

Abstract
Genome editing holds great promise for experimental biology and potential clinical use. To successfully utilize genome editing, it is critical to sensitively detect and quantify its outcomes: homology-directed repair (HDR) and nonhomologous end joining (NHEJ). This has been difficult at endogenous gene loci and instead is frequently done using artificial reporter systems. Here, we describe a droplet digital PCR (ddPCR)-based method to simultaneously measure HDR and NHEJ at endogenous gene loci. This highly sensitive and quantitative method may significantly contribute to a better understanding of DNA repair mechanisms underlying genome editing and to the improvement of genome editing technology by allowing for efficient and systematic testing of many genome editing conditions in parallel.
Keywords: Genome editing, TALEN, CRISPR/Cas9, HDR, NHEJ, ddPCR
1. Introduction
Genome editing allows for manipulation of genomes at will in any cell type. This can be used to precisely alter a single base, to insert a sequence of interest at a precise location or to introduce random insertions and deletions which can disrupt gene function. The tools for genome editing are sequence-specific nucleases that induce a double strand break (DSB) or nicks at targeted genomic regions. The induced DNA damage activates two major DNA repair pathways to repair the damage. One is nonhomologous end-joining (NHEJ), in which the broken DNA ends are joined together without any templates. NHEJ often generates random insertions and deletions at the site of repair. The other pathway is homology-directed repair (HDR), in which cells use homologous DNA as a repair template to precisely repair DNA. By delivering homologous donor DNA with a new DNA sequence along with editing reagents, the genome can be precisely modified via donor-dependent repair [1].
When precise donor-dependent HDR is required, it is important to measure genome editing outcomes—HDR and NHEJ—for successful application of genome editing, since NHEJ often predominates. This task has typically been done by using artificial reporter systems, deep sequencing, or gel-based assays, or by detecting only NHEJ as a surrogate for HDR because of technical limitations [2–5]. None of these methods are suitable for high-throughput screening. To overcome this hurdle, the method we describe here utilizes a combination of allele-specific hydrolysis probes and a new digital PCR technology called Droplet Digital™ PCR(ddPCR™) [6]. Our original ddPCR-based assay with probes for the wild-type (WT) and HDR alleles was designed to detect only HDR-mediated point mutagenesis without measuring NHEJ [7]. (In this protocol, we describe the original allele before genome editing as “WT,” but the same strategy can be applied to correct mutations.) The method we describe here [8] contains additional NHEJ probes to allow simultaneous quantification of both HDR-mediated point mutagenesis and NHEJ-mediated introduction of insertions and deletions (Figs. 1 and 2). ddPCR partitions a reaction into 20,000 nanoliter-scale water-in-oil droplets, so that each droplet contains from zero to ~10 copies of the genome targets which are analyzed by HDR and NHEJ allele-specific hydrolysis probes in individual droplet reactions. The allele-specific combination of fluorescent signals results in droplets that contain either HDR, NHEJ, or WT alleles, or combinations of these; these droplets in turn occupy distinct locations on the two-dimensional plot, allowing absolute quantification of discrete alleles (e.g., Fig. 1c). Therefore, HDR and NHEJ events can be detected in a highly sensitive and quantitative manner.
Fig. 1.
