side-reactions with other reactive moieties present in a protein or peptide, and proceed with complete conversion. “Bio-orthogonal click chemistry”16 —the reaction of chemically unique functional groups that are orthogonal to those that exist in nature, that are fast and proceed with complete conversion with no undesired side-reactions— offers a versatile approach for the synthesis of site-specific and stoichiometric protein/peptide-polymer conjugates.8,11,13–16 It is important to note, however, that some bio-orthogonal reactions may require catalysts, which may pose a risk for clinical use of therapeutic conjugates due to possible contamination by these frequently toxic moieties, and which may necessitate laborious purification steps to get rid of the catalyst. Table 1 shows a brief list of the bio-orthogonal click reactions available for the synthesis of therapeutic protein/peptide-polymer conjugates.
Table 1.
Bio-orthogonal click reactions available for the synthesis of therapeutic protein/peptide- polymer conjugates.
| Bio-orthogonal Chemistry |
Bio-orthogonal Group 1 |
Bio-orthogonal Group 2 |
Bio-orthogonal Reaction Product |
Reaction Rate (x 10−3 M−1.s−1) |
References |
|---|---|---|---|---|---|
| Copper-mediated Huisgen azide alkyne 1,3-dipolar cycloaddition |
 |
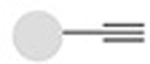 |
 |
0.01−107 | 16,17 |
| Strain-promoted azide alkyne 1,3-dipolar cycloaddition |
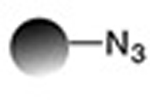 |
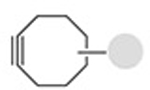 |
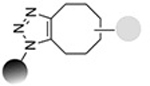 |
0.9−109 | 16,18,19 |
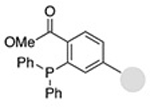
| Staudinger ligation (Nontraceless) |
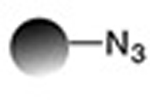 |
 |
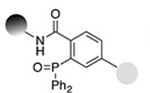 |
0.83−3.8 | 19 |
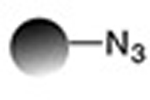
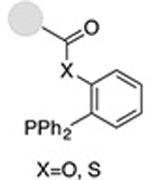
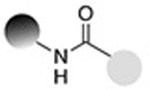
| Staudinger ligation (traceless) |
 |
 |
 |
0.12−7.7 | 19,20 |
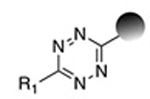
| Inverse- electron-demand Diels-Alder reaction (TCO-Tetrazine) |
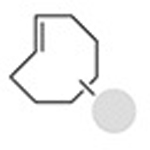
 |
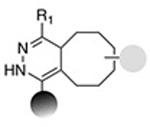
 |
 |
107−108 | 17,21 |
| Inverse- electron-demand Diels-Alder reaction (Vinylboronic acid- Tetrazine) |
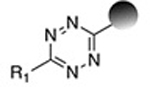 |
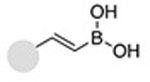 |
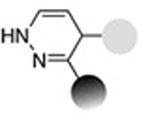 |
130−27000 | 22 |
Implementing bio-orthogonal click chemistry for polymer conjugation to proteins or peptides requires the site-specific installation of complementary bio-orthogonal clickable reactive groups —handles— on the polymer and the peptide/protein of interest. Compared to the installation of a bio-orthogonal group into polymers, typically at the chain end, which though non-trivial, is relatively easy to accomplish, the far greater challenge is the site-specific and high-yield installation of a complementary bio-orthogonal handle into recombinantly expressed proteins and peptides. For peptides that can be synthesized by solid phase peptide synthesis23, the incorporation of an unnatural amino acid (UAA) that has a bio-orthogonal side-chain is fairly straightforward. Incorporation of bio-orthogonal UAA into a recombinantly expressed peptide or protein, in contrast, is non-trivial because the native translational machinery of the common hosts used to express peptide and proteins — E. coli, yeast and mammalian cells— have to be reprogrammed to accept UAAs.8
The most powerful and general approach for site-specific UAA incorporation was pioneered by Schultz and co-workers24,25 (Figure 1).26 This approach requires: (1) a supply of the UAA that has the bio-orthogonal handle on its side-chain that must be added to the culture medium; (2) an orthogonal aminoacyl-tRNA synthetase/tRNA pair that is encoded by the host cell, and wherein the tRNA synthetase solely recognizes the UAA and charges it to the orthogonal tRNA and where conversely the native tRNA synthetases in the cell do not recognize the UAA; and (3) a unique codon in the gene of the peptide/protein that the bio-orthogonal tRNA is reprogrammed to recognize, typically the TAA —amber— stop codon, so that the bio-orthogonal UAA is incorporated at that stop codon during translation.25,27 Utilizing amber stop codons as UAA-encoding codons is the most common approach for site-selective incorporation of a single UAA, and other methods are being developed for multiple incorporation of more than one type of UAA into a peptide or protein.17,28
Figure 1.
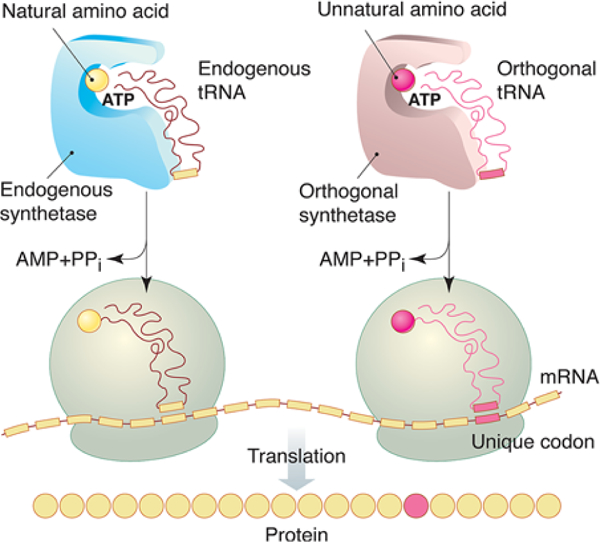
Mechanism of genetically encoded unnatural amino acid incorporation into recombinant peptides and proteins.29 Reprinted with permission from AAAS.
With advances in understanding and engineering cellular machinery to incorporate UAAs, and the chemical synthesis of UAAs bearing a variety of bio-orthogonal handles (Figure 2), the application of UAA incorporation technology to synthesize site-selective and stoichiometric peptide/protein-stealth polymer conjugates is poised to significantly expand.15 However, this method is nontrivial; it can involve long optimization procedures of the expression system, the culture medium, gene induction procedure, and the time and temperature of culture, and despite optimization, the yield of the modified peptide/protein can be significantly lower, even for peptides/proteins that normally have high expression levels.8
Figure 2.
