

Abstract
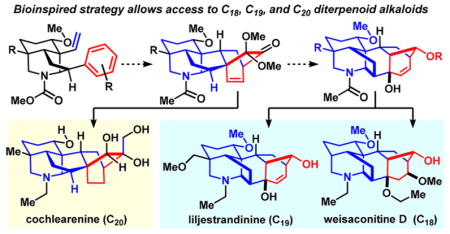
The secondary metabolites that comprise the diterpenoid alkaloids are categorized into C18, C19, and C20 families depending on the number of contiguous carbon atoms that constitute their central framework. Herein, we detail our efforts to prepare these molecules by chemical synthesis, including a photochemical approach, and ultimately a bioinspired strategy that has resulted in the development of a unifying synthesis of one C18 (weisaconitine D), one C19 (liljestrandinine), and three C20 (cochlearenine, paniculamine, and N-ethyl-1α-hydroxy-17-veratroyldictyzine) natural products from a common intermediate.
Graphical Abstract
INTRODUCTION
The diterpenoid alkaloids are complex, bridged, polycyclic natural products that possess analgesic, anti-inflammatory, antiarrhythmic, and other pharmacological activities.1–3 The Aconitum, Consolidum, and Delphinium plants from which these pseudoalkaloids are isolated have long been used in traditional medicine for the treatment of pain and cardiovascular diseases. In fact, guan-fu base A (5, Figure 1) is in phase IV clinical trials as an antiarrhythmic agent in China,4 while the hydrobromide salt of lappaconitine (1) is approved and marketed in both China and Russia as a pain and arrhythmia treatment.5 Perhaps most intriguing is the ability of some diterpenoid alkaloids to modulate Na+ and/or K+ ion channels.6 For example, aconitine (3) is a highly toxic Na+ channel activator with an estimated lethal dose of 1–2 mg in humans.7 However, structurally related lappaconitine (1) is a Na+ channel blocker that has therapeutic value.6 The potential for these molecules to act as subtype-selective ion channel modulators has significant implications for treating channelopathies without significant undesirable side effect profiles.8,9 While over 1200 members make up this class of secondary metabolites, few have been studied for their function due to inadequate accessibility. Developing synthesis strategies that allow access to a variety of these natural products and related unnatural analogs would facilitate structure-activity-relationship studies and provide myriad opportunities for clinical applications.
Figure 1.

Examples of diterpenoid alkaloids.
Attracted by the challenges associated with the architectural complexity and the opportunities to learn about the biological function of these natural products, we embarked on a total synthesis campaign targeting members of the C18-, C19-, and C20-diterpenoid alkaloids 5,10,11 with varying substitution and oxygenation patterns decorating their framework. These efforts have led to the first syntheses of weisaconitine D (C18, 2), liljestrandinine (C19, 4), cochlearenine (C20, 6), N-ethyl-1α-hydroxy-17-veratroyldictyzine (C20, 7), and paniculamine (C20, 8).12,13
RESULTS AND DISCUSSION
The assembly of these structurally complex molecules has challenged synthetic chemists since the 1960s, beginning with Nagata, Masamune, and Wiesner’s formal syntheses14 of C20-members atisine,15 garryine,16,17 and viatchine.17,18 While lengthy, these contributions set the stage for the field and provided inspiration even to modern synthetic approaches, including our own (see Scheme 7).19,20 Recently, the Baran and Xu groups have devised unified strategies toward various diterpenoid alkaloids but particularly natural products within the C20 family.21,22 Our group demonstrated conversion of the C20 hetidine core to the atisine framework,23,24 and the Fukuyama group subsequently showed that an advanced intermediate used en-route to the C20 natural product (–)-lepenine25 can also be used to synthesize the C19 alkaloid (–)-cardiopetaline.26 Here, we describe our studies on an approach that unifies the syntheses of secondary metabolites belonging to each of the C18, C19, and C20 families.
Scheme 7.

Wagner–Meerwein Approaches to the [3.2.1]-Bicyclic Motif (BCD Ring System)
Retrosynthesis
For bridged, polycyclic molecules with complexity as daunting as that of the diterpenoid alkaloids, network analysis, an approach formalized by Corey,27 provides an objective starting point and guide for retrosynthetic analyses. In a complex molecular setting containing a network of fused and bridged-ring systems, the latter are generally perceived to be more challenging to assemble. However, significant simplification to the network can be achieved by iteratively cleaving bonds associated with the maximally bridged ring(s), which is a key element of network analysis.
In applying network analysis to the hexacyclic aconitine framework, as exemplified by weisaconitine D (Scheme 1), each of the individual rings can be analyzed for the number of bridging atoms (or bridgehead sites) that they contain. In this case, the B-ring contains five bridging atoms, the E- and F-rings four bridging atoms (F-ring not shown, see SI), the C-ring contains three bridging atoms, the D-ring three bridging atoms (not shown), and the A-ring two bridging atoms (not shown). Having identified the B-ring as the maximally bridged ring, we envisioned a bicyclization transform that effectively simplifies the target to tricyclic vinyl arene 9. In the forward sense, this operation forges two bonds and three new rings in a single step.28 Similarly, applying network analysis to intermediate 9 yields the piperidine E-ring as the maximally bridged system with four bridging atoms. Since the piperidine ring also contains a nitrogen atom (a functional group), we reasoned that its disconnection would provide the greatest simplification, thus leading back to fused AF-ring system 10 that contains no bridging elements. The protected amine functionality of 10 can arise from a hydration and Hofmann rearrangement sequence on nitrile 11, which in turn can be prepared from vinyl nitrile 12 through a conjugate arylation reaction and transformation of the methyl ester. This intermediate can be further simplified to hydrindenone 13, which can be accessed in >20 g quantities using a Diels–Alder reaction.13
Scheme 1. Network-Analysis-Guided Retrosynthetic Analysis of Aconitine-Type Natural Productsa.

aThe red dots indicate bridging atoms in the network analyses.
Correctly identifying the maximally bridged ring systems in complex polycycles manually is inherently time-consuming.29,30 Therefore, to facilitate the use of network analysis in retrosynthetic planning,31 we developed a computer algorithm that correctly identifies the maximally bridged ring(s) for any molecule using the Chemistry Development Kit software library.12,32–34 We envision this freely-available, web-based deterministic program to be a useful tool in both the research laboratory and classroom for retrosynthesis and chemical education.
A Photochemical Approach to the [3.2.1] CD-Bicyclic Ring System
Using network analysis, we determined that a bicyclization reaction to form the BCD-ring systems would be most effective in directly assembling the target hexacyclic structure. To achieve this, we imagined a late-stage meta-π-arene photocycloaddition between the alkene and arene groups of vinyl arene 14 (Scheme 2). Photoexcitation of the arene component with UV irradiation was expected to lead to formation of an exciplex where concerted σ-bond formations would occur to generate diradical 15. Spontaneous collapse of the diradical intermediate would form vinyl cyclopropane 16.35 Subsequent oxidation of the alkene to epoxide 17 could be followed by an acid-mediated cyclopropane fragmentation and epoxide ring-opening to furnish diketone 18.36,37 This approach would be unlike the proposed biosynthesis of the lappaconitine and aconitine-type diterpenoid alkaloids that contain the [3.2.1] CD bicyclic structure and would provide a more direct route to the desired carbon framework.
Scheme 2.
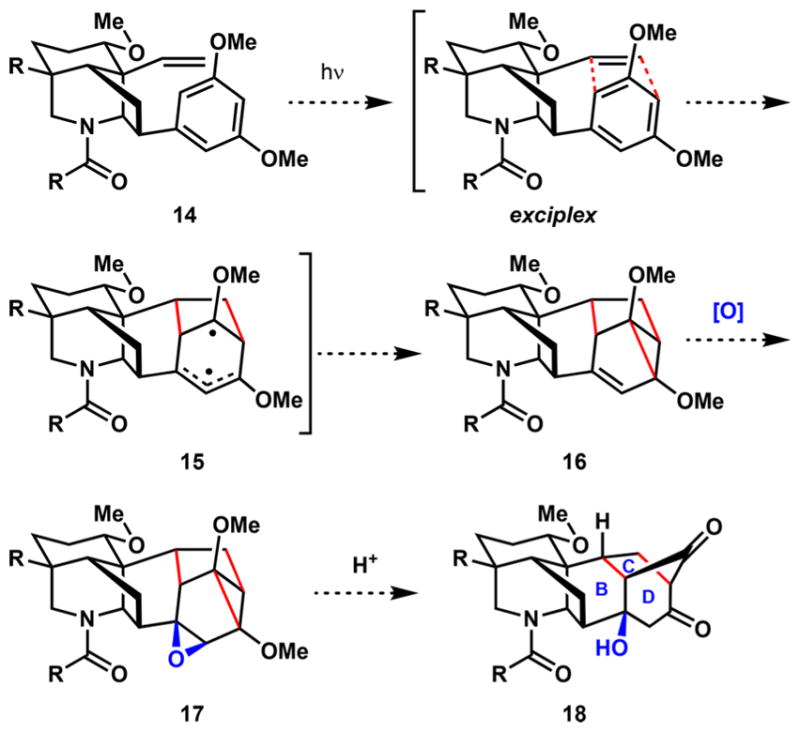
Proposed meta-π-Arene Photocycloaddition Approach to the BCD Ring System
We chose to introduce a methoxy-substituted aromatic ring (see 14) because the arene oxygens would be native to many of our target natural products. In addition, electron-rich methoxy groups direct meta-π-functionalization to sites ortho to them, which would permit us to take maximum advantage of the substituents inherent to the target structures, thereby streamlining synthesis.
