

Abstract
Tetrahydroisoquinoline (THIQ) 6-O-sulfamate-based anticancer agents, inspired by the endogenous steroid 2-methoxyestradiol and its sulfamate derivatives, are further explored for antiproliferative and microtubule disruptor activity. Based on recently designed C3-methyl C7-methoxy-substituted THIQ derivatives, compounds with mono- and dichloro-substitutions on the pendant N-benzyl ring were synthesized and evaluated. Although improved antiproliferative activity was observed, for example, 4a versus 4b and 4b versus 8c, it was relatively modest. Compound 8c, a 2′,5′-dichlorobenzyl derivative was, however, identified as a promising antiproliferative agent with in vitro activities exceeding that of the parent steroid (e.g., GI50 90 nM in DU-145 cells) and was highly potent against a range of tumor cell lines (e.g., GI50 26 nM for OVCAR-3). 8c inhibited the polymerization of tubulin in vitro with an IC50 only twofold less potent than combretastatin A-4 and inhibited colchicine binding to tubulin. Tubulin polymerization assays showed the parent THIQ 4a to be only a very weak inhibitor, but a striking potency difference was seen between compounds with C2′ methoxy and chloro substituents, whereas this was much smaller when these substituents were positioned at C5′. To confirm the target in atomic detail and because 8c is a racemic mixture, an achiral parent THIQ 6-O-sulfamate derivative 10 was successfully cocrystallized with the αβ-tubulin heterodimer. The derivative 10 binds at the colchicine site on tubulin, the first example of this compound class investigated in such detail, with its sulfamate group interacting with residues beyond the reach of colchicine itself, similar to a recently reported quinazolinone sulfamate derivative, 6a. The structure also suggests that for racemic C3-methyl-substituted THIQ derivatives, such as 8c, the (S)-enantiomer is likely to be preferentially accommodated within the colchicine site for steric reasons. The results further confirm the potential of nonsteroidal THIQ sulfamate derivatives for oncology and suggest that the mechanism of microtubule destabilization for the THIQ compound class is to prevent the curved-to-straight conformational transition of tubulin required for polymerization.
Introduction
In previous studies, we described N-benzyl-substituted tetrahydroisoquinoline (THIQ) derivatives as novel microtubule disruptors with potential therapeutic application for the treatment of cancer.1−5 These compounds were designed to mimic the 2-substituted estratriene class of microtubule disruptors derived from the endogenous steroid 2-methoxyestradiol (2ME) and, in particular, its sulfamate derivatives (e.g., 2a (STX140), Figure 1).6−10 Incorporation of a phenolic 3-O-sulfamate group is generally observed to be highly beneficial for both activity and oral bioavailability, and STX140 is an optimized anticancer agent with both 3-O- and 17-O-sulfamate groups.11 Agents possessing such sulfamate esters have reached multiple clinical trials in oncology and elsewhere, primarily for hormone-dependent diseases.11,12 The sulfamoyl group can serve in diverse roles.12
Figure 1.

Design of dichlorobenzyl-substituted THIQ-based microtubule disruptors 7 and 8.
STX140 was developed primarily for hormone-independent cancer applications, and the nonsteroidal THIQ core was used as a mimic of the steroidal AB-ring system from which steroidomimetics were constructed. Such compounds have markedly better physicochemical properties than 2ME and even STX140 but can maintain in vivo potency. Substitution of the THIQ nucleus at C6 and C7, with those groups requisite for activity in the steroidal series, was thus desirable. Attachment at N2 of a group projecting into the area of space occupied by the steroidal D-ring and bearing an H-bond acceptor known to be required for optimal activity completed the prototypical steroidomimetic design.1,2,11 Further optimization led to THIQs methylated at C3 forcing a more favorable conformation of the steroidal D-ring mimic by steric repulsion (THIQs of type 3 and 4, Figure 1).3 Related THIQ core chimeric microtubule disruptors possessing the trimethoxy aryl motif common to many tubulin colchicine site binders were designed and then further optimized.4,5 As a result, the best compounds in both series displayed nanomolar in vitro activities, making them equipotent or better than the steroidal derivatives that underpinned their design.3−5 Most recently, we designed another steroidal AB-ring mimic by using a dihydroquinazolinone (DHQ) core structure.13 The most potent DHQs derived from this series were also sulfamates of type 6 (Figure 1) with the best compounds again displaying nanomolar in vitro activities.
Like the steroids, the most potent heterocyclic derivatives were found to disrupt the polymerization of tubulin and inhibit the binding of [3H]colchicine to tubulin. THIQ sulfamates also inhibit carbonic anhydrases (CA) II,4 an interaction believed to contribute to the high oral bioavailability observed for the steroid derivatives,14,15 and CAIX,16 which affords extra targeting of hypoxic tumors. In addition, the activity of nonsteroidal THIQ derivatives mirrors that of the steroidal series, with compounds capable of inhibiting the growth of taxane-resistant cancer cells17−19 and human umbilical vein endothelial cell proliferation (a commonly used marker for antiangiogenic activity),20,21 thus supporting the idea that these small-molecule agents work in a similar manner to 2ME and its bis-O-sulfamate derivative 2a (STX140).
Very recently, the mechanism of these heterocyclic sulfamate esters in the destabilization process of microtubule polymerization was investigated for the first time in atomic detail. The crystal structure of the most potent DHQ sulfamate of type 6, the N-(2′,5′-dimethoxybenzyl) derivative 6a, in complex with tubulin revealed molecular details to support further strategies for structure-based optimization. Interestingly, the sulfamate group was shown to play a role in binding at the colchicine site of tubulin. The mechanism of microtubule destabilization, at least for this particular DHQ 6a, was established as preventing the curved-to-straight tubulin conformational transition required for microtubule formation.13,22
DHQ derivatives, although highly effective, are not charged at the linking N-atom. THIQ derivatives likely have more developmental potential, their charged nature in particular affording better solubility via salt formation at the N2 position. Moreover, they have been shown to be orally active in vivo when dosed in an aqueous buffer, with some being as potent as STX140.4 It was thus of interest to explore the potential for further optimization of such derivatives via new substitutions in the pendant aryl ring and, moreover, if this THIQ class could also be cocrystallized with the αβ-tubulin heterodimer, such that atomic level information could be obtained, for further structure–activity relationship (SAR) development. In the present work, we explore the replacement of methoxy groups present in the steroidal D-ring mimic of C3-methylated THIQs with chloro substituents to determine whether such less polar and more lipophilic H-bond acceptors improve microtubule disruptor potency (Figure 1). Therefore, we introduced 2′,3′-, 2′,4′-, and 2′,5′-dichlorobenzyl groups at N2 to explore SAR in a focused series of phenols and their sulfamate esters and explored their antiproliferative and antitubulin activities.
Results and Discussion
Chemistry
The candidate THIQs were synthesized in two steps starting from (±)-6-hydroxy-7-methoxy-3-methyl-1,2,3,4-THIQ 9.3N-Benzylation using dichlorobenzyl halides, diisopropylethylamine (DIPEA) in N,N-dimethylformamide (DMF) at 140 °C for 18 h gave phenols 7a–c in moderate yields. The target sulfamates 8a–c were synthesized using sulfamoyl chloride as a solution in N,N-dimethylacetamide (DMA)23 (Scheme 1).
Scheme 1. Synthesis of Dichloro-tetrahydroisoquinoline-Based Microtubule Disruptors.

Reagents and conditions: (i) ArCH2X (X = Cl or Br), NaBr (for X = Cl), DIPEA, DMF, 140 °C, 18 h; (ii) H2NSO2Cl, DMA, rt.