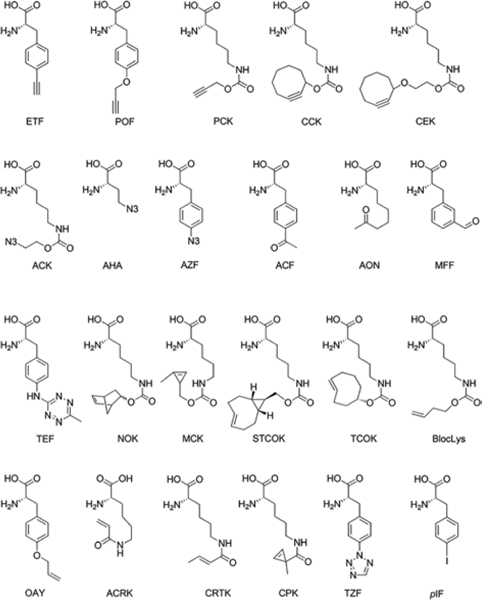
Unnatural amino acids with bio-orthogonal side-chains suitable for bioconjugation. Reproduced from Jung and Kwon17 with permission from The Royal Society of Chemistry.
The bio-orthogonal handle can also be attached to peptides and proteins using site-specific enzymatic ligation methods, though these methods are more restrictive in the site of incorporation, compared to the genetically encoded incorporation of UAAs. Many enzymes can catalyze the modification of peptides/proteins with bio-orthogonal handles, but only a few fulfill the requirement of site-selectivity.30 N-myristoyl transferase (NMT) enzyme recognizes the N-terminal GXXXS/T sequence, where X is any amino acid, and catalyzes acyl transfer from myristoyl-coA to the amino group of the N-terminal glycine within the GXXXS/T sequence, yielding an amide bond. This reaction has been utilized to incorporate azide and alkyne bio-orthogonal groups into proteins in vitro and in vivo.31–33 Protein farnesyl transferase (PFTase) is another enzyme that recognizes a CAAX motif at the C-terminus of peptides/proteins, where A represents an aliphatic (non-polar and hydrophobic) amino acid and X is any amino acid. PFTase catalyzes the transfer of a farnesyl group to the C-terminal cysteine within the CAAX motif from farnesyl pyrophosphate (FPP). PFTase can tolerate a variety of FPP substrate analogs and many CAAX motifs without compromising its activity. Azide34,35 and alkyne35,36 bio-orthogonal groups have been successfully installed in proteins using PFTase. It is important to note, however, that NMT and PFTase both incorporate extra moieties, a myristate (14-carbon fatty acid32) and a farnesyl group (15-carbon isoprenoid ((2Z, 6Z)-3,7,11-trimethyldodeca-2,6,10-trien-1-ol)37 respectively, along with the desired bio-orthogonal handle, which in many cases is not needed to create a site-specific conjugate, and is likely to change in the in vivo activity of the peptide/protein.30
Rush and Bertozzi38 reported the site-selective incorporation of a clickable handle into a protein, catalyzed by formylglycine generating enzyme (FGE), which recognizes the CXPXR sequence, where X is any amino acid except proline within a protein of interest, and oxidizes the cysteine residue (C) within the CXPXR sequence to formylglycine (fG), yielding an aldehyde moiety that can subsequently be reacted with aminooxy or hydrazide groups.38,39 Finally, Sortase A (SrtA), a transpeptidase derived from S. aureus, has been widely used for synthesis of many bioconjugates. SrtA recognizes the LPXTG sequence on the C-terminus of proteins/peptides and catalyzes a transpeptidation reaction between the LPXTG sequence and an oligoglycine substrate bearing a bio-orthogonal handle, yielding a native peptide bond (Figure 3).8,40,41 SrtA was used to catalyze efficient, site-selective, and stoichiometric attachment of an azide moiety to Exendin-4, peptide drug for treatment of type 2 diabetes, and to install and an ATRP initiator at the C-terminus of green fluorescent protein (GFP).8,14 It is important to note that the canonical SrtA function allows site-specific attachment of only one bio-orthogonal handle to the C-terminus of a target protein or peptide. Bellucci et al.40 discovered an alternative “non-canonical” SrtA reaction that involves the attachment of a LPXTG motif to the ε-amino groups of lysine residues (K) in a short four amino acid YKPH motif through an isopeptide bond. This method allows attachment of more than one bio-orthogonal motif to a protein or peptide of interest by incorporating an oligomer of the YKPH motif in the peptide/protein, since the ligation site is the ε-amino group of each individual K residue in the YPKH motif. The exquisite level of positional and stoichiometric control achieved via both canonical and non-canonical activities of SrtA provides a valuable toolbox for synthesis of therapeutic peptides/proteins that are reactive to polymers at a specific site. A potential concern related to utilizing both SrtA and FGE-mediated bio-orthogonal handle attachment for therapeutic applications is the incorporation of an extra CXPXR or LPXT(G)n sequence into peptides and proteins along with the bio-orthogonal moiety of interest, which may induce an undesirable immunological response.14
Figure 3.
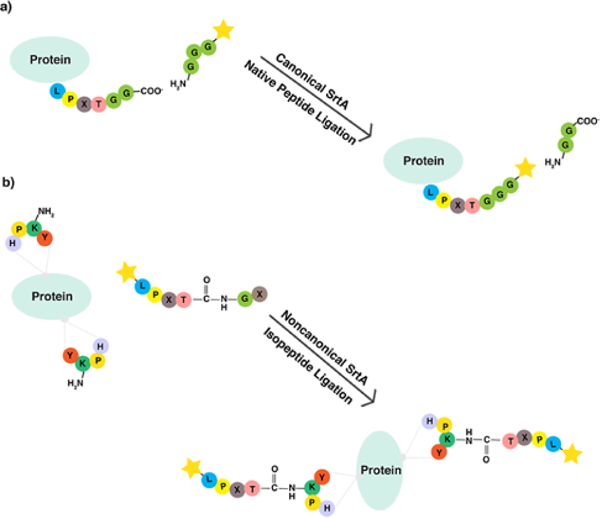
Site-specific installation of a bio-orthogonal handle in a protein by a) canonical SrtA native peptide ligation and b) non-canonical isopeptide ligation.
3. POST-POLYMERIZATION CONJUGATION
Post-polymerization conjugation involves the attachment of one or more pre-synthesized polymer chain(s) to a peptide or protein of interest.12,42 Stealth polymers bearing a bio-orthogonal clickable moiety that enable click reaction with a peptide or protein of interest can be synthesized via two approaches: (1) post-polymerization modification of the polymer with a small molecule bearing the bio-orthogonal handle of interest or (2) using polymerization initiators or chain transfer agents carrying a bio-orthogonal handle of interest (Figure 4).
