

Abstract
The avidity of prostate cancer cells for copper sensitizes them to cytotoxicity by disulfiram, a dithiocarbamate-containing drug approved for alcohol aversion therapy. While disulfiram has received attention as a possible therapy for various cancers, its lack of cancer specificity is a liability that limits this promise. Here we present a prodrug approach to direct copper-dependent cytotoxicity of dithiocarbamate pharmacophores to prostate cancer cells. The prochelator GGTDTC requires activation by gamma-glutamyl transferase (GGT) to release the metal chelator diethyldithiocarbamate from a linker that masks the thiol reactivity and metal binding properties of the pharmacophore prior to activation. In vitro studies demonstrated successful masking of its copper binding properties as well as clean liberation of the chelator by GGT. GGTDTC was found to be stable to non-specific degradation when incubated with a series of prostate cancer and normal cell lines, with selective release of diethyldithiocarbamate only occurring in cells with measurable GGT activity. The antiproliferative efficacy of the prochelator correlated with cellular GGT activity, with 24 h half maximal inhibitory concentrations ranging from 800 nM in prostate cancer lines 22Rv1 and LNCaP to over 15 μM in normal prostate PWR-1E cells. These findings underscore a new strategy to leverage the amplified copper metabolism of prostate cancer by conditional activation of a metal-binding pharmacophore.
Keywords: prochelator, copper, γ-glutamyl transpeptidase, dithiocarbamate, cancer
Chelator in disguise:
Dithiocarbamate, a metal ion chelator is masked for its ability to bind to Cu(II) with a γ-glutamyl transpeptidase (GGT) enzyme trigger. Activated by GGT overexpressed in cancer cells, the chelator is unveiled to form cytotoxic Cu complexes in situ selectively in GGT-expressing cancer cells.
Most deaths related to prostate cancer are due to metastatic disease that progresses despite therapy. While the androgen receptor remains the major drug target, resistance to androgen deprivation often leads to aggressive and lethal metastatic castration-resistant prostate cancer (mCRPC).[1] The relatively poor prognosis and limited therapy options for patients with this disease state underscore the need for innovative therapies directed to targets other than the androgen receptor.
Observations of increased copper (Cu) uptake by prostate cancer cells have raised the intriguing concept of targeting Cu metabolism for diagnosis and treatment.[2] The proclivity of prostate tumors to accumulate Cu is so prominent that 64CuCl2 has shown promise as a PET tracer for imaging prostate cancer.[3] Recent preclinical work has further shown that castration-resistant prostate cancer cells increase expression of several Cu trafficking proteins, a response that is accentuated upon activation of the androgen receptor.[4] This cancer-specific remodelling of the cell’s Cu biology creates local Cu reserves that sensitize prostate cancer cells to cytotoxicity by disulfiram (DSF), a known drug approved for alcohol aversion therapy.[4]
Disulfiram is a disulfide dimer that upon reduction yields dithiocarbamate (DTC) moieties that contain reactive thiol nucleophiles and are effective metal chelators. Further metabolism in the liver leads to sulfoxide and sulfone metabolites that ultimately inhibit aldehyde dehydrogenase, the eventual target that causes an aversive accumulation of acetaldehyde in patients who consume ethanol.[5] While most of these reactions and metabolites are counterproductive in terms of cancer therapy, DTC’s ability to form Cu complexes is a key facet of its anticancer properties.[6] The mechanism of Cu- assisted disulfiram toxicity is not fully understood, although many reports provide evidence that reactive oxygen species mediate cell death.[4, 7] Recently, the p-97-NPL4-UFDI protein complex was identified as a likely target of cu(DTC)2, ultimately resulting in accumulation of ubiquitinated proteins and induction of a heat shock response.[8] Furthermore, cu(DTC)2 complexes have been detected in mice fed DSF and in human plasma of patients undergoing DSF treatment, validating the concept that DSF transforms in vivo to form Cu-DTC complexes.[8]
While disulfiram has been touted as an interesting compound for various cancers,[9] significant hurdles hinder its implementation for cancer treatment.[10] Notably, a phase 2 clinical trial of disulfiram in mCRPC patients demonstrated significant toxicity without improvement in efficacy.[11] The failure of DSF in the clinical setting could be attributed to its lack of specificity along with its unwanted metabolites.[5b] While attempts have been made to improve formulation and drug delivery strategies,[12] little has been done to optimize the molecule itself for application to cancer.
