

Abstract
Nanoscale carriers with an acid-labile linker between the carrier and drug are commonly used for drug delivery. However, their efficacy is potentially limited by inefficient linker cleavage, and lysosomal entrapment of drugs. To address these critical issues, we developed a new imaging method that spatially overlays the location of a nanoparticle and the released drug from the nanoparticle, on a map of the local intracellular pH that delineates individual endosomes and lysosomes, and the therapeutic intracellular target of the drug—the nucleus. We used this method to quantitatively map the intracellular fate of micelles of a recombinant polypeptide conjugated with doxorubicin via an acid-labile hydrazone linker as a function of local pH and time within live cells. We found that hydrolysis of the acid-labile linker is incomplete because the pH range of 4–7 in the endosomes and lysosomes does not provide complete cleavage of the drug from the nanoparticle, but that once cleaved, the drug escapes the acidic endo-lysosomal compartment into the cytosol and traffics to its therapeutic destination—the nucleus. This study also demonstrated that unlike free drug, which enters the cytosol directly through the cell membrane and then traffics into the nucleus, the nanoparticle-loaded drug almost exclusively traffics into endosomes and lysosomes upon intracellular uptake, and only reaches the nucleus after acid-triggered drug release in the endo-lysosomes. This methodology provides a better and more quantitative understanding of the intracellular behavior of drug-loaded nanoparticles, and provides insights for the design of the next-generation of nanoscale drug delivery systems.
Keywords: Ratiometric fluorescence imaging, Fluorescence de-quenching, Quantitative image analysis, Nanoparticle drug delivery, Acid-labile linker
1. Introduction
Nanoscale drug delivery systems are a common strategy to enhance the delivery of anti-cancer drugs to tumors [1,2]. These systems—typically involving a carrier such as a polymeric nanocarrier [3] or an inorganic nanoparticle [4]—often incorporate acid-labile linkers between the drug and the carrier to enable intracellular controlled drug release in endo-lysosomes [5–8]. However, two critical issues related to the intracellular fate of pH-responsive drug delivery systems have not been quantitatively investigated in live cells. Because most drugs act in locations outside endo-lysosomes in cells and the susceptibility of various acid-labile linkers to acid-sensitive hydrolysis is different, the first question relates to the cleavage efficiency of acid-labile linkers within endo-lysosomes [9,10]. A second and related issue is the mechanism and kinetics of transport of the drug within the cell and especially out of the endo-lysosomal compartment into the cytosol [11,12]. This is because nanoparticle-delivered drugs usually enter cells by endocytosis and are then trafficked to acidic endosomes and lysosomes [13], where the conjugated drug may be trapped. The issue of lysosomal entrapment is an even greater concern with weakly basic drugs, such as doxorubicin, as they are ionized in the acidic endosomes and lysosomes, and studies have shown that the ionized form of the drug cannot diffuse across the endo-lysosomal membrane to the cytosol as freely as the nonionized form of the drug [14,15]. Furthermore, the pH of endosomes and lysosomes has a broad distribution, so that it is critical to investigate drug release from nanoparticles as a function of pH within live cells at a spatial resolution that captures the dynamics of the intracellular trafficking of the drug-loaded nanoparticles and release of the drug from nanoparticles at the level of individual endosomes and lysosomes. Answering these questions is critical for the rational design of nanoparticle based drug delivery systems, and provides the motivation for this study.
Previous studies have attempted to address these issues using various fluorescence techniques, such as fluorescence resonance energy transfer (FRET) [16], fluorescence-lifetime imaging microscopy (FLIM) [17] and fluorescence de-quenching [18,19]. These studies arrived at the general conclusions that acid-labile linkers can be successfully cleaved in acidic endo-lysosomes, and that the released drug can escape from endo-lysosomes to the cytosol. However, none of these studies quantified the cleavage of acid-labile linkers across the pH range that exists in endo-lysosomes nor did they quantitatively spatially map the intracellular trafficking and distribution of drugs and carriers associated with local pH.
To address these questions quantitatively, we have developed a new method to quantify the intracellular distribution of drug-loaded pH-responsive nanoparticles in endosomes and lysosomes associated with local pH in live cells. This method involves pixel-by-pixel analysis of multi-color fluorescence images of: (1) ratiometric fluorescence imaging of a pH-sensitive dye to map the intracellular pH [20], (2) fluorescence imaging of a nanoparticle carrier, and (3) fluorescence de-quenching of a fluorescent drug—doxorubicin—to spatially map the intracellular distribution of drug-loaded nanoparticles, and release of the drug from the nanoparticle as a function of pH and time within live cells. Although each technique used in this paper has been reported separately in previous papers [21–23], these techniques have, to the best of our knowledge, never been combined to spatially and—quantitatively—map the intracellular fate of pH-responsive drug-loaded nanoparticles.
We used this methodology to track the spatial distribution, trafficking and drug release in live cells of a well-characterized, drug-loaded nanoparticle with an acid-triggered drug release mechanism. The nanoparticles are spherical micelles of a chimeric polypeptide (CP)-doxorubicin (Dox) conjugate. The CP consists of a hydrophilic elastin-like polypeptide (ELP) and a short peptide trailer that contains multiple cysteine residues to which Dox is attached by an acid-labile hydrazone linker. CP-Dox self-assembles into nanoscale spherical micelles in aqueous solution. Compared with the free drug, pH-sensitive CP-Dox micelles have significantly longer plasma half-life, enhanced tumor accumulation and higher therapeutic efficacy in vivo, though the precise mechanism of drug release within cells has not been elucidated [24–26]. Because this nanoparticle is a precise molecularly engineered delivery system with demonstrated in vivo efficacy across multiple tumor models and multiple drugs such as doxorubicin and more recently paclitaxel [27], it provides a well-characterized system to examine the intracellular fate of drugs that are covalently conjugated and loaded into nanoparticles through acid-labile linkers.
We successfully quantified the uptake and intracellular trafficking of doxorubicin-loaded polypeptide nanoparticles and showed that unlike free drug, which can enter the cytosol directly through the cell membrane and then traffic into the nucleus, Dox-loaded nanoparticles are almost exclusively trafficked into endosomes and lysosomes upon intracellular entry. We quantified the kinetics of drug release in endo-lysosomes as a function of pH in live cells and demonstrated the pH-dependent cleavage of acid-labile hydrazone linker below pH 6 in endo-lysosomes. We found that the weakly basic drug released from the nanoparticle can successfully escape the acidic endo-lysosomes, and then enters the cytosol and traffics to the nucleus. This more circuitous route retards the kinetics of cytosolic and nuclear accumulation of the drug that is delivered by the nanoparticle compared to delivery of free drug, but does not change the final intracellular distribution of the drug between different organelles compared with free drug. Our study also points out that an efficient mechanism of acid-triggered drug release with a pH optimum between pH 4 and 7 is ideal to enable maximum release of sequestered drug from the nanoparticle.
2. Materials and methods
2.1. Materials
Lysosensor yellow/blue DND-160 and Alexa Fluor 488 NHS ester were purchased from Life Technology. BHQ2 amine was from Biosearch Technology. Dox-SMCC was from MedKoo Biosciences. TCEP, BMPH, sulfo-SMCC were from Thermo Scientific. Chloroquine and all other reagents were from Sigma.
2.2. Cell culture
Human pharynx squamous cell carcinoma (FaDu) cells were grown in Eagle’ s Minimum Essential Medium supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 100 U/ml penicillin and 100 μg/ml streptomycin. Cells were maintained at 37 °C with 5% CO2.
2.3. Chemical conjugation and micelle preparation
The CP consists of two segments: the first segment is an elastin-like polypeptide (ELP). ELPs are a class of artificial peptide polymers composed of a XGVPG pentapeptide repeat derived from human tropoelastin, where the guest residue, X, can be any amino acid except proline [28]. The specific ELP we used herein consists of 160 repeats of XGVPG, where X = V: A: G [1: 8: 7]. The second—drug attachment segment—is a WPC(GGC)7 peptide that is embedded at the C-terminus of the CP, where the cysteine residues provide a unique site for conjugation of drugs. In this study, a CP with the amino acid sequence—SKGPG(XGVPG)160WPC(GGC)7—where X = V: A: G [1: 8:7]) was synthesized and purified as previously described [24].
2.3.1. CP-hyd-Dox
The pH-sensitive CP-hyd-Dox conjugate (hyd: hydrazone linker) (Fig.S1A) was synthesized by conjugating Dox to the cysteine residues in the CP using an N-[β-maleimidopropionic acid] hydrazide (BMPH) crosslinker, as described previously [24]. First, the hydrazide group in BMPH was reacted with the ketone group of Dox to form an acid-labile hydrazone bond, and then the maleimide moiety of the product was reacted with the free thiol of the cysteine residues in the CP to form a thioether bond. Briefly, 400 μmol Dox was first reacted with 360 μmol BMPH in anhydrous methanol (containing 100 μl TFA) for 16 h at room temperature. The activated Dox was then coupled to 9 μmol TCEP-reduced CP, which corresponds to 72 μmol free cysteine residues, by continuously stirring at room temperature for another 16 h. The CP-hyd-Dox conjugate was then separated from unreacted Dox by ultra-centrifugation (Amicon Ultra-15 Centrifugal Filter Units, 10 K MWCO, Millipore) using 30% acetonitrile and 70% PBS. The purity of the CP-hyd-Dox conjugate was verified by size exclusion high pressure liquid chromatography (SE-HPLC) (HPLC: Shimadzu; column: Shodex SB-804HQ, Phenomenex) using 30% acetonitrile and 70% PBS as the elution buffer. The HPLC profile of purified CP-hyd-Dox is shown in Fig.S1B. CP and Dox concentrations were calculated by the following equations: C(CP, M) = A280 nm - 0.918 × A494 nm / 5690, where 5690 cm−1M−1 is the molar extinction coefficient of the CP; C(Dox, M) = A494 nm / 8030, where 8030 cm−1M−1 is the molar extinction coefficient of the Dox. The conjugation ratio of CP-hyd-Dox was ~3.2 Dox molecules per CP.