Figure 4.
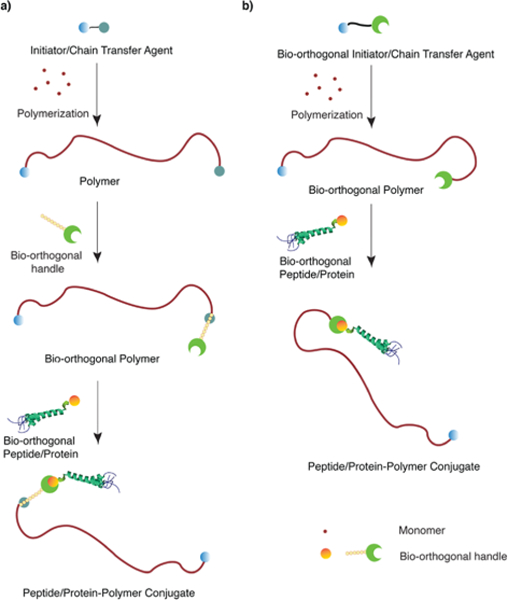
Synthesis of stealth polymer-peptide/protein conjugates. a) Post-polymerization modification of a polymer with reactive end-group that is converted to a bio-orthogonal moiety by chemical derivatization. b) Employing polymerization initiators or chain transfer agents that carry a bio-orthogonal handle of interest to synthesize polymer chains bearing the bio-orthogonal handle.
Pang et al.8 developed a post-polymerization conjugation approach that exploits the versatility of catalyst-free strain-promoted azide-alkyne click chemistry (SPAAC) and showed, for the first time, the site-selective (C-terminal) and stoichiometric (1:1) attachment of Exendin-4, a peptide drug for treatment of type 2 diabetes, to a zwitterionic polymer with ~100% yield in less than 1 h. An exendin-4 fusion was recombinantly expressed with a C-terminal elastin-like polypeptide (ELP) bearing the SrtA recognition sequence —LPETG— located between Exendin and the ELP. An azide bio-orthogonal handle was then installed at the C-terminus of Exendin-4 by exploiting the canonical native peptide ligation feature of SrtA, where SrtA cleaves between the T and G in the LPETG motif and covalently ligates an oligoglycine bearing a clickable azide handle (Figure 5a). In a separate step, a zwitterionic polymer, poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC), with a clickable DBCO handle at the α end-group of the polymer was synthesized by atom transfer radical polymerization (ATRP) (Figure 5b). Finally, the Exendin-4 derivative with an azide group and PMPC with a DBCO end-group were conjugated via SPAAC in aqueous environment yielding a site-specific and stoichiometric (1:1) Exendin-4-PMPC peptide conjugate. The same chemistry was also applied to synthesize Exendin-4-PEG conjugates to compare the stealth effect of the PMPC conjugate with the PEG conjugate. In vitro GLP-1R (Glucagon-like peptide 1 receptor) binding/activation studies revealed a slightly decreased potency (i.e. increased EC50) of Exendin-4 conjugates compared to the native peptide, probably due to the steric hindrance to receptor binding caused by the attached polymer. A single s.c. dose of the conjugate provided glucose control for up to 3 days in six-week-old male C57BL/6J diabetic mice that were kept on a 60 kCal % fat diet. In contrast, the unmodified peptide only afforded up to 4 h of glucose control from a single s.c. injection. These results clearly show that the zwitterionic “stealth” polymer conjugate led to improved therapeutic efficacy of Exendin-4 for treatment of type 2 diabetes. Importantly, the PMPC conjugates were found to be at least as effective as PEG conjugates, suggesting an alternative to the “gold standard” stealth polymer, PEG (Figure 5 c-d). This conjugation methodology was also used to conjugate an azide-functionalized GFP to a PEG-like polycarbonate, a nucleic acid-like polyphosphoester, and a phospholipid-like zwitterionic polymethacrylate separately to highlight its versatility. A potential concern related to this methodology is incomplete conversion of the polymer end-groups to bio-orthogonal handles, which would yield a mixture of polymer chains, although the unmodified polymer can be easily separated by downstream purification after conjugation of the bio-orthogonal-end group modified polymer to the peptide/protein.
Figure 5.
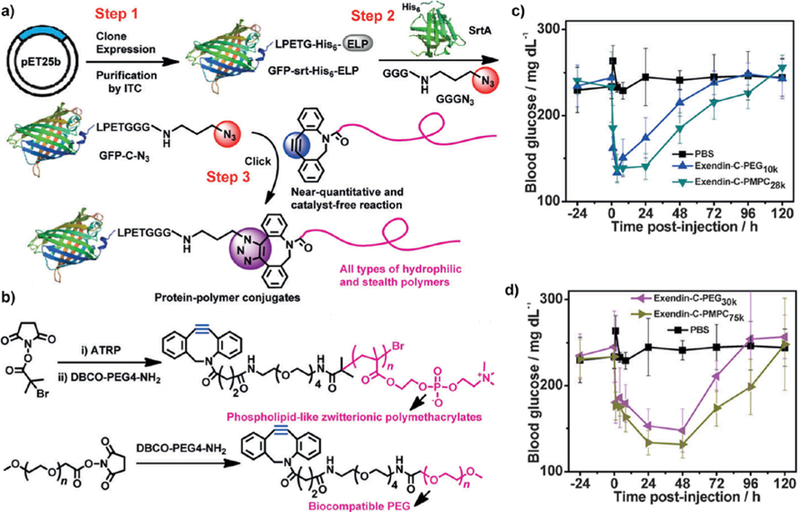
Site-specific post-polymerization conjugation of DBCO-bearing stealth polymers to azide-functionalized GFP and Exendin-4. a) SPAAC-mediated bioconjugation of DBCO-bearing stealth polymers to azide-functionalized GFP after recombinant expression of ELP-GFP and SrtA mediated cleavage of ELP and attachment of an azide at the C-terminus of GFP. b) Synthesis and DBCO end-functionalization of a variety of hydrophilic stealth polymers. Blood glucose levels of fed mice, which were kept on a 60 kcal% fat diet, before and after a single s.c. injection of c) PBS, Exendin-C-PEG10K and Exendin-C-PMPC28K, d) PBS, Exendin-C-PEG30K and Exendin-C-PMPC75K. From Pang et al.8
With the emergence of bio-orthogonal click chemistry as a tool in the bioconjugation toolbox, many clickable initiators and chain transfer agents bearing alpha (α) or omega (ω) end-group bio-orthogonal functionalities, especially azide43–46, trimethylsilyl (TMS)-protected47,48 or unprotected alkyne49 and dibenzocylooctyne (DBCO)50 have enabled the facile synthesis of polymers with clickable moieties (Figure 6). More detailed information on the synthesis of a variety of clickable initiators and chain transfer agents can be found in a recent review paper.51 Moatsou et al.52 utilized this approach to synthesize site-selective and quantitative (1:1 or 1:2) POEGMA conjugates of superfolder green fluorescence protein (sfGFP). To do so, alkyne-functionalized POEGMA was synthesized via Reversible Addition Fragmentation Chain Transfer (RAFT) polymerization using an alkyne-functionalized chain transfer agent and the resulting polymers were conjugated to sfGFP bearing an azide-containing unnatural amino acid via copper-mediated azide-alkyne click chemistry (CuAAC).