Biology
All six compounds were evaluated at the US National Cancer Institute (NCI) in the full 60-cell-line assay that allows activity across a wide range of cancer types to be assessed (Tables 1 and 2). The in vitro activities of these compounds against DU-145 prostate and MDA MB-231 breast cancer cell line proliferation are shown in Table 1. Data from both cell lines are in strong agreement. Overall, compounds in this small series of N2-dichlorobenzyl C3-methyl-substituted THIQs exhibit activities in the micromolar and nanomolar ranges. Phenols 7b–c are only about 3–5-fold less active than their corresponding sulfamates 8b–c against the proliferation of DU-145 prostate cancer cells and MDA MB-231 breast cancer cells. The exception is phenol 7a that proved to be more than 10-fold less active than its corresponding sulfamate, 8a. GI50 values of the sulfamates 8a–c range between 90 nM (8c; DU-145) and 551 nM (8a; MDA MB-231). The best THIQ 8c of this small SAR screening set proved slightly more potent or very similar to previously reported THIQs, 4a–b.3 However, data from the NCI-60-cell-line panel for 4a–b against DU-145 and MDA-MB 231 cells suggested that 8c is significantly more potent than 4a, but only about as potent as 4b. Overall, the antiproliferative activity was significantly improved when the 2′-methoxy group was replaced with a 2′-chloro substituent. However, no further enhancement was observed when the 5′-methoxy group too was replaced with a 5′-chloro substituent. As these (±)-C3 methyl-substituted THIQs adopt a steroid-like conformation with the substituent at C5′ pointing into the same area of space as the steroidal C17 β-hydroxyl group,1−3 it seems likely that the substituent at C2′ will sterically interact with the two hydrogens at C1. Therefore, only relatively small one-atom substituents such as chlorine seem well tolerated at the C2′ position. Note that the hydrogen and fluoro substituents at C2′ in combination with a C5′ methoxy group were also very well tolerated.3 It seems that the only marginally larger C2′ methoxy group could already force too much of an angle between the THIQ B-ring and the pendant N2-benzyl ring, resulting in a less favorable overall conformation, leading to a decrease in activity (4a; MDA MB-231: 2.1 μM). However, there seem to be less restrictions for the second H-bond acceptor at C5′ and more space might be available around that binding site. Note, of course, that in all cases here, the activities of racemic mixtures are being compared, with all usual caveats, but vide infra for further discussion on this point.
Table 1. Antiproliferative Activity of Racemic THIQ Derivatives Against DU-145 Human Prostate and MDA MB-231 Human Breast Cancer Cells in Vitro from the NCI-60 Cell Line Panela.
| GI50 (μM) |
|||||||
|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | R3 | R4 | R5 | DU-145 | MDA MB-231 |
| 4a | SO2NH2 | OMe | H | H | OMe | 0.371 | 2.1 |
| 0.189b | 0.16b | ||||||
| 4b | SO2NH2 | Cl | H | H | OMe | 0.085 | 0.299 |
| 0.4b | 0.4b | ||||||
| 7a | H | Cl | Cl | H | H | 4.18 | 7.05 |
| 8a | SO2NH2 | Cl | Cl | H | H | 0.301 | 0.551 |
| 7b | H | Cl | H | Cl | H | 0.261 | 0.465 |
| 8b | SO2NH2 | Cl | H | Cl | H | 0.195 | 0.365 |
| 7c | H | Cl | H | H | Cl | 0.241 | 0.542 |
| 8c | SO2NH2 | Cl | H | H | Cl | 0.090 | 0.324 |
GI50 figures represent mean values from triplicate experiments. All compounds of type 4, 7, and 8 are racemic mixtures.
GI50 values for 4a–b are additionally taken from the literature.3
Table 2. Antiproliferative Activity of N-Dichlorobenzyl-Substituted THIQs against Various Other Cancer Cell Lines in Vitro from the NCI-60 Cell Line Panela,b.
| GI50 (μM) | |||||||
|---|---|---|---|---|---|---|---|
| compd | lung HOP-62 | colon HCT-116 | CNS SF-539 | melanoma UACC-62 | ovarian OVCAR-3 | renal SN12-C | MGM |
| 4a | 0.504 | 0.446 | 0.331 | 0.584 | 0.298 | 1.64 | 0.562 |
| 4b | 0.075 | 0.048 | 0.042 | 0.069 | 0.039 | 0.372 | 0.102 |
| 7a | 3.69 | 4.61 | 4.39 | 4.37 | 2.64 | 5.83 | 5.01 |
| 8a | 0.387 | 0.226 | 0.227 | 0.119 | 0.137 | 0.933 | 0.316 |
| 7b | 0.427 | 0.34 | 0.235 | 0.596 | 0.201 | 0.668 | 0.457 |
| 8b | 0.183 | 0.054 | 0.106 | 0.050 | 0.045 | 0.657 | 0.151 |
| 7c | 0.33 | 0.26 | 0.132 | 0.448 | 0.17 | 0.706 | 0.324 |
| 8c | 0.055 | 0.073 | 0.046 | 0.056 | 0.026 | 0.345 | 0.129 |
GI50 figures are mean values from triplicate experiments. All compounds of type 4,7, and 8 are racemic mixtures. MGM represents the mean concentration that caused 50% growth inhibition in all 60 cell lines.
GI50 values for 4a–b are taken from the literature.3
Additionally, data from six more tumor cell lines are presented along with the mean activity across the whole panel (MGM value; Table 2). The trend that emerged for these THIQ derivatives against the proliferation of DU-145 prostate cancer cells and MDA MB-231 breast cancer cells in vitro was essentially confirmed across the whole NCI 60-cell-line assay. It also confirms the potential of these N-dichlorobenzyl compounds against a broad range of cancer phenotypes with 8c, in particular, proving highly active (e.g., 26 nM in OVCAR-3; MGM = 129 nM). Over the whole 60-cell-line panel, compound 4b was slightly more potent (MGM = 102 nM; Table 2). Additionally, the trend mentioned above with regard to 4a–b and 8c seems to hold well. Overall 4b and 8c were about 4–5 times more potent than 4a.
To confirm the target for these, we established the activity of the sulfamoylated compounds, 8a–c as microtubule disruptors, alongside the established potent clinical microtubule disruptor, combretastatin A-4 (CA-4), initially in tubulin polymerization assays and alongside the previously reported THIQ sulfamate, 10(5) (Table 3). Gratifyingly, the 2′,5′-dichlorobenzyl derivative 8c very effectively inhibited the assembly of tubulin, with an IC50 of 0.98 μM, being only about 2-fold less potent than CA-4. The IC50 in these tubulin-based assays, as is typical with antitubulin agents with potent cytotoxic activity, far exceeds the antiproliferative GI50 dose, at least in part because the GI50 values refer to the concentration in the media, not in the cells. Inhibition of colchicine binding to tubulin for these compounds was also determined relative to CA-4 for further proof of targeting, with 8c being the overall best THIQ derivative to date showing 67% inhibition at 5 μM (Table 3). It is reasonable to propose that these novel THIQs can, at least partially, be assumed to disrupt normal dynamic tubulin polymerization by interacting at the colchicine site. Also, this provides further evidence that methoxy groups as polar H-bond acceptors are not an essential requirement in the D-ring mimic. Overall, the tubulin assays showed a striking difference in potency between compounds with methoxy and chloro as the C2′ substituents (4a vs 4b), whereas the difference in potency is much smaller when these substituents are positioned at C5′ (4b vs 8c). But also, here, the C5′ chloro compound 8c displayed an improved IC50 and inhibition of colchicine binding than the corresponding C5′ methoxy compound, 4b.
Table 3. Activity of N-Dichlorobenzyl-Substituted THIQs as Tubulin Polymerization Inhibitors and [3H]Colchicine Binding (5 μM Inhibitor) to Tubulina.
| tubulin assembly | colchicine binding | |
|---|---|---|
| compd | IC50 (μM) | (% inhibition) |
| CA-4 | 0.54 ± 0.06 | 98 ± 0.1 |
| 4a | >20 | 8.2 ± 2 |
| 4b | 1.7 ± 0.2 | 55 ± 5 |
| 8a | 2.1 ± 0.2 | 35 ± 1 |
| 8b | 1.4 ± 0.2 | 47 ± 2 |
| 8c | 0.98 ± 0.06 | 67 ± 2 |
| 10 | 1.3 ± 0.01 | 49 ± 3 |
Values are the mean ± SD of at least two determinations. All compounds of type 4 and 8 are racemic mixtures.