2.3.2. CP-ami-Dox
The pH-insensitive CP-ami-Dox conjugate (ami: amide linker) (Fig.S2A) was synthesized by conjugating Dox-SMCC to the cysteine residues in the CP to form a thioether bond. 24 μmol Dox-SMCC was reacted with 1 μmol TCEP-reduced CP, which corresponds to 8 μmol cysteine residues, in 75% methanol and 25% PBS by continuously stirring for 16 h at room temperature. Purification and purity evaluation of the CP-ami-Dox conjugate was carried out identically to the CP-hyd-Dox conjugate. The HPLC profile of purified CP-ami-Dox is shown in Fig.S2B. The conjugation ratio of CP-ami-Dox was ~3.2 Dox molecules per CP.
2.3.3. CP-BHQ2
The pH-insensitive CP-BHQ2 conjugate (Fig.S3A) was synthesized by conjugating BHQ2 to the cysteine residues in the CP using a sulfosuccinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate (sulfo-SMCC) crosslinker. The NHS ester in sulfo-SMCC was reacted with the primary amine of BHQ2 to form a pH-insensitive amide bond, and then the purified product was reacted with the free thiol of the cysteine residues in the CP to form a thioether bond. In the first step, 20 μmol BHQ2 was stirred with 350 μmol sulfo-SMCC in 5 ml DMSO at room temperature for 1 h. The BHQ2-SMCC was then separated from un-reacted BHQ2 and sulfo-SMCC by silica gel column chromatography using 86% chloroform and 14% methanol as the elution buffer. In the second step, 16 μmol BHQ2-SMCC was reacted with 1 μmol TCEP-reduced CP in the reacting buffer (5% DMSO, 70% methanol, and 25% PBS) by continuously stirring for 16 h at room temperature. After the reaction, the CP-BHQ2 conjugate was purified in the same way as the CP-hyd-Dox conjugate, and its purity, measured by HPLC, is shown in Fig.S3B. CP and BHQ2 concentrations were calculated by the following equations: C(CP, M) = A280 nm - 0.413× A579 nm/5690, where 5690 cm−1M−1 is the molar extinction coefficient of the CP; C(BHQ2, M) = A579 nm/38,000, where 38,000 cm−1M−1 is the molar extinction coefficient of the BHQ2. The conjugation ratio of CP-SMCC-BHQ2 was ~1.6 BHQ2 molecules per CP.
2.3.4. AF488-labeled CP-Dox
Alexa Fluor 488 (AF488) NHS was reacted with the primary amine on the N-terminus of CP-hyd-Dox or CP-ami-Dox as follows: 2 μmol AF488 NHS ester in 100 μl DMSO and 0.4 μmol CP-Dox in 1 ml reaction buffer (70% 0.1 M NaHCO3, 30% acetonitrile) were mixed and stirred at room temperature for 1 h in the dark. The conjugate was then separated from unreacted AF488 NHS ester by gel filtration chromatography (Sephadex G-25 column, Thermo Scientific) using 30% acetonitrile and 70% PBS as the elution buffer.
2.3.5. Micelle preparation and dynamic light scattering measurement
CP-hyd-Dox, CP-ami-Dox and CP-BHQ2 were collected from the purification step in 30% acetonitrile and 70% PBS, conditions that not permit self-assembly of the CP conjugates into micelles. When the buffer was replaced with PBS by ultracentrifugation (Amicon Ultra-15 centrifugal filter units, 10 K MWCO, Millipore), each construct self-assembles into ~40 nm hydrodynamic radius (Rh) micelles (Fig.S4). To prepare pH-sensitive mixed micelles, CP-hyd-Dox and CP-BHQ2 conjugates were mixed in 30% acetonitrile and 70% PBS at a molar ratio of 2:1 (CP concentration). After vortexing, the buffer was replaced with PBS by ultracentrifugation (Amicon Ultra-15 centrifugal filter units). pH-insensitive CP-ami-Dox/CP-BHQ2 mixed micelles were prepared similarly. To prepare AF488-labeled mixed micelles, AF488-labeled CPDox was mixed with CP-Dox/CP-BHQ in 30% acetonitrile and 70% PBS at a molar ratio of 1:20 (CP concentration), then the buffer was replaced with PBS by ultracentrifugation (Amicon Ultra-15 centrifugal filter units). The size of the CP-Dox micelles or mixed micelles was measured by dynamic light scattering (DLS) (Wyatt Technology) at 20 μM (CP concentration) at 37 °C, and the data is shown in Fig.S4.
2.4. Dox release from CP-Dox/CP-BHQ2 mixed micelles
Dox release from pH-sensitive CP-hyd-Dox/CP-BHQ2 mixed micelles or pH-insensitive CP-ami-Dox/CP-BHQ2 mixed micelles in sodium phosphate buffer was studied by fluorescence spectroscopy and fluorescence detection on a HPLC. In the mixed micelles, Dox fluorescence is quenched by BHQ2, whose absorption spectrum overlaps the fluorescence emission spectrum of Dox from 530 nm to 670 nm (Fig.S5). When Dox is released from micelles, separation of released Dox from BHQ2 enables the recovery of Dox fluorescence. In these experiments, CP-Dox and CP-BHQ2 were mixed at a 2:1 molar ratio at a total CP concentration of 20 μM to create CP-Dox/CP-BHQ2 mixed micelles with a Dox concentration of ~5.3 μM. 20 μM of micelles in 1 ml sodium phosphate buffer were incubated at 37 °C at different pH. At a given time point, 2 μl of sample was loaded on a Nanodrop 3300 fluorescence spectrometer (Thermo Scientific) and the fluorescence emission spectrum of Dox was measured at an excitation wavelength of 470 nm. The cumulative drug release ratio was calculated by dividing the fluorescence intensity of the sample at 590 nm, which was a mixture of CP-Dox/CP-BHQ2 mixed micelles and released Dox, by the fluorescence intensity of ~5.3 μM free Dox. SEC (Shodex SB-804HQ, Phenomenex) on an HPLC (Shimadzu) was used to separate CP-Dox/CPBHQ2 from released Dox, and the Dox fluorescence emission intensity at 590 nm with excitation at 490 nm was recorded. The elution buffer was 30% acetonitrile and 70% PBS. The elution times of CP-Dox/CPBHQ2 and released free Dox start at ~5.5 min and ~13.7 min, respectively.
2.5. Ratiometric fluorescence imaging and analysis
100,000 cells were seeded onto 35 mm glass bottom dishes (MatTek) and incubated at 37 °C overnight to allow cell attachment to the dish bottom. Cells were then incubated with 20 μM (CP concentration) of AF488-labeled CP-Dox/CP-BHQ2 mixed micelles (5.3 μM Dox equivalent) at 37 °C, and at the desired time point, cells were rinsed with PBS thrice and incubated with 2 μM of Lysosensor at 37 °C for 30 min. Cells were then rinsed with PBS thrice and imaged immediately thereafter.
A spinning disk confocal microscope (XD revolution, Andor) equipped with a high sensitivity electron multiplier charged coupled device (EMCCD) camera was used to image the fluorescence of Lysosensor, AF488 and Dox in live cells. The EMCCD camera allows quantitative measurement of fluorescence emission intensities. Ex = 405 nm/blue filter (Ex: excitation) and Ex = 405 nm/green filter were used to measure Lysosensor’s two fluorescence emission peaks at 440 nm and 530 nm, respectively. Ex = 488 nm/green filter and Ex = 488 nm/red filter were used to measure AF488 and Dox, respectively. The blue, green and red emission filters were 447 ± 30 nm, 525 ± 15 nm and 607 ± 18 nm, respectively. The images were acquired at a resolution of 512 pixels × 512 pixels and analyzed by ImageJ and Matlab (Mathworks) software. Prior to observation, cells stained with Lysosensor, AF488 and Dox, respectively, were measured at these four channels, which were 405 nm/blue filter, 405 nm/green filter, 488 nm/green filter and 488 nm/red filter, and the results con-firmed that no fluorescence bleed-through occurred among these three fluorophores in the four channels (Fig.S6). To obtain average data, for each sample, at least 10 images were acquired and every image included 10 to 20 cells. Every experiment was repeated at least three times.