Figure 6.
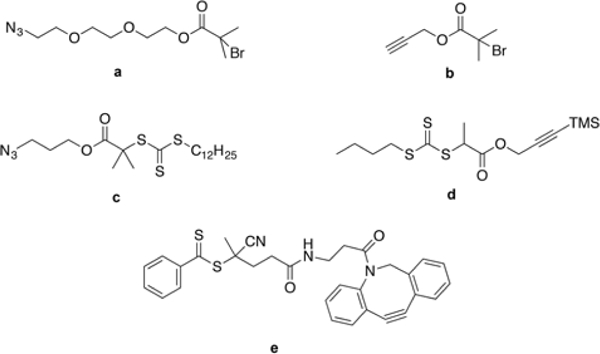
Clickable a) azide, b) alkyne-bearing ATRP initiators, and c) azide, d) TMS-protected alkyne and e) DBCO-functionalized chain transfer agents for RAFT polymerization.
A potential concern related to using functional initiators or chain transfer agents with clickable handles for synthesizing clickable polymers is the possibility of losing the handle due to alternative reactions, which may occur during polymerization. For example, azide groups undergo 1,3-cycloaddition with the double bond of vinyl monomers, especially at elevated temperatures, resulting in loss of bio-orthogonality at the chain end.47–50,53,54 Alkynes, on the other hand, can react with thiols by a thiol-yne click reaction.51 In addition, alkyne-functional initiators and chain transfer agents may not mediate the radical polymerization at desired levels and can be consumed during the reaction, which may increase the polydispersity of the resulting polymer. As a result, to prevent loss of bio-orthogonal handle of interest, side-reactions and the inherent properties initiators or chain transfer agents with clickable handles should be taken into account when designing polymerization and conjugation methodologies.
The versatility of UAA incorporation and click chemistry methodologies was also exploited for the post-polymerization conjugation of several therapeutic proteins with PEG.55–59 Upon conjugation of interferon-β1b (IFN-β1b) bearing an azide-functional UAA at its N-terminus with an alkyne-functionalized linear PEG (10K, 20K, 30K and 40K) and branched PEG 40K via CuAAC, the resulting conjugates, specifically that of branched PEG, showed significantly prolonged plasma half-life in rats and monkeys. Even though the activity of the PEG-IFN-β1b conjugate was found to be slightly lower than that of the native protein, possibly due to the steric hindrance imposed by PEG on receptor binding, the conjugate was more effective in vivo in reducing tumor size than unmodified IFN-β1b.57
Interferon-α2b (IFN-α2b) is another therapeutic protein that was modified to present an azide handle at distinct locations of the protein and was “clicked” on to DBCO-functionalized linear (5K, 10K and 20K) or branched (40K) PEG chains via catalyst-free SPAAC. The resulting IFN-α2b-PEG conjugates were reported to show prolonged plasma half-life but reduced activity.60 The first phase IIB clinical study of a therapeutic protein-polymer conjugate —of human growth hormone (hGH)-PEG conjugates— bearing a bio-orthogonal UAA was launched in 2014.17 To synthesize this conjugate, first, a bio-orthogonal UAA, p-acetylphenylalanine (pAcF), was selectively incorporated into hGH at 20 distinct locations, yielding hGH with a bio-orthogonal reactive handle, which was then conjugated to an aminooxy PEG 30K via oxime ligation. The pre-clinical studies of the hGH-PEG conjugates showed enhanced blood circulation time in GH-deficient rodents and humans compared to unmodified hGH.55
Post-polymerization bioconjugation in combination with bio-orthogonal click chemistry provides fast and versatile synthesis of therapeutic protein/peptide-stealth polymer conjugates with an exquisite level of positional and quantitative control. Compared to in situ polymer conjugation, better characterization of conjugates is possible because the polymer can be extensively characterized by techniques developed for polymer characterization, prior to attachment to the peptide or protein.42,61 Post-polymerization conjugation may also allow easier scale-up, which is a significant advantage for the ultimate clinical end-use of therapeutic conjugates. A potential concern related to this methodology, however, is the need for downstream purification techniques to separate the conjugate from free polymer and peptide/protein.61
4. IN SITU POLYMER CONJUGATION
In situ polymer conjugation involves polymerization from a specific site on a peptide or protein, which serves as an initiator or a chain transfer agent depending on the polymerization methodology.12,62 In situ polymer conjugation typically employs living radical polymerization (LRP) techniques such as ATRP, RAFT polymerization, SET-LRP and Nitroxide-Mediated Radical Polymerization (NMP), as they can be carried out under conditions that are not deleterious to the structure and hence activity of most peptides or proteins. As a detailed discussion of polymerization techniques is not within the scope of this review, we only provide some necessary information related to design of conjugates as discussing the literature studies below.
Qi et al.63 synthesized poly(oligoethylene glycol)methyl ether methacrylate) (POEGMA) —a PEG-like polymer— conjugate of Exendin-4 by in situ Activator Regenerated by Electron Transfer (ARGET) ATRP (Figure 7). They site-specifically attached an ATRP initiator to the C-terminus of the peptide via SrtA-mediated native peptide ligation at the C-terminus of Exendin-4, yielding an Exendin-4 ATRP macroinitiator (Figure 7a). The reaction product was characterized by tandem mass spectroscopy (MS/MS) to confirm site-selective and stoichiometric (1:1) installation of the ATRP initiator. The macroinitiator was then used to synthesize well-defined POEGMA conjugates with controlled molecular weight and low polydispersity (<1.2) by in situ ATRP (Figure 7b) in aqueous buffer containing up to 30% methanol. An in vitro GLP-1 receptor activation assay of the Exendin-4-POEGMA conjugates showed that POEGMA attachment slightly reduced the potency of Exendin-4 in a molecular weight dependent manner, possibly due to steric hindrance imposed on receptor binding by the POEGMA chain. The therapeutic efficacy of the conjugates, as defined by their blood glucose lowering effect in male C57BL/6J diabetic mice that were kept on a 60 kCal % fat diet lasted up to 120 hours, which was twenty-fold longer than that of native Exendin-4 (Figure 7c). The in vitro antigenicity of the conjugates with ~9 side-chain ethylene glycol (EG) repeating units was found to be much lower than that of two FDA-approved PEGylated protein drugs. Shortening the ethylene glycol chains to 3 EG repeating units was found to eliminate the antigenicity of conjugates towards pre-existing anti-PEG antibodies without compromising the in vivo efficacy of the peptide (Figure 7d).