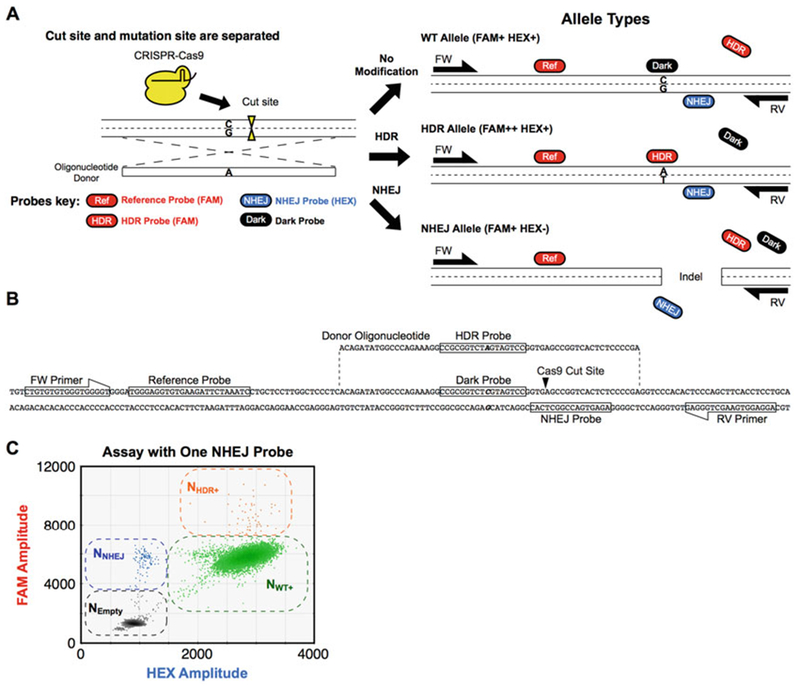
Separated cut and mutation sites: Assay design, example target sequence, and 2D droplet plot of edited genomic DNA. Depending on the editing strategy and the relative positions of cut site and edit site, the positions of assay probes and the need for a competitive blocking (dark) probe vary. The reference and HDR probes are constant regardless of the location or number of cut sites. (a) Assay design when cut site and mutation site are separated. Outcomes for three different alleles are shown (retention of WT sequence, alteration of a single base via HDR, or indel via NHEJ). The HDR and NHEJ probes are located on nonoverlapping mutation and cut sites, respectively. To prevent nonspecific binding of the HDR probe to the original “WT” sequence, a nonfluorescent (dark) “WT” probe is included that competes with the HDR probe for WT allele binding (see Figs. 2, 4, and 5 for other cases). (b) Assay design when cut site and mutation site overlap. See Fig. 4 legend for details. (c) Assay design when two cut sites are introduced. See Fig. 5 legend for details
Fig. 2.
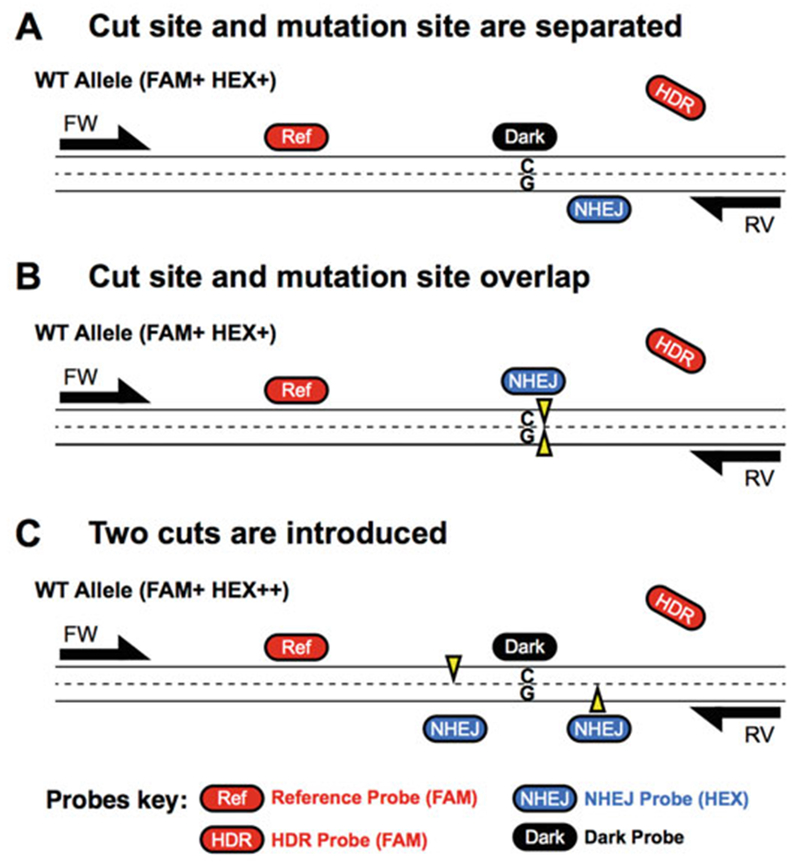
Comparison of three ddPCR assay designs for three different editing designs. (a) Assay design when cut site and mutation site are separated. See Fig. 1a legend for details. (b) Assay design when two cut sites are introduced. See Fig. 4 legend for details. (c) Assay design when cut site and mutation site overlap. See Fig. 5 legend for detail
Measuring genome editing outcomes is critical in evaluating genome editing tools (ZFN, TALEN, CRISPR/Cas9, etc.) and conditions (concentration of nucleases, target sequences, donor types, etc.). This protocol provides researchers with a rapid way to measure HDR- and NHEJ-inducing activities of genome editing conditions.