Image analysis then proceeded in the following five steps: (1) The spatial overlap of Lysosensor with fluorescent nanoparticles and fluorescent drugs was evaluated to confirm the endocytosis of nanoparticles, release of drugs, and their accumulation in endo-lysosomes. (2) Fluorescent pixels in the endo-lysosomal compartment were identified and distinguished from the largely non-fluorescent cytosol by setting an intensity threshold in ImageJ software. All fluorescent pixels identified as belonging to the endo-lysosomal compartment were subject to further analysis. (3) Lysosensor’s IBlue/IGreen ratio at each pixel throughout the endo-lysosomal compartment was obtained using image calculator in ImageJ software. (4) Lysosensor’s IBlue/IGreen ratio at each pixel was converted to a pH value according to the calibration between IBlue/IGreen ratio and pH obtained through live cell experiments explained below. (5) Pixels with the same pH value throughout the endo-lysosomal compartment were binned, and the corresponding AF488 fluorescence intensity of the nanoparticles or the Dox fluorescence intensity at these pixels was averaged. This average AF488 intensity or Dox intensity or Dox/AF488 intensity ratio was plotted as a function of pH by Matlab software (see the Supporting Code). The detailed image analysis process is shown as a flowchart in Fig.S7.
Dual-emission ratiometric measurements of Lysosensor allow the conversion from the Lysosensor’s IBlue/IGreen ratio to a pH value according to a calibration curve. To plot the IBlue/IGreen versus pH calibration curve, we measured endo-lysosomal pH in a series of calibration buffers by a fluorescence spectrophotometer (Cary Eclipse) as described before [29]. FaDu live cells were first incubated with 2 μM Lysosensor at 37 °C for 30 min, and then washed twice with ice-cold 2-(N-morpholino)ethanesulfonic acid (MES) calibration buffers at different pH (from pH 4.0 to pH 7.5), equilibrated for 2 min, and then the blue and green fluorescence of Lysosensor in live cells were immediately measured by the spinning disk confocal microscope. The MES buffers contain 5 mM NaCl, 115 mM KCl, 1.2 mM MgSO4, 25 mM MES, 10 μM monensin and 10 μM nigericin. After image acquisition, the fluorescent pixels which represented the endo-lysosomal compartment in FaDu cancer cells were identified and distinguished from the largely non-fluorescent cytosol by setting the intensity threshold in ImageJ, and then Lysosensor’s IBlue/IGreen ratio in the endo-lysosomal compartment was plotted as a function of buffer pH.
3. Results
3.1. Spatially mapping endo-lysosomal pH in live cells using lysosensor is feasible and reliable
To study drug release from nanoparticles in endo-lysosomes as a function of pH, we needed to first spatially map the endo-lysosomal pH, and then correlate the local pH with the amount of drug and carrier accumulated in endo-lysosomes. To map the endo-lysosomal pH in live cells, we used Lysosensor yellow/blue DND-160 (hereafter referred to as Lysosensor) and performed pixel-by-pixel analysis of the fluorescence emission ratiometric images of Lysosensor to spatially quantify the pH of individual endosomes and lysosomes. Lysosensor is a fluorescent pH indicator that partitions into the acidic endo-lysosomal compartment in cells [30]. It exhibits dual pH-dependent emission peaks at 440 nm and 530 nm (Fig. 1A), where the ratio of these two peaks shows a linear relation with pH in sodium phosphate buffer (Fig. 1B, top). We chose this specific Lysosensor variant of the many available, because it can track a broad range of acidic pH between 4 and 7 [30], which spans the expected pH range in endosomes and lysosomes [31]. To confirm Lysosensor’s pH-dependent emission in live cells, we imaged Lysosensor-treated cells in a series of calibration buffers with pH ranging from 4.0 to 7.5 by a spinning disk confocal microscope, and measured the two emission peaks of Lysosensor with a blue filter (447 ± 30 nm) and a green filter (525 ± 15 nm) (Fig. 1C). The ratio of these two fluorescence emission peaks of Lysosensor in endo-lysosomes, IBlue/IGreen(I: fluorescence intensity), also shows a linear relation against buffer pH with R2 = 0.995 (Fig. 1B, bottom), which suggests that measuring the endo-lysosomal pH using Lysosensor in live cells is feasible.
Fig. 1.
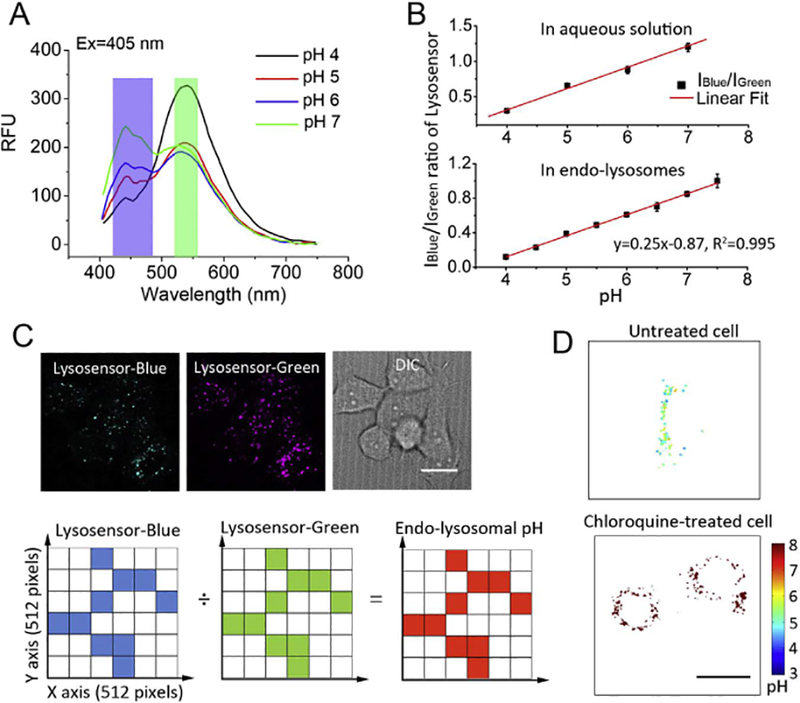
Method of spatially mapping endo-lysosomal pH in live cells by fluorescence dual-emission ratiometric imaging of Lysosensor and pixel-by-pixel analysis. (A) Lysosensor exhibits pH-dependent fluorescence emission peaks at 440 nm and 530 nm. To measure Lysosensor’s two fluorescence emission peaks in live cells, the Lysosensor’s emission was separately measured with a blue filter (blue bar) and a green filter (green bar) with excitation at 405 nm by a spinning disk confocal microscope. (B) (Top) In aqueous solution, the ratio of Lysosensor’s two emission peaks IBlue/IGreen (I: fluorescence intensity, herein IBlue/IGreen corresponds to I440 nm/I530 nm) shows a linear relation with pH. (Bottom) In live cells equilibrated with a series of calibration buffers ranging from pH 4.0 to pH 7.5, the ratio of Lysosensor’s two emission peaks IBlue/IGreen in endo-lysosomes (herein IBlue/IGreen corresponds to I447 ± 30nm/I525 ± 15nm) also shows a linear dependence as a function of pH (R2 = 0.995). (C) (Top) Lysosensor-treated live cells were imaged with a spinning disk confocal microscope, and the blue and green fluorescence images of Lysosensor and the differential interference contrast (DIC) image of cells were acquired in the same region. The white scale bar indicates 2 μm. (Bottom) In the analysis, every image was analyzed as a 512 pixels × 512 pixels grid where the origin (0, 0) was set at the bottom-left and every pixel was assigned a coordinate (x, y). Fluorescent endo-lysosomal areas (shown in color) were identified and distinguished from the largely non-fluorescent cytosol, and then the IBlue/IGreen intensity ratio at each pixel throughout the endo-lysosomal compartment was converted to a local pH value by the calibration provided by Fig.1B, bottom. Finally, the spatial endo-lysosomal pH map in live cells was plotted. (D) Untreated cells show endo-lysosomal pH ranging from pH 4 to pH 7 while chloroquine-treated cells show increased endo-lysosomal pH. The black scale bar indicates 2 μm and the color bar indicates the pH value.
To spatially map the endo-lysosomal pH in live cells, we analyzed the blue and the green fluorescence images of Lysosensor-treated cells at a resolution of 512 pixels × 512 pixels (Fig. 1C). The origin of the coordinates (0, 0) in the grid was set at the bottom-left, and every pixel was defined by a coordinate (x, y), and assigned blue and green fluorescence intensity values from two images. Image analysis then proceeded in the following three steps: (1) The fluorescent endo-lysosomal compartment was identified and distinguished from the largely non-fluorescent cytosol by setting a fluorescence intensity threshold in ImageJ software. All fluorescent pixels identified as belonging to the endo-lysosomal compartment were subject to further analysis. (2) The IBlue/IGreen intensity ratio at each fluorescent pixel was converted to a pH value according to the calibration curve (Fig. 1B, bottom) using Matlab software. (3) Based on the pH value and the coordinate of each pixel, cellular endo-lysosomal pH map was plotted by Matlab software (Fig. 1D). To test whether this method of spatially mapping the endolysosomal pH in live cells is reliable, we compared the spatial pH map of untreated cells with the spatial pH map of cells treated with chloroquine, which prevents endosomal acidification and raises the lysosomal pH [32] Consistent with the known effects of chloroquine, the endo-lysosomal pH in chloroquine-treated cells was close to or above pH 7, while the endo-lysosomal pH in untreated cells ranged from pH 4 to pH 7 (Fig. 1D). The distinctly different endo-lysosomal pH between untreated cells and chloroquine-treated cells strongly suggested that this is a sensitive and reliable method to spatially map endo-lysosomal pH in live cells.