Figure 7.
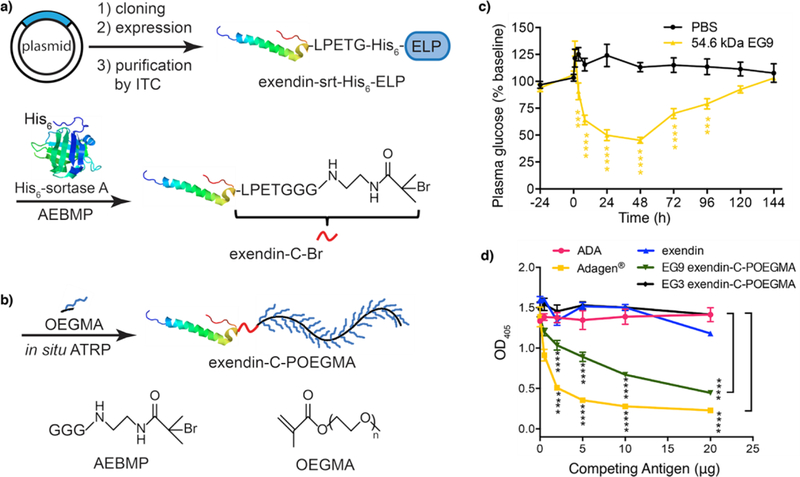
Grafting POEGMA from a therapeutic peptide — Exendin-4. a) Recombinant expression of ELP fusion of Exendin-4 peptide, and following ATRP initiator (AEBMP) attachment via canonical SrtA mediated native peptide ligation to C-terminus. b) In situ ARGET ATRP of OEGMA yielding Exendin-C-POEGMA. c) Blood glucose levels of fed mice, which were kept 60 kCal % fat diet before and after s.c. injections of PBS and 54.6 kDa Exendin-C-POEGMA (EG9). d) Comparison of anti-PEG reactivity of native Exendin-4 and adenosine deaminase (ADA), EG3 and EG9 Exendin-C-POEGMA conjugates and PEGylated ADA, Adagen™, via competitive ELISA. Adapted by permission from Macmillan Publishers Ltd: Nature Biomedical Engineering63 copyright 2016.
UAAs bearing a polymerization initiator can also be used as macroinitiators for in situ synthesis of site-specific and quantitative protein/peptide-polymer conjugates. To best of our knowledge, to date, only green fluorescent protein (GFP) with a UAA at single site that presents an ATRP initiator on its side chain has been recombinantly expressed and used as an ATRP macroinitiator (Figure 8). The GFP macroinitiator was then used to synthesize POEGMA conjugates of GFP, with no adverse impact on the structure of the protein.64
Figure 8.
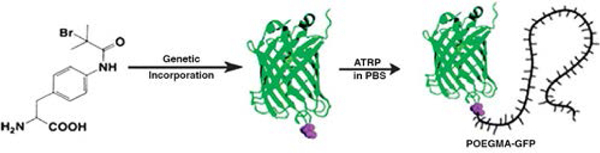
Genetic incorporation of an UAA initiator into GFP followed by in situ ATRP to obtain a GFP-POEGMA conjugate. Adapted with permission from Peeler et al.64 Copyright 2016 American Chemical Society.
In situ RAFT polymerization is another method that offers positional control over the site of conjugation of a polymer. In situ RAFT requires the synthesis of a macroRAFT agent, which is a chain transfer agent that is appended to a peptide or protein of interest. MacroRAFT agents can be attached to the peptide or protein either at the R or Z end-group and upon RAFT polymerization, leading to a conjugate that has α-or ω-terminal polymer chains grafted to the biomolecule, respectively (Figure 9).42 This unprecedented level of control over the location of chain end functionality is specific to RAFT polymerization.65 However, it is important to note that the labile -C(=S)-S bond between the biomolecule and polymer in conjugates synthesized via the Z end-group approach can be cleaved in the presence of primary amines, which might be a potential concern depending on the application of the conjugate12,42 and may limit the applicability of in situ RAFT polymerization employing unprotected peptides or proteins as macroRAFT agents.65–67
Figure 9.
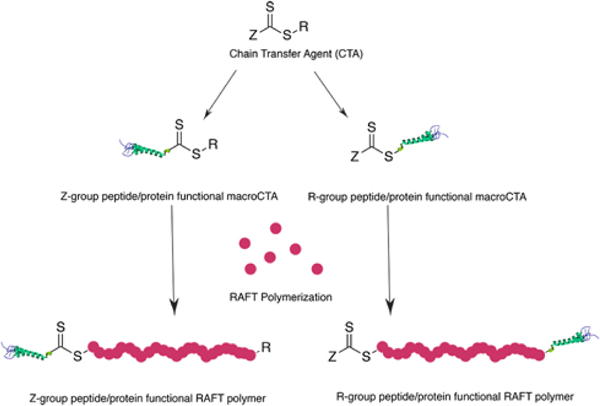
Installation of a peptide or protein at the R or Z end-group of a RAFT chain transfer agent, yielding α-or ω-terminal polymer chains grafted to the biomolecule (Protein Data Bank ID: 1JRJ).
Zhao et al.65 synthesized Z end-group glutathione tripeptide (GSH) functional macroRAFT agents and used them in the RAFT polymerization of several vinyl monomers. The resulting conjugates were found to have low polydispersity and controlled molecular weight demonstrating the utility of a Z end group macroRAFT agent for synthesis of a peptide-polymer conjugate. The living character of the GSH-poly(N-isopropylacrylamide) (GSH-PNIPAAM) and GSH-poly(N,N-dimethylacrylamide) (GSH-PDMA) conjugates was demonstrated by subsequent polymerization of a second block with other vinyl monomers through the thiocarbonylthio bond that is retained at the ω-terminal of polymer chains.
The advantage of in situ polymer conjugation compared to post-polymerization conjugation is that it offers simpler purification, because the polymer solely grows from the peptide or protein, so that only the unreacted peptide or protein needs to be separated from the conjugate which is relatively easy to do, given the difference between the size and physicochemical properties of the peptide/protein and conjugate.12 A potential disadvantage of this methodology compared to post-polymerization conjugation, however, is the need to individually optimize the polymerization conditions for every biomolecule.