We hypothesized that the efficacy of disulfiram in cancer settings could be improved by targeting cytotoxic Cu(DTC)2complexes selectively to cancer cells. To that end, we reasoned that a prochelator approach that leverages a cancer-specific activation mechanism could preferentially release DTCs in situ to take advantage of the amplified Cu metabolism of prostate cancer cells for conditional cytotoxicity, while minimizing the offtarget pathways that impede disulfiram’s anticancer promise. As a type of prodrug, prochelators are designed to hinder the metal-binding properties of a compound until activation by a desired stimulus initiates metal-dependent bioactivity.[13] Here, we introduce GGTDTC as an anticancer prochelator that requires activation by gamma-glutamyl transferase (GGT) to release DTC from a self-immolative linker that serves to mask the thiol reactivity and metal binding properties of DTC prior to activation (Scheme 1).
Scheme 1.
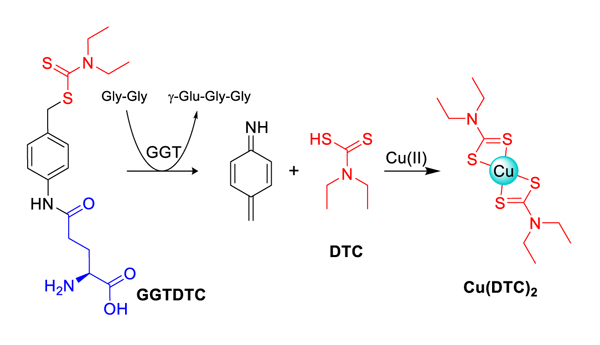
Prochelator GGTDTC uses a p-amidobenzyl linker (PAB, black) to connect a dithiocarbamate (DTC, red) pharmacophore to a γ-linked glutamic acid. Recognition by the GGT enzyme results in transpeptidation to release γ-GluGlyGly and via 1,6-benzyl elimination of the linker, DTC that is then available to bind copper.
GGT is a cell surface enzyme normally localized along the luminal surfaces of epithelial cells. It catalyzes the extracellular cleavage of the γ-glutamyl bond of glutathione and other γ-glutamyl amides to transfer the γ-glutamyl group to water (hydrolysis) or to amino acid or peptide substrates (transpeptidation).[14] As most cells cannot import glutathione, GGT provides a key step for recycling cysteine to enable intracellular biosynthesis of glutathione, a major component of cellular redox homeostasis. Interestingly, many types of cancer cells over-express GGT along their entire cell surface, and clinical studies have found that GGT expression levels in human tumors correlate with poor patient survival.[14a] Whereas normal GGT-expressing cells access γ-glutamate substrates in ductal fluids, GGT-positive tumor cells can access substrates in interstitial fluids and in blood.[14a] This difference in location and activity level suggests that strategies targeting GGT activity are promising therapeutic avenues. On this front, there has been significant work developing GGT inhibitors, γ-glutamyl drug conjugates, and fluorescent substrates for the potential treatment and visualization of a variety of GGT-positive cancers.[15]
Our design for a γ-glutamyl prodrug uses γ-glutamate (γ-GIu) as a selective recognition and trigger moiety for prochelator activation. In principle, this strategy leverages two different processes to differentially target cancer cells: their overexpression of GGT and their altered copper biology. To maintain the required γ-glutamyl amide bond for GGT recognition, DTC was conjugated to γ-glutamate via a self-immolative p-aminobenzyl (PAB) linker. Once the amide bond is hydrolyzed by GGT, 1,6 benzyl elimination of the PAB linker is expected to release DTC specifically in the vicinity of cells expressing GGT, where it is free to bind available metal ions (Scheme 1). The iminoquinone methide released as an intermediate is highly reactive and is expected to undergo hydrolysis.[16]
GGTDTC was synthesized by conjugating commercially available Boc-L-glutamic acid α-tertbutyl ester to p-aminobenzylalcohol using HBTU coupling to generate compound 2 (Scheme 2). Reaction of 2 with PBr3 converted the alcohol functionality to bromide while simultaneously removing the Boc and t-butyl protecting groups to give 3, which was subsequently reacted with sodium diethyldithiocarbamate to yield the final GGTDTC after purification by HPLC.