3.2. Hydrazone linkers are efficiently cleaved in acidic solution
Next, we used this method to study the pH-dependent release of Dox from CP-Dox nanoparticles in endo-lysosomes of live cells by cleavage of an acid-labile linker that links the drug to the carrier. Before commencing live cell experiments, we quantified the Dox release from pH-sensitive CP-hyd-Dox/CP-BHQ2 mixed micelles (hyd: hydrazone linker; BHQ: black hole quencher; BHQ2 is a quencher of Dox) in aqueous solution using fluorescence de-quenching to evaluate the cleavage efficiency of the acid-labile hydrazone linkers under idealized buffer conditions, with the view that that these data could serve as a positive control for comparison with the data obtained from subsequent live cell experiments.
We selected fluorescence de-quenching to quantitatively measure Dox release from CP-Dox micelles, due to this technique’s ability to sensitively detect the release of fluorescent drugs from quenched micelles [33,34]. Although Dox partially self-quenches when confined to the core of a micelle (Fig. 2A), we sought to further improve the sensitivity of detection by developing fully quenched micelles. To achieve this, we chose BHQ2 for its ability to efficiently quench doxorubicin fluorescence, as seen by a broad overlap of BHQ2’s absorbance spectrum with Dox’s emission spectrum (Fig.S5) [35,36] pH-sensitive CP-hyd-Dox/CP-BHQ2 mixed micelles were prepared as described in the methods section, in which Dox was attached to the CP by an acid-labile hydrazone linker while BHQ2 was attached to the CP by a pH-insensitive amide bond. A mixture of CP-hyd-Dox/CP-BHQ2 self-assembles into spherical micelles with a ~40 nm hydrodynamic radius (Rh) in aqueous solution, with a size that is almost identical to the size of CP-hyd-Dox micelles (Fig.S4). As expected, compared with free Dox, the Dox fluorescence is partly quenched in CP-hyd-Dox micelles because of self-quenching, while the residual fluorescence of Dox is completely quenched in CP-hyd-Dox/CP-BHQ2 mixed micelles because of contact quenching of Dox by BHQ2 in the micelle core (Fig. 2A).
Fig. 2.
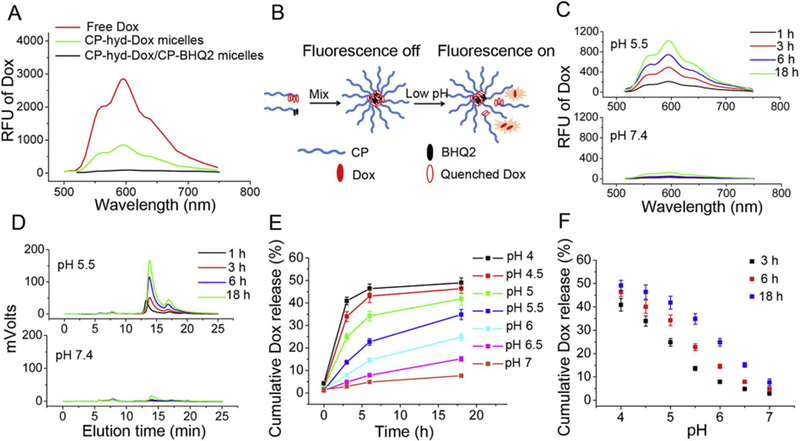
Acid triggers Dox release from pH-sensitive CP-hyd-Dox/CP-BHQ2 mixed micelles (hyd: hydrazone linker) in sodium phosphate buffer studied by Dox fluorescence de-quenching. (A) The Dox fluorescence emission spectra of ~5.3 μM (Dox concentration) free Dox, CP-hyd-Dox micelles and CP-hyd-Dox/CP-BHQ2 mixed micelles excited at 470 nm. Dox fluorescence is partly quenched in CP-hyd-Dox micelles and completely quenched in CP-hyd-Dox/CP-BHQ2 mixed micelles. (B) Scheme of measuring Dox release from micelles by fluorescence de-quenching. CP-hyd-Dox/CP-BHQ2 self-assembles into micelles in aqueous solution and Dox is completely quenched by BHQ2 in the micelle core. When Dox is released from the micelles upon acid-triggered cleavage of hydrazone linkers, Dox separates from BHQ2 and recovers its red fluorescence. (C) Dox release studied by fluorescence spectroscopy. CP-hyd-Dox/CPBHQ2 mixed micelles were incubated in aqueous solution at pH 5.5 or pH 7.4. (D) Dox release studied by SE-HPLC. CP-hyd-Dox/CP-BHQ2 was separated from released free Dox and Dox fluorescence was measured. The retention times of CP-hyd-Dox/CP-BHQ2 and released Dox start at ~5.5 min and ~13.7 min, respectively. (E, F) Time- and pH-dependent Dox release from CP-hyd-Dox/CP-BHQ2 mixed micelles measured by fluorescence spectroscopy.
When Dox is released from CP-hyd-Dox/CP-BHQ2 mixed micelles upon acid-triggered cleavage of the hydrazone linker, the Dox fluorescence recovers (Fig. 2B). Comparing the recovered Dox fluorescence in the mixed micelles with that of free Dox at the same concentration enables us to quantify the Dox release in aqueous solution. To first validate the utility of using fluorescence de-quenching to quantify Dox release, we compared Dox release in an acidic solution of pH 5.5 with a neutral solution at pH 7.4 by fluorescence spectroscopy, and found Dox fluorescence increased with incubation time at pH 5.5, but did not change over time at pH 7.4 (Fig. 2C). To independently confirm these results, size-exclusion high performance liquid chromatography (SEHPLC) was used to separate the CP-hyd-Dox/CP-BHQ2 from the released Dox, and the Dox fluorescence was then measured by a fluorescence detector on the HPLC. In the SE-HPLC elution buffer of 30% acetonitrile in PBS, the mixed micelles disassembled into unimers, and the fluorescence peak of the CP-hyd-Dox and CP-BHQ2 unimers starting at ~5.5 min was small and did not change with time, while the fluorescence peak of released free Dox starting at ~13.7 min increased with time at pH 5.5 (Fig. 2D). In contrast, both peaks were small and did not change with time at pH 7.4. These HPLC data suggested that hydrolysis of the hydrazone bond occurs at low pH, and that the disassembled CP-Dox shows much smaller fluorescence peak due to self-quenching compared with free Dox (Fig. 2D, pH 5.5). The observed Dox fluorescence in SE-HPLC, and by inference within cells, is largely derived from released free Dox, so that the contribution of CP-Dox unimers to the fluorescence signal is negligible and hence ignored.
Having shown the feasibility of the fluorescence de-quenching technique for measurement of Dox release, we applied it to quantitatively measure Dox release from CP-hyd-Dox/CP-BHQ2 mixed micelles as a function of pH between pH 4 and 7 and time in aqueous solution. We deliberately chose this range of pH because the endo-lysosomal pH of cells locates in this range [31]. Drug release, which occurs upon hydrolysis of the hydrazone linker, was both time- and pH-dependent (Fig. 2E, Fig. 2F). At pH 4, hydrolysis was fast in the first 6 h but slowed down afterwards and reached a steady state level of ~50%. In contrast, the rate of hydrolysis was slower at higher pH, and the maximum level of drug release after 18 h scaled inversely with pH. After 18 h incubation, about 50%, 40%, 22% and 5% of hydrazone linkers were cleaved at pH 4, pH 5, pH 6 and pH 7, respectively. Taken together, these results clearly show that hydrazone linkers are cleaved at acidic pH, but even at a pH of 4, which corresponds to the lowest pH likely to be seen in lysosomes, only half of the hydrazone linkers are cleaved.
3.3. Hydrazone linkers are efficiently cleaved in endo-lysosomes below pH 6
Next, we studied pH-triggered drug release from CP-hyd-Dox/CPBHQ2 mixed micelles in live cells as a function of pH and time. To track and quantify the carrier (CP) within cells independently of the drug, an Alexa Fluor 488 (AF488) was attached to the hydrophilic, solvent exposed terminus of CP-hyd-Dox by a pH-insensitive amide linker. These micelles then contain three spectroscopic tags—AF488 fluorophore that decorates the corona of the micelles, and fluorescent Dox and the Dox quencher BHQ2 that is attached to different polymer chains in the micelles, and is in close proximity to Dox in the core of the micelles. When Dox is released from AF488-labeled CP-hyd-Dox/CP-BHQ2 mixed micelles, Dox fluorescence recovers while AF488 fluorescence is invariant, so that the drug release rate is determined by calculating the intensity ratio of Dox red fluorescence to AF488 green fluorescence.
In these experiments, FaDu cancer cells were first incubated with AF488-labeled CP-hyd-Dox/CP-BHQ2 mixed micelles at 37 °C for the desired time, and then stained with Lysosensor, rinsed and observed under a spinning disk confocal microscope by taking four images: (1) blue and (2) green fluorescence of Lysosensor excited at 405 nm, (3) green fluorescence of AF488 excited at 488 nm, and (4) red fluorescence of Dox excited at 488 nm (Fig. 3A). No fluorescence bleed-through occurred between these four fluorescence images (Fig.S6). After pixel-by-pixel analysis, the distributions of AF488-labeled CP, released Dox, and the Dox/AF488 intensity ratio in endo-lysosomes as a function of pH were plotted by Matlab software.