The synthesis begins with dimethylation of 1,4-butynediol (19, Scheme 3) followed by NaNH2-promoted elimination/ isomerization to generate enyne 20 in 60% yield.38 A two-step sequence involving alkylation of the terminal alkyne with paraformaldehyde and subsequent reduction of the propargyl alkyne with LiAlH4, followed by TBS-protection of the primary alcohol yields diene 21,39 which can be prepared on 54 g scale in a single pass. Heating an excess of diene 21 with dienophile 22 (prepared from α-selenation of β-ketoester 23, followed by selenoxide elimination from 24)40 results in formation of the desired endo-cycloadduct (13) as the major product in 71% yield, along with the exo-diastereomer in 4% yield.
Scheme 3. Decagram-Scale Synthesis of Diene 20 and Cycloadducts 22a.
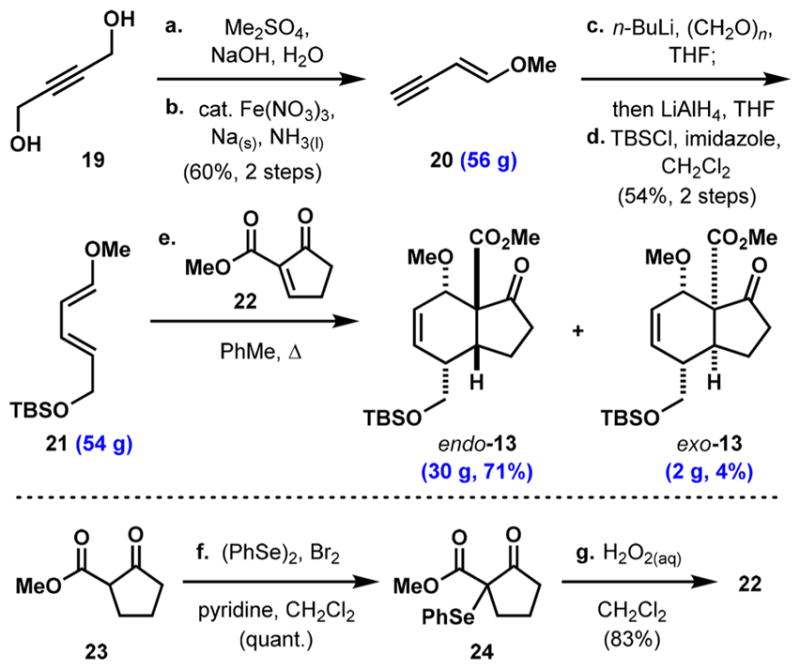
aReaction conditions: (a) Me2SO4, NaOH, H2O, 90–100 °C; (b) NH3(I), Fe(NO3)3·9H2O (cat.), Na(s), 60% over two steps; (c) n-BuLi, THF, –78 to 23 °C, then (CH2O)n, 0–23 °C, then LiAlH4, 0–23 °C; (d) TBSCl, imidazole, CH2Cl2, 23 °C, 54% over two steps; (e) diene 21, dienophile 22, PhMe, 100 °C, 71% endo-13 + 4% exo-13, (f) (PhSe)2, Br2, pyridine, CH2Cl2, quant; (g) H2O2(aq), CH2Cl2, 0–23 °C, 83%. TBSCl = tert-butyldimethylsilyl chloride.
The alkene group in hydrindenone endo-13 is hydrogenated with Pd/C as the catalyst, and the ketone group converted to the vinyl triflate by treatment with LiHMDS and PhNTf2 in 88% yield over the two steps (Scheme 4). The resulting vinyl triflate is then converted to vinyl nitrile 12 in 78% yield by a Pd- and Cu-catalyzed cyanation protocol using NaCN.41 Introducing the electron-rich aromatic group onto the vinyl nitrile is accomplished using a Rh-catalyzed 1,4-addition reaction developed by the Hayashi group, providing trans-β-aryl nitrile 25 in 69% yield (78% brsm).42 The ester moiety is selectively reduced to the primary alcohol with Red-Al, leaving the nitrile intact (77%). The primary hydroxyl is oxidized to aldehyde 26 (89%), which is subsequently methylenated to alkene 27 (98%). In the presence of acid-labile functionalities such as the silyl ether, hydration of the nitrile to afford primary amide 29 proceeds in 65–75% yield under anhydrous conditions employing Wilkinson’s catalyst and acetaldoxime (28) as the water equivalent.43 Hypervalent iodine-mediated Hofmann rearrangement of 29 in methanol solvent forms the corresponding methyl carbamate, which is then subjected to TBAF-mediated deprotection to reveal alcohol 30 in 94% yield over two steps. Successive activation of the alcohol as the mesylate and treatment with KH in DMF results in cyclization to piperidine 31 (86% yield over two steps), which served as our substrate for photocycloaddition studies.
Scheme 4. Synthesis of Vinyl Arene Intermediate 31 for Photocycloadditiona.

aReaction conditions: (a) H2, Pd/C (cat.), EtOAc, 23 °C; (b) LiHMDS, then PhNTf2, THF, –78 to 23 °C, 88% over two steps; (c) NaCN, CuI (cat.), Pd(PPh3)4 (cat.), MeCN, 78%; (d) aryl boronic ester (prepared from the corresponding ArBr, n-BuLi, Et2O, –78 to 23 °C; then B(OMe)3, –40 °C); then 12, [Rh(cod)OH]2 (cat.), H2O, 1,4-dioxane, 100 °C, 69%; (e) Red-Al, CH2Cl2, 0–23 °C, 77%; (f) DMP, NaHCO3, CH2Cl2, 89%; (g) PPh3MeBr, LiHMDS, THF, 70 °C; then 26, 0–23 °C, 98%; (h) 28, [Rh(PPh3)3Cl] (cat.), PhMe, 130 °C, 65–75%; (i) PhI(OAc)2, KOH, MeOH, 94%; (j) TBAF, THF, 23 °C, 94% over two steps; (k) MsCl, CH2Cl2; (l) KH, DMF, 60 °C, 86% over two steps. TBAF = tetrabutylammonium fluoride. DMP = Dess Martin periodinane.
Irradiation of a solution of vinyl arene 31 in C6D6 with 254 nm UV light results in the formation of diastereomeric [2 + 2]-cycloadducts 32 and 33 (Scheme 5a) via ortho-π-arene photocycloaddition. Using 310 nm UV light gives a similar outcome by 1H NMR analysis. Attempts to isolate these cycloadducts by silica gel chromatography resulted in their decomposition to a mixture of hydrolyzed products. However, reconstituting this mixture of 32 and 33 in methanol with p-TsOH fully converts the bis-enol ether intermediates to an isomeric mixture of hexacyclic, vinylogous esters 34, 35, and 36 in 12%, 28%, and 8% yields, respectively. These cycloadducts were unambiguously characterized by single-crystal X-ray crystallography. Products arising from formation of exciplex 37 and the desired (3 + 2) meta-π-arene photocycloaddition pathway were not observed (e.g., 38). UV irradiation in other nonpolar solvents such as cyclohexane and toluene at room temperature or 4 °C has minimal effect on reactivity, and the use of methanol as solvent partially facilitates hydrolysis of photocycloadducts 32 and 33. The lack of (3 + 2) reactivity is likely due to the developing strain that is imposed by tethering the alkene meta to the methoxy-directing groups, a substitution pattern that has not been demonstrated to undergo meta-π-arene photocycloaddition.35 To help alleviate strain, bicyclic vinyl arenes 27 and 39 (precursors to alcohol 30) were subjected to 254 nm UV irradiation (Scheme 5b). However, reducing strain by not having the piperidine ring in place results in no reactivity, likely due to the system being unable to adopt an energetically accessible, reactive conformation. Based on similar reasoning, a less constrained vinyl arene (40) lacking the C7–C17 bond was prepared and irradiated with 254 nm UV light (Scheme 5c). While reactive, this substrate gives rise to a complex mixture of products that could not be easily purified. To test whether the lack of (3 + 2) reactivity could be due to a high barrier in forming a strained six-membered B-ring, we designed substrate 41 where the alkene is tethered ortho to the methoxy-group (Scheme 5d). In this case, if a (3 + 2) photocycloaddition were to occur through exciplex 42 and diradical 43, a more favorable five-membered B-ring (highlighted in red) would form. This hypothesis proved correct, providing (3 + 2) cycloadduct 44 as the major product in 44% yield. The [2 + 2] cycloaddition pathway is also operative to a small extent, affording cyclobutene-bearing 47 in 16% yield. This product differs from the previously observed ortho-photocycloaddition reactions (cf. Scheme 5a) in that cyclo-butane intermediate 45 bearing a tertiary methoxy group formed from the initial photochemical [2 + 2]-cycloaddition undergoes spontaneous 6π-electrocyclic ring opening to cyclooctatriene 46, followed by a final photochemical 4π-electrocyclic ring closing process to arrive at 47.44 These products, however, could not be easily advanced to the target natural products.
Scheme 5.