Data for 4a are taken from the literature.3
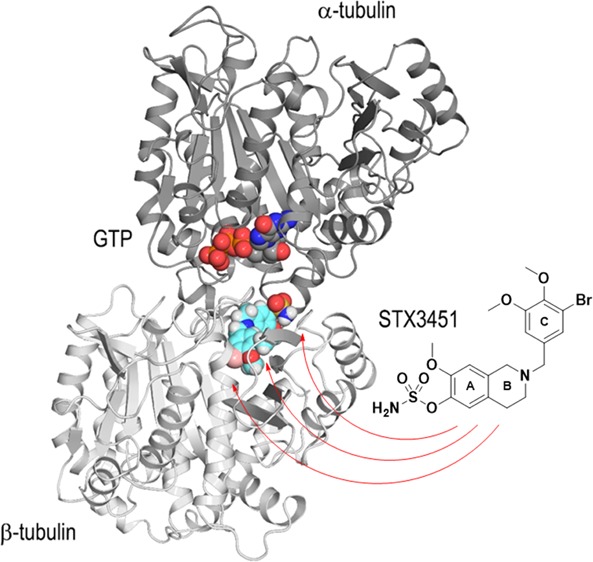
As in previously published work on DHQs,13 we employed X-ray crystallography to determine the atomic level binding mode of a THIQ derivative within the αβ-tubulin heterodimer (Figure 2A). To avoid complications that would arise from using the racemic sulfamate, 8c, we chose a highly potent nonracemic THIQ, [2-(3′-bromo-4′,5′-dimethoxybenzyl)-7-methoxy-6-sulfamoyloxy-1,2,3,4-THIQ—STX3451, 10], without the C3-methyl group chiral center that had been optimized earlier5 and which is not too dissimilar to 8c in its antitubulin activities (Table 3). STX3451 exerts antiproliferative and antimitotic effects, inducing apoptosis and involving autophagic processes in MDA-MB-231 metastatic breast and A549 epithelial lung carcinoma cells24 and also induces cell death effectively in NF1 tumor cell lines.25 It is therefore somewhat more attractive as a potential development candidate than 8c. Compound 10 was soaked into crystals formed by a protein complex composed of two αβ-tubulin heterodimers, the stathmin-like protein RB3 and tubulin tyrosine ligase (termed T2R-TTL),26 and we solved the T2R-TTL-10 complex structure using X-ray crystallography to 2.4 Å resolution (Figures 2 and 3B, 4A; Table 4) (PDB ID 6HX8). STX3451 binds to the tubulin colchicine site22 at the intradimer interface created by residues from the strands βS8 and βS9 the helix βH8 of β-tubulin, and the loop αT5 of α-tubulin (Figure 4A). STX3451 is well accommodated within the binding site, displaying an extended “steroid-like” conformation. These data now provide the second cocrystal structure of an antitubulin agent that possesses a sulfamate group and the first one where the steroidal AB-ring system is mimicked by a THIQ core. Figure 4B shows a comparison of the new THIQ sulfamate binding mode within the colchicine site with that of the earlier DHQ sulfamate 6a.13
Figure 2.
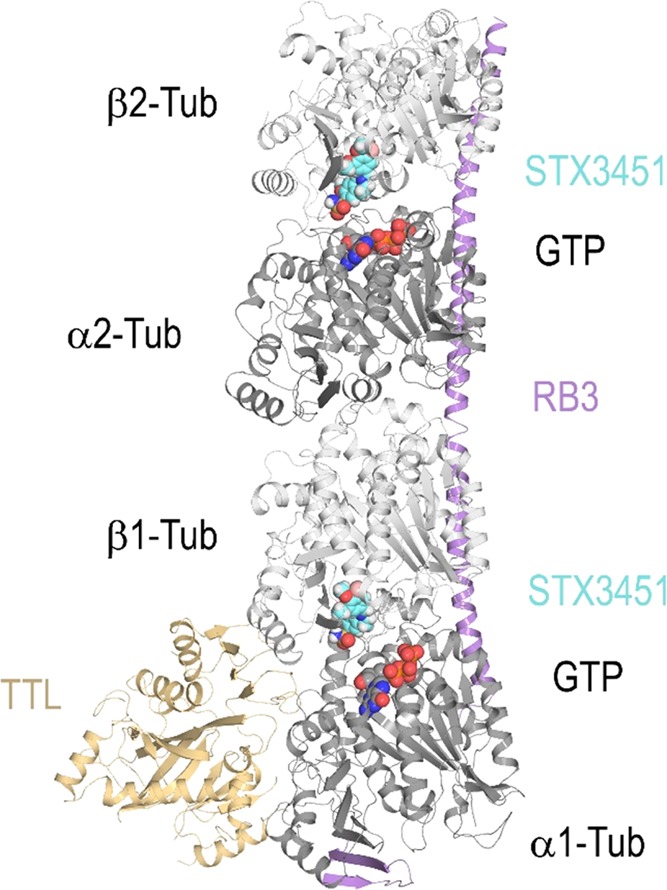
Overall view of the T2R-TTL-STX3451 complex structure. The α- and β-tubulin chains are in dark and light grey, respectively, TTL is in yellow/orange, and RB3 is in violet ribbon representation. The tubulin-bound STX3451 and the nonexchangeable guanosine 5′-triphosphate (GTP) molecules bound to α-tubulin are shown as sphere representations. The carbon atoms of STX3451 are colored in cyan.
Figure 3.
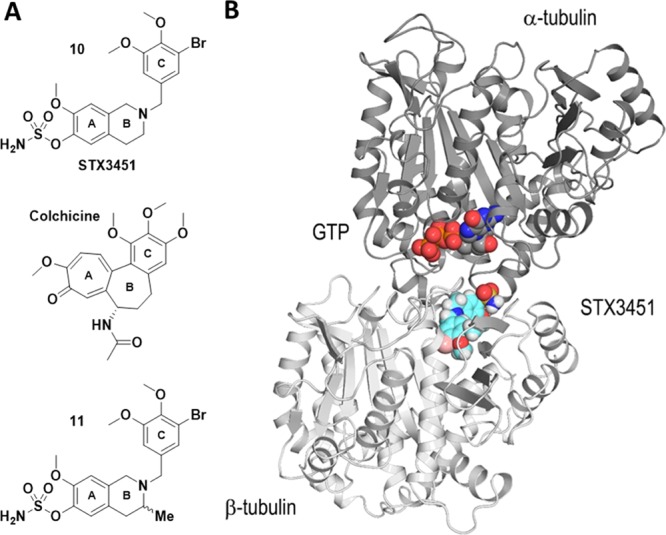
Crystal structure of the complex of tubulin with STX3451. (A) Structures of 10 (STX3451), colchicine, and 11. (B) STX3451 binding mode within the αβ-tubulin heterodimer. The α- and β-tubulin subunits are represented as dark and light gray ribbons, respectively. STX3451 carbon atoms and the nonexchangeable GTP molecule bound to α-tubulin are shown as cyan and orange spheres, respectively.
Table 4. Refinement Statistics of T2R-TTL-STX3451a.
| Data Collectiona | |
|---|---|
| space group | P212121 |
| Cell Dimensions | |
| a, b, c (Å) | 104.0, 155.6, 180.9 |
| resolution (Å) | 48.4–2.4 (2.46–2.40) |
| Rmerge (%) | 9.3 (266.0) |
| Rmeas (%) | 10.1 (288.9) |
| Rpim (%) | 4.2 (115.8) |
| I/σI | 16.7 (0.8) |
| CC1/2b | 99.9 (29.4) |
| completeness (%) | 100 (100) |
| redundancy | 6.8 (6.6) |
| Refinement | |
|---|---|
| resolution (Å) | 48.4–2.4 |
| no. unique reflections | 115 121 |
| Rwork/Rfree | 19.5/24.0 |
| No. Atoms | |
| protein | 17 324 |
| ligand | 104 |
| water | 303 |
| AverageB-Factors(Å2) | |
| protein | 91,3 |
| ligand (chain B/D) | 66.3/114.3 |
| water | 74.3 |
| Wilson B-factor | 63.9 |
| R.m.s. Deviations | |
| bond lengths (Å) | 0.002 |
| bond angles (deg) | 0.535 |
| Ramachandran Statisticsc | |
| favored regions (%) | 97.1 |
| allowed regions (%) | 2.9 |
| outliers (%) | 0 |
Figure 4.