Fig. 3.
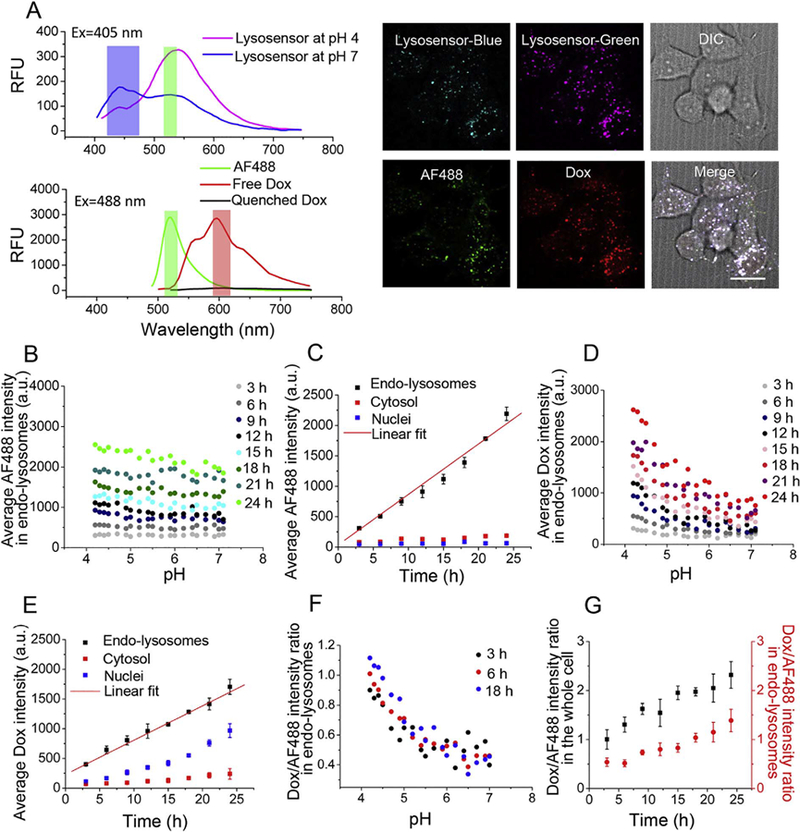
Acid triggers Dox release from pH-sensitive AF488-labeled CP-hyd-Dox/CP-BHQ2 mixed micelles (hyd: hydrazone linker) in endo-lysosomes. (A) The top panel shows the fluorescence emission spectrum of Lysosensor at pH 4 and pH 7 excited at 405 nm, and the bottom panel shows the fluorescence emission spectra of AF488, free Dox and quenched Dox excited at 488 nm. The blue, green and red bars indicate the emission filters used for fluorescence emission measurements. Live cells were incubated with AF488-labeled CP-hyd-Dox/CPBHQ2 mixed micelles and Lysosensor, and then were imaged by a spinning disk confocal microscope. The panel on the right shows representative blue and green fluorescence images of Lysosensor, a green fluorescence image of AF488, a red fluorescence image of Dox and a DIC image of cells. The white scale bar indicates 2 μm. (B) Average AF488 intensity in endolysosomes as a function of pH over time. Endo-lysosomal distribution of AF488-labeled CP is invariant across all pH but increases with time. (C) Average AF488 intensity in endo-lysosomes, cytosol and nuclei over time. AF488-labeled CP accumulates in endo-lysosomes with linear kinetics and does not traffic to the cytosol and nuclei. (D) Average Dox intensity in endo-lysosomes against pH over time. Dox fluorescence, which derives from the released drug, shows preferred accumulation in the low pH environment of endo-lysosomes after 6 h. (E) Average Dox intensity in endo-lysosomes, cytosol and nuclei over time. Released Dox accumulates in endo-lysosomes with linear kinetics, and it diffuses to the cytosol and accumulates in the nuclei over time, with minimal accumulation in the cytosol. (F) Dox/AF488 intensity ratio in endo-lysosomes as a function of pH at different time points changes inversely with pH below pH 6. (G) Dox/AF488 intensity ratio in endo-lysosomes and in the whole cell increases over time.
The endo-lysosomal distribution of the average AF488 fluorescence intensity, which represents the CP, was invariant across all pH but increased with time (Fig. 3B). Quantification of the accumulation of AF488-labeled CP in different organelles over time shows that CP accumulated in endo-lysosomes with linear kinetics and was trapped there, as there was minimal trafficking to the cytosol and nuclei (Fig. 3C). The endo-lysosomal fluorescence of Dox also increased with time, but shows a pronounced increase in endo-lysosomes at pH < 6 after 6 h (Fig. 3D). This asymmetric distribution of Dox fluorescence in endo-lysosomes at lower pH indicates that the cleavage of the hydrazone linker is far more efficient at lower pH. In contrast to the CP, the released Dox diffused from endo-lysosomes to the cytosol and trafficked into cell nuclei, as seen by the increased Dox fluorescence in the nucleus over time (Fig. 3E). The Dox/AF488 intensity ratio in endolysosomes increased as the pH decreased from pH 6 to 4 (Fig. 3F), indicating that the cleavage of hydrazone linkers initiates below pH 6 and is more efficient at lower pH. This result agrees with our study performed in aqueous solution, and a previous report which comprehensively studied the cleavage efficiency of hydrazone linkers in aqueous solution [37] The Dox/AF488 intensity ratio in the whole cell better represents drug release rate because the portion of Dox in the cytosol and nuclei that diffuse out of endo-lysosomes is taken into account. A plot of this ratio versus time shows that regardless of the complex trafficking of drug-loaded micelles within cells, continuous Dox release caused by cleavage of the hydrazone linker was observed without reaching a plateau in 24 h (Fig. 3G).
3.4. Amide linkers are not cleaved in acidic endo-lysosomes
To confirm that drug release in endo-lysosomes requires an acid-labile linker, we carried out control experiments with CP-Dox nano-particles where the Dox was attached to the CP through an acid-insensitive amide (ami) linker, and then prepared CP-ami-Dox/CP-BHQ2 mixed micelles. CP-ami-Dox/CP-BHQ2 mixed micelles have the same ~40 nm Rh as CP-ami-Dox micelles (Fig.S4), and the Dox fluorescence is partly quenched in CP-ami-Dox micelles but is completely quenched in CP-ami-Dox/CP-BHQ2 mixed micelles (Fig.S8).
When pH-insensitive CP-ami-Dox/CP-BHQ2 micelles were incubated in aqueous solution, no Dox was released at either pH 4 or pH 7 (Fig. 4A), suggesting that acid cannot trigger the cleavage of amide linkers. In live cell experiments with AF488-labeled CP-ami-Dox/CPBHQ2 mixed micelles, we found the endo-lysosomal distribution of AF488-labeled CP was independent of local pH (Fig. 4B) and the accumulation of the CP in endo-lysosomes increased over time without leaking to the cytosol and nuclei (Fig. 4C), a trend similar to the spatial distribution and accumulation kinetics of the pH-sensitive CP-hyd-Dox/CP-BHQ2 mixed micelles. However, Dox fluorescence remained invariant in endo-lysosomes, cytosol and nuclei, regardless of local pH and incubation time (Fig. 4D, Fig. 4E). This result suggests that Dox was not released from micelles due to lack of cleavage of the amide linker in endo-lysosomes, and it also implies that disassembly of micelles, which can in principle occur even if the drug remains attached to the CP, is not sufficient to restore Dox fluorescence. The increased accumulation of AF488-labeled CP and the low fluorescence of the quenched and bound Dox within the CP-ami-Dox micelles resulted in a decrease in the Dox/AF488 intensity ratio in the whole cell and endo-lysosomes over time (Fig. 4F). Taken together, these results provide compelling evidence that amide linkers are not cleaved in the acidic environment of endolysosomes, and combined with the results with CP-hyd-Dox/CP-BHQ2 mixed micelles, prove that an acid-labile linker between the drug and the CP is critical to achieve drug release and escape from endo-lysosomes.
Fig. 4.