Scheme 2.
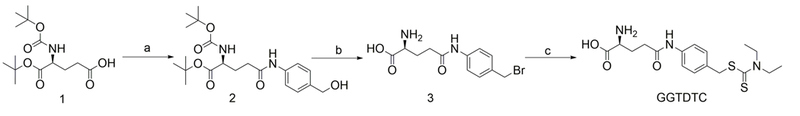
Synthesis of GGTDTC. a) p-Aminobenzyl alcohol, HBTU, 4% NMM in DMF, 3 h, rt; b) PBr3, dry THF, 1 h, 0 °C; c) Sodium diethyldithiocarbamate, dry ACN, 2 h, rt. Overall yield 11.6%
To test our hypothesis that masking DTC impedes its ability to bind Cu(II), we used a fluorescence competition assay in which the fluorescence of calcein is quenched upon Cu(II) binding. As shown in Fig. 1, addition of DTC caused an immediate recovery of fluorescence, indicating that it readily competes with calcein for chelating Cu(II), whereas the prodrug GGTDTC alone did not compete with calcein for Cu(II). However, exposure of the prochelator to the GGT enzyme resulted in recovery of calcein fluorescence over time, indicating that reaction with GGT releases a product capable of chelating Cu(II). Indeed, the presence of cu(DTC)2 as a product was confirmed by HPLC and mass spectral analysis of solutions following exposure of GGTDTC to GGT enzyme in the presence of Cu(II) (Fig. S2). Together, these results validate the prochelator concept of masking metal-chelating competency in GGTDTC and support our molecular mechanism that GGT activation induces DTC release.
Figure 1.
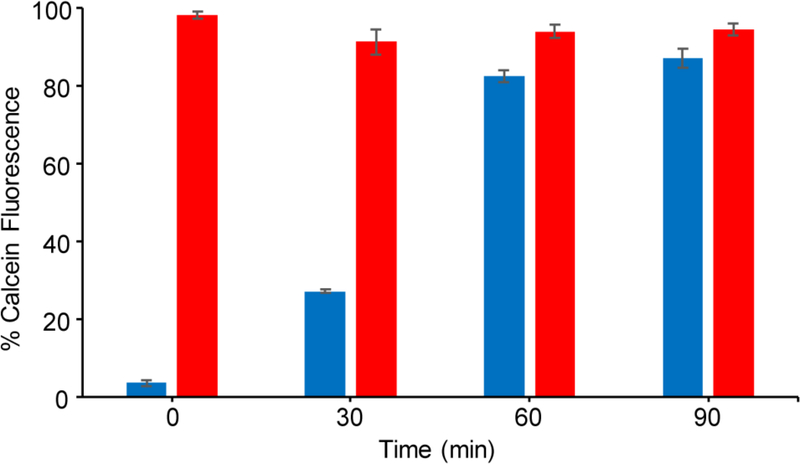
Copper-calcein competition assay showing recovery of calcein fluorescence as a function of added chelator or prochelator in the presence of GGT enzyme over time. Solutions contained 1μM CuSO4, 1 μM calcein, 20 U/L of GGT enzyme, and 12.5 μM of either GGTDTC (blue) or DTC (red) in PBS buffer pH 7.4 with 1 mM Gly-Gly; 37 °C.
To probe the reactivity of GGTDTC in comparison to the enzyme’s natural substrate GSH, competition experiments were conducted against the colorimetric substrate L-glutamic acid γ-(p-nitroanilide) [E-pNA] (Fig S3). The colorimetric assay involves monitoring p-nitroaniline release by absorbance at 405 nm upon reaction of the probe substrate E-pNA with GGT in the presence of an inhibiting substrate, either GGTDTC or GSH. Ki for GSH was found to be 99 ± 7 μM under our experimental conditions (PBS, pH 7.4, 1 mM GlyGly) and similar to prior reports (73 ± 6 μM).[17] With a Ki of 23 ± 5 μM, GGTDTC was found to have greater affinity for GGT than glutathione. This finding shows that GGTDTC should be preferentially cleaved over GSH. The assay also confirmed that the presence of DTC does not affect enzyme kinetics.