Detailed interaction of the tubulin-STX3451 complex. (A) Detailed view of the interaction network seen between STX3451 (aquamarine) and tubulin (gray). Residues of tubulin that interact are labeled and shown in stick representation. Oxygen and nitrogen atoms are colored red and blue, respectively, and carbon atoms are represented as aquamarine (STX3451) or gray (tubulin). Hydrogen bonds are shown as black dashed lines, and secondary structural elements of tubulin are labeled in blue. For simplicity, only α-tubulin residues and secondary structural elements are marked with an α. (B) Same close-up view as in (A) with the superimposed DHQ, 6a (pale yellow; PDB ID 5OSK; root-mean-square deviation of 0.20 Å over 409 Cα-atoms) structure. (C) Same close-up view as in (A) with the final 2mFo-DFc (blue) and mFo-DFc (green and red) electron density maps contoured at 1.0σ and ±3.0σ, respectively. The green electron density blob marked with an arrow suggests an alternate conformation of the 5′-methoxy group of the C-ring. (D) Sigma A-weighted mFo-DFc (light green mesh) simulated annealing electron-density omit map contoured at 3.0σ. The map was calculated excluding the atoms corresponding to STX3451 only. Both the STX3451 molecule (aquamarine sticks) and the two water molecules (red spheres, HOH) of the final refined structure are superimposed to highlight the quality of the map.
The C-ring of STX3451 is buried into a hydrophobic pocket formed by the β-tubulin residues: βVal238, βCys241, βLeu242, βLeu255, βMet259, βAla316, βIle318, βAla354, βThr376, and βIle378 (Figure 4A). Moreover, the 4′-methoxy oxygen of the C-ring forms a water-mediated hydrogen bond to the main chain amide and carbonyls of βCys241, βGly237, and βVal238. The A-ring of STX3451 is stacked between the side chains of βAsn258 and βLys352, and further stabilization is provided by hydrogen bonds between the sulfamate moiety and the side chains of βLys352, βAsn349, and αSer178, the main chain amide of αVal181, and the main chain carbonyl group of βAsn349. Compared to the DHQ derivative 6a(13) (PDB ID 5OSK), which possesses an amine group in the B-ring, no water-mediated interaction to the carbonyl groups of the side chain and main chain of αAsn101 and αThr179 is observed for STX3451 (Figure 4B). Moreover, the 3′-bromo substituent of the C-ring in STX3451 forms a water-mediated hydrogen bond to the main chain carbonyl of βVal238, a space that is otherwise occupied by the 2′-methoxy group in 6a (Figure 4B).
Recently, THIQ compound (±)-11 (Figure 3A), which is the same as 10 but with an additional (±)-C3 methyl group as in the present 8c compound series, was synthesized and evaluated.5 As it was found to be similar to 10 (STX3451) in antitubulin assays (IC50 2.4 μM, 50% inhibition of colchicine binding5 vs IC50 1.3 μM and 49%, respectively) (Table 3, but note the very slightly different assay conditions), we would suggest that the additional (±)-C3 methyl group of the present series should, in principle, be well accommodated, as presumably also the 2′,5′-dichloroaryl motif. This former feature was not an issue in the DHQ 6a instance (see Figure 4B) because a planar carbonyl group occupies the C3 position. A closer inspection of the C3 environment in the tubulin-STX3451 crystal structure reveals that there may be enough space to accommodate both enantiomers of compound 11 (Figure 3A). However, we modeled compound 11 in both the (R)- and (S)-configurations into the STX3451 structure without energy minimization and inspected the C3 environment of both the enantiomers (Figure 5). The model suggests that the (S)-enantiomer likely binds without any structural rearrangements, whereas the (R)-enantiomer would clash into helix βH8, thereby requiring structural adaptations to get accommodated into the pocket or even preventing binding. For these reasons, we expect that administration of the racemate would preferentially select for the (S)-enantiomer binding to tubulin. It is tempting to view the respective antitubulin IC50 values of (±)-11 and 10 in the light of this (2.4 and 1.3 μM) and speculate that (R)-11 might be a very weak inhibitor or even totally inactive. The antitubulin activity of racemic 3-methyl-substituted THIQs, such as 8c, may therefore actually be an underestimate if the (R)-enantiomer is not accommodated or only binds weakly in the colchicine site because of the structural adaptations necessary. Exploration of this possibility must however await the resolution of the isomers but, given the greater development potential of the achiral THIQ sulfamate 10, this has not been pursued. Further development of these THIQs through medicinal chemistry might include the introduction of a (±)-C4 hydroxyl group as it could attract a water-mediated interaction to the carbonyl groups of the side chain and main chain of αAsn101 and αThr179, as was observed for DHIQ, 6a.13
Figure 5.
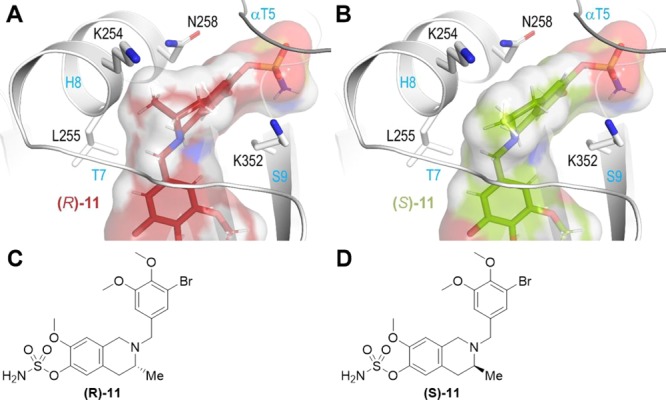
Molecular models of compound 11 in (A) (R)- and (B) (S)-configuration. Both enantiomers are shown in stick and semi-transparent surface representation. Carbon atoms are colored in red (A) and green (B). The tubulin backbone is in white and grey ribbon representation. Selected residue sidechains are in stick representation and are labeled. Secondary structural elements are labeled in blue. Chemical structures of compound 11 in (C) (R)- and (D) (S)-configuration.
We further compared the binding mode of STX3451 10 with that of colchicine by superimposition of both β-tubulin chains of their tubulin-ligand complexes (Figure 6; PDB ID 4O2B; rmsd of 0.428 Å over 393 Cα-atoms of chains B). Tubulin undergoes a “curved-to-straight” conformational change when polymerized into microtubules,22,27 involving an overall compaction of the colchicine binding site through mainly the βT7 loop and βS8 strand of the β-tubulin chain.22 As already observed for the DHQ derivative 6a,13 both compound poses show similarity and reveal that the respective A rings and associated methoxy groups of STX3451 and colchicine are very well superimposed, whereas the more flexible C ring of STX3451 is more extended. Both compounds, STX3451 and 6a, share a unique and common polar interaction with the sulfamoyl group of the main chain of αVal181. As a result of the different shapes of the molecules (STX3451 vs colchicine), major conformational changes of both the αT5 and βT7 loops are observed (Figure 6). In summary, the results have established STX3451 10 as a tubulin-binding ligand at the colchicine site in atomic detail. They also imply that 10, similar to other colchicine-site ligands,22,28 achieves microtubule destabilization mechanistically by preventing the curved-to-straight conformational transition.
Figure 6.