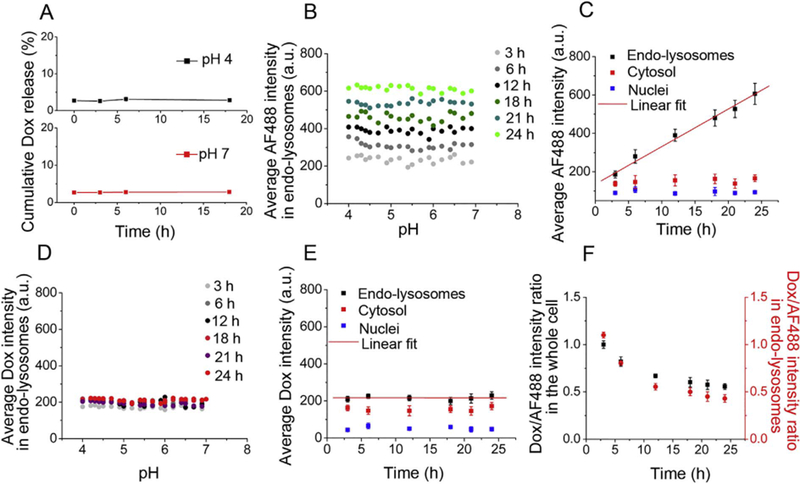
Acid does not trigger Dox release from AF488-labeled CP-ami-Dox/CP-BHQ2 mixed micelles (ami: amide linker) in live cells. (A) No Dox is released from CP-ami-Dox/CP-BHQ2 mixed micelles in aqueous solution at pH4 or pH7 in sodium phosphate buffer as a function of time post-incubation. (B) Average AF488 intensity in endo-lysosomes as a function of pH over time. Endo-lysosomal distribution of AF488-labeled CP is invariant across all pH but increases with time. (C) Average AF488 intensity in endo-lysosomes, cytosol and nuclei over time. AF488-labeled CP accumulates in endo-lysosomes with linear kinetics, but does not traffic to the cytosol and nuclei. (D) Average Dox intensity in endo-lysosomes as a function of pH over time. Dox fluorescence is invariant across all pH and does not change over time. (E) Average Dox intensity in endo-lysosomes, cytosol and nuclei over time. Because Dox is not released from micelles, Dox fluorescence does not change inside or outside endo-lysosomes. (F) Dox/AF488 intensity ratio within the entire cell and within endo-lysosomes over time. Increased AF488 fluorescence and unchanged Dox fluorescence within cells and endo-lysosomes result in decreased Dox/AF488 intensity ratio over time.
3.5. Dox delivered by CP-hyd-Dox nanoparticle has similar distribution to free Dox at steady state
Dox release from CP-hyd-Dox micelles is triggered at low pH in endo-lysosomes. At low pH, weakly basic drugs such as doxorubicin exist predominately in an ionized form, which could result in endolysosomal trapping due to the lower membrane permeability of ionized molecules [14,15]. This could reduce the efficacy of nanoparticle-delivered Dox, as it must reach the nuclei to intercalate DNA, inhibit topoisomerase II, and inhibit DNA replication [38]. To address this question, we compared the intracellular distribution and trafficking of CP-hyd-Dox micelles with free Dox in endo-lysosomes, cytosol and nuclei.
Free Dox first diffuses across the cell membrane into the cytosol, and then distributes to nuclei and endo-lysosomes (Fig. 5A, left) [39]. In live cells incubated with free Dox, we found Dox fluorescence first appeared in the nuclei, suggesting cytosol → nuclei trafficking was faster than cytosol → endo-lysosomes trafficking at as early as 3 h; with time however, Dox fluorescence in endo-lysosomes became brighter and exceeded that in nuclei (Fig. 5B, top panel), as seen by the increasing Average Dox intensity(endo-lysosomes)/Average Dox intensity(nuclei) ratio increased from 0.4 at 3 h to 1.4 at 24 h (Fig. 5C, left). In contrast to free Dox, upon cellular uptake, micelle Dox is largely accumulated in endo-lysosomes, and upon acid-triggered drug release in endo-lysosomes, the released Dox traffics into the cytosol and then to nuclei (Fig. 5A, right) [13]. This scenario is consistent with the images in Fig. 5B (bottom panel), where Dox fluorescence first appeared in endolysosomes in the cells incubated with CP-hyd-Dox/CP-BHQ2 mixed micelles, and the Dox fluorescence in nuclei only became significant after 12 h. At 3 h, the Average Dox intensity(endo-lysosomes)/Average Dox intensity(nuclei) ratio was 3.5. As the Dox began to accumulate in nuclei, this ratio decreased over time and dropped to 1.5 at 24 h (Fig. 5C, right). Although there are distinct differences between the free drug and micelle-delivered drug in their internalization and trafficking pathways, which is reflected in the opposing trends in their Average Dox intensity(endo-lysosomes)/Average Dox intensity(nuclei) ratio, at 24 h the ratio in these two groups approached each other (Fig. 5C).
Fig. 5.
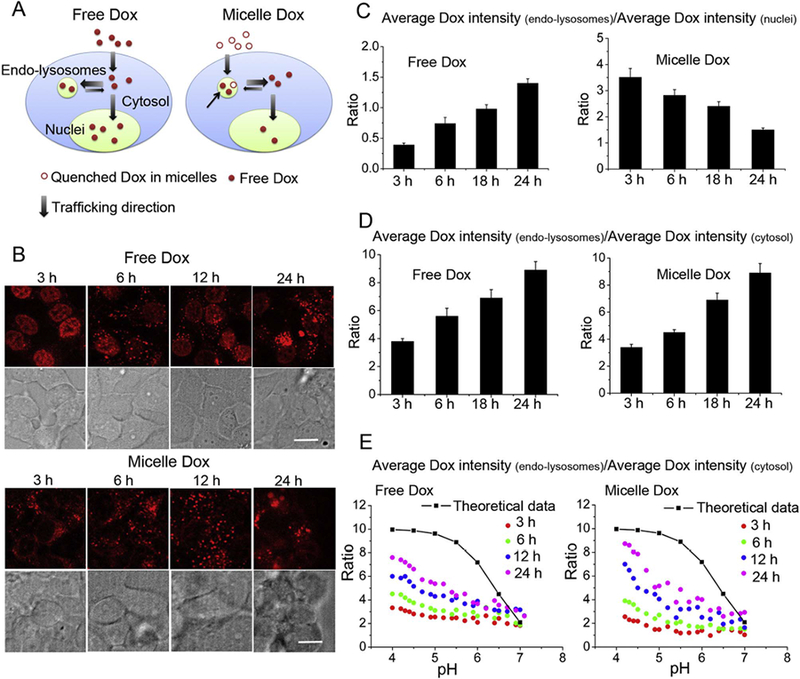
Intracellular trafficking and distribution of released Dox from CP-hyd-Dox/CP-BHQ2 mixed micelles and its comparison with free Dox (control) as a function of time. (A) Schematic illustration of internalization and intracellular trafficking of free Dox and Dox delivered by CP-hyd-Dox/CP-BHQ2 mixed micelles (micelle Dox). Free Dox diffuses across the cell membrane to the cytosol and then distributes to endo-lysosomes and nuclei. Micelle Dox is internalized by endocytosis to endo-lysosomes, and after linker cleavage at low pH (black arrow), Dox diffuses out of endo-lysosomes to the cytosol and then enters nuclei. (B) Intracellular localization of released Dox from micelles and free Dox at different time points. In the free Dox group, nuclei show obvious Dox fluorescence at 3 h; afterwards, Dox fluorescence in endo-lysosomes becomes bright and exceeds that in nuclei. In the micelle Dox group, Dox fluorescence first appears in endo-lysosomes, and after 12 h, Dox fluorescence in nuclei is notable. The white scale bar indicates 2 μm. (C) Comparison of Dox accumulation between endolysosomes and nuclei, as reflected by the Average Dox intensity(endo-lysosomes)/Average Dox intensity(nuclei) ratio as a function of time. The ratio in the free Dox group increases over time while the ratio in the micelle Dox group decreases over time. At 24 h, the ratios in these two groups are similar to each other. (D) The trafficking of Dox between endo-lysosomes and cytosol, as reflected by the Average Dox intensity(endo-lysosomes)/Average Dox intensity(cytosol) ratio as a function of time.The ratios in both free Dox group and micelle Dox group increase over time and the rates are similar. (E) Distribution of Dox in endo-lysosomes as a function of pH at different time points, as reflected by the Average Dox intensity(endo-lysosomes)/Average Dox intensity(cytosol) ratio plotted as a function of local pH. Irrespective of different internalization pathways, both free Dox and released Dox from micelles gradually approach the theoretical distribution equilibrium between endo-lysosomes and the cytosol as indicated by the black line and squares. The calculation of the theoretical distribution is described in the text.
Another parameter, the Average Dox intensity(endo-lysosomes)/Average Dox Intensity(cytosol) ratio, reflects the trafficking of Dox between the endo-lysosomes and the cytosol. As shown in Fig. 5D, the Average Dox intensity(endo-lysosomes)/Average Dox Intensity(cytosol) ratios in both groups increased over time at similar rates, increasing from about 3.8 at 3 h to 9 at 24 h. The equation shown below was developed to calculate the theoretical lysosome/cytosol concentration ratio of weakly basic lysosomotropic drugs at steady state [40],
| (1) |
where Ka denotes the dissociation constant of the weak base, and the pKa of Dox is 8.4, and [H+] denotes the proton concentration. The term alpha (α) denotes the ratio of lysosomal membrane permeability for the ionized base divided by that of the non-ionized species, and α = 0.01 was used based on a previous measurement [41]. The theoretical prediction (black line with squares) and the experimental results (colored dots) of free Dox and micelle-delivered Dox are shown in Fig. 5E. The asymmetric distribution of Dox became more pronounced over time in both groups. Specifically, the[lysosome]/[cytosol] value of micelle Dox changed from 1 to 3 over 24 h at pH 7 while the ratio increased from 2.5 to 8.7 at pH 4. The change of these ratios was very similar in the free Dox group. Comparing the theoretical data with our experimental results, we find both delivery methods gradually approach—but do not quite reach—the theoretical steady state value over 24 h.