To test the stability of GGTDTC and the selectivity of its activation in human cells, an HPLC assay was performed on cell culture supernatants after incubation with GGTDTC. Fig. 2 shows representative examples from two cell lines: 22Rv1, an aggressive prostate cancer cell line that tested positively for GGT activity, and PWR-1E, a normal prostate epithelial line which tested negatively for GGT activity under these conditions (vide infra). As shown in Fig. 2, exposure of 100 μM GGTDTC to 22Rv1 cells caused a significant loss of detectable GGTDTC. Co-exposure with the GGT inhibitor Acivicin, however, completely reversed this trend and GGTDTC was found to be stable for at least 24 h. The level of intact GGTDTC was also found to be unchanged in the GGT(−) PWR-1E cells. Together, these results provide evidence for the selectivity of the prochelator for GGT activation and its stability against nonspecific degradative processes in cellular environments.
Figure 2.
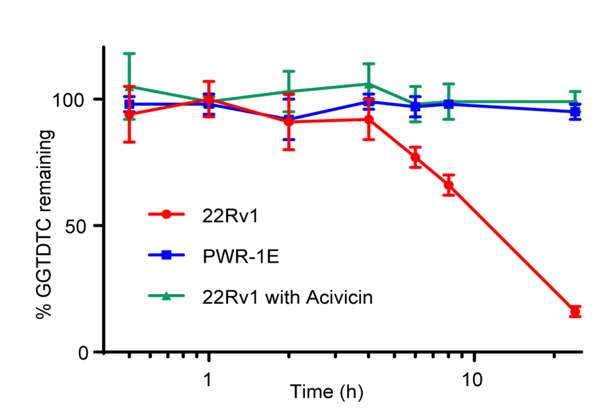
Levels of intact GGTDTC observed by HPLC analysis of supernatants of prostate cell lines incubated with 100 μM GGTDTC for 24 h at 37°C in medium supplemented with 1 mM GlyGly. GGTDTC depletion was observed over 24 h in prostate cancer 22Rv1 cells, whereas GGTDTC levels were unchanged in 22Rv1 treated with GGT-inhibitor Acivicin or in control prostate PWR-1E cells
In order to evaluate the potential anticancer activity of GGTDTC, the prodrug and DSF were tested for their ability to inhibit cell growth of prostate cancer cells 22Rv1, LNCaP, and PC3, as well as PWR-1E prostate epithelial cells, and MCF-7 breast cancer cells. Because Cu is known to be required for disulfiram’s antiproliferative activity, we performed checkerboard assays to establish the Cu dependence of DSF and GGTDTC for inhibition of cell viability under our conditions. Pilot studies in 22Rv1 cells revealed that antiproliferative activity of both DSF and GGTDTC required as low as 300 nM Cu(II) added to the growth medium, with 1 μM Cu providing a robust response for both compounds (Fig S4). No difference in activity was observed on using either CuCl2 or CuSO4; subsequent studies were conducted in medium supplemented with 1 μM CuSO4. Representative dose-response curves for GGTDTC with and without supplemental Cu are shown in Fig. 3a for LNCaP cells and 3b for PWR-1E cells, with IC50 values for all cell lines in the Cu-supplemented condition shown in Fig. 3c. The data in Fig. 3 were collected at 24 h, where the difference in the antiproliferative activity of GGTDTC across the cell lines is already evident, with IC50 values ranging from 800 nM in 22Rv1 and LNCaP cancer cell lines to over 15 μM in normal prostate PWR-1E cells. The difference in antiproliferative activity of GGTDTC across these cell lines narrows but persists after 72 h continuous exposure, with IC50 values ranging from 300 nM in 22Rv1 to 1.5 μM in PWR-1E cells. (Table S1). In contrast, DSF was robustly effective across all cell lines, with IC50 values in the 40–150 nM range at 72 h, consistent with prior studies (Table S1).[4] Since DSF is a dimer of DTC, the IC50 per monomer is therefore 80–300 nM at 72 h.
Figure 3.