2. Materials
Prepare all solutions using ultrapure water (prepared by purifying deionized water to attain a sensitivity of 18 MΩ cm at 25 °C) and analytical grade reagents. Use filtered tips and compatible pipetters (such as RANIN) to avoid contamination of reagents in sample preparation and droplet generation. Prepare and store all reagents at −20 °C (unless indicated otherwise). Diligently follow all waste disposal regulations when disposing of waste materials.
2.1. Reagents
20× FAM and HEX hydrolysis probe and primer assay mixtures in water. Components are shown in Table 1.
WT genomic DNA in water adjusted to 100–150 ng/μl.
-
Synthetic double-stranded DNA (gBlocks, Integrated DNA Technologies) that contains the point mutation at the desired edit site (HDR control) or a 2-bp deletion at the predicted nuclease cut site (NHEJ control). Lyophilized gBlocks are resuspended in 250 μl TE + 100 ng/μl polyA carrier. Two additional 200-fold dilutions in TE + polyA result in a master stock of around 40,000 copies/μl that is maintained in LoBind tubes (Eppendorf) (see Note 1). Dilute the master stock by 20-fold to make a solution of around 2000 copies/μl for use.
Note that these are concentrations estimates and can be off by tenfold in either direction.
Restriction enzyme that does not cut within the amplicon. HindIII-HF, CviQI, MseI, AluI, HaeIII restriction enzymes (NEB) have all been validated to work directly in the ddPCR Supermix. 2–4 U restriction enzyme can be directly added to a 20 μl ddPCR reaction without additional incubation.
ddPCR™ Supermix for Probes (No dUTP) (Bio-Rad 186-3024).
ddPCR™ Buffer Control Kit (Bio-Rad 186-3052).
Droplet Generation Oil for Probes (stored at room temperature) (Bio-Rad 186-3005).
DG8™ Gaskets for QX200™ Droplet Generator (Bio-Rad 186-3009).
DG8™ Cartridges for Droplet Generator (Bio-Rad 186-4008).
Pierceable Foil Heat Seal (Bio-Rad 181-4040).
Eppendorf twin.tec 96-well plates, semiskirted (Fisher 951020346).
Rainin filter pipette tips (see Note 2).
Genomic DNA in distilled water or TE isolated from cells treated with genome editing tools to induce HDR and/or NHEJ.
Table 1.
Components of 20× assay mixtures
| Component | Concentration (μM) |
|---|---|
| Forward primer | 18 |
| Reverse primer | 18 |
| Reference probe (FAM) | 5 |
| HDR probe (FAM) | 5 |
| NHEJ probe(s) (HEX/VIC) | 5 (each, if multiple probes are used) |
| Dark probe (3′ phosphate is added) | 10 (depending on the assay) |
2.2. Equipment:
QX100™ or QX200™ Droplet Digital™ PCR system (Bio-Rad 186-4001).
PX1™ PCR Plate Sealer (Bio-Rad 181-4000).
DG8™ Cartridge Holder (Bio-Rad 186-3051).
96-well thermocycler (such as Bio-Rad C1000 that has a uniform block with deep wells, 2 °C/s ramp rate, and temperature gradient capability).
Rainin 20 μl eight-channel pipette.
Rainin 50 μl eight-channel pipette (see Note 3).