These results clearly demonstrate the distinct uptake pathways and intracellular trafficking of free Dox and micelle Dox. Free Dox first diffuses across the cell membrane into the cytosol of cells, and then most of the drug accumulates in nuclei at early time points. The accumulation of free Dox in endo-lysosomes is much slower than its accumulation in nuclei (Fig. 5A, left). In contrast, micelle Dox is first taken up by endocytosis, which forms internalizing vesicles, and then these internalizing vesicles traffic to endo-lysosomes. Upon acid-triggered drug release in endo-lysosomes, released Dox diffuses across the endo-lysosomal membrane to the cytosol and then reaches the nuclei (Fig. 5A, right). Despite the differences in the internalization pathways and primary route of intracellular trafficking and its kinetics, we found that the released Dox from micelles, where Dox is conjugated by an acid-labile linker, has a similar intracellular distribution as free Dox at steady state. This finding indicates that lysosomal entrapment of weakly basic drugs delivered by nanoparticles is unlikely to impede their therapeutic effect.
4. Discussion
There are two important findings from this study. The first finding addresses the question of the efficiency of cleavage of an acid-labile linker in live cells. We find that acid-labile linkers between carriers and drugs are cleaved in endo-lysosomes in live cells with an efficiency that approaches in vitro conditions of a low pH aqueous solution, so that an acid-labile hydrazone linker can achieve localized drug release within endosomes and lysosomes. However, the acid-labile linkers are not fully hydrolyzed in endo-lysosomes, consistent with a previous report that a pH of ~3 is required for complete hydrolysis of hydrazone linkers in aqueous solution [37]. This finding may help explain why nanoparticle-delivered Dox consistently shows a somewhat higher IC50 than free Dox in the in vitro cytotoxicity studies [24], and it also explains why a polymer-drug conjugate with a pH-sensitive hydrazone linker has significantly higher cytotoxicity than a conjugate wherein the drug is conjugated to the polymer via a pH-insensitive amide linker [42]. These results also suggest that linkers that exhibit complete cleavage between a pH of 4 and 7 may be optimal to maximize the therapeutic efficacy of the drug when it is attached to a carrier via an acid-labile linker.
The second notable finding is that upon endocytosis, CP-Dox nanoparticles are initially solely trafficked into endo-lysosomes with no appreciable direct transport to the cytosol. For nanoparticles wherein the drug is conjugated through an acid-labile hydrazone linker, the drug does not remain permanently sequestered there. Cleavage of the drug from the polypeptide carrier triggers its escape from the endo-lysosomes and its traffic into the cytosol and subsequently into the nuclei. The acid-labile linker is critical for endo-lysosomal escape, as Dox that is conjugated through a pH-insensitive amide linker remains attached to the CP and therefore CP-ami-Dox is permanently sequestered within the endo-lysosomes. In contrast, free drug directly enters the cytosol by diffusion across the cell membrane and then reaches the nuclei. Without the step of acid-triggered linker cleavage in endo-lysosomes and the step of slow diffusion from endo-lysosomes to the cytosol, the initial accumulation of free drug at its pharmacological site of action—the nucleus—is hence faster than for nanoparticle-delivered drug. However, the weakly basic character of doxorubicin does not impede its escape from the endo-lysosomes upon its release from CPDox nanoparticles.
Finally, we emphasize that an attractive feature of this imaging method is that it can simultaneously measure the intracellular distribution of one or more fluorophores and overlay it on a quantitative spatial map of the local pH within live cells [20]. By doing so, it allows individual endosomes and lysosomes to be identified with their corresponding local pH, and allows quantitative interrogation of intracellular events related to the local pH in these compartments. This imaging method provides an easy and fast alternative to the traditional methods which visualize endosomes and lysosomes by transfecting cells with recombinant fluorescent protein-tagged markers or by staining the fixed cells with fluorohpor-conjugated antibodies [43,44]. While we demonstrated the utility of this methodology to quantify the release of a fluorescent drug from a pH-sensitive nanoparticle, it can be used more broadly to investigate the intracellular trafficking of other drug delivery systems, such as antibody-drug conjugates in which the drug is attached to the carrier through a pH-sensitive linker, or short interfering RNA (siRNA) encapsulated in lipid nanoparticle [43,44].
While this study focused on nanoparticles that are non-specifically taken up by cells, the delivery efficiency of such nanoparticles can be enhanced by appending ligands that target specific cell surface receptors that are overexpressed by cancer cells or by cell penetrating peptides [45,46]. Recently, we studied the intracellular trafficking of ELP nanoparticles that present cell-penetrating peptides (CPPs) at the nanoparticle-water interface by a similar fluorescence imaging method, and showed that arginine-rich CPPs greatly increase the cell uptake and accelerate endosome-to-lysosome trafficking of CPP-functionalized ELP nanoparticles compared with the unfunctionalized nanoparticles [20]. In conclusion, the spatially resolved and quantitative information offered by this imaging methodology can provide us a better understanding of intracellular fate of nanoparticles and useful insights for the design of next-generation drug delivery systems.
5. Conclusions
In this study, we combined multiple fluorescence imaging techniques with quantitative pixel-by-pixel analysis to spatially track the release of the drug doxorubicin from pH-responsive doxorubicin-poly-peptide nanoparticles as a function of time and local pH within live cells. We used this newly developed method to address two important issues in drug delivery: (1) the extent to which the acid-labile linker between drug and carriers is cleaved in individual endosomes and lysosomes as a function of their local pH, and (2) the extent to which the released drug can escape from endo-lysosomes and traffic within the cytosol to reach its therapeutic destination, the nucleus. We show that unlike free drug, which can diffuse into the cytosol directly across the cell membrane and then traffic into the nucleus, doxorubicin-loaded nanoparticles are almost exclusively trafficked into endosomes and lysosomes upon cellular entry, following which only ~50% of the drug is released from the nanoparticle upon acid-triggered linker cleavage, as the lowest pH in the endo-lysosomes of ~4 do not enable complete cleavage of the linker. The released drug then enters the cytosol and finally the nucleus. This more circuitous route retards the kinetics of cytosolic and nuclear accumulation of the drug but does not change the final intracellular distribution of the drug between different organelles. The spatially resolved and quantitative information uncovered by this newly developed method provides us a quantitative understanding on the intracellular behavior of drug-loaded nanoparticles, which will help improve the design of the next-generation of nanoparticle drug delivery systems.
Supplementary Material
Acknowledgments
This work was supported by NIH grants R01 EB-00188 and R01 EB-007205 to A.C. We thank Dr. Yasheng Gao and Dr. Sam Johnson at Duke University’s Light Microscopy Core Facility for assistance with confocal microscopy.
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jconrel.2017.05.032.