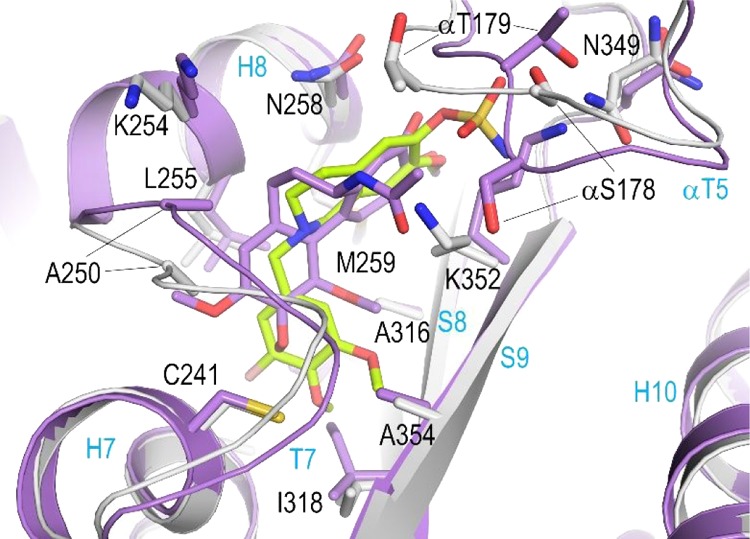
Superimposition of the tubulin-STX3451 (lime/grey) and the tubulin-colchicine (violet; PDB ID 4O2B) complex structure in the same orientation and representation as in Figure 4A. The structures were superimposed onto their β1-tubulin chains.
Conclusions
THIQ sulfamate-based microtubule disruptors have been further explored. Racemic N2-dichlorobenzyl C3-methyl-substituted THIQs showed excellent in vitro activities in the NCI-60-cell-line assays and antitubulin properties. Chlorine substitution for methoxy was found to be effective, and 8c in particular exhibits antiproliferative activity in the 90 nM range, inhibiting tubulin assembly and interfering effectively with the colchicine site. To explore tubulin binding at the atomic level without the complication of chirality, a parent THIQ STX3451 10 was cocrystallized successfully with the αβ-tubulin heterodimer. STX3451 binds more deeply in the colchicine site than colchicine itself, and the sulfamate group is involved in binding through specific interactions with β-tubulin residues beyond those accessed by colchicine. This is the first example of a THIQ derivative bearing a sulfamate ester bound to tubulin to be explored in such detail. With subtle differences, 10 adopts a broadly similar binding pose to a related DHQ derivative. Structural implications derived from the ligand are that C3 methyl group substitutions, as in the dichlorobenzyl derivatives, can be accommodated in the colchicine site. Modeling of (R)-11 and (S)-11 into the STX3451-tubulin structure further revealed that the (S)-enantiomer might be mainly responsible for the tubulin activity of such racemic C3 methyl-THIQ sulfamates as the C3 methyl group of the (R)-enantiomer would sterically clash with tubulin residues. Most likely, mechanistically microtubule destabilization by STX3451 is via prevention of the curved-to-straight conformational transition of tubulin, as shown for colchicine and the STX3451-related quinazolinone DHQ derivative. With the usual caveats with regard to chirality, such compounds of this THIQ class are worthy development candidates for oncology.
Experimental Section
Tubulin Assays
Bovine brain tubulin was prepared as previously described31 and used in the studies presented. Assembly IC50’s were determined as fully described elsewhere.32 In brief, 1.0 mg/mL (10 μM) tubulin was preincubated with varying compound concentrations without GTP for 15 min at 30 °C. Reaction mixtures were placed on ice, and GTP (final concentration, 0.4 mM) was added. Mixtures were transferred to cuvettes held at 0 °C in a recording spectrophotometer. Baselines were established at 0 °C and increase in turbidity was followed for 20 min following a rapid (<30 s) jump to 30 °C. Concentrations of compound required to reduce the turbidity increase by 50% were determined. Inhibition of the binding of [3H]colchicine to tubulin was described fully previously.33 Reaction mixtures contained 0.1 mg/mL (1.0 μM) tubulin, 5.0 μM [3H]colchicine, and a potential inhibitor at 5.0 μM. Compounds were compared to CA-4, a particularly potent inhibitor of colchicine binding to tubulin.34 Mixtures were incubated for 10 min at 37 °C, a time point at which colchicine binding in control reaction mixtures is generally 40–60% complete.
Crystallization, Data Collection, and Structure Solution
Crystals of T2R-TTL were generated as previously described.25,35 Crystals were soaked for 3 h at 20 °C in a reservoir solution (10% PEG 4K, 16% glycerol, 30 mM MgCl2, 30 mM CaCl2, and 0.1 M 2-(N-morpholino)ethanesulfonic acid/imidazole pH 6.7) containing 5 mM of compound 10 (STX3451) and subsequently transferred into a reservoir supplemented with 20% glycerol before cryo-cooling in liquid nitrogen. All data were collected at the Swiss Light Source (beamline X06DA, Paul Scherrer Institut, Villingen PSI, Switzerland) with images indexed and processed using XDS.36 Structure solution using the difference Fourier method and refinement were performed using PHENIX37 and model building was carried out iteratively using Coot software.38 The atomic coordinates and structure factors have been deposited in the Protein Data Bank (www.rcsb.org) under accession number 6HX8 (T2R-TTL-STX3451).
Structural Analysis and Figure Preparation
Molecular graphics and analyses were carried out using PyMol (The PyMOL Molecular Graphics System, Version 1.8.6.2. Schrödinger, LLC).
Chemistry
All chemicals were either purchased from Aldrich Chemical Co. (Gillingham, UK) or Alfa Aesar (Heysham, UK). Organic solvents of A.R. grade (PE, EtOAc, CHCl3, acetone, and CH2Cl2) were supplied by Fisher Scientific (Loughborough, UK) and were used as supplied. Petroleum ether (PE) used for crystallization was of fractions 40–60 °C. DMA and DMF were purchased from Aldrich and stored under a positive pressure of N2 after use. Sulfamoyl chloride was prepared using an adaptation of Appel and Berger methodology39 and was stored in the refrigerator as a solution in toluene under a positive pressure of N2, as described by Woo et al.40 An appropriate volume was freshly concentrated in vacuo immediately before use. Compound 9 was synthesized according to a literature procedure.3 Reactions were performed at room temperature unless stated otherwise. Flash column chromatography was carried out on silica gel (MatrexC60). 1H NMR spectra were recorded with a Varian Mercury VX 400 NMR spectrometer and a Bruker AVIII HD 400 spectrometer at 400 MHz. 13C NMR spectra were recorded with a Bruker AVIII HD 400 spectrometer at 100 MHz. Chemical shifts are reported in parts per million (ppm) relative to the solvent residual peaks as internal standards. 1H NMR: 7.26 ppm (CDCl3); 2.05 ppm (acetone-d6). 13C NMR: 29.84 ppm (acetone-d6). High resolution mass spectrometry was performed using a Bruker microTOF electrospray ionization mass spectrometer. Compounds were ≥96% pure by reversed-phase HPLC run with CH3CN/H2O or MeOH/H2O (Sunfire C18 column, 4.6 × 150 mm, 3.5 μm pore size).
(±)-2-(2,3-Dichlorobenzyl)-6-hydroxy-7-methoxy-3-methyl-1,2,3,4-tetrahydroisoquinoline 7a.
Compound 9 (386 mg, 2.0 mmol) was treated with 2,3-dichlorobenzyl bromide (506 mg, 2.1 mmol) and DIPEA (1.041 g, 8.0 mmol) in DMF (4.0 mL) at 140 °C for 18 h. After cooling to room temperature (RT), the reaction mixture was evaporated, then treated with H2O (100 mL) and NH4Cl (saturated, 10 mL) before extracting into EtOAc (2 × 100 mL). The combined organic layers were dried (NaCl), filtered, and evaporated. Purification by flash column chromatography (CHCl3/acetone 9:1 → 9:1 and 2% MeOH) afforded compound 7a as a yellow solid (202 mg, 28%). 1H NMR (400 MHz, CDCl3): δ 1.14 (3H, d, J = 6.6), 2.52 (1H, dd, J = 16.1, 5.7), 2.94 (1H, dd, J = 15.9, 4.8), 3.14 (1H, sext, J 6.0), 3.56–3.90 (4H, m), 3.81 (3H, s), 5.42 (1H, s, br), 6.45 (1H, s), 6.66 (1H, s), 7.17 (1H, t, J 7.8), 7.35 (1H, dd, J 8.0, 1.3), and 7.50 (1H, d, J 7.0). LC/MS (ES+) m/z: 352.0 (M+ + H). HRMS (ES+) m/z: found 352.0872; C18H20Cl2NO2+, (M+ + H) requires 352.0866.