3. Methods
To detect HDR and NHEJ at the same time, four different kinds of probes are designed within a single amplicon [8]. The first is a FAM (reference) probe that is nonoverlapping with the cut site and which always binds to the genomic DNA. It provides a positive FAM amplitude for counting total genome copies in the sample (contributing to a FAM+ or FAM++ signal, Fig. 1). The second is a HEX (NHEJ) probe that binds at the cut or nick site of the genomic locus. It has a wild type sequence, so in the case where NHEJ occurs and results in either insertion or deletion of sequences at the cut site, the probe will no longer be able to bind resulting in a loss of HEX signal. Loss of HEX signal with retention of the reference probe FAM signal (FAM+, HEX−) identifies molecules that have undergone NHEJ (Fig. 1b, c). The third probe is another FAM probe (HDR probe), which will bind to the DNA when precise edits have occurred indicating an HDR event. In the case of HDR, the FAM amplitude will further increase separating the HDR population from the wild type (FAM++, HEX+). The fourth probe is a nonfluorescent (“dark”) probe having “WT” sequence which prevents non-specific-binding of the HDR probe to the unaltered “WT” allele (see Fig. 2, assay designs a and c).
3.1. Design of Hydrolysis Probes and Primers
Modify the settings of Primer3Plus (http://primer3plus.com) compatible with the master mix: 50 mM monovalent cations, 3.0 mM divalent cations, 0 mM dNTPs, SantaLucia 1998 thermodynamic and salt correction parameters (see ref. 9 and further guidance below).
For the target region of interest, position the predicted nuclease cut sites mid-amplicon, with 75–125 bp flanking either side up to and including the primer binding sites (see Note 4).
Set melting temperatures for primers, reference probes, NHEJ probes, HDR probes, and Dark probes as 55 ± 1 °C, 60 °C, 57 ± 1 °C, 55 °C, and 57 °C, respectively.
At least one primer must be positioned outside the donor molecule sequence to detect integrated edits (Fig. 1b).
Reference probe and primers are designed distant from the cut site to avoid loss of binding sites by NHEJ.
In some cases, a dark, nonextendible oligonucleotide (3′ phosphorylation) is designed to block cross-reactivity of the HDR probe and the WT sequence. This is generally required if the HDR edit site and nuclease cut site do not closely overlap (Figs. 1a and 2a, c). If they do directly overlap, the NHEJ probe can serve as the competitive blocker (Fig. 2b). When measuring NHEJ induced by dual nuclease systems, two NHEJ probes should be designed for the two cut/nick sites (Fig. 2c).
Probe position and number vary depending on the relative positions of the cut site(s) and edit site (Figs. 1 and 2). Our assay for RBM20 is shown as an example (Fig. 1b).
3.2. Assay Validation
Mix the reagents shown in Table 2 in a well of a 96-well plate to make a 25-μl reaction (see Note 5).
Carefully apply 20 μl of the mixture into each of the 8 “sample” wells of a DG8 Droplet Generator Cartridge (see Note 6).
Apply 70 μl of Droplet Generation Oil for Probes into each of the 8 “oil” wells of the DG8 Droplet Generator Cartridge (see Note 7).
Hook a DG8 Gasket for QX200 Droplet Generator onto the DG8™ Cartridge Holder.
Put the holder in the droplet generator to generate droplets in 8 wells (in ~2 min).
Transfer droplets into a semiskirted Eppendorf twin.tec 96-well plate by using an eight-channel pipette set to 45 μl (see Note 8).
Seal the plate with a Pierceable Foil Heat Seal using the PX1™ PCR Plate Sealer set to 180 °C, 5 s.
For assay validation, a two-step thermal cycling with a 50–60 ° C gradient is recommended (Table 3). If assay amplicon exceeds 150 bp in length, a three step thermal cycling protocol with a discrete 72 °C extension step can be helpful (Table 4).
Analyze the droplets by the Droplet Reader.
To determine the limit of detection of an assay, analyze 100–150 ng of WT genomic DNA samples spiked with different amounts of HDR or NHEJ gBlock. The limit of detection is the lowest amount of gBlock that gives positive signal significantly higher than the background noise by comparison of results with and without gBlock.
Table 2.
Reagents for assay validation
| Reagent | Amount per well |
|---|---|
| ddPCR Supermix for Probes (no dUTP) | 12.5 μl |
| 20× assay mixture | 1.25 μl |
| Restriction enzyme of choice | 2–4 U |
| WT genomic DNA in water | Up to 150 ng |
| HDR and NHEJ gBlock control solutions with 2000 copies/μl | 1 μl each |
| Water | To give 25 μl final reaction volume |
Table 3.