References
- [1].Maeda H, Wu J, Sawa T, Matsumura Y, Hori K, Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review, J. Control. Release 65 (2000) 271–284. [DOI] [PubMed] [Google Scholar]
- [2].Petros RA, DeSimone JM, Strategies in the design of nanoparticles for therapeutic applications, Nat. Rev. Drug Discov 9 (2010) 615–627. [DOI] [PubMed] [Google Scholar]
- [3].Duncan R, The dawning era of polymer therapeutics, Nat. Rev. Drug Discov 2 (2003) 347–360. [DOI] [PubMed] [Google Scholar]
- [4].Ojea-Jimenez I, Comenge J, Garcia-Fernandez L, Megson ZA, Casals E, Puntes VF, Engineered inorganic nanoparticles for drug delivery applications, Curr. Drug Metab 14 (2013) 518–530. [DOI] [PubMed] [Google Scholar]
- [5].Ulbrich K, Šubr V, Polymeric anticancer drugs with pH-controlled activation, Adv. Drug Deliv. Rev 56 (2004) 1023–1050. [DOI] [PubMed] [Google Scholar]
- [6].Liu R, Zhang Y, Zhao X, Agarwal A, Mueller LJ, Feng P, pH-Responsive nanogated ensemble based on gold-capped mesoporous silica through an acid-labile acetal linker, J. Am. Chem. Soc 132 (2010) 1500–1501. [DOI] [PubMed] [Google Scholar]
- [7].Cheng R, Meng F, Deng C, Klok H-A, Zhong Z, Dual and multi-stimuli responsive polymeric nanoparticles for programmed site-specific drug delivery, Biomaterials 34 (2013) 3647–3657. [DOI] [PubMed] [Google Scholar]
- [8].Meng F, Zhong Y, Cheng R, Deng C, Zhong Z, pH-Sensitive polymeric nanoparticles for tumor-targeting doxorubicin delivery: concept and recent advances, Nanomedicine 9 (2014) 487–499. [DOI] [PubMed] [Google Scholar]
- [9].Kratz F, Beyer U, Schutte MT, Drug-polymer conjugates containing acid-cleavable bonds, Crit. Rev. Ther. Drug Carrier Syst 16 (1999) 245–288. [DOI] [PubMed] [Google Scholar]
- [10].Liu J, Huang Y, Kumar A, Tan A, Jin S, Mozhi A, Liang X-J, pH-Sensitive nano-systems for drug delivery in cancer therapy, Biotechnol. Adv 32 (2014) 693–710. [DOI] [PubMed] [Google Scholar]
- [11].Watson P, Jones AT, Stephens DJ, Intracellular trafficking pathways and drug delivery: fluorescence imaging of living and fixed cells, Adv. Drug Deliv. Rev 57 (2005) 43–61. [DOI] [PubMed] [Google Scholar]
- [12].Chen K-J, Chiu Y-L, Chen Y-M, Ho Y-C, Sung H-W, Intracellularly monitoring/imaging the release of doxorubicin from pH-responsive nanoparticles using Förster resonance energy transfer, Biomaterials 32 (2011) 2586–2592. [DOI] [PubMed] [Google Scholar]
- [13].Hillaireau H, Couvreur P, Nanocarriers’ entry into the cell: relevance to drug delivery, Cell. Mol. Life Sci. 66 (2009) 2873–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kirtane AR, Kalscheuer SM, Panyam J, Exploiting nanotechnology to overcome tumor drug resistance: challenges and opportunities, Adv. Drug Deliv. Rev 65 (2013) 1731–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ndolo RA, Luan Y, Duan S, Forrest ML, Krise JP, Lysosomotropic properties of weakly basic anticancer agents promote cancer cell selectivity in vitro, PloS One 7 (2012) e49366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Krüger HR, Schütz I, Justies A, Licha K, Welker P, Haucke V, Calderón M, Imaging of doxorubicin release from theranostic macromolecular prodrugs via fluorescence resonance energy transfer, J. Control. Release 194 (2014) 189–196. [DOI] [PubMed] [Google Scholar]
- [17].Basuki JS, Duong HT, Macmillan A, Erlich RB, Esser L, Akerfeldt MC, Whan RM, Kavallaris M, Boyer C, Davis TP, Using fluorescence lifetime imaging microscopy to monitor theranostic nanoparticle uptake and intracellular doxorubicin release, ACS Nano 7 (2013) 10175–10189. [DOI] [PubMed] [Google Scholar]
- [18].Wang F, Wang Y-C, Dou S, Xiong M-H, Sun T-M, Wang J, Doxorubicintethered responsive gold nanoparticles facilitate intracellular drug delivery for overcoming multidrug resistance in cancer cells, ACS Nano 5 (2011) 3679–3692. [DOI] [PubMed] [Google Scholar]
- [19].Li S-Y, Liu L-H, Jia H-Z, Qiu W-X, Rong L, Cheng H, Zhang X-Z, A pH-responsive prodrug for real-time drug release monitoring and targeted cancer therapy, Chem. Commun 50 (2014) 11852–11855. [DOI] [PubMed] [Google Scholar]
- [20].Wang J, MacEwan SR, Chilkoti A, Quantitative mapping of the spatial distribution of nanoparticles in endo-lysosomes by local pH, Nano Lett. 17 (2017) 1226–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li K, Su Q, Yuan W, Tian B, Shen B, Li Y, Feng W, Li F, Ratiometric monitoring of intracellular drug release by an upconversion drug delivery nano-system, ACS Appl. Mater. Interfaces 7 (2015) 12278–12286. [DOI] [PubMed] [Google Scholar]
- [22].Wozniak AL, Griffin S, Rowlands D, Harris M, Yi M, Lemon SM, Weinman SA, Intracellular proton conductance of the hepatitis C virus p7 protein and Its contribution to infectious virus production, PLoS Pathog 6 (2010) e1001087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Abràmoff MD, Magalh aes PJ, Ram SJ, Image processing with ImageJ, Biophoton. Int 11 (2004) 36–42. [Google Scholar]
- [24].MacKay JA, Chen M, McDaniel JR, Liu W, Simnick AJ, Chilkoti A, Self-assembling chimeric polypeptide-doxorubicin conjugate nanoparticles that abolish tumours after a single injection, Nat. Mater 8 (2009) 993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].McDaniel JR, Bhattacharyya J, Vargo KB, Hassouneh W, Hammer DA, Chilkoti A, Self-assembly of thermally responsive nanoparticles of a genetically encoded peptide polymer by drug conjugation, Angew. Chem. Int. Ed 52 (2013) 1683–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mastria EM, Chen M, McDaniel JR, Li X, Hyun J, Dewhirst MW, Chilkoti A, Doxorubicin-conjugated polypeptide nanoparticles inhibit metastasis in two murine models of carcinoma, J. Control. Release 208 (2015) 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bhattacharyya J, Bellucci JJ, Weitzhandler I, McDaniel JR, Spasojevic I, Li X, Lin C-C, Chi J-TA, Chilkoti A, A paclitaxel-loaded recombinant polypeptide nanoparticle outperforms abraxane in multiple murine cancer models, Nat. Commun 6 (2015) 7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Meyer DE, Chilkoti A, Purification of recombinant proteins by fusion with thermally-responsive polypeptides, Nat. Biotechnol 17 (1999) 1112–1115. [DOI] [PubMed] [Google Scholar]
- [29].Wang Z, Zhang J, Wang Y, Xing R, Yi C, Zhu H, Chen X, Guo J, Guo W, Li W, Matrine A novel autophagy inhibitor, blocks trafficking and the proteolytic activation of lysosomal proteases, Carcinogenesis 34 (2013) 128–138. [DOI] [PubMed] [Google Scholar]
- [30].Diwu Z, Chen C-S, Zhang C, Klaubert DH, Haugland RP, A novel acidotropic pH indicator and its potential application in labeling acidic organelles of live cells, Chem. Biol 6 (1999) 411–418. [DOI] [PubMed] [Google Scholar]
- [31].Sorkin A, von Zastrow M, Signal transduction and endocytosis: close encounters of many kinds, Nat. Rev. Mol. Cell Biol 3 (2002) 600–614. [DOI] [PubMed] [Google Scholar]
- [32].Misinzo G, Delputte PL, Nauwynck HJ, Inhibition of endosome-lysosome system acidification enhances porcine circovirus 2 infection of porcine epithelial cells, J. Virol 82 (2008) 1128–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhao Y, Alakhova DY, Kim JO, Bronich TK, Kabanov AV, A simple way to enhance Doxil® therapy: drug release from liposomes at the tumor site by amphiphilic block copolymer, J. Control. Release 168 (2013) 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Santra S, Kaittanis C, Santiesteban OJ, Perez JM, Cell-specific, activatable, and theranostic prodrug for dual-targeted cancer imaging and therapy, J. Am. Chem. Soc 133 (2011) 16680–16688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Marras SA, Selection of fluorophore and quencher pairs for fluorescent nucleic acid hybridization probes, Fluorescent Energy Transfer Nucleic Acid Probes: Designs and Protocols (2006) 3–16. [DOI] [PubMed] [Google Scholar]
- [36].Mu CJ, LaVan DA, Langer RS, Zetter BR, Self-assembled gold nanoparticle molecular probes for detecting proteolytic activity in vivo, ACS Nano 4 (2010) 1511–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bae Y, Nishiyama N, Fukushima S, Koyama H, Yasuhiro M, Kataoka K, Preparation and biological characterization of polymeric micelle drug carriers with intracellular pH-triggered drug release property: tumor permeability, controlled subcellular drug distribution, and enhanced in vivo antitumor efficacy, Bioconjug. Chem 16 (2005) 122–130. [DOI] [PubMed] [Google Scholar]
- [38].Tacar O, Sriamornsak P, Dass CR, Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems, J. Pharm. Pharmacol 65 (2013) 157–170. [DOI] [PubMed] [Google Scholar]
- [39].Wang J, Wang Y, Liang W, Delivery of drugs to cell membranes by encapsulation in PEG-PE micelles, J. Control. Release 160 (2012) 637–651. [DOI] [PubMed] [Google Scholar]
- [40].Duvvuri M, Krise JP, A novel assay reveals that weakly basic model compounds concentrate in lysosomes to an extent greater than pH-partitioning theory would predict, Mol. Pharm 2 (2005) 440–448. [DOI] [PubMed] [Google Scholar]
- [41].Duvvuri M, Gong Y, Chatterji D, Krise JP, Weak base permeability characteristics influence the intracellular sequestration site in the multidrug-resistant human leukemic cell line HL-60, J. Biol. Chem 279 (2004) 32367–32372. [DOI] [PubMed] [Google Scholar]
- [42].Dong D-W, Tong S-W, Qi X-R, Comparative studies of polyethyleniminedoxorubicin conjugates with pH-sensitive and pH-insensitive linkers, J. Biomed. Mater. Res. A 101 (2013) 1336–1344. [DOI] [PubMed] [Google Scholar]
- [43].Sahay G, Querbes W, Alabi C, Eltoukhy A, Sarkar S, Zurenko C, Karagiannis E, Love K, Chen D, Zoncu R, Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling, Nat. Biotechnol 31 (2013) 653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wittrup A, Ai A, Liu X, Hamar P, Trifonova R, Charisse K, Manoharan M, Kirchhausen T, Lieberman J, Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown, Nat. Biotechnol 33 (2015) 870–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].van der Meel R, Vehmeijer LJ, Kok RJ, Storm G, van Gaal EV, Ligand-targeted particulate nanomedicines undergoing clinical evaluation: current status, Adv. Drug Deliv. Rev 65 (2013) 1284–1298. [DOI] [PubMed] [Google Scholar]
- [46].Sudimack J, Lee RJ, Targeted drug delivery via the folate receptor, Adv. Drug Deliv. Rev 41 (2000) 147–162. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.