Exploring Alkene-Arene Photocycloadditions
A Bioinspired Wagner–Meerwein Rearrangement Approach to the BCD-Ring System
An alternative bicyclization disconnection that still retrosynthetically cleaves the maximally bridged B-ring of the aconitine-type framework involves a [4 + 2]-cycloaddition between the alkene and the arene system following oxidative dearomatization (Scheme 6). The product will contain a bicyclo[2.2.2]octane system characteristic of the denudatine-type molecules that will need to be rearranged to give the bicyclo[3.2.1] CD-ring system. Since nature employs such an isomerization in the biosynthesis of numerous diterpenes by rearranging the diterpene skeleton of denudatine alkaloids to that of the aconite skeleton,45–48 we reasoned that this approach could offer promising opportunities for developing a divergent synthesis. In fact, Wagner–Meerwein-type rearrangement of a bicyclo[2.2.2]octane to the corresponding bicyclo[3.2.1]octane was first reported in 194949,50 by Doering et al. and successfully applied to early syntheses of the diterpenoid alkaloids talatisamine51 and 13-desoxydelphonine52 by Wiesner and co-workers (Scheme 7a,b). More recently, Fukuyama and co-workers disclosed an unusual solvolytic rearrangement of sulfonyloxirane 50, followed by reduction of the resultant ketone to alcohol 51 (Scheme 7c). We were particularly interested in implementing a process reported by Xu and Wang in 2012,53 in which an oxidative Wagner–Meerwein-type rearrangement on the bicyclo[2.2.2]-octene system of 52 furnishes the bicyclo[3.2.1] system of 53 with concomitant introduction of a new oxygen atom (Scheme 7d). The alkene group in 53 also presents additional opportunities for functionalization, which we envisioned to be useful for generating diversity when planning natural and unnatural product synthesis. We thus targeted hexacycle 54, which exhibits the denudatine framework, with the intention of rearranging it to 55, which exhibits the aconitine framework, via an oxidative Wagner–Meerwein rearrangement (Scheme 7e).
Scheme 6.
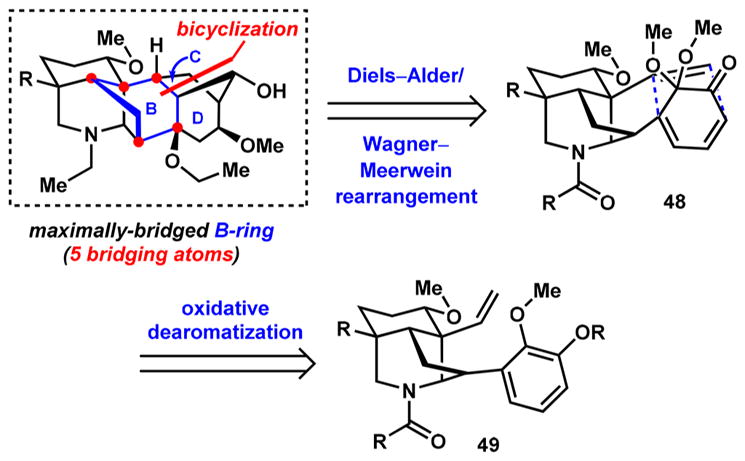
Revised Retrosynthetic Plan
The synthesis of the denudatine framework commences with the 1,4-arylation of vinyl nitrile 12 using aryl boronate ester 56 to form β-aryl nitrile 57 in 60–75% yields (Scheme 8). Similar to the sequence illustrated in Scheme 4, selective reduction of the methyl ester to its corresponding primary alcohol (81–87% yields), followed by oxidation (76–82% yields) and Wittig olefination (85–95% yields) provides alkene 58. Subjecting 58 to Rh-catalyzed nitrile hydration gives the desired primary amide (59) in 70% yield, with the remainder of the mass balance accounted for by unreacted starting material that can be resubjected to the same reaction conditions. The primary amide is effectively converted to its corresponding methyl carbamate by treatment with PhI(OAc)2 in methanol and the primary alcohol deprotected with TBAF to yield 60 in 96% over the two steps. Mesylation of the alcohol and cyclization of the piperidine ring occur by reacting 60 with MsCl/Et3N and KOt-Bu, respectively, to give tricycle 61 in 73% yield over the two steps. Subsequent cleavage of the MOM protecting group with HCl and oxidative dearomatization with PhI(OAc)2 in methanol furnishes dienone 62, the immediate precursor to intramolecular Diels–Alder cycloaddition, in 98% yield over two steps. Heating a solution of 62 in p-xylene to 150 °C in a sealed flask induces the intramolecular [4 + 2]-cycloaddition to provide the desired hexacycle (63) as a single diastereomer in 77% yield. Reduction of the ketone carbonyl of 63 occurs exclusively from the more sterically encumbered β-face, which is then followed by acid-catalyzed cleavage of the dimethyl ketal to reveal α-hydroxyketone 64 in 87% yield. This secondary hydroxy group is masked as the MOM ether by treatment with NaH and MOMCl, and a NaBH4-mediated reduction of the remaining carbonyl group again occurs exclusively from the β-face, generating α-disposed alcohol 65 in 87% yield over two steps. Xu and Wang observed a similar β-selective NaBH4 reduction in their synthesis of model substrate 52 (see Scheme 7d); however, they stated that a rationalization for the observed selectivity could not be provided. We postulate that the exclusive approach from the β-face of the molecule arises from a torsional steering effect: reduction from the seemingly less-hindered α-face would give rise to an unfavorable eclipsing interaction in the transition state, whereas reduction from the seemingly more-hindered β-face would lead directly to an energetically favorable transition state with a staggered conformation (Scheme 9).
Scheme 8. Formation of the Key Hexacyclic Intermediate Containing the [2.2.2] Bicyclea.

aReaction conditions: (a) 56 (prepared from the corresponding ArBr), n-BuLi, Et2O, –78 to 23 °C; then B(OMe)3, –40 °C; then 12, [Rh(cod)OH]2 (cat.), H2O, 1,4-dioxane, 100 °C, 60–75%; (b) Red-Al, CH2Cl2, 0–23 °C, 81–87%; (c) DMP, NaHCO3, CH2Cl2, 76–82%; (d) PPh3MeBr, LiHMDS, THF, 70 °C; then aldehyde, 0–23 °C, 85–95%; (e) 28, [Rh(PPh3)3Cl] (cat), PhMe, 130 °C, 70% (>95% brsm); (f) PhI(OAc)2, KOH, MeOH; (g) TBAF, THF, 23 °C, 96% over 2 steps; (h) MsCl, Et3N, CH2Cl2; (i) KOt-Bu, THF, 73% over two steps; (j) HCl, i-PrOH/THF; (k) PhI(OAc)2, NaHCO3, MeOH, 98% over two steps; (l) p-xylene, 150 °C, 77%; (m) NaBH4, MeOH; (n) TFA, H2O, 87% over two steps; (o) NaH, MOMCl, CH2Cl2; (p) NaBH4, MeOH, 87% over 2 steps.
Scheme 9.
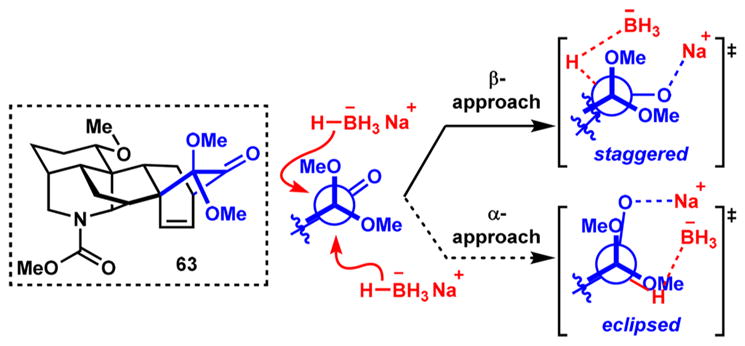
Torsional Steering Controls NaBH4 Reduction
Activating alcohol 65 with triflic anhydride and then heating the newly formed triflate (66) in a mixture of DBU and DMSO resulted in the formation of allylic alcohol 69 as the only observable product (Scheme 10). Enone 70, which was anticipated on the basis of the precedent by Xu and Wang, was not formed to any appreciable extent. One possible pathway involves a thermally induced 1,2-alkyl shift leading to the formation of tertiary, allylic carbocation 67, which is trapped by the DMSO solvent and ultimately affords allylic alcohol 69 through an elimination pathway (Scheme 10a). In contrast, enone 70 would arise through an SN2′ addition of DMSO to the less hindered position of allylic carbocation 67, followed by oxidation of the allylic hydroxyl. Presumably, the strain imposed by the alkene group of the enone precludes the formation of 70.54 Alternatively, a dyotropic rearrangement55 involving simultaneous, concerted migration of the alkyl and triflate groups through the intermediacy of allylic triflate 71 can be invoked to explain the observed selectivity for 69 (Scheme 10b).
Scheme 10. Wagner–Meerwein Rearrangement and Possible Mechanismsa.

aReaction conditions: (a) Tf2O, pyridine, CH2Cl2, –78 to 23 °C; (b) DBU, DMSO, 120 °C, 55% over two steps. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene.
Synthesis of C18 Weisaconitine D
Allylic alcohol 69, although unanticipated, bears the desired bicyclo[3.2.1]octene system that can be advanced to weisaconitine D (2).56 Alcohol 69 also possesses the framework of other C18 and C19 diterpenoid alkaloids such as the lappaconitines, ranaconitines, and aconitines.57 Epoxidation of 69 yields epoxy alcohol 72 as a single diastereomer (Scheme 11). X-ray crystallographic analysis of this intermediate provided unambiguous confirma-tion of its structure and stereochemistry. The alcohol group was alkylated to give epoxy ether 73 in 78% yield over two steps. A titanium(III)-mediated epoxide ring opening using water as the hydrogen atom source58 occurs regioselectively, and the hydroxyl obtained in this manner was methylated in 66% yield over two steps with an isomeric ratio of 14:1 (the minor isomer has the methoxy group at C15, not shown). To introduce the N-ethyl substituent, the carbamate of 74 was first cleaved by base-mediated hydrolysis, and the unveiled piperidine nitrogen acetylated with Ac2O and reduced with LiAlH4. As discussed later, installing the ethyl group in a more direct fashion would not yield a more efficient synthesis (Scheme 15). A final deprotection with aqueous HCl cleaved the MOM protecting group and accomplished the first synthesis of weisaconitine D (54% yield over the last four steps). Derivatizing 2 to p-nitrobenzoate ester 75 allowed us to acquire a single crystal X-ray structure to support the characterization of the natural product.