Two-step thermal cycling for ddPCR (all the steps ramped by 2 °C/s)
| Step | Temperature (°C) | Duration and repeat |
|---|---|---|
| 1 | 95 | 10 min |
| 2 | 94 | 30 s |
| 3 | 59 | 1 min, (repeat steps 2 and 3, 39 more times) |
| 4 | 98 | 10 min |
| 5 | 12 | Hold |
Table 4.
Three-step thermal cycling for ddPCR (all the steps ramped by 2 °C/s)
| Step | Temperature (°C) | Duration and repeat |
|---|---|---|
| 1 | 95 | 10 min |
| 2 | 94 | 30 s |
| 3 | 58 | 1 min |
| 4 | 72 | 2 min, (repeat steps 2, 3, and 4, 39 more times) |
| 5 | 98 | 10 min |
| 6 | 12 | Hold |
3.3. Digital PCR Detection of HDR and NHEJ Events in Genomic DNA Samples
Dilute genomic DNA samples to 100–150 ng/μl in distilled water or TE (see Note 9).
Assemble the master mix below on ice by mixing the reagents shown in Table 5 (volumes shown are for one reaction to which DNA must still be added). Multiply these by the number of samples, a distilled water only negative control, and the positive control mixture (WT genomic DNA with synthetic alleles as made in Subheading 3.2, above).
Aliquot 25—X μl of master mix into eight-tube PCR strips or a 96-well plate (any 96-well plate).
Add X μl (for a total of 100–150 ng) of genomic DNA per sample. Also, add controls to tubes or wells designated for the controls.
If the total number of samples to be analyzed is not a multiple of 8, the remaining empty tubes of the PCR strip must be filled with 25 μl of ddPCR™ Buffer Control Kit diluted to 1× by distilled water.
Briefly spin the PCR tubes or plates down.
Gently pipette up and down 7–8 times to mix reactions by a 20 μl eight-channel pipette and carefully apply the mixtures into “sample” wells.
Generate droplets as in Subheading 3.2, steps 2–6.
Repeat steps 4–8 until droplets are generated for all the samples.
Seal the plate with foil using the PX1™ PCR Plate Sealer set to 180 °C, 5 s.
Perform a thermal cycling with the best temperature found in Subheading 3.2.
Analyze the droplets by the ddPCR analyzer in the same way as Subheading 3.2.
Table 5.
Reagents for quantification of HDR and NHEJ in genomic DNA samples (X μl (100–150 ng) of a genomic DNA sample later)
| Reagent | Volume per well |
|---|---|
| ddPCR Supermix for Probes (no dUTP) | 12.5 μl |
| 20× assay mixture | 1.25 μl |
| Restriction enzyme of choice | 2–4 U |
| Water | Up to 25—X μl |
3.4. Analysis of Droplet Digital PCR Data
Go to “Analyze” and then “2-D Amplitude” in the QuantaSoft software.
In a successful assay, distinct negative, WT, NHEJ, and HDR populations should be seen in the 2D-plot (Figs. 3, 4, and 5).
If four distinct populations cannot be detected, the probes and/or primers must be redesigned.
By using the lariat tool, gate the droplets without any template (NEmpty), droplets only with NHEJ allele (NNHEJ), droplets with WT allele and both WT and NHEJ alleles (NWT+), and other droplets with HDR allele (NHDR+) as black, blue, green, and orange populations, respectively (Figs. 3, 4, and 5).
To obtain the numbers of droplets in each population, export the data as a CSV file by clicking “Export CSV” icon in the QuantaSoft software. In the CSV file, the Ch1+Ch2+, Ch1+Ch2−, Ch1−Ch2+, and Ch1−Ch2− columns are NHDR+, NNHEJ, NWT+, and NEmpty, respectively. Use formulas below to calculate the frequencies of WT, HDR, and NHEJ alleles from these numbers.
-
The standard formula for ddPCR quantification is:
Where
c = copies per microliter of initial reaction Nneg = the number of droplets that do not contain the species of interest.
Ntotal = the total number of droplets.
Vdroplet = the volume of an individual droplet (0.85 nl with current reaction conditions).