(±)-2-(2,4-Dichlorobenzyl)-6-hydroxy-7-methoxy-3-methyl-1,2,3,4-tetrahydroisoquinoline 7b
Compound 9 (387 mg, 2.0 mmol), 2,4-dichlorobenzyl chloride (415 mg, 2.1 mmol), sodium bromide (41 mg, 0.4 mmol), and DIPEA (1.038 g, 8.0 mmol) in DMF (4.0 mL) were reacted at 140 °C for 18 h, as described for the synthesis of 7a. Purification by flash column chromatography (CHCl3/acetone 9:1 → 9:1 and 2% MeOH) afforded compound 7b as a yellow solid (341 mg, 48%). 1H NMR (400 MHz, CDCl3): δ 1.14 (3H, d, J = 6.5), 2.51 (1H, dd, J = 16.1, 5.8), 2.92 (1H, dd, J = 16.2, 4.8), 3.12 (1H, sext, J 6.1), 3.53–3.80 (4H, m), 3.81 (3H, s), 5.42 (1H, s, br), 6.44 (1H, s), 6.65 (1H, s), 7.21 (1H, dd, J = 8.3, 2.1), 7.36 (1H, d, J 2.1), and 7.51 (1H, d, J 8.3). LC/MS (ES+) m/z: 352.0 (M+ + H). HRMS (ES+) m/z: found 352.0869; C18H20Cl2NO2+, (M+ + H) requires 352.0866.
(±)-2-(2,5-Dichlorobenzyl)-6-hydroxy-7-methoxy-3-methyl-1,2,3,4-tetrahydroisoquinoline 7c
Compound 9 (387 mg, 2.0 mmol), 2,5-dichlorobenzyl chloride (413 mg, 2.1 mmol), sodium bromide (41 mg, 0.4 mmol), and DIPEA (1.035 g, 8.0 mmol) in DMF (4.0 mL) were reacted at 140 °C for 18 h, as described for the synthesis of 7a. Purification by flash column chromatography (CHCl3/acetone 9:1 → 9:1 and 2% MeOH) afforded compound 7c as a yellow solid (275 mg, 39%). 1H NMR (400 MHz, CDCl3): δ 1.14 (3H, d, J = 6.6), 2.52 (1H, dd, J = 16.1, 5.9), 2.94 (1H, dd, J = 16.0, 5.0), 3.13 (1H, sext, J 6.1), 3.57–3.83 (4H, m), 3.82 (3H, s), 5.43 (1H, s, br), 6.46 (1H, s), 6.66 (1H, s), 7.14 (1H, dd, J = 8.5, 2.6), 7.26 (1H, d, J = 8.4), and 7.59 (1H, d, J = 2.5). LC/MS (ES+) m/z: 352.0 (M+ + H). HRMS (ES+) m/z: found 352.0875; C18H20Cl2NO2+, (M+ + H) requires 352.0866.
(±)-2-(2,3-Dichlorobenzyl)-7-methoxy-3-methyl-6-sulfamoyloxy-1,2,3,4-tetrahydroisoquinoline 8a
Sulfamoyl chloride (0.63 M in toluene, 2.4 mL, 1.5 mmol) was concentrated in vacuo and cooled to 0 °C until the reagent solidified. DMA (2.0 mL) was added, and the resulting solution was added directly to 7a (176 mg, 0.5 mmol) at 0 °C. The reaction mixture was stirred at RT for 2 h. Sodium bicarbonate (saturated, 50 mL) was added, and the mixture was extracted with EtOAc (100 mL). The organic layer was washed repeatedly with water (8 × 100 mL), then brine (2 × 5 mL), and then dried (MgSO4) and evaporated. The residue was stirred in PE/CH2Cl2 (∼20 mL, ∼4:1) to afford compound 8a as a pale yellow amorphous powder (150 mg, 69%). 1H NMR (400 MHz, acetone-d6): δ 1.17 (3H, d, J 6.5), 2.59 (1H, dd, J = 16.1, 5.5), 2.99 (1H, dd, J = 16.1, 5.1), 3.22 (1H, sext, J = 6.0), 3.67 (1H, d, J 16.0), 3.75 (1H, d, J 16.0), 3.78 (3H, s), 3.84 (1H, d, J = 14.6), 3.97 (1H, d, J = 14.6), 6.79 (1H, s), 6.90 (2H, s, br), 7.07 (1H, s), 7.35 (1H, t, J = 7.8), 7.50 (1H, dd, J = 8.0, 1.6), and 7.53 (1H, dd, J = 7.9, 1.6). 13C NMR (100 MHz, acetone-d6): δ 15.1 (CH3), 35.2 (CH2), 51.8 (CH2), 53.6 (CH), 55.6 (CH2), 56.2 (CH3), 111.7 (CH), 124.8 (CH), 126.6 (C), 128.5 (CH), 129.9 (CH), 130.1 (CH), 132.6 (C), 133.3 (C), 134.0 (C), 138.7 (C), 140.4 (C), and 151.1 (C). LC/MS (ES+) m/z: 431.1 (M+ + H). HRMS (ES+) m/z: found 431.0602; C18H21Cl2N2O4S+, (M+ + H) requires 431.0594.
(±)-2-(2,4-Dichlorobenzyl)-7-methoxy-3-methyl-6-sulfamoyloxy-1,2,3,4-tetrahydroisoquinoline 8b
The same method as for 8a was followed using compound 7b (317 mg, 0.9 mmol) and sulfamoyl chloride (0.63 M in toluene, 4.3 mL, 2.7 mmol) in DMA (3.6 mL) at RT for 2 h. The residue was stirred in PE/CH2Cl2 (∼20 mL, ∼4:1) to afford compound 8b as a pale yellow amorphous powder (240 mg, 61%). 1H NMR (400 MHz, acetone-d6): δ 1.15 (3H, d, J = 6.5), 2.57 (1H, dd, J = 16.1, 5.5), 2.97 (1H, dd, J = 16.1, 5.0), 3.17 (1H, sext, J = 6.0), 3.63 (1H, d, J = 16.0), 3.72 (1H, d, J = 16.0), 3.75 (1H, d, J = 14.5), 3.78 (3H, s), 3.90 (1H, d, J = 14.5), 6.79 (1H, s), 6.89 (2H, s, br), 7.06 (1H, s), 7.37 (1H, dd, J = 8.3, 2.2), 7.47 (1H, d, J = 2.2), and 7.63 (1H, d, J = 8.3). 13C NMR (100 MHz, acetone-d6): δ 15.1 (CH3), 35.3 (CH2), 51.8 (CH2), 53.4 (CH), 54.4 (CH2), 56.2 (CH3), 111.6 (CH), 124.8 (CH), 126.7 (C), 128.0 (CH), 129.7 (CH), 132.9 (CH), 133.5 (C), 134.2 (C), 135.4 (C), 137.0 (C), 138.6 (C), and 151.1 (C). LC/MS (ES+) m/z: 431.1 (M+ + H). HRMS (ES+) m/z: found 431.0599; C18H21Cl2N2O4S+, (M+ + H) requires 431.0594.
(±)-2-(2,5-Dichlorobenzyl)-7-methoxy-3-methyl-6-sulfamoyloxy-1,2,3,4-tetrahydroisoquinoline 8c
The same method as for 8a was followed using compound 7c (247 mg, 0.7 mmol) and sulfamoyl chloride (0.63 M in toluene, 3.4 mL, 2.1 mmol) in DMA (2.8 mL) at RT for 2 h. The residue was stirred in PE/CH2Cl2 (∼20 mL, ∼4:1) to afford compound 8c as a pale yellow amorphous powder (185 mg, 61%). 1H NMR (400 MHz, acetone-d6): δ 1.17 (3H, d, J = 6.5), 2.59 (1H, dd, J = 16.2, 5.6), 3.00 (1H, dd, J = 16.1, 4.9), 3.21 (1H, sext, J = 6.0), 3.68 (1H, J = 16.0), 3.76 (1H, J = 16.0), 3.78 (3H, s), 3.79 (1H, J = 15.0), 3.92 (1H, J = 15.0), 6.81 (1H, s), 6.90 (2H, s, br), 7.08 (1H, s), 7.32 (1H, dd, J = 8.5, 2.7), 7.43 (1H, d, J = 8.5), and 7.67 (1H, d, J = 2.7). 13C NMR (100 MHz, acetone-d6): δ 15.2 (CH3), 35.2 (CH2), 51.9 (CH2), 53.6 (CH), 54.6 (CH2), 56.2 (CH3), 111.7 (CH), 124.9 (CH), 126.6 (C), 129.1 (CH), 131.0 (CH), 131.7 (CH), 133.0 (C), 133.3 (C), 134.0 (C), 138.6 (C), 140.1 (C), and 151.1 (C). LC/MS (ES+) m/z: 431.1 (M+ + H). HRMS (ES+) m/z: found 431.0605; C18H21Cl2N2O4S+, (M+ + H) requires 431.0594.