Scheme 11. Completion of Weisaconitine D and Confirmation of Chemical Structure by X-ray Crystallographya.

aReaction conditions: (a) m-CPBA, CH2Cl2, 0–23 °C; (b) NaH, EtI, THF, 0–40 °C, 78% over two steps; (c) Cp2TiCl2, Mn, H2O, THF, 23 °C; (d) NaH, Me2SO4, THF, 66% over 2 steps, rr = 14:1; (e) KOH(aq), (HOCH2)2; (f) Ac2O, pyridine, CH2Cl2, 0–23 °C; (g) LiAlH4, Et2O, 0–40 °C; (h) 2 N HCl(aq), THF, 54% over four steps; (i) 4-O2NC6H4COCl, Et3N, DMAP, CH2Cl2, 0–23 °C, 58%. m-CPBA = meta-chloroperbenzoic acid, DMAP = 4-(dimethylamino)pyridine.
Scheme 15. Synthesis of Cochlearenine, N-Ethyl-1α-hydroxy-17-veratroyldictyzine, and Paniculaminea.

aReagents and conditions: (a) NaI, Zn0, DME, 105 °C; (b) HCl, THF/i-PrOH, 23 °C; (c) PhI(OAc)2, NaHCO3, MeOH, 23 °C, 61% over three steps; (d) p-xylene, 150 °C, 80–87%; (e) Me2S(O)=CH2, THF/DMSO, 0–21 °C, 85%; (f) Ph3P=CH2 (from Ph3PMeBr, LiHMDS, 70 °C), 0–40 °C, then 1 M HCl(aq), 96%; (g) Ph3CO2Li (from Ph3CO2H, MeLi), THF, 0–40 °C, 57% (64% brsm); (h) HBr, AcOH, microwave (110 °C), 50 min, then 2 M NaOH(aq); (i) Ac2O, pyridine, CH2Cl2, 0–23 °C, ~5% 100 + 28% 101 + 22% 102 + 29% 103 over two steps (84% combined yield); (j) KOAc, AcOH, 120 °C; (k) H2 (100 psi), Pd/C (cat.), MeOH, 23 °C; (l) LiAlH4, THF, 0–23 °C, three steps (63% from 102, 77% from 103); (m) H2O2, MeOH/H2O, 60 °C, 50%; (n) veratroyl chloride, polymer-supported DMAP, CH2Cl2, 0–23 °C, 25% (34% brsm); (o) TFA, CDCl3, quant. DMAP = 4-(dimethylamino)pyridine; TFA = trifluoroacetic acid.
Synthesis of C19 Liljestrandinine.59
All of the C19- and C20-diterpenoid alkaloids contain an additional carbon atom (C18) at C4 with respect to the C18 natural products (see Figure 1). Using an intermediate (60, Scheme 8) from the synthesis of weisaconitine D (2), we initially sought to introduce this one-carbon unit through an enolate alkylation reaction (Scheme 12a). Thus, alcohol 60 was oxidized to aldehyde 76 in 95% yield under typical Swern conditions. We expected that exposure of 76 to base and a methylating agent would effect alkylation from the convex face of the bicycle. Treating 76 with excess KOt-Bu leads to formation of bicyclic enolate 77 with accompanying deprotonation of the methyl carbamate. However, reacting this enolate with iodomethane did not result in the desired alkylation from the convex face to give β-methyl aldehyde 78, but instead alkylation occurred exclusively from the concave face of the bicycle to yield α-methyl aldehyde 79 in 54% yield. The stereochemistry of α-79 was confirmed by its conversion to mesylate 80 and acquiring single crystal X-ray diffraction data. Unfortunately, the aldehyde of 79 is not correctly disposed for cyclization to yield the requisite piperidine structure. To gain insight into the observed alkylation selectivity, we modeled the intermediate (77) arising from the double deprotonation of aldehyde 76 by DFT at the B3LYP/6-31+G(d,p) level of theory (Scheme 12b). In the two views of the minimized structure, the vinylic proton (shown in blue) protrudes directly over the reactive p-orbital of the enolate, effectively shielding approach of the methyl electro-phile from the convex face. In contrast, approach of the electrophile from the concave face is relatively unhindered, providing a rationale for the exclusive α-selectivity that is observed.
Scheme 12. α-Alkylation Generates Only the Undesired Diastereomera.

aReaction conditions: (a) (COCl)2, Et3N, CH2Cl2, –78 to 23 °C, 95%; (b) KOt-Bu, THF, 0 °C, then MeI, 54% 79.
On the basis of this analysis, any type of electrophile should approach the enolate from the undesired concave face. Inspired by the precedent of Wiesner, transforming aldehyde 76 to diol 81 using an aldol-Cannizzaro reaction,60 which we accomplished in 97% yield, obviates the need for a diastereoselective alkylation (Scheme 13). Attempts at selective deoxygenation directly from diol 81 using Morandi’s protocol (Ph2SiH2/ Et3SiH and catalytic B(C6F5)3)61 or through a more traditional stannane-mediated deoxygenation62 of thiocarbonate 83 gave complex mixtures of products. In these reactions, the desired monodeoxygenated products 82 and 84 were not formed in significant amounts. We next sought to synthesize bis-mesylate 85. This was achieved through an optimized three-step sequence from alcohol 60 in 71% overall yield, involving Swern oxidation, an aldol-Cannizarro reaction, and mesylation (Scheme 14). We reasoned that the carbamate nucleophile should react selectively with the mesylate on the same face. Thus, addition of KOt-Bu to 85 results in displacement of the α-mesylate to form the piperidine ring and ensuing addition of NaOMe displaces the remaining mesylate to form the methoxymethyl group. However, these basic conditions also led to undesired cleavage of the N-carbamoyl group, which was reintroduced by treating the crude mixture with methyl chloroformate. An acidic workup procedure ultimately yields tricycle 86 with the free phenolic group in 26% yield over the three steps (see Table 1 for an optimization of this process and its application to the syntheses of the C20 natural products). Conversion of guaiacol derivative 86 to dienone 87 with hypervalent iodine occurs smoothly in 89% yield and sets the stage for an intramolecular Diels–Alder reaction. Heating to 150 °C effectively transforms 87 into the Diels–Alder cycloadduct, and its ketone carbonyl is reduced stereo-selectively with NaBH4 to afford hexacyclic alcohol 88 in 60% yield over two steps. The ketal hydrolysis and MOM protection occur in 90% yield over two steps. Similar to the protocol adopted for the synthesis of weisaconitine D (2, Scheme 8), the ketone group in 89 is stereoselectively reduced to the corresponding secondary alcohol from the desired β-face, and the resulting alcohol activated for skeletal rearrangement by triflation. Heating the resultant triflate with DBU and DMSO affords allylic alcohol 90, which contains the desired aconitine core, in 62% yield over three steps. A final four-step sequence involving piperidine deprotection, N-acetylation, amide reduction, and MOM cleavage was executed in 57% yield to complete the first synthesis of liljestrandinine (4).
Scheme 13. Alternative Attempts at Installing the C4-Methyl Groupa.
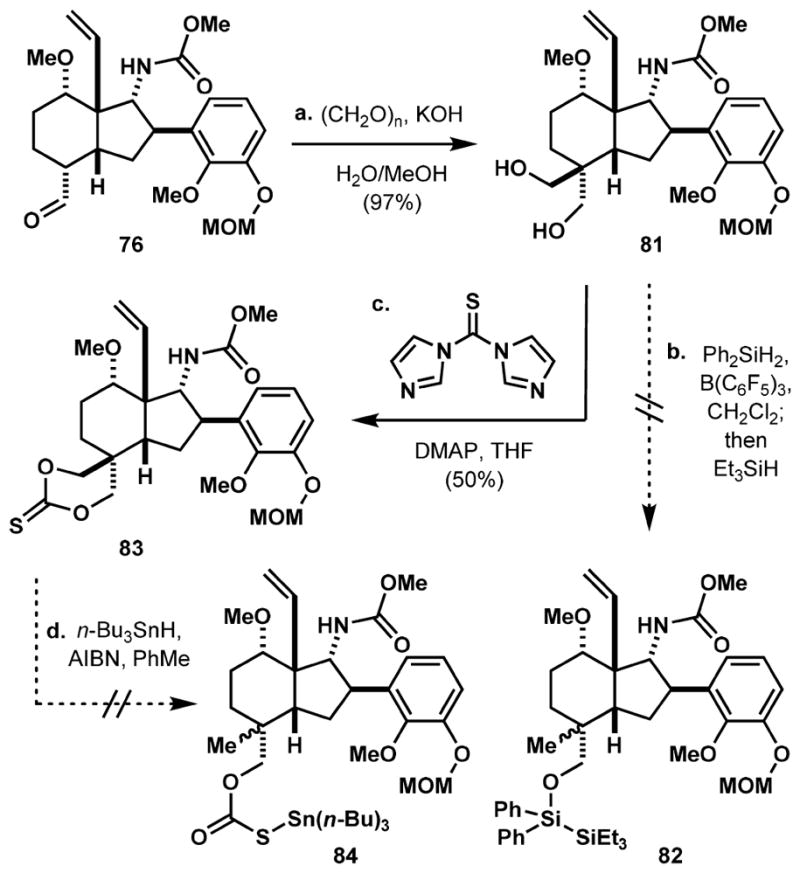
aReaction conditions: (a) (CH2O)n, KOH, H2O/MeOH, 97%; (b) B(C6F5)3 (cat.), Ph2SiH2, CH2Cl2, then Et3SiH; (c) thiocarbonyldii-midazole, DMAP, THF, 23–60 °C, 50%; (d) n-Bu3SnH, AIBN, PhMe. DMAP = 4-(dimethylamino)pyridine AIBN = azobis(isobutyronitrile)
Scheme 14. Synthesis of C19-Diterpenoid Alkaloid Liljestrandininea.