CONCLUSION
Therapeutic applications of proteins and peptides have significantly expanded due to their potency, specificity and lack of toxicity. However, the pharmacokinetic profiles of most peptide and proteins are unsatisfactory, and need to be improved to make them useful as drugs. Efforts to overcome this drawback have led to the conjugation of therapeutic proteins and peptides with stealth polymers that confer long —and tunable— pharmacokinetics.
A review of the recent literature on therapeutic protein/peptide-polymer conjugates, as discussed in this review, reveals that much progress has been made in the past decade by exploiting advances in genetically encoded bio-orthogonal amino acid incorporation into peptide/proteins, enzyme catalyzed conjugation, and polymer chemistry for the synthesis of stealth polymer conjugates of peptides and proteins with precise control over the site and stoichiometry of the polymer conjugate and control over the physicochemical properties of the stealth polymers, such as their sequence, architecture, length and polydispersity. Considering the exceptional benefits of peptide and protein therapeutics and progress in the precision synthesis of stealth polymers, it is expected that stealth polymer conjugates of peptide and protein drugs will remain —for the foreseeable future— an important class of therapeutic agents for the treatment of a variety of diseases.
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health (R01-DK092665) to A.C.
ABBREVIATIONS
Abbreviations of unnatural amino acids with bio-orthogonal side-chains suitable for bioconjugation in Figure 2:
- (ETF)
p-ethynyl-L-phenylalanine
- (POF)
p-propargyloxy-L-phenylalanine
- (PCK)
N-6-[(2-propynyloxy)carbonyl]-L-lysine
- (CCK)
N-ε-(cyclooct-2-yn-1-yloxy)carbonyl-L-lysine
- (CEK)
N-ε-(2-(cyclooct-2-yn-1-yloxy)ethyl)car-bonyl-L-lysine
- (ACK)
N-6-[(2-azidoethoxy) carbonyl]-L-lysine
- (AHA)
4-azido-L-homoalanine
- (AZF)
p-azido-L-phenylalanine
- (ACF)
p-acetyl-L-phenylalanine
- (AON)
2-amino-8-oxononanoic acid
- (MFF)
m-formyl-L-phenylalanine
- (TEF)
4-(6-methyl-s-tetrazin-3-yl)amino-L-phenylalanine
- (NOK)
N-ε-5-norbornene-2-yloxycarbonyl-L-lysine
- (MCK)
N-ε-[((2-methyl-cycloprop-2-en-1-yl)methoxy)carbonyl]-L-lysine
- (STCOK)
(2S)-2-amino-6-[({[(1R,4E,8S,9S)-bicyclo[6.1.0]non-4-en-9-yl]methoxy}carbonyl)amino]-hexanoic acid
- (TCOK)
(2S)-2-amino-6-({[(1S,4E)-cyclooct-4-en-1-yloxy]-carbonyl}amino)hexanoic acid
- (OAY)
O-allyl-L-tyrosine
- (ACRK)
N-ε-acry-loyl-L-lysine
- (CRTK)
N-ε-crotonyl-L-lysine
- (CPK)
N-ε-(1-methylcycloprop-2-enecarboxamido)-L-lysine
- (TZF)
p-tetrazole-L-phenylalanine
- (BlocLys)
2-amino-6-[(but-3-enyloxy)carbonylamino]hexanoic acid
- (pIF)
p-iodo-L-phenylalanine
REFERENCES
- 1.Fosgerau K, and Hoffmann T (2015) Peptide therapeutics: current status and future directions. Drug Discovery Today 20, 122–8. [DOI] [PubMed] [Google Scholar]
- 2.Kaspar AA, and Reichert JM (2013) Future directions for peptide therapeutics development. Drug Discovery Today 18, 807–17. [DOI] [PubMed] [Google Scholar]
- 3.Vlieghe P, Lisowski V, Martinez J, and Khrestchatisky M (2010) Synthetic therapeutic peptides: science and market. Drug Discovery Today 15, 40–56. [DOI] [PubMed] [Google Scholar]
- 4.THPdb: A Database of FDA Approved Therapeutic Peptides and Proteins. http://crdd.osdd.net/raghava/thpdb/index.html (Accessed: Nov 4, 2016).
- 5.FDA Approved Drug Products. https://www.accessdata.fda.gov/scripts/cder/drugsatfda. (Accessed: Nov 2, 2016).
- 6.Sawant SG, Fielden MR, and Black KA (2014) Evaluation of genotoxicity testing of FDA approved large molecule therapeutics. Regul. Toxicol. Pharmacol. 70, 87–97. [DOI] [PubMed] [Google Scholar]
- 7.Global Markets for Bioengineered Protein Drugs http://www.prnewswire.com/news-releases/global-markets-for-bioengineered-protein-drugs-300011924.html. (Accessed: Nov 4, 2016).