-
In this ddPCR-based assay, some of the droplet populations cannot easily be separated, such as the droplet group containing WT and NHEJ + WT droplets (NWT+) or the droplet group containing HDR and HDR + WT droplets (NHDR+). To quantify in this case, an appropriate subset of droplets is used to calculate Nneg and Ntotal (see Note 10).
Definitions
Nempty = number of droplets in the double-negative cluster labeled “empty.”
NNHEJ = number of droplets in the cluster labeled “NHEJ.”
NWT+ = number of droplets in the clusters labeled “WT+”.
NHDR+ = number of droplets in the clusters labeled “HDR+”.
-
For NHEJ quantification
Nneg = Nempty
Ntotal = Nempty + NNHEJ
-
For HDR quantification
Nneg = Nempty + NNHEJ + NWT+
Ntotal = Nempty + NNHEJ + NWT+ + NHDR+
-
For WT quantification
Nneg = Nempty + NNHEJ + NHDR+
Ntotal = Nempty + NNHEJ + NWT+ + NHDR+
-
Frequencies of WT, HDR, and NHEJ are calculated as:
FWT = cWT × 100/(cWT + cNHEJ + cHDR)
FHDR = cHDR × 100/(cWT + cNHEJ + cHDR)
FNHEJ = cNEHJ × 100/(cWT + cNHEJ + cHDR)
where
F = allelic frequency (%).
cWT = the WT allelic concentration.
cHDR = the HDR allelic concentration.
cNHEJ = the NEHJ allelic concentration.
Fig. 3.
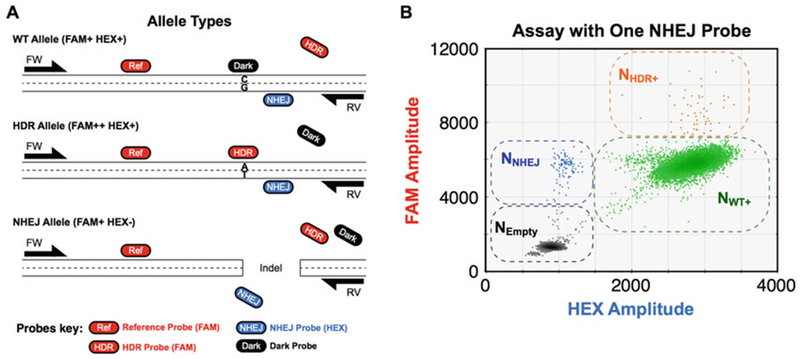
Separated cut and mutation sites, with one NHEJ probe: Assay design and 2D droplet plot with droplet group definitions for analysis. Details as in Fig. 1a, c
Fig. 4.
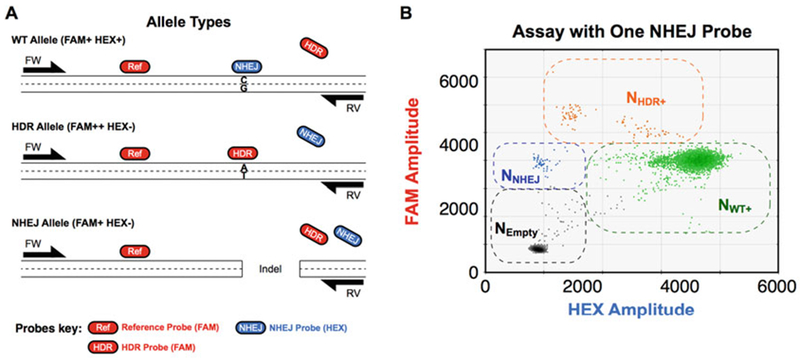
Cut site and mutation site overlap. (a) Assay design when cut site and mutation site overlap. In these cases, the NHEJ probe overlaps with and is on the same strand as the HDR probe. The NHEJ probe thus competes with the HDR probe for WT binding and a dark probe is not necessary. Also, the HDR allele becomes FAM++ and HEX−. (b) Other droplet populations defined as in Fig. 1c
Fig. 5.