aReaction conditions: (a) DMSO, (COCl)2, Et3N, CH2Cl2, –78 to 23 °C; (b) (CH2O)n, KOH, H2O/MeOH, 23 °C; (c) MsCl, pyridine, 0–23 °C, 71% over three steps; (d) KOt-Bu, THF, 50 °C; (e) 0.5 M NaOMe in MeOH, 120 °C; (f) ClCO2Me, K2CO3, acetone, reflux; then 2 N HCl, i-PrOH, 23 °C, 26% over three steps; (g) PhI(OAc)2, NaHCO3, MeOH, 0 °C, 89%; (h) p-xylene, 150 °C; (i) NaBH4, MeOH, 0–23 °C, 60% over two steps; (j) TFA, H2O/CHCl3, 0 °C, quant.; (k) MOMCl, i-Pr2NEt, THF, 0–23 °C, 90%; (l) NaBH4, MeOH, 0 °C; (m) Tf2O, pyridine, CH2Cl2, –78 to 23 °C; (n) DBU, DMSO, 120 °C, 62% over three steps; (o) KOH(aq), (HOCH2)2, 100 °C; (p) Ac2O, pyridine, CH2Cl2, 0–23 °C; (q) LiAlH4, Et2O, CH2Cl2, 0–40 °C; (r) 12 N HCl(aq), THF, 23 °C, 57% over four steps. TFA = trifluoroacetic acid; DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene.
Table 1.
Optimization of E-Ring Piperidine formation
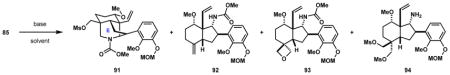
| |||||
|---|---|---|---|---|---|
| entry | conditions | 91 | 92 | 93 | 94 |
| 1 | KOt-Bu (3 equiv), THF, 50 °C | 30% | 0% | 0% | 39% |
| 2 | KH (1.6 equiv), THF, 55–60 °C | 62% | 8% | 8% | trace |
| 3 | KH (1.6 equiv), DMF, rt | 83% | 0% | 0% | 0% |
Synthesis of C20-Diterpenoid Alkaloids
In developing syntheses of weisaconitine D (2) and liljestrandinine (4), we prepared intermediates that possess the denudatine framework and solvolytically rearranged these scaffolds to molecules that have the aconitine core structure. We thus aimed to take advantage of these intermediates to access denudatine-type natural products. Like liljestrandinine, the C20-diterpenoid alkaloids contain a carbon atom at C4, but in the form of a methyl group (C18, see Figure 1). To install this carbon, we surmised that a sequence similar to that used to install the methoxymethyl group of liljestrandinine (Scheme 14, steps a–f) would be most effective. However, the low yield (26%) associated with forming the piperidine ring and transforming the second mesylate needed to be improved in order for this sequence of reactions to be truly useful in synthesizing any of the C20-alkaloids.
The formation of the piperidine E-ring was thus reinvestigated. In contrast to the mesylation and subsequent KOt-Bu-mediated cyclization of alcohol 60 (Scheme 8), which occurs in good yield, the analogous KOt-Bu-mediated cyclization of bis-mesylate 85 occurs in a low 30% yield, with the majority of the mass balance accounted for by free amine 94, which is isolated in 39% yield (Table 1, entry 1). A similar cleavage of carbamates using KOt-Bu in THF has been reported in the literature.63 By switching to KH in THF, the isolated yield for piperidine 91 increases to 62%. Under these conditions, exocyclic alkene 92 and oxetane 93 are each produced in 8% yields, while the decarbamoylation side product 94 is formed in only trace quantities (LC-MS analysis; entry 2). As a consequence of the poor solubility of bis-mesylate 85 in THF, elevated temperatures between 50–60 °C are necessary to facilitate dissolution of the substrate and achieve reactivity. Presumably, the high temperatures are responsible for some of the undesired side reactions that were observed. Performing the same reaction with DMF as the solvent at room temperature results in exclusive formation of desired piperidine 91 in 83% yield (entry 3).
With robust conditions established for installing the piperidine moiety, we next turned our attention to transforming the methylene mesylate group in 91 to a methyl group (Scheme 15). Attempts to reduce 91 with lithium triethylborohydride64 and the CuCl2·2H2O/Li/catalytic 4,4′-di-t-butylbiphenyl system65 failed, returning only unreacted starting material. The lack of reactivity is presumably due to the steric congestion at the neopentyl mesylate site. The desired reduction, however, is accomplished using a combination of NaI and Zn and holding the reacting mixture at reflux in DME. Successive acidic cleavage of the MOM protecting group and oxidative dearomatization generates masked o-benzoquinone 95 (61% yield over three steps), which was then heated to 150 °C in p-xylene to furnish Diels–Alder cycloadduct 96 (80–87% yields). To complete the C20 denudatine core, a final one-carbon unit needed to be delivered to the ketone group of 96. We opted to use the Corey–Chaykovsky reagent,66 which would provide an epoxide that can be conveniently elaborated to the target natural products. Contrary to the stereoselectivity observed for nucleophilic addition with NaBH4 (Scheme 9) which we postulate to be governed by torsional steering and results in nucleophilic approach from the β-face, dimethylsul-foxonium ylide approaches ketone 96 from the opposite, less sterically demanding α-face to yield epoxide 97 as a single, albeit the undesired, diastereomer in 85% yield. To obtain the desired epoxide diastereomer that can be advanced to the C20-targets, 96 was subjected to a Wittig olefination, which after an acidic workup produces enone 98 in 96% yield. We hypothesized that delivery of an oxygen atom can occur from the α-face provided a sufficiently large oxidant is used. As postulated, a Weitz–Scheffer epoxidation67 with trityl hydro-peroxide forges the desired α-epoxide (99) in 57% yield (64% brsm) as the sole diastereomer. To cleave the methyl ether and methyl carbamate, 99 is heated in a mixture of HBr in AcOH under microwave conditions, and then basified with aqueous NaOH.68 Ensuing treatment with Ac2O and pyridine acetylates the free amine and leads to the isolation of elimination product 100 (~5% yield), fragmentation product 101 (28% yield), and the desired demethylated products 102 and 103 (51% combined yield). LC-MS analyses suggest that the 1-acetoxy group of 103 is introduced during the dealkylation step with HBr in AcOH and not during the subsequent acetylation step with Ac2O. Epoxide ring-opening was effected in the presence of KOAc in AcOH, affording α-hydroxyketone 104. This was followed by hydrogenation of the strained alkene using Pd/C and global reduction with LiAlH4 in 63–77% yield over three steps, which accomplished the first synthesis of cochlearenine (6).
We found density functional theory (DFT) to be critical in confirming the structures of the natural products.69 Cochlear-enine (6) has been isolated and characterized twice in the literature, albeit with significant discrepancies between the two sets of data.70,71 The spectroscopic data obtained by us for synthetic 6 are consistent with those reported by Wada et al. apart from one 13C signal, raising the concern that the C15-epimer of the natural product might have formed during the global reduction step (Scheme 15, step l). We thus computed the 1H and 13C NMR data for both cochlearenine (6) and its C15-epimer and found that our experimental data closely agree with the computed data for the natural product with a computed mean absolute deviation (CMAD) of 0.14 ppm for the 1H NMR data and 1.5 ppm for the 13C NMR data (see SI). This analysis also allowed us to discount the possibility of having formed the C15-epimer, which exhibits larger CMADs of 0.21 and 2.9 ppm for the 1H NMR and 13C NMR data, respectively, compared to the synthetic material.
The primary hydroxyl (C17 position) of cochlearenine (6) is veratroylated by exposure to veratroyl chloride and polymer-supported DMAP to give 7 in 25% yield (34% brsm). In this case, the acquired spectroscopic data for the neutral amine bears no resemblance to that reported in the isolation disclosure.72 An attempt to acidify 7 with anhydrous HCl in diethyl ether resulted in a species with a 1H NMR spectrum that exhibits broad signals differing significantly from those reported by Díaz et al. However, titrating 7 with trifluoroacetic acid distinctly converts the neutral species to its protonated state, 7·TFA (see SI), the spectroscopic data of which is fully consistent with the reported data. This observation also supports our assignment of cochlearenine (6).
Finally, paniculamine (8) was prepared as a single diastereomer in 50% yield by treating 6 with aqueous H2O2. Because the 1H NMR data for 8 are only partially reported in the isolation study and the 13C NMR data not reported at all,73 we used DFT to confirm the identity of our synthetic material. The computed 1H and 13C spectra for 8 match closely our experimental data with CMADs of 0.23 and 1.5 ppm, respectively. In contrast, larger discrepancies with CMADs of 0.27 ppm for the 1H NMR data and 2.5 ppm for the 13C NMR data were found for N-epi-8 compared to those of the synthesized compound. Altogether, the results suggest that the synthesized material is consistent with the structure of 8. The natural product is also found to be 9.8 kcal/mol lower in energy than its N-epimer, presumably in large part due to a favorable enthalpic interaction between the oxygen of the N-oxide and the 1-hydroxy group (Figure 2). Thus, the oxidation to form 8 is likely both thermodynamically and kinetically favored, with the 1-hydroxy group directing oxidation from the back face of the piperidine ring.