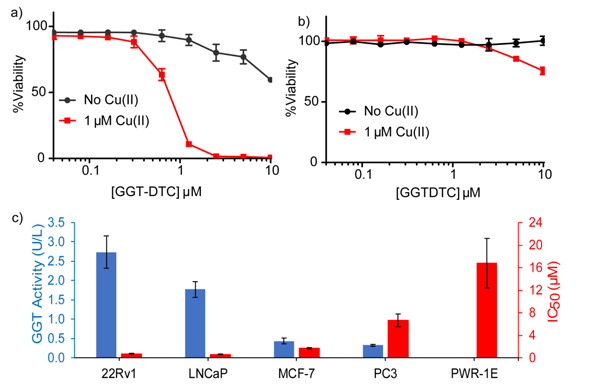
Representative dose-response curves for GGTDTC with and without supplemental CuSO4 for a) prostate cancer LNCaP and b) prostate normal epithelial PWR-1E cell lines, as determined by a resazurin viability assay measured at 24 h. c) GGT activity (blue) measured by E-pNA colorimetric assay, and IC50 values of GGTDTC (red) across multiple cell lines in FBS-free medium with 1 μM CuSO4 for 24 h. Antiproliferative activity of GGTDTC correlates with levels of GGT enzyme activity.
In parallel to the cell viability assays, the GGT activity of each cell line was measured by using the colorimetric test substrate E-pNA. The prostate cancer lines 22Rv1 and LNCaP showed high GGT activity, consistent with expectations of overactive GGT activity in these cells (Fig 3). In contrast, MCF-7 and PC3 revealed measurable but lower activity, while PWR-1E showed no detectable activity at 24 h, with some activity emerging by 72 h (Fig S5). The emergence of GGT activity over long incubation times for these cells explains the reduction in IC50 of GGTDTC in PWR-1E cells at 72 h (Table S1). Importantly, the trend in GGT activity correlates with antiproliferative efficacy of GGTDTC: the more active the cell line, the lower the IC50 for GGTDTC.
CONCLUSIONS
The data communicated herein show that the dithiocarbamate-releasing prochelator GGTDTC is activated by GGT both in vitro and in select cancer cells, with Cu-dependent cytotoxicity correlating with the cells’ level of GGT enzyme activity. Interestingly, disulfiram itself must undergo reduction prior to formation of Cu complexes, a process that may involve additional redox steps relevant to its cytotoxic mechanism of action.[6a, 6b] GGTDTC, on the other hand, releases dithiocarbamate in situ directly available for metal coordination, making it an interesting tool to investigate further the molecular mechanisms underlying the Cu-dependent cytotoxicity of these kinds of agents. While the efficacy of GGTDTC awaits in vivo validation, the current data substantiate the concept that using an activatable targeting group to mask metal-binding functionality of a pharmacophore like dithiocarbamate provides a promising strategy for engendering disease specificity for metal-interacting drugs. A potential complication while translating GGTDTC in vivo could be presented by GGT expressed in the liver. We note that the versatile synthetic strategy reported here for GGTDTC can be readily modified to target different cancers or diseases with their own unique biomarkers, an objective we are pursuing.
Supplementary Material
ACKNOWLEDGEMENTS
We gratefully acknowledge support from the National Science Foundation (NSF CHE-1152054), the Duke Cancer Institute P30 Cancer Center Support Grant (NIH CA014236), and a Foerster-Bernstein postdoctoral fellowship for S.B.
