

Abstract
Duramycin is a heavily post-translationally modified peptide that binds phosphatidylethanolamine. It has been investigated as an antibiotic, inhibitor of viral entry, therapeutic for cystic fibrosis, and tumor and vasculature imaging agent. Duramycin contains a β-hydroxylated Asp (Hya) and four macrocycles, including an essential lysinoalanine (Lal) crosslink. The mechanism of Lal formation is not known. We here show that Lal is installed stereospecifically by DurN via addition of Lys19 to a dehydroalanine. The structure of DurN reveals an unusual dimer with a new fold. Surprisingly, in the structure of duramycin bound to DurN, no residues of the enzyme are near the Lal crosslink. Instead, Hya15 of the substrate makes interactions with Lal, suggesting it acts as a base to deprotonate Lys19 during catalysis. Biochemical data suggest that DurN preorganizes the reactive conformation of the substrate, such that the Hya15 of the substrate can serve as the catalytic base for Lal formation.
Graphical Abstract
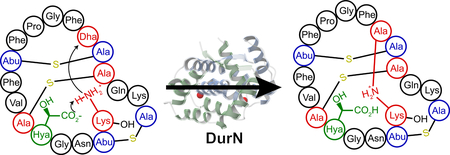
Duramycin and closely related compounds are ribosomally synthesized and post-translationally modified peptides (RiPPs) produced by various actinomycetes1–5 and marine symbionts6. Duramycin binds phosphatidylethanolamine (PE), a major structural phospholipid in mammalian and microbial cell membranes7, 8. Duramycin contains 19 amino acids and five posttranslational modifications (PTMs): one lanthionine (Lan), two methyllanthionines (MeLan), an erythro-3-hydroxy-L-aspartic acid (Hya) resulting from the hydroxylation of Asp15, and one (2S,9S)-Lal (Fig. 1a). The carboxylate and hydroxyl groups of Hya15 make key interactions with the ethanolamine head group of PE, providing nanomolar affinity and high specificity8, 9. As such, duramycin has been investigated as an antibiotic1–3, inhibitor of viral entry10, therapeutic for cystic fibrosis11, and tumor and vasculature imaging agent12, 13. Recently, divamide A, a close homolog of duramycin discovered from the symbiotic microbiome of small marine animals, was reported to display potent anti-HIV activity6.
Figure 1: Duramycin biosynthesis.

a, Co-expression of a minimal biosynthetic gene cassette and in vitro proteolytic removal of the LP from peptide 2 produces duramycin. b, Peptide 3 is converted to 4 by DurN in vitro (blue). Formation of the Lal is monitored by reaction with DTT, which provides an adduct (5) if Lal is not generated (yellow). The characteristic y1 fragment ion is marked with an orange line; c, Compound 3 was reacted with MBP-DurN (top) or MBP (bottom), treated with DTT, and analyzed by LC-MS (Supplementary Fig. 2). Expected and observed masses are listed in Supplementary Table 2. Experiment was performed independently three times with similar results. d, LC-MS/MS analysis of the products in c. Because of the extensive crosslinking, the only fragments observed in addition to the y1 ion, are a likely a2 ion at m/z 191, and a phenyl fragment at m/z 120.
Previous studies have identified the biosynthetic genes (dur) encoding the enzymes that introduce these PTMs in Streptomyces cinnamoneus ATCC12686 (ref.14). The precursor peptide DurA is synthesized with an N-terminal leader peptide (LP), which is removed after maturation of a C-terminal core peptide (CP) (Fig. 1a). Serine and threonine residues in the CP region of DurA are dehydrated by DurM to form dehydroalanine (Dha) and dehydrobutyrine (Dhb) residues. Subsequently, DurM catalyzes addition of cysteine thiols onto the β-carbon of Dha or Dhb to produce Lan or MeLan, respectively (Fig. 1a). The final two PTMs are hydroxylation of Asp15 by DurX to generate Hya15 and formation of (2S,9S)-Lal, putatively by DurN-catalyzed addition of Lys19 to Dha6 (Fig. 1a)14. A previous study demonstrated that duramycin lacking Lal does not have antimicrobial activity14, indicating that the Lal linkage is critical to set up the PE binding pocket. DurN and its orthologs have no sequence homology with any characterized proteins, their mechanism of Lal formation is not known, and previous attempts to reconstitute activity of orthologs in vitro were unsuccessful15, 16. Instead, prior studies have resorted to non-enzymatic base-catalyzed formation of Lal6, 15.
In this study, we demonstrate DurN activity in vitro and present the co-crystal structures of the protein with a substrate analog and product. We compared Lal formation under enzymatic and non-enzymatic conditions, indicating that only DurN catalyzes stereospecific and diastereoselective Lal formation. Our data suggest that DurN promotes Lal formation via an unusual substrate-assisted mechanism.
RESULTS
In vitro DurN activity
In previous studies, the DurN orthologs CinN and MatN were expressed as His6-tagged proteins and were devoid of enzymatic activity. In this work, DurN was expressed in Escherichia coli as an N-terminal fusion with maltose binding protein (MBP-DurN; Supplementary Fig. 1). To prepare its putative substrate, the precursor peptide DurA was converted in E. coli to an intermediate 1 (Fig. 1a) containing Lan, MeLan, and Hya residues, but lacking Lal by co-expression with DurM and DurX14. We also generated the desired product 2 by coexpression of DurA with DurM, DurX, and DurN in E. coli. To test the requirement of the leader peptide, the majority of the leader peptide was removed from purified peptide 1 by endoproteinase Glu-C, yielding compound 3 with three N-terminal residues (AlaPheAla) originating from the leader peptide (Fig. 1b). Similarly, Glu-C treatment of peptide 2 resulted in compound 4 containing all the PTMs found in duramycin (Fig. 1b)14. Because addition of Lys19 to Dha6 to form the desired Lal does not lead to a change in mass nor a change in retention time, we adopted an indirect assay to follow Lal formation. Peptides 1 and 3 contain an electrophilic Dha, whereas peptides 2 and 4 do not. Hence, dithiothreitol (DTT) was used to monitor unreacted peptide by liquid chromatography-mass spectrometry (LC-MS) (Supplementary Fig. 2).
We first reacted 1 with MBP-DurN in vitro followed by DTT treatment; a DTT adduct was not observed, suggesting that DurN performed the desired activity. We then reacted the truncated peptide 3 with MBP-DurN and again the product was not reactive towards DTT, whereas adduct 5 was formed nearly quantitatively in control assays (Fig. 1c). The products were also analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS), demonstrating that the y1 ion is observed for 3 and 5, but not for the product of the MBP-DurN reaction because of the formation of the Lal ring (Fig. 1d; Supplementary Fig. 2b). Finally, as discussed in more detail in a later section, we hydrolyzed the product peptide of the MBP-DurN reaction and observed the presence of Lal by gas chromatography coupled to mass spectrometry (GC-MS). Thus, MBP-DurN catalyzes in vitro Lal formation in peptide 3 to generate product 4, thereby also demonstrating that the leader peptide is not required for its activity.
The structure of DurN with a substrate analog
We removed the MBP tag from MBP-DurN (see Online Methods) and obtained crystal structures of DurN in complex with a substrate analog and in complex with duramycin. We generated an unreactive variant of 3 in which Dha6 (originating from Ser6 in DurA) was mutated to Ala (6, Supplementary Fig. 3a). The structure of DurN with 6 was solved to 1.90 Å resolution by single wavelength anomalous dispersion (SAD) methods using an iodide-soaked crystal. DurN also crystallized in the apo form and the structure was solved to 2.15 Å by molecular replacement using the SAD-derived coordinates (Fig. 2a). The overall structure of DurN consists of an interlaced homodimer where α-helix 1 of each monomer is engaged with residues from α-helices 2–6 in the adjacent monomer. The α-helices are followed by a single β-strand that forms an intermolecular, antiparallel interaction with its neighboring β-strand (Fig. 2a,b). A query against the PDB17 using the DALI server18 shows that DurN does not confidently resemble any known domain, indicating that DurN contains a new protein fold and dimerization mode.
Figure 2: Structural analysis of DurN.