Figure 2.
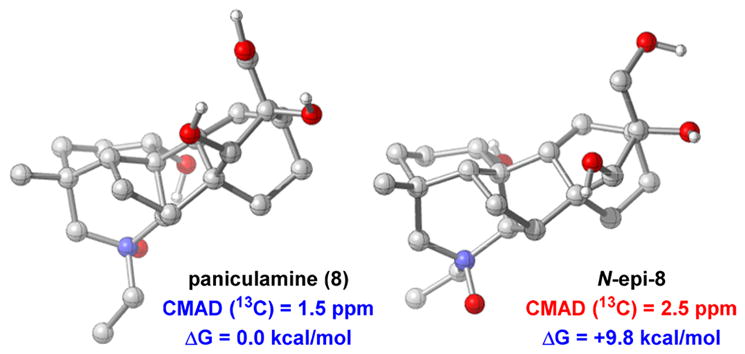
Optimized geometries for paniculamine (8) and its epimer computed at the M06-2X/6-31+G(d,p)/IEFPCM (H2O) level of theory.
Early Installation of an N-Ethyl Substituent
Because all of the natural products that we have targeted bear an N-ethyl substituent, the use of the N-carbamate as a protecting group does not provide the most direct approach since it requires three additional steps (decarbamoylation, acetylation, amide reduction) to transform it into the N-ethyl group. In addition, hydrolysis of carbamates 74 (Scheme 11, step e) and 90 (Scheme 14, step o) is slow and requires harsh conditions (4 M KOH, 100 °C, 60–120 h). We thus explored the possibility of introducing the N-ethyl substituent early on. Instead of submitting primary amide 59 to Hofmann rearrangement conditions in MeOH (Scheme 8), performing the reaction in CH3CN solvent forms the isocyanate intermediate that can be trapped with methyl magnesium bromide to form alcohol 105 (Scheme 16). This material was advanced to masked ortho-benzoquinone 107, the precursor for the intramolecular Diels–Alder cycloaddition, using the same sequence developed for the synthesis of denudatine natural products 6–8.
Scheme 16. Studying the Feasibility of Protecting the E-Ring Piperidine with an Acetyl Groupa.

aReaction conditions: (a) PhI(OAc)2, CH3CN, 0–23 °C; (b) MeMgBr, Et2O, 0 °C; (c) TBAF, THF, 23 °C, 67% over three steps; (d) formalin, KOH(aq), MeOH, 23 °C, 87%; (e) MsCl, pyridine, 0–23 °C, 75%; (f) KOt-Bu, THF, 50 °C, 93%; (g) NaI, Zn0, DME, 105 °C; (h) HCl(aq), THF/i-PrOH, 23 °C; (i) PhI(OAc)2, NaHCO3, 23 °C, MeOH, 67% over three steps; (j) p-xylene, 150 °C, 35% exo-108 + 40% endo-108.
Surprisingly, the Diels–Alder reaction of 107 proceeds to yield a mixture of the undesired exo-108 in 35% yield and the desired endo-108 in 40% yield. This is in stark contrast to dienones 62 (Scheme 8), 87 (Scheme 14), and 95 (Scheme 15), which all bear carbamate protected piperidine structures and undergo [4 + 2] cycloadditions with complete diaster-eocontrol for the desired endo-cycloadducts. A possible rationale for this observation is that the transition state leading to the exo-cycloadduct experiences substantial repulsive interactions between the oxygens of the carbamate and the exo-disposed dimethyl acetal (Figure 3a), an interaction that is absent in the endo transition state (TS). In the case of the N-acetylated substrate (107), its exo-TS, while more sterically encumbered, can orient the oxygen atom of the amide away from the dimethyl acetal and reduce some of the unfavorable repulsive interactions (Figure 3b). The endo-TS experiences less of the unfavorable interactions and is still formed as the major product. Regardless, the poor diastereoselectivity observed in this key transformation precluded the use of the N-acetyl group in the syntheses.
Figure 3.
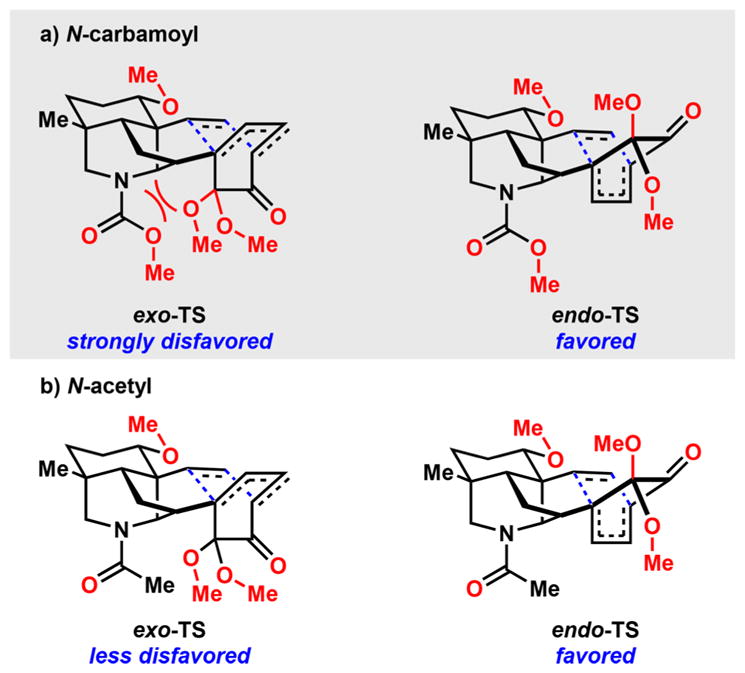
Proposed rationale for selective, intramolecular, Diels–Alder cycloaddition.
Enantioselective Synthesis of endo-Diels–Alder Cy-cloadduct 13
To synthesize weisaconitine D (2), liljestrandi-nine (4), cochlearenine (6), N-ethyl-1α-hydroxy-17-veratroyl-dictyzine (7), and paniculamine (8) in enantioenriched form, an initial enantioselective Diels–Alder cycloaddition to access endo-13 is necessary. We initially sought to directly combine diene 21 and dienophile 22 enantioselectively by employing a cationic ruthenium(II) center coordinated to a chiral, tetradentate PNNP ligand, (1S,2S)-N,N′-bis[o-(diphenylphos-phino)-benzylidene]cyclohexane-1,2-diamine,74,75 or a copper-(II) center ligated to a chiral bisoxazoline. When these complexes were applied as catalysts in our system, significant decomposition of diene 21 and minimal formation of the desired cycloadduct (13) were observed. The small amount of product obtained was of low ee, suggesting inadequate binding of the ketoester component of 22 to the catalyst. Inspired by the use of β-oxo imides in Cu-catalyzed Mukaiyama–Michael additions,76 [4 + 2]-cycloadditions, and Hosomi–Sakurai reactions,77 we hypothesized that incorporating the β-oxo imide motif into our dienophile could enhance binding of the dienophile to a Lewis-acid catalyst (relative to a β-ketoester) and facilitate Diels–Alder reactivity and selectivity. A scalable synthesis of β-oxo imide containing dienophile 112 was thus developed starting from vinyl bromide 109 (Scheme 17a). Lithium-halogen exchange, followed by nucleophilic addition to ethyl isocyanoformate 110 provides ketal 111 in 62% yield. Ketal hydrolysis using TsOH reveals β-oxo imide 112 in 97% yield. Upon subjecting a mixture of diene 21 and dienophile 112 to [Cu-((S,S)-t-Bu-box)](OTf)2 catalysis, enantioselective [4 + 2]-cycloaddition proceeds to furnish endo-cycloadduct 113 in 67% yield (>20:1 dr) and 92% ee. Related copper catalysts modified with different BOX-ligands generally resulted in lower yields and ee (Scheme 17b).
Scheme 17.

Development of an Enantioselective Diels–Alder Reaction
To achieve the synthesis of enantioenriched 13, selective methanolysis of the imide functionality of 113 at the “amide carbonyl” over the “carbamate carbonyl” is necessary. Methanolysis under standard conditions using NaOMe in MeOH resulted in a 2:1 mixture of products in favor of the undesired primary amide (114, Table 2, entry 1). We turned to Lewis acids with the intention of reversing the selectivity. AgOTf did not promote methanolysis even after extended reaction times (entry 2). Oxophilic di- and trivalent Lewis acids were more reactive. Cu(OTf)2, Sn(OTf)2, Eu(OTf)3, and Sm(OTf)3 were all capable of transforming 113 to mixtures of products favoring amide 114 over ester 115 in 2.5:1 to 4.8:1 selectivities (entries 3–6), with Sn(OTf)2 simultaneously cleaving the silyl group. Ga(OTf)3 completely favors the formation of 114 and results in deprotection of the alcohol (entry 7). While Yb(OTf)3 is unreactive (entry 8), Bi(OTf)3 desirably and effectively methanolyses the imide at the more hindered carbonyl to yield ester 115 with high site selectivity (entry 9). We postulate that this mode of reactivity may result from coordination of bismuth to the ketone oxygen and the proximal “amide carbonyl,” effectively activating it for nucleophilic substitution. Because this Lewis acid also cleaves the silyl group, a final step to resilylate the alcohol group was required, producing the desired enantioenriched 13 in 63% yield. This enantioenriched bicycle can be used to access enantioenriched diterpenoid alkaloids.