Acknowledgments
Synthetic work was supported by Sterix Ltd., a member of the Ipsen Group. We thank the NCI DTP for providing in vitro screening resources. X-Ray data were collected at the Swiss Light Source (beamline X06DA, Paul Scherrer Institut, Villigen PSI, Switzerland). This work was supported by a grant from the Swiss National Science Foundation (31003A_166608 to M.O.S.). We thank Dr M. P. Leese for early discussions.
Glossary
Abbreviations
- THIQ
tetrahydroisoquinoline
- 2ME
2-methoxyestradiol
- HUVEC
human umbilical vein endothelial cell
- DHQ
dihydroquinazolinone
- SAR
structure–activity relationship
- DIPEA
diisopropylethylamine
- DMF
N,N-dimethylformamide
- DMA
N,N-dimethylacetamide
- MGM
mean graph midpoint
- GTP
guanosine 5′-triphosphate
Author Present Address
# Biotrial, 35000 Rennes, France.
Author Contributions
W.D. and A.E.P. contributed equally. All authors have given their approval to the final version of the manuscript.
The authors declare no competing financial interest.
Notes
The content of this paper is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health.
Notes
The structure of 10 (STX3451) bound to the T2R-TTL complex has PDB access code 6HX8. Authors will release the atomic coordinates upon article publication
References
- Leese M. P.; Jourdan F.; Dohle W.; Kimberley M. R.; Thomas M. P.; Bai R.; Hamel M. R.; Ferrandis E.; Potter B. V. L. Steroidomimetic tetrahydroisoquinolines for the design of new microtubule disruptors. ACS Med. Chem. Lett. 2011, 3, 5–9. 10.1021/ml200232c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leese M. P.; Jourdan F. L.; Major M. R.; Dohle W.; Hamel E.; Ferrandis E.; Fiore A.; Kasprzyk P. G.; Potter B. V. L. Tetrahydroisoquinoline-based steroidomimetic and chimeric microtubule disruptors. ChemMedChem 2013, 9, 85–108. 10.1002/cmdc.201300261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohle W.; Leese M. P.; Jourdan F. L.; Major M. R.; Bai R.; Hamel E.; Ferrandis E.; Kasprzyk P. G.; Fiore A.; Newman S. P.; Purohit A.; Potter B. V. L. Synthesis, anti-tubulin and anti-proliferative SAR of C-3/C-1 substituted tetrahydroisoquinolines. ChemMedChem 2014, 9, 350–370. 10.1002/cmdc.201300412. [DOI] [PubMed] [Google Scholar]
- Leese M. P.; Jourdan F.; Kimberley M. R.; Cozier G. E.; Thiyagarajan N.; Stengel C.; Regis-Lydi S.; Foster P. A.; Newman S. P.; Acharya K. R.; Ferrandis E.; Purohit A.; Reed M. J.; Potter B. V. L. Chimeric microtubule disruptors. Chem. Commun. 2010, 46, 2907–2909. 10.1039/c002558e. [DOI] [PubMed] [Google Scholar]
- Dohle W.; Leese M. P.; Jourdan F. L.; Chapman C. J.; Hamel E.; Ferrandis E.; Potter B. V. L. Optimisation of tetrahydroisoquinoline-based chimeric microtubule disruptors. ChemMedChem 2014, 9, 1783–1793. 10.1002/cmdc.201402025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leese M. P.; Hejaz H. A. M.; Mahon M. F.; Newman S. P.; Purohit A.; Reed M. J.; Potter B. V. L. A-ring-substituted estrogen-3-O-sulfamates: Potent multitargeted anticancer agents. J. Med. Chem. 2005, 48, 5243–5256. 10.1021/jm050066a. [DOI] [PubMed] [Google Scholar]
- Bubert C.; Leese M. P.; Mahon M. F.; Ferrandis E.; Regis-Lydi S.; Kasprzyk P. G.; Newman S. P.; Ho Y. T.; Purohit A.; Reed M. J.; Potter B. V. L. 3,17-Disubstituted 2-alkylestra-1,3,5(10)-trien-3-ol derivatives: Synthesis, in vitro and in vivo anticancer activity. J. Med. Chem. 2007, 50, 4431–4443. 10.1021/jm070405v. [DOI] [PubMed] [Google Scholar]
- Jourdan F.; Bubert C.; Leese M. P.; Smith A.; Ferrandis E.; Regis-Lydi S.; Newman S. P.; Purohit A.; Reed M. J.; Potter B. V. L. ffects of C-17 heterocyclic substituents on the anticancer activity of 2-ethylestra-1,3,5(10)-triene-3-O-sulfamates: Synthesis, in vitro evaluation and computational modelling. Org. Biomol. Chem. 2008, 6, 4108–4119. 10.1039/b810300c. [DOI] [PubMed] [Google Scholar]
- Leese M. P.; Leblond B.; Smith A.; Newman S. P.; Di Fiore A.; De Simone G.; Supuran C. T.; Purohit A.; Reed M. J.; Potter B. V. L. 2-Substituted estradiol bis-sulfamates, multitargeted antitumor agents: Synthesis, in vitro SAR, protein crystallography, and in vivo activity. J. Med. Chem. 2006, 49, 7683–7696. 10.1021/jm060705x. [DOI] [PubMed] [Google Scholar]
- Leese M.; Newman S. P.; Purohit A.; Reed M. J.; Potter B. V. L. 2-Alkylsulfanyl estrogen derivatives: Synthesis of a novel class of multi-targeted anti-tumour agents. Bioorg. Med. Chem. Lett. 2004, 14, 3135–3138. 10.1016/j.bmcl.2004.04.027. [DOI] [PubMed] [Google Scholar]
- Thomas M. P.; Potter B. V. L. Discovery and development of the aryl O-sulfamate pharmacophore for oncology and women’s health. J. Med. Chem. 2015, 58, 7634–7658. 10.1021/acs.jmedchem.5b00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter B. V. L. Steroid sulphatase inhibition via aryl sulphamates: clinic progress, mechanism and future prospects. J. Mol. Endocrinol. 2018, 61, T233–T252. 10.1530/jme-18-0045. [DOI] [PubMed] [Google Scholar]
- Dohle W.; Jourdan F. L.; Menchon G.; Prota A. E.; Foster P. A.; Mannion P.; Hamel E.; Thomas M. P.; Kasprzyk P. G.; Ferrandis E.; Steinmetz M. O.; Leese M. P.; Potter B. V. L. Quinazolinone-based anticancer agents: Synthesis, antiproliferative SAR, antitubulin activity, and tubulin co-crystal structure. J. Med. Chem. 2018, 61, 1031–1044. 10.1021/acs.jmedchem.7b01474. [DOI] [PubMed] [Google Scholar]
- Elger W.; Schwarz S.; Hedden A.; Reddersen G.; Schneider B. Sulfamates of various estrogens are prodrugs with increased systemic and reduced hepatic estrogenicity at oral application. J. Steroid Biochem. Mol. Biol. 1995, 55, 395–403. 10.1016/0960-0760(95)00214-6. [DOI] [PubMed] [Google Scholar]
- Ireson C. R.; Chander S. K.; Purohit A.; Parish D. C.; Woo L. W. L.; Potter B. V. L.; Reed M. J. Pharmacokinetics of the nonsteroidal steroid sulphatase inhibitor 667 COUMATE and its sequestration into red blood cells in rats. Br. J. Cancer 2004, 91, 1399–1404. 10.1038/sj.bjc.6602130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andring J. T.; Dohle W.; Chingkuang T.; Potter B. V. L.; McKenna R., STX140 and Non-Steroidal Sulfamate Derivatives Inhibit Carbonic Anhydrase IX: Structure Activity Optimization of Isoform Selectivity. J. Med. Chem. 2019, in revision. [DOI] [PubMed] [Google Scholar]