REFERENCES
- [1].a)Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BV, Varambally S, Tomlins SA, Nanus DM, Tagawa ST, Van Allen EM, Elemento O, Sboner A, Garraway LA, Rubin MA, Demichelis F, Nat Med 2016, 22, 298–305; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Aggarwal RR, Feng FY, Small EJ, Oncology 2017, 31, 467–474. [PubMed] [Google Scholar]
- [2].a)Cater MA, Pearson HB, Wolyniec K, Klaver P, Bilandzic M, Paterson BM, Bush AI, Humbert PO, La Fontaine S, Donnelly PS, Haupt Y, ACS Chem Biol 2013, 8, 1621–1631; [DOI] [PubMed] [Google Scholar]; b)Denoyer D, Pearson HB, Clatworthy SAS, Smith ZM, Francis PS, Llanos RM, Volitakis I, Phillips WA, Meggyesy PM, Masaldan S, Cater MA, Oncotarget 2016, 7, 37064–37080; [DOI] [PMC free article] [PubMed] [Google Scholar]; c)Wehbe M, Leung AWY, Abrams MJ, Orvig C, Bally MB, Dalton Trans. 2017, 46, 10758–10773. [DOI] [PubMed] [Google Scholar]
- [3].a)Piccardo A, Paparo F, Puntoni M, Righi S, Bottoni G, Bacigalupo L, Zanardi S, DeCensi A, Ferrarazzo G, Gambaro M, Ruggieri FG, Campodonico F, Tomasello L, Timossi L, Sola S, Lopci E, Cabria M, J Nucl Med 2018, 59, 444–451; [DOI] [PubMed] [Google Scholar]; b)Cai H, Wu J.-s., Muzik O, Hsieh J-T, Lee RJ, Peng F, J Nucl Med 2014, 55, 622–628; [DOI] [PMC free article] [PubMed] [Google Scholar]; c)Capasso E, Durzu S, Piras S, Zandieh S, Knoll P, Haug A, Hacker M, Meleddu C, Mirzaei S, Ann Nucl Med 2015, 29, 482–488; [DOI] [PubMed] [Google Scholar]; d)Peng F, Lu X, Janisse J, Muzik O, Shields AF, J Nucl Med 2006, 47, 1649–1652. [PubMed] [Google Scholar]
- [4].Safi R, Nelson ER, Chitneni SK, Franz KJ, George DJ, Zalutsky MR, McDonnell DP, Cancer Res 2014, 74, 5819–5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a)Koppaka V, Thompson DC, Chen Y, Ellermann M, Nicolaou KC, Juvonen RO, Petersen D, Deitrich RA, Hurley TD, Vasiliou V, Pharmacol Rev 2012, 64, 520–539; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Johansson B, Acta Psychiatr Scand Suppl 1992, 369, 15–26. [DOI] [PubMed] [Google Scholar]
- [6].a)Cen D, Brayton D, Shahandeh B, Meyskens FL, Farmer PJ, J Med Chem 2004, 47, 6914–6920; [DOI] [PubMed] [Google Scholar]; b)Lewis DJ, Deshmukh P, Tedstone AA, Tuna F, O’Brien P, Chem Commun 2014, 50, 13334–13337; [DOI] [PubMed] [Google Scholar]; c)Tawari PE, Wang Z, Najlah M, Tsang CW, Kannappan V, Liu P, McConville C, He B, Armesilla AL, Wang W, Toxicol Res 2015, 4, 1439–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a)Cen D, Gonzalez RI, Buckmeier JA, Kahlon RS, Tohidian NB, Meyskens FL, Mol Cancer Therap 2002, 1, 197; [PubMed] [Google Scholar]; b)Conticello C, Martinetti D, Adamo L, Buccheri S, Giuffrida R, Parrinello N, Lombardo L, Anastasi G, Amato G, Cavalli M, Chiarenza A, De Maria R, Giustolisi R, Gulisano M, Di Raimondo F, Intl J Cancer 2012, 131, 2197–2203. [DOI] [PubMed] [Google Scholar]
- [8].Skrott Z, Mistrik M, Andersen KK, Friis S, Majera D, Gursky J, Ozdian T, Bartkova J, Turi Z, Moudry P, Kraus M, Michalova M, Vaclavkova J, Dzubak P, Vrobel I, Pouckova P, Sedlacek J, Miklovicova A, Kutt A, Li J, Mattova J, Driessen C, Dou QP, Olsen J, Hajduch M, Cvek B, Deshaies RJ, Bartek J, Nature 2017, 552, 194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a)Iljin K, Ketola K, Vainio P, Halonen P, Kohonen P, Fey V, Grafström RC, Perälä M, Kallioniemi O, Clin Cancer Res 2009. 15, 6070–6078; [DOI] [PubMed] [Google Scholar]; b)Wiggins HL, Wymant JM, Solfa F, Hiscox SE, Taylor KM, Westwell AD, Jones AT, Biochem Pharmacol 2015, 93, 332–342; [DOI] [PMC free article] [PubMed] [Google Scholar]; c)Allensworth JL, Evans MK, Bertucci F, Aldrich AJ, Festa RA, Finetti P, Ueno NT, Safi R, McDonnell DP, Thiele DJ, Van Laere S, Devi GR, Mol Oncol 2015, 9, 1155–1168; [DOI] [PMC free article] [PubMed] [Google Scholar]; d)Morrison BW, Doudican NA, Patel KR, Orlow SJ, Melanoma Res 2010, 20, 11–20. [DOI] [PubMed] [Google Scholar]