Table 2.
Exploring Reagents for Selective Imide Methanolysis
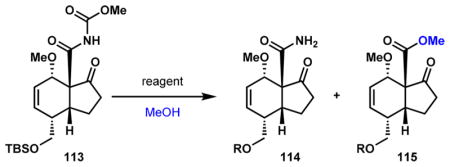
| ||||
|---|---|---|---|---|
| entry | reagent | time (h) | R | 114/115c |
| 1a | NaOMe | 4 | TBS | 2:1 |
| 2b | AgOTf | 48 | TBS | no conv. |
| 3b | Cu(OTf)2 | 2 | TBS | 2.9:1 |
| 4b | Sn(OTf)2 | >48 | H | 2.5:1 |
| 5b | Eu(OTf)3 | 0.5 | TBS | 4.8:1 |
| 6b | Sm(OTf)3 | 0.5 | TBS | 3.8:1 |
| 7b | Ga(OTf)3 | 0.5 | H | only 114 |
| 8b | Yb(OTf)3 | 24 | TBS | no conv. |
| 9b | Bi(OTf)3 | 5.5 | H | 1:10 |
18 equiv NaOMe used, [113] = 0.1 M, 0–23 °C.
10 mol % Lewis acid used, [113] = 0.1 M, 28 °C.
Ratios determined by 1H NMR analysis of the crude reaction mixtures.
CONCLUSION
Guided by network analysis, we accomplished a unified strategy to synthesize natural products belonging to each of the C18, C19, and C20 families of diterpenoid alkaloids. Specifically, aconitine-type molecules weisaconitine D (2) and liljestrandi-nine (4) are synthesized in 30 and 29 steps, respectively, beginning from the initial Diels–Alder reaction between diene 21 and dienophile 22. Denudatine-type cochlearenine (6), N-ethyl-1α-hydroxy-17-veratroyldictyzine (7), and paniculamine (8) are synthesized in 25, 26, and 26 steps, respectively, from hydrindenone 13, which is readily prepared on 30 g scale in a single pass. Advanced intermediate 60 is common to the synthesis of these natural products and is prepared on multigram scale per run. We have also developed a gram-scale enantioselective approach to 13 that should allow for the preparation of enantioenriched natural products. In doing so, we discovered a bismuth-catalyzed methanolysis of imide 113 that exclusively targets the more sterically encumbered carbonyl. Motivated by the effectiveness of this network analysis approach, we have developed a freely accessible web-based program that facilitates quick and accurate application of this concept by anyone in the synthetic community designing retrosyntheses.
Supplementary Material
Acknowledgments
This project is supported by award no. R01 GM084906 from the National Institute of General Medical Sciences. K.G.M.K. and T.P.L. thank NSERC for postdoctoral fellowships. We are grateful to the NSF GRFP for graduate fellowships to C.J.M. and N.A.D. (DGE 1106400). G.M.G. acknowledges the NIH (5F31GM095238) for a graduate fellowship. X-ray crystallography instrumentation is supported by NIH Shared Instrumentation Grant S10-RR027172. The AV-600, AV-500, DRX-500, AVQ-400, and AVB-400 NMR spectrometers are partially supported by NIH grants SRR023679A and 1S10RR016634-01 and NSF grants CHE-9633007 and CHE-0130862. The 900 MHz NMR instrument is funded by NIH grant GM68933. The Molecular Graphics and Computation Facility is funded by NIH S10OD023532. We thank Dr. Kathleen Durkin and Dr. Yinka Olatunji-Ojo for assistance with DFT calculations and Dr. Antonio DiPasquale and Nicholas Settineri for single crystal X-ray diffraction studies.
Footnotes
ORCID
Kevin G. M. Kou: 0000-0002-4676-8846
Richmond Sarpong: 0000-0002-0028-6323
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b07706.
Experimental procedures, characterization data for new compounds and the natural products, density functional theory energies and geometries (PDF)
Crystallographic data for 34 (CIF)
Crystallographic data for 35 (CIF)
Crystallographic data for 36 (CIF)
Crystallographic data for 44 (CIF)
Crystallographic data for 47 (CIF)
Crystallographic data for endo-108 (CIF)
References
- 1.Rahman Au, Choudhary MI. Nat Prod Rep. 1999;16:619. doi: 10.1039/a705715f. [DOI] [PubMed] [Google Scholar]
- 2.Wang FP, Chen QH, Liu XY. Nat Prod Rep. 2010;27:529. doi: 10.1039/b916679c. [DOI] [PubMed] [Google Scholar]
- 3.Wang XW, Xie H. Drugs Future. 1999;24:877. [Google Scholar]
- 4.Sun J, Peng Y, Wu H, Zhang X, Zhong Y, Xiao Y, Zhang F, Qi H, Shang L, Zhu J, Sun Y, Liu K, Liu J, AJ, Ho RJY, Wang G. Drug Metab Dispos. 2015;43:713. doi: 10.1124/dmd.114.060905. [DOI] [PubMed] [Google Scholar]
- 5.Cordell GA, editor. The Alkaloids: Chemistry and Biology. Vol. 67. Academic Press; San Diego: 2009. The C18-Diterpenoid Alkaloids; pp. 1–78. [DOI] [PubMed] [Google Scholar]
- 6.Ameri A. Prog Neurobiol. 1998;56:211. doi: 10.1016/s0301-0082(98)00037-9. [DOI] [PubMed] [Google Scholar]
- 7.Fujita Y, Terui K, Fujita M, Kakizaki A, Sato N, Oikawa K, Aoki H, Takahashi K, Endo S. J Anal Toxicol. 2007;31:132. doi: 10.1093/jat/31.3.132. [DOI] [PubMed] [Google Scholar]
- 8.Stevens M, Peigneur S, Tytgat J. Front Pharmacol. 2011;2:71. doi: 10.3389/fphar.2011.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borcsa B, Fodor L, Csupor D, Forgo P, Molnár A, Hohmann J. Planta Med. 2014;80:231. doi: 10.1055/s-0033-1360278. [DOI] [PubMed] [Google Scholar]
- 10.Wang F-P, Chen Q-H. The C19-Diterpenoid Alkaloids. In: Cordell GA, editor. The Alkaloids: Chemistry and Biology. Vol. 69. Academic Press; San Diego: 2010. pp. 1–577. [DOI] [PubMed] [Google Scholar]
- 11.Wang F-P, Liang X-TC. 20-Diterpenoid Alkaloids. In: Cordell GA, editor. The Alkaloids: Chemistry and Biology. Vol. 59. Academic Press; San Diego: 2002. pp. 1–280. [DOI] [PubMed] [Google Scholar]
- 12.Marth CJ, Gallego GM, Lee JC, Lebold TP, Kulyk S, Kou KGM, Qin J, Lilien R, Sarpong R. Nature. 2015;528:493. doi: 10.1038/nature16440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kou KGM, Li BX, Lee JC, Gallego GM, Lebold TP, DiPasquale AG, Sarpong R. J Am Chem Soc. 2016;138:10830. doi: 10.1021/jacs.6b07268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pelletier SW, Parthasarathy PC. Tetrahedron Lett. 1963;4:205. [Google Scholar]
- 15.For early formal syntheses of atisine, see: Nagata W, Sugasawa T, Narisada M, Wakabayashi T, Hayase Y. J Am Chem Soc. 1963;85:2342.Nagata W, Sugasawa T, Narisada M, Wakabayashi T, Hayase Y. J Am Chem Soc. 1967;89:1483.Masamune S. J Am Chem Soc. 1964;86:291.Guthrie RW, Valenta Z, Wiesner K. Tetrahedron Lett. 1966;7:4645. doi: 10.1016/s0040-4039(01)89239-x.
- 16.Masamune S. J Am Chem Soc. 1964;86:290. [Google Scholar]
- 17.(a) Nagata W, Narisada M, Wakabayashi T, Sugasawa T. J Am Chem Soc. 1964;86:929. [Google Scholar]; (b) Nagata W, Narisada M, Wakabayashi T, Sugasawa T. J Am Chem Soc. 1967;89:1499. [Google Scholar]
- 18.(a) Wiesner K, Uyeo S, Philipp A, Valenta Z. Tetrahedron Lett. 1968;9:6279. doi: 10.1016/s0040-4039(00)75452-9. [DOI] [PubMed] [Google Scholar]; (b) Wiesner K, Komlossy ZI, Philipp A, Valenta Z. Experientia. 1970;26:471. doi: 10.1007/BF01898446. [DOI] [PubMed] [Google Scholar]
- 19.For a review on the total syntheses of diterpenoid alkaloids, see: Zhu G, Liu R, Liu B. Synthesis. 2015;47:2691.
- 20.For a review on the use of the oxidative dearomatization/Diels–Alder cycloaddition sequence for the synthesis of diterpenes and diterpenoid alkaloids, see : Liu X-Y, Qin Y. Nat Prod Rep. 2017 doi: 10.1039/C7NP00033B. Advance Article.