- Suzuki R. N.; Newman S. P.; Purohit A.; Leese M. P.; Potter B. V. L.; Reed M. J. Growth inhibition of multi-drug-resistant breast cancer cells by 2-methoxyoestradiol-bis-sulphamate and 2-ethyloestradiol-bis-sulphamate. J. Steroid Biochem. Mol. Biol. 2003, 84, 269–278. 10.1016/s0960-0760(03)00035-9. [DOI] [PubMed] [Google Scholar]
- Newman S. P.; Foster P. A.; Stengel C.; Day J. M.; Ho Y. T.; Judde J.-G.; Lassalle M.; Prevost G.; Leese M. P.; Potter B. V. L.; Reed M. J.; Purohit A. STX140 is efficacious in vitro and in vivo in taxane-resistant breast carcinoma cells. Clin. Cancer Res. 2008, 14, 597–606. 10.1158/1078-0432.ccr-07-1717. [DOI] [PubMed] [Google Scholar]
- Day J. M.; Foster P. A.; Tutill H. J.; Newman S. P.; Ho Y. T.; Leese M. P.; Potter B. V. L.; Reed M. J.; Purohit A. BCRP expression does not result in resistance to STX140 in vivo, despite the increased expression of BCRP in A2780 cells in vitro after long-term STX140 exposure. Br. J. Cancer 2009, 100, 476–486. 10.1038/sj.bjc.6604873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman S. P.; Leese M. P.; Purohit A.; James D. R. C.; Rennie C. E.; Potter B. V. L.; Reed M. J. Inhibition of in vitro angiogenesis by 2-methoxy- and 2-ethyl-estrogen sulfamates. Int. J. Cancer 2004, 109, 533–540. 10.1002/ijc.20045. [DOI] [PubMed] [Google Scholar]
- Chander S. K.; Foster P. A.; Leese M. P.; Newman S. P.; Potter B. V. L.; Purohit A.; Reed M. J. In vivo inhibition of angiogenesis by sulphamoylated derivatives of 2-methoxyoestradiol. Br. J. Cancer 2007, 96, 1368–1376. 10.1038/sj.bjc.6603727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravelli R. B. G.; Gigant B.; Curmi P. A.; Jourdain I.; Lachkar S.; Sobel A.; Knossow M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- Okada M.; Iwashita S.; Koizumi N. Efficient general method for sulfamoylation of a hydroxyl group. Tetrahedron Lett. 2000, 41, 7047–7051. 10.1016/s0040-4039(00)01130-8. [DOI] [Google Scholar]
- Nel M.; Joubert A.; Dohle W.; Potter B. V. L.; Theron A. Modes of cell death induced by tetrahydroisoquinoline-based analogs in MDA-MB-231 breast and A549 lung cancer cell lines. Drug Des. Dev. Ther. 2018, 12, 1881–1904. 10.2147/dddt.s152718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.-C.; Upadhyayula R.; Cevallos S.; Messick R. J.; Hsia T.; Leese M. P.; Jewett D. M.; Ferrer-Torres D.; Roth T. M.; Dohle W.; Potter B. V. L.; Barald K. F. Targeted NF1 cancer therapeutics with multiple modes of action: Small molecule hormone-like agents resembling the natural anti-cancer metabolite, 2-methoxyestradiol. Br. J. Cancer 2015, 113, 1158–1167. 10.1038/bjc.2015.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Prota A. E.; Bargsten K.; Zurwerra D.; Field J. J.; Diaz J. F.; Altmann K.-H.; Steinmetz M. O. Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science 2013, 339, 587–590. 10.1126/science.1230582. [DOI] [PubMed] [Google Scholar]; b Gaspari R.; Prota A. E.; Bargsten K.; Cavalli A.; Steinmetz M. O. Structural basis of cis- and trans-combretastatin binding to tubulin. Chem 2017, 2, 102–113. 10.1016/j.chempr.2016.12.005. [DOI] [Google Scholar]; c Zhou P.; Liu Y.; Zhou L.; Zhu K.; Feng K.; Zhang H.; Liang Y.; Jiang H.; Luo C.; Liu M.; Wang Y. Potent antitumor activities and structure basis of the chiral β-lactam bridged analogue of combretastatin A-4 binding to tubulin. J. Med. Chem. 2016, 59, 10329–10334. 10.1021/acs.jmedchem.6b01268. [DOI] [PubMed] [Google Scholar]
- Brouhard G. J.; Rice L. M. The contribution of αβ-tubulin curvature to microtubule dynamics. J. Cell Biol. 2014, 207, 323–334. 10.1083/jcb.201407095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prota A. E.; Danel F.; Bachmann F.; Bargsten K.; Buey R. M.; Pohlmann J.; Reinelt S.; Lane H.; Steinmetz M. O. The novel microtubule-destablizing drug BAL27862 binds to the colchicine site of tubulin with distinct effects on microtubule organization. J. Mol. Biol. 2014, 426, 1848–1860. 10.1016/j.jmb.2014.02.005. [DOI] [PubMed] [Google Scholar]
- Karplus P. A.; Diederichs K. Linking crystallographic model and data quality. Science 2012, 336, 1030–1033. 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis I. W.; Murray L. W.; Richardson J. S.; Richardson D. C. MOLPROBITY: Structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004, 32, W615–W619. 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel E.; Lin C. M. Separation of active tubulin and microtubule-associated proteins by ultracentrifugation and isolation of a component causing the formation of microtubule bundles. Biochemistry 1984, 23, 4173–4184. 10.1021/bi00313a026. [DOI] [PubMed] [Google Scholar]
- Hamel E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell Biochem. Biophys. 2003, 38, 1–22. 10.1385/cbb:38:1:1. [DOI] [PubMed] [Google Scholar]
- Verdier-Pinard P.; Lai J.-Y.; Yoo H.-D.; Yu J.; Marquez B.; Nagle D. G.; Nambu M.; White J. D.; Falck J. R.; Gerwick W. H.; Day B. W.; Hamel E. Structure-activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells. Mol. Pharmacol. 1998, 53, 62–76. 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
- Lin C. M.; Ho H. H.; Pettit G. R.; Hamel E. Antimitotic natural products combretastatin A-4 and combretastatin A-2: Studies on the mechanism of their inhibition of the binding of colchicine to tubulin. Biochemistry 1989, 28, 6984–6991. 10.1021/bi00443a031. [DOI] [PubMed] [Google Scholar]
- Prota A. E.; Magiera M. M.; Kuijpers M.; Bargsten K.; Frey D.; Wieser M.; Jaussi R.; Hoogenraad C. C.; Kammerer R. A.; Janke C.; Steinmetz M. O. Structural basis of tubulin tyrosination by tubulin tyrosine ligase. J. Cell Biol. 2013, 200, 259–270. 10.1083/jcb.201211017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. 10.1107/s0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Phyton-based system for macromolecular structure solution. Acta Crystallogr. 2010, 66, 213–221. 10.1107/s0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004, 60, 2126–2132. 10.1107/s0907444904019158. [DOI] [PubMed] [Google Scholar]
- Appel R.; Berger G. Über das hydrazidosulfamid (On hydrazidosulfamide). Chem. Ber. 1958, 91, 1339–1341. 10.1002/cber.19580910633. [DOI] [Google Scholar]
- Woo L. W. L.; Lightowler M.; Purohit A.; Reed M. J.; Potter B. V. L. Heteroatom-substituted analogues of the active-site directed inhibitor estra-1,3,5(10)-trien-17-one-3-sulphamate inhibit estrone sulphatase by a different mechanism. J. Steroid Biochem. Mol. Biol. 1996, 57, 79–88. 10.1016/0960-0760(95)00244-8. [DOI] [PubMed] [Google Scholar]