- [10].a)Mohapatra S, Sahoo MR, Rath N, Gen Hosp Psychiatry 2015, 37, 97.e95–97.e96; [DOI] [PubMed] [Google Scholar]; b)Farci AMG, Piras S, Murgia M, Chessa A, Restivo A, Gessa GL, Agabio R, Eat Behav 2015, 16, 84–87; [DOI] [PubMed] [Google Scholar]; c)Kumar KK, Bondade S, Sattar FA, Singh N, Indian J Psychol Med 2016, 38, 344–347; [DOI] [PMC free article] [PubMed] [Google Scholar]; d)Tran AT, Rison RA, Beydoun SR, J Med Case Rep 2016, 10, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Schweizer MT, Lin J, Blackford A, Bardia A, King S, Armstrong AJ, Rudek MA, Yegnasubramanian S, Carducci MA, Prostate Cancer Prostatic Dis 2013, 16, 357–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a)Fasehee H, Dinarvand R, Ghavamzadeh A, Esfandyari- Manesh M, Moradian H, Faghihi S, Ghaffari SH, J Nanobiotech 2016, 14, 32; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Wang Z, Tan J, McConville C, Kannappan V, Tawari PE, Brown J, Ding J, Armesilla AL, Irache JM, Mei Q-B, Tan Y, Liu Y, Jiang W, Bian X-W, Wang W, Nanomed: NBM 2017, 13, 641–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a)Wang Q, Franz KJ, Acc. Chem. Res 2016, 49, 2468–2477; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Zaengle-Barone JM, Jackson AC, Besse DM, Becken B, Arshad M, Seed PC, Franz KJ, ACS Infect Dis 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]; c)Haškova P, Jansova H, Bureš J, Machaček M, Jirkovska A, Franz KJ, Kovařikova P, Šimůnek T, Toxicol 2016, 371, 17–28; [DOI] [PMC free article] [PubMed] [Google Scholar]; d)Pujol AM, Cuillel M, Jullien AS, Lebrun C, Cassio D, Mintz E, Gateau C, Delangle P, Angew Chem 2012, 51, 7445–7448; [DOI] [PubMed] [Google Scholar]; e)Akam EA, Utterback RD, Marcero JR, Dailey HA, Tomat E, J Inorg Biochem 2018,180, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a)Hanigan MH, Adv. Cancer Res 2014, 122, 103–141; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Corti A, Franzini M, Paolichhi A, Pompella A, Anticancer Res 2010. 30, 1169–1181. [PubMed] [Google Scholar]
- [15].a)King JB, West MB, Cook PF, Hanigan MH, J Biol Chem 2009, 284, 9059–9065; [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Hanigan MH, Macdonald T, (Ed.: V. The University of Virginia; (Charlottesville), United States, 1998; [Google Scholar]; c)Ramsay EE, Dilda PJ, Front Pharmacol 2014, 5; [DOI] [PMC free article] [PubMed] [Google Scholar]; d)Miyata Y, Ishizawa T, Kamiya M, Yamashita S, Hasegawa K, Ushiku A, Shibahara J, Fukayama M, Urano Y, Kokudo N, Sci Rep 2017, 7, 3542; [DOI] [PMC free article] [PubMed] [Google Scholar]; e)Luo Z, Feng L, An R, Duan G, Yan R, Shi H, He J, Zhou Z, Ji C, Chen HY, Ye D, Chem. Eur. J. 2017, 23, 14778–14785. [DOI] [PubMed] [Google Scholar]
- [16].a)Carl PL, Chakravarty PK, Katzenellenbogen JA, J Med Chem 1981, 24, 479–480; [DOI] [PubMed] [Google Scholar]; b)Hagen H, Marzenell P, Jentzsch E, Wenz F, Veldwijk MR, Mokhir A, J Med Chem 2012, 55, 924–934; [DOI] [PubMed] [Google Scholar]; c)Kielar F, Helsel ME, Wang Q, Franz KJ, Metallomics 2012, 4, 899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wickham S, Regan N, West Matthew B., Thai J, Cook Paul F., Terzyan Simon S., Li Pui K., Hanigan Marie H., Biochem. J. 2013, 450, 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.