- 21.Cherney EC, Lopchuk JM, Green JC, Baran PS. J Am Chem Soc. 2014;136:12592. doi: 10.1021/ja507321j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng H, Zeng FH, Yang X, Meng YJ, Xu L, Wang FP. Angew Chem, Int Ed. 2016;55:392. doi: 10.1002/anie.201508996. [DOI] [PubMed] [Google Scholar]
- 23.Hamlin AM, Cortez F, de J, Lapointe D, Sarpong R. Angew Chem, Int Ed. 2013;52:4854. doi: 10.1002/anie.201209030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamlin AM, Lapointe D, Owens K, Sarpong R. J Org Chem. 2014;79:6783. doi: 10.1021/jo501214b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishiyama Y, Han-ya Y, Yokoshima S, Fukuyama T. J Am Chem Soc. 2014;136:6598. doi: 10.1021/ja503023h. [DOI] [PubMed] [Google Scholar]
- 26.Nishiyama Y, Yokoshima S, Fukuyama T. Org Lett. 2016;18:2359. doi: 10.1021/acs.orglett.6b00789. [DOI] [PubMed] [Google Scholar]
- 27.Corey EJ, Howe WJ, Orf HW, Pensak DA, Petersson G. J Am Chem Soc. 1975;97:6116. [Google Scholar]
- 28.For a book chapter on connectivity analysis, see: Wender PA, Miller BL. Toward the Ideal Synthesis: Connectivity Analysis and Multi-Bond Forming Processes. In: Hudlický T, editor. Organic Synthesis: Theory and Applications. Vol. 2. JAI Press Inc; Greenwich, CT: 1993. pp. 27–66.
- 29.Corey EJ, Cheng X-M. The Logic of Chemical Synthesis. Wiley; Hoboken, NJ: 1989. pp. 43–44. [Google Scholar]
- 30.Hoffmann RW. Elements of Synthesis Planning. Springer-Verlag; Berlin: 2009. [Google Scholar]
- 31.See SI for an example with weisaconitine D (2).
- 32.Steinbeck C, Han Y, Kuhn S, Horlacher O, Luttmann E, Willighagen EL. J Chem Inf Comput Sci. 2003;43:493. doi: 10.1021/ci025584y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steinbeck C, Hoppe C, Kuhn S, Floris M, Guha R, Willighagen EL. Curr Pharm Des. 2006;12:2111. doi: 10.2174/138161206777585274. [DOI] [PubMed] [Google Scholar]
- 34.The web-based graphing program that we developed is available at http://www.cadrerl.com/maxbridge.
- 35.Streit U, Bochet CG. Beilstein J Org Chem. 2011;7:525. doi: 10.3762/bjoc.7.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Avent AG, Byrne PW, Penkett CS. Org Lett. 1999;1:2073. [Google Scholar]
- 37.Wang Q, Chen C. Org Lett. 2008;10:1223. doi: 10.1021/ol800111j. [DOI] [PubMed] [Google Scholar]
- 38.Adams H, Anderson JC, Bell R, Jones DN, Peel MR, Tomkinson NC. J Chem Soc, Perkin Trans. 1;1998:3967. [Google Scholar]
- 39.Prabhakaran J, Lhermitte H, Das J, Sasi-Kumar TK, Grierson DS. Synlett. 2000:658. [Google Scholar]
- 40.Lebold TP, Gallego GM, Marth CJ, Sarpong R. Org Lett. 2012;14:2110. doi: 10.1021/ol300647k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bandyopadhyaya AK, Manion BD, Benz A, Taylor A, Rath NP, Evers AS, Zorumski CF, Mennerick S, Covey DF. Bioorg Med Chem Lett. 2010;20:6680. doi: 10.1016/j.bmcl.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takaya Y, Ogasawara M, Hayashi T. Tetrahedron Lett. 1999;40:6957. [Google Scholar]
- 43.Lee J, Kim M, Chang S, Lee HY. Org Lett. 2009;11:5598. doi: 10.1021/ol902309z. [DOI] [PubMed] [Google Scholar]
- 44.Nuss JM, Chinn JP, Murphy MM. J Am Chem Soc. 1995;117:6801. [Google Scholar]
- 45.Wenkert E. Chem Ind (London) 1955:282. [Google Scholar]
- 46.Coates RM, Bertram EF. J Org Chem. 1971;36:3722. [Google Scholar]
- 47.Adams MR, Bu’Lock JD. J Chem Soc, Chem Commun. 1975:389. [Google Scholar]
- 48.Lupi A, Patamia M, Grgurina I, Bettolo RM, Leo OD, Gioia P, Antonaroli S. Helv Chim Acta. 1984;67:2261. [Google Scholar]
- 49.Doering WvE, Farber M. J Am Chem Soc. 1949;71:1514. [Google Scholar]
- 50.For a review on the synthesis of bicyclo[3.2.1]octanes, see: Filippini M-H, Rodriguez J. Chem Rev. 1999;99:27. doi: 10.1021/cr970029u.
- 51.Wiesner K, Tsai TYR, Huber K, Bolton SE. J Am Chem Soc. 1974;96:4990. [Google Scholar]
- 52.Wiesner K, Tsai TYR, Nambiar KP. Can J Chem. 1978;56:1451. [Google Scholar]
- 53.Cheng H, Xu L, Chen DL, Chen QH, Wang FP. Tetrahedron. 2012;68:1171. [Google Scholar]
- 54.DFT calculations suggest that the observed DMSO-adduct is 5.0–6.4 kcal/mmol lower in energy than the alternative DMSO-adducts formed via an SN2′ addition onto the carbocation intermediate. See SI for details.
- 55.(a) Fernández I, Cossío FP, Sierra MA. Chem Rev. 2009;109:6687. doi: 10.1021/cr900209c. [DOI] [PubMed] [Google Scholar]; (b) Hugelshofer CL, Magauer T. Nat Prod Rep. 2017;34:228. doi: 10.1039/c7np00005g. [DOI] [PubMed] [Google Scholar]; (c) Santalla H, Faza ON, Gómez G, Fall Y, López CS. Org Lett. 2017;19:3648. doi: 10.1021/acs.orglett.7b01621. [DOI] [PubMed] [Google Scholar]
- 56.Zhao DK, Ai HL, Zi SH, Zhang LM, Yang SC, Guo HC, Shen Y, Chen YP, Chen JJ. Fitoterapia. 2013;91:280. doi: 10.1016/j.fitote.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 57.Wang FP, Chen QH, Liu XY. Nat Prod Rep. 2010;27:529. doi: 10.1039/b916679c. [DOI] [PubMed] [Google Scholar]
- 58.Cuerva JM, Campaña AG, Justicia J, Rosales A, Oller-Löpez JL, Robles R, Cárdenas DJ, Buñuel E, Oltra JE. Angew Chem, Int Ed. 2006;45:5522. doi: 10.1002/anie.200600831. [DOI] [PubMed] [Google Scholar]
- 59.Xie GB, Chen QH, Chen DL, Jian XX, Wang FP. Heterocycles. 2003;60:631. [Google Scholar]
- 60.A related, two-step, aldol-NaBH(OAc)3 reduction sequence was used to install a hydroxymethyl group at the C4 position in a study toward acochlearine: Fujioka K, Miyamoto N, Toya H, Okano K, Tokuyama H. Synlett. 2016;27:621.
- 61.Drosos N, Morandi B. Angew Chem, Int Ed. 2015;54:8814. doi: 10.1002/anie.201503172. [DOI] [PubMed] [Google Scholar]
- 62.Barton DHR, Subramanian R. J Chem Soc, Perkin Trans. 1;1977:1718. [PubMed] [Google Scholar]
- 63.Tom NJ, Simon WM, Frost HN, Ewing M. Tetrahedron Lett. 2004;45:905. [Google Scholar]
- 64.Holder RW, Matturro MG. J Org Chem. 1977;42:2166. [Google Scholar]
- 65.Radivoy G, Alonso F, Moglie Y, Vitale C, Yus M. Tetrahedron. 2005;61:3859. [Google Scholar]
- 66.Corey EJ, Chaykovsky M. J Am Chem Soc. 1965;87:1353. [Google Scholar]
- 67.Weitz E, Scheffer A. Ber Dtsch Chem Ges B. 1921;54:2327. [Google Scholar]
- 68.Tang P, Chen QF, Wang L, Chen QH, Jian XX, Wang FP. Tetrahedron. 2012;68:5668. [Google Scholar]
- 69.Willoughby PH, Jansma MJ, Hoye TR. Nat Protoc. 2014;9:643. doi: 10.1038/nprot.2014.042. [DOI] [PubMed] [Google Scholar]
- 70.Kolak U, Öztürk M, Özgökçe F, Ulubelen A. Phytochemistry. 2006;67:2170. doi: 10.1016/j.phytochem.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 71.Wada K, Kawahara N. Helv Chim Acta. 2009;92:629. [Google Scholar]
- 72.Díaz JG, Ruiza JG, Herz W. Phytochemistry. 2005;66:837. doi: 10.1016/j.phytochem.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 73.Yusupova IM, Bessonova IA, Tashkodzhaev B. Chem Nat Compd. 1995;31:228. [Google Scholar]
- 74.Schotes C, Mezzetti A. J Am Chem Soc. 2010;132:3652. doi: 10.1021/ja910039e. [DOI] [PubMed] [Google Scholar]
- 75.Schotes C, Althaus M, Aardoom R, Mezzetti A. J Am Chem Soc. 2012;134:1331. doi: 10.1021/ja210372u. [DOI] [PubMed] [Google Scholar]
- 76.Oyama H, Orimoto K, Niwa T, Nakada M. Tetrahedron: Asymmetry. 2015;26:262. [Google Scholar]
- 77.Orimoto K, Oyama H, Namera Y, Niwa T, Nakada M. Org Lett. 2013;15:768. doi: 10.1021/ol303381c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.