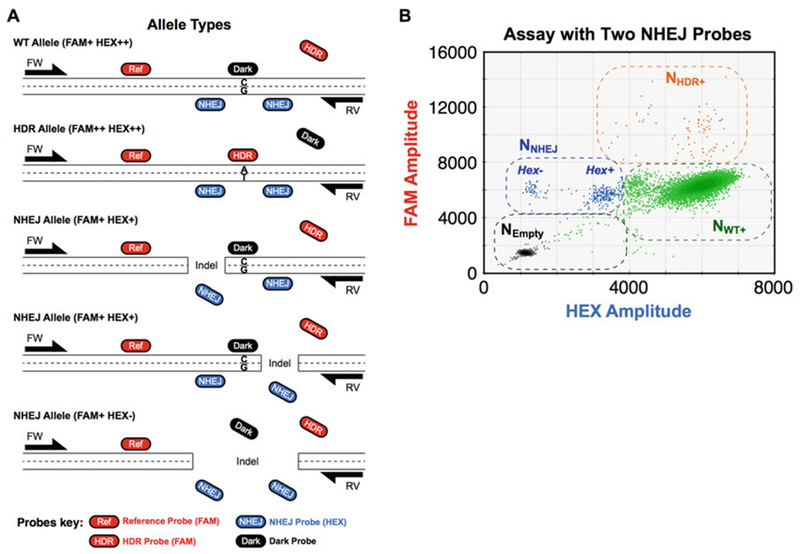
Two cut sites are introduced: ddPCR assay design and 2D droplet plot. (a) Assay design when two cut sites are introduced. Because dual Cas9 systems introduce two cuts, two NHEJ probes are included in the assay to detect possible indels on either side of the mutation site. When neither NHEJ probe competes with the HDR probe—as shown here—a dark probe is also designed to avoid binding of the HDR probe to the WT allele. Because two NHEJ probes are included in this assay, the WT allele is detected as FAM+ and HEX++, and the NHEJ alleles are detected as FAM+ and HEX+, or FAM+ and HEX−. (b) Two-dimensional plot of an assay with two NHEJ probes. Definitions of NEmpty and NHDR+ populations are the same as in Fig. 1c. However, because two NHEJ probes are included in this assay, there are two droplet populations containing only NHEJ alleles—one that lost one of the two NHEJ probe binding sites of NHEJ probes (FAM+ HEX+) and one that lost both (FAM+ HEX−). All other droplets were gated as NWT+ population (FAM+ HEX++). These definitions are used to calculate the HDR and NHEJ allelic frequencies (see Subheading 3.4)
An example of analysis is shown in Fig. 6.
Fig. 6.
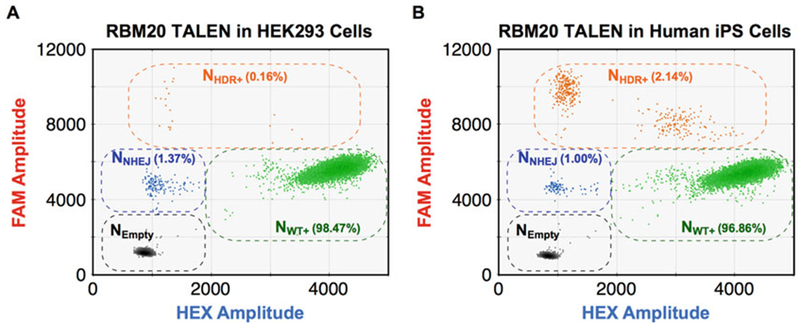
Different HDR and NHEJ frequencies induced by TALEN in RBM20 in HEK293 cells and human induced pluripotent stem cells. (a and b) HDR and NHEJ allelic frequencies induced by the same TALENs targeting RBM20 in HEK293 cells (a) and human induced pluripotent stem cells (b). The NHEJ and HDR probes directly compete to each other, so NHDR+ population was FAM++ HEX− as in Fig. 4. The frequencies are shown in the two-dimensional plots. The two different cell types showed different HDR and NHEJ frequencies
Acknowledgment
We thank Jennifer R. Berman, Samantha B. Cooper, Bin Zhang, and George A. Karlin-Neumann (Bio-Rad) for technical help and helpful discussions. This work was supported by the National Institutes of Health (U01-HL100406, U01-GM09614, R01-HL108677, U01-HL098179, U01-HL099997, P01-HL089707, and R01-HL060664 to B.R.C.); the UCSF Liver Center to B.R.C., the Bluefield Project to Cure Frontotemporal Dementia to B.R.C., the Uehara Memorial Foundation Research Fellowship to Y.M., and Gladstone-CIRM Fellowship to Y.M.