- 8.Pang Y, Liu J, Qi Y, Li X, and Chilkoti A (2016) A Modular Method for the High-Yield Synthesis of Site-Specific Protein–Polymer Therapeutics. Angew. Chem., Int. Ed. 128, 10452–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chames P, Van Regenmortel M, Weiss E, and Baty D (2009) Therapeutic antibodies: successes, limitations and hopes for the future. Br. J. Pharmacol. 157, 220–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parrott MC, and DeSimone JM (2012) Drug delivery: Relieving PEGylation. Nat Chem 4, 13–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turecek PL, Bossard MJ, Schoetens F, and Ivens IA (2016) PEGylation of Biopharmaceuticals: A Review of Chemistry and Nonclinical Safety Information of Approved Drugs. J. Pharm. Sci. 105, 460–75. [DOI] [PubMed] [Google Scholar]
- 12.Pelegri-O’Day EM, Lin EW, and Maynard HD (2014) Therapeutic protein-polymer conjugates: advancing beyond PEGylation. J. Am. Chem. Soc. 136, 14323–32. [DOI] [PubMed] [Google Scholar]
- 13.Qi Y, Amiram M, Gao W, McCafferty DG, and Chilkoti A (2013) Sortase-Catalyzed Initiator Attachment Enables High Yield Growth of a Stealth Polymer from the C Terminus of a Protein. Macromol. Rapid Commun. 34, 1256–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qi YZ, and Chilkoti A (2014) Growing polymers from peptides and proteins: a biomedical perspective. Polym. Chem. 5, 266–76. [Google Scholar]
- 15.Spicer CD, and Davis BG (2014) Selective chemical protein modification. Nat. Commun. 5, 4740–54. [DOI] [PubMed] [Google Scholar]
- 16.McKay CS, and Finn MG (2014) Click chemistry in complex mixtures: bioorthogonal bioconjugation. Chem. Biol. 21, 1075–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jung S, and Kwon I (2016) Expansion of bioorthogonal chemistries towards site-specific polymer-protein conjugation. Polym. Chem. 7, 4584–98. [Google Scholar]
- 18.Karver MR, Weissleder R, and Hilderbrand SA (2012) Bioorthogonal Reaction Pairs Enable Simultaneous, Selective, Multi-Target Imaging. Angew. Chem., Int. Ed. 51, 920–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knight JC, and Cornelissen B (2014) Bioorthogonal chemistry: implications for pretargeted nuclear (PET/SPECT) imaging and therapy. Am. J. Nucl. Med. Mol. Imaging 4, 96–113. [PMC free article] [PubMed] [Google Scholar]
- 20.Soellner MB, Nilsson BL, and Raines RT (2006) Reaction Mechanism and Kinetics of the Traceless Staudinger Ligation. J. Am. Chem.Soc. 128, 8820–28. [DOI] [PubMed] [Google Scholar]
- 21.Lorenzo MM, Decker CG, Kahveci MU, Paluck SJ, and Maynard HD (2016) Homodimeric Protein–Polymer Conjugates via the Tetrazine–trans-Cyclooctene Ligation. Macromolecules 49, 30–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eising S, Lelivelt F, and Bonger KM (2016) Vinylboronic Acids as Fast Reacting, Synthetically Accessible, and Stable Bioorthogonal Reactants in the Carboni–Lindsey Reaction. Angew. Chem., Int. Ed. 55, 12243–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang W, and Becker ML (2014) “Click” reactions: a versatile toolbox for the synthesis of peptide-conjugates. Chem. Soc. Rev. 43, 7013–39. [DOI] [PubMed] [Google Scholar]
- 24.Wang L, Magliery TJ, Liu DR, and Schultz PG (2000) A New Functional Suppressor tRNA/Aminoacyl−tRNA Synthetase Pair for the in Vivo Incorporation of Unnatural Amino Acids into Proteins. J. Am. Chem. Soc. 122, 5010–11. [Google Scholar]
- 25.Wang L, Brock A, Herberich B, and Schultz PG (2001) Expanding the genetic code of Escherichia coli. Science 292, 498–500. [DOI] [PubMed] [Google Scholar]
- 26.Jewett MC, and Noireaux V (2016) Synthetic biology: Tailor-made genetic codes. Nat. Chem. 8, 291–92. doi: 10.1038/nchem.2484. [DOI] [PubMed] [Google Scholar]
- 27.Davis L, and Chin JW (2012) Designer proteins: applications of genetic code expansion in cell biology. Nat. Rev. Mol. Cell Biol. 13, 168–82. [DOI] [PubMed] [Google Scholar]
- 28.Lee BS, Shin S, Jeon JY, Jang KS, Lee BY, Choi S, and Yoo TH (2015) Incorporation of Unnatural Amino Acids in Response to the AGG Codon. ACS Chem. Biol. 10, 1648–53. [DOI] [PubMed] [Google Scholar]
- 29.Wang L (2003) Expanding the genetic code. Science 302, 584–85. [DOI] [PubMed] [Google Scholar]
- 30.Rashidian M, Dozier JK, and Distefano MD (2013) Chemoenzymatic Labeling of Proteins: Techniques and Approaches. Bioconjugate Chem. 24, 1277–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heal WP, Wickramasinghe SR, Bowyer PW, Holder AA, Smith DF, Leatherbarrow RJ, and Tate EW (2008) Site-specific N-terminal labelling of proteins in vitro and in vivo using N-myristoyl transferase and bioorthogonal ligation chemistry. Chem. Commun. (Cambridge, U.K.) 4, 480–82. [DOI] [PubMed] [Google Scholar]
- 32.Heal WP, Wright MH, Thinon E, and Tate EW (2012) Multifunctional protein labeling via enzymatic N-terminal tagging and elaboration by click chemistry. Nat. Protoc. 7, 105–17. [DOI] [PubMed] [Google Scholar]
- 33.Heal WP, Wickramasinghe SR, Leatherbarrow RJ, and Tate EW (2008) N-Myristoyl transferase-mediated protein labelling in vivo. Org. Biomol. Chem. 6, 2308–15. [DOI] [PubMed] [Google Scholar]
- 34.Kho Y et al. (2004) A tagging-via-substrate technology for detection and proteomics of farnesylated proteins. Proc. Natl. Acad. Sci. U.S.A. 101, 12479–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Labadie GR, Viswanathan R, and Poulter CD (2007) Farnesyl Diphosphate Analogues with ω-Bioorthogonal Azide and Alkyne Functional Groups for PFTase-Catalyzed Ligation Reactions. J. Org. Chem. 72, 9291–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duckworth BP, Zhang ZY, Hosokawa A, and Distefano MD (2007) Selective labeling of proteins by using protein farnesyltransferase. ChemBioChem 8, 98–105. [DOI] [PubMed] [Google Scholar]
- 37.DrugBank: Farnesol http://www.drugbank.ca/drugs/DB0250. (Accessed: 11-07-2016).