Footnotes
Master high-copy gBlock stocks should be kept in a post-PCR environment to avoid contamination. The control gBlock stocks must be handled with utmost care to avoid contamination, only bringing highly dilute solutions into the assay setup area.
Filtered tips must be used to avoid contamination.
In order to slowly pipette droplets to avoid shearing of them, a 50 μl pipette is recommended rather than a 200 μl pipette.
Predicted cut sites are 3 bp upstream of PAM for CRISPR and equidistant between DNA binding domains for TALEN or FokI-dCas9.
Handle positive control mixtures carefully (e.g., careful opening of the control stock tubes and changing gloves after handling the control stock). If assay contamination is suspected, remake the 20× assay.
Avoid creating bubbles in the DG8 sample wells. Bubbles floating on the surface of the sample may not affect droplet generation, but bubbles in the bottom of the well will disrupt droplet generation. Spinning prepared samples before droplet generation in tubes or plates removes bubbles. Take note of orientation of the cartridge with respect to sample order to ensure correct loading of the final Eppendorf twin.tec 96-well plate.
Samples should be loaded onto the chip prior to droplet generation oil.
Do not press the pipette tightly to the bottom of the cartridge or pipette too vigorously as this will shear the droplets. Cover the PCR plate with the foil sheet immediately after transfer to reduce the risk of contamination.
With too much input DNA, each droplet would have too many genomic DNA copies, which interferes with proper separation of the four distinct populations and thus accurate estimation of allelic concentrations. With too little input DNA, the number of genomic copies analyzed by the assay would not be enough to have high sensitivity. Therefore, the amount of genomic DNA input should not exceed 150 ng per ddPCR reaction.
For NHEJ quantification, only NHEJ single positives and the Empty (double-negative) droplets were used. For WT and for HDR quantification, all droplets are used.
References
- 1.Gaj T, Gersbach CA, Barbas CF III (2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31(7):397–405. 10.1016/j.tibtech.2013.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Certo MT, Ryu BY, Annis JE, Garibov M, Jarjour J, Rawlings DJ, Scharenberg AM (2011) Tracking genome engineering outcome at individual DNA breakpoints. Nat Methods 8 (8):671–676. 10.1038/nmeth.1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154 (6):1380–1389. 10.1016/j.cell.2013.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hendel A, Kildebeck EJ, Fine EJ, Clark JT, Punjya N, Sebastiano V, Bao G, Porteus MH (2014) Quantifying genome-editing outcomes at endogenous loci with SMRT sequencing. Cell Rep 7(1):293–305. 10.1016/j.celrep.2014.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin S, Staahl BT, Alla RK, Doudna JA (2014) Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. elife 3:e04766 10.7554/eLife.04766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, Kitano TK, Hodel MR, Petersen JF, Wyatt PW, Steenblock ER, Shah PH, Bousse LJ, Troup CB, Mellen JC, Wittmann DK, Erndt NG, Cauley TH, Koehler RT, So AP, Dube S, Rose KA, Montesclaros L, Wang S, Stumbo DP, Hodges SP, Romine S, Milanovich FP, White HE, Regan JF, Karlin-Neumann GA, Hindson CM, Saxonov S, Colston BW (2011) High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem 83(22):8604–8610. 10.1021/ac202028g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyaoka Y, Chan AH, Judge LM, Yoo J, Huang M, Nguyen TD, Lizarraga PP, So PL, Conklin BR (2014) Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat Methods 11(3):291–293. 10.1038/nmeth.2840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miyaoka Y, Berman JR, Cooper SB, Mayerl SJ, Chan AH, Zhang B, Karlin-Neumann GA, Conklin BR (2016) Systematic quantification of HDR and NHEJ reveals effects of locus, nuclease, and cell type on genome-editing. Sci Rep Mar 31(6):23549 PMID: , PMC4814844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bio-Rad Bulletin 6407, Droplet Digital Application guide [Google Scholar]