- 38.Rush JS, and Bertozzi CR (2008) New Aldehyde Tag Sequences Identified by Screening Formylglycine Generating Enzymes in Vitro and in Vivo. J. Am. Chem. Soc. 130, 12240–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.York D, Baker J, Holder PG, Jones LC, Drake PM, Barfield RM, Bleck GT, and Rabuka D (2016) Generating aldehyde-tagged antibodies with high titers and high formylglycine yields by supplementing culture media with copper(II). BMC Biotechnol. 16, 23–34. doi: 10.1186/s12896-016-0254-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bellucci JJ, Bhattacharyya J, and Chilkoti A (2015) A noncanonical function of sortase enables site-specific conjugation of small molecules to lysine residues in proteins. Angew. Chem., Int. Ed. 54, 441–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen L et al. (2016) Improved variants of SrtA for site-specific conjugation on antibodies and proteins with high efficiency. Sci. Rep. 6, 31899–911. doi: 10.1038/srep31899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bulmus V (2011) RAFT polymerization mediated bioconjugation strategies. Polym. Chem. 2, 1463–72. [Google Scholar]
- 43.Boyer C, Liu J, Bulmus V, Davis TP, Barner-Kowollik C, and Stenzel MH (2008) Direct synthesis of well-defined heterotelechelic polymers for bioconjugations. Macromolecules 41, 5641–50. [Google Scholar]
- 44.Gondi SR, Vogt AP, and Sumerlin BS (2007) Versatile pathway to functional telechelics via RAFT polymerization and click chemistry. Macromolecules 40, 474–81. [Google Scholar]
- 45.De P, Gondi SR, and Sumerlin BS (2008) Folate-conjugated thermoresponsive block copolymers: Highly efficient conjugation and solution self-assembly. Biomacromolecules 9, 1064–70. [DOI] [PubMed] [Google Scholar]
- 46.Sen Gupta S, Raja KS, Kaltgrad E, Strable E, and Finn MG (2005) Virus-glycopolymer conjugates by copper(I) catalysis of atom transfer radical polymerization and azide-alkyne cycloaddition. Chem. Commun. (Cambridge, U.K.) 4315–7. [DOI] [PubMed] [Google Scholar]
- 47.Akeroyd N, and Klumperman B (2011) The combination of living radical polymerization and click chemistry for the synthesis of advanced macromolecular architectures. Eur. Polym. J. 47, 1207–31. [Google Scholar]
- 48.Kakwere H, Chun CKY, Jolliffe KA, Payne RJ, and Perrier S (2010) Polymer-peptide chimeras for the multivalent display of immunogenic peptides. Chem. Commun. (Cambridge, U.K.) 46, 2188–90. [DOI] [PubMed] [Google Scholar]
- 49.Dehn S, Castelletto V, Hamley IW, and Perrier S (2012) Altering peptide fibrillization by polymer conjugation. Biomacromolecules 13, 2739–47. [DOI] [PubMed] [Google Scholar]
- 50.Pola R, Braunova A, Laga R, Pechar M, and Ulbrich K (2014) Click chemistry as a powerful and chemoselective tool for the attachment of targeting ligands to polymer drug carriers. Polym. Chem. 5, 1340–1350. [Google Scholar]
- 51.Mansfeld U, Pietsch C, Hoogenboom R, Becer CR, and Schubert US (2010) Clickable initiators, monomers and polymers in controlled radical polymerizations - a prospective combination in polymer science. Polym. Chem. 1, 1560–98. [Google Scholar]
- 52.Moatsou D, Li J, Ranji A, Pitto-Barry A, Ntai I, Jewett MC, and O’Reilly RK (2015) Self-Assembly of Temperature-Responsive Protein–Polymer Bioconjugates. Bioconjugate Chem. 26, 1890–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tao L, Mantovani G, Lecolley F, and Haddleton DM (2004) α-Aldehyde Terminally Functional Methacrylic Polymers from Living Radical Polymerization: Application in Protein Conjugation “Pegylation”. J. Am. Chem. Soc. 126, 13220–21. [DOI] [PubMed] [Google Scholar]
- 54.Ladmiral V, Legge TM, Zhao YL, and Perrier S (2008) “Click” chemistry and radical polymerization: Potential loss of orthogonality. Macromolecules 41, 6728–32. [Google Scholar]
- 55.Cho H et al. (2011) Optimized clinical performance of growth hormone with an expanded genetic code. Proc. Natl. Acad. Sci. U.S.A. 108, 9060–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mu J et al. (2012) FGF21 Analogs of Sustained Action Enabled by Orthogonal Biosynthesis Demonstrate Enhanced Antidiabetic Pharmacology in Rodents. Diabetes 61, 505–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nairn NW, Shanebeck KD, Wang A, Graddis TJ, VanBrunt MP, Thornton KC, and Grabstein K (2012) Development of Copper-Catalyzed Azide–Alkyne Cycloaddition for Increased in Vivo Efficacy of Interferon β−1b by Site-Specific PEGylation. Bioconjugate Chem. 23, 2087–97. [DOI] [PubMed] [Google Scholar]
- 58.Debets MF, van Berkel SS, Schoffelen S, Rutjes F, van Hest JCM, and van Delft FL (2010) Aza-dibenzocyclooctynes for fast and efficient enzyme PEGylation via copper-free (3+2) cycloaddition. Chem. Commun. (Cambridge, U.K.) 46, 97–9. [DOI] [PubMed] [Google Scholar]
- 59.Hoffmann E, Streichert K, Nischan N, Seitz C, Brunner T, Schwagerus S, Hackenberger CPR, and Rubini M (2016) Stabilization of bacterially expressed erythropoietin by single site-specific introduction of short branched PEG chains at naturally occurring glycosylation sites. Mol. BioSyst. 12, 1750–55. doi: 10.1039/C5MB00857C. [DOI] [PubMed] [Google Scholar]
- 60.Zhang B, Xu H, Chen J, Zheng Y, Wu Y, Si L, Wu L, Zhang C, Xia G, Zhang L, and Zhou D (2015) Development of next generation of therapeutic IFN-α2b via genetic code expansion. Acta Biomater.19, 100–11. [DOI] [PubMed] [Google Scholar]
- 61.Broyer RM, Grover GN, and Maynard HD (2011) Emerging synthetic approaches for protein-polymer conjugations. Chem. Commun. (Cambridge, U.K.) 47, 2212–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cobo I, Li M, Sumerlin BS, and Perrier S (2015) Smart hybrid materials by conjugation of responsive polymers to biomacromolecules. Nat. Mater. 14, 143–59. [DOI] [PubMed] [Google Scholar]
- 63.Qi Y et al. (2016) A C-terminal PEG-like brush polymer conjugate of exendin-4 reduces blood glucose for up to five days and eliminates PEG antigenicity. Nat. Biomed. Eng. 1, 0002. doi: 10.1038/s41551-016-0002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peeler JC, Woodman BF, Averick S, Milyake-Stoner SJ, Stokes AL, Hess KR, Matyjaszewski K, and Mehl RA (2010) Genetically encoded initiator for polymer growth from proteins. J. Am. Chem. Soc. 132, 13575–77. [DOI] [PubMed] [Google Scholar]
- 65.Zhao Y, and Perrier S (2007) Synthesis of well-defined conjugated copolymers by RAFT polymerization using cysteine and glutathione-based chain transfer agents. Chem. Commun. (Cambridge, U.K.) 4294–96. [DOI] [PubMed] [Google Scholar]
- 66.Boyer C, Bulmus V, Liu J, Davis TP, Stenzel MH, and Barner-Kowollik C (2007) Well-defined protein-polymer conjugates via in situ RAFT polymerization. J. Am. Chem. Soc. 129, 7145–54. [DOI] [PubMed] [Google Scholar]
- 67.Liu J, Bulmus V, Herlambang DL, Barner-Kowollik C, Stenzel MH, and Davis TP (2007) In situ formation of protein-polymer conjugates through reversible addition fragmentation chain transfer polymerization. Angew. Chem., Int. Ed. 46, 3099–103. [DOI] [PubMed] [Google Scholar]