a, Apo-DurN with the two monomers represented as green and blue ribbons. Two potassium ions are depicted as red spheres. b, Secondary structure mapped onto the DurN homodimer. For clarity, only the green monomer is labeled. c, DurN bound to 6 (black sticks). A difference Fourier map (Fo – Fc) contoured at 2.5σ is superimposed and shows electron density for portions of the ligand including one Lan, one MeLan, and Hya15. Critical catalytic DurN residues are represented as green and blue sticks. d, DurN bound to duramycin (orange sticks) with a difference Fourier map (Fo – Fc) contoured at 2.5σ revealing occupancy for nearly the entire ligand. e, Two representations of the DurN homodimer featuring two molecules of duramycin related by a 90° rotational translation about the axis depicted by the black arrow. f, Superimposition of 6 and duramycin-bound structures of DurN. Phe7 in DurA is not modeled in the ligand structures (c, d, and f) because of insufficient electron density. g, Close-up view of the DurN–Hya15 interactions (interatomic distances are calculated averages between two ligand binding sites comprising chains C and G in PDB 6C0Y). For residue numbering, see Figure 1a.
The homodimer contains two structural ions that are related by the pseudo two-fold symmetry axis of the DurN homodimer. Our data are consistent with potassium as the bound metal ions (Supplementary Figs. 4 and 5). DurN coordinates potassium at both sites via an octahedral array of oxygen ligands derived from amides in the backbone (Supplementary Fig. 4). In addition to potassium, we also considered the possibility of magnesium ions bound at the ion binding sites and carried out equivalent rounds of refinement in REFMAC519 after modeling either potassium or magnesium ions independently. Whereas no positive signal was observed in the difference Fourier map (Fo – Fc) at 3σ for the corresponding potassium-bound model, a positive difference signal was observed for the magnesium-bound model (Supplementary Fig. 4c). Additional support for structural potassium ions was gathered by performing differential scanning fluorimetry (DSF), whereby DurN melting curves were obtained in the presence of various potassium or magnesium ion concentrations. While potassium ions conferred an increase in thermal stability of DurN as a function of ion concentration, magnesium ions had no effect on DurN thermal stability (Supplementary Fig. 4a,b). Molecular dynamics (MD) simulations also favor a structural potassium ion over magnesium (Supplementary Fig. 5), and we needed potassium in our assays for enzymatic activity and to prevent precipitation.
Analysis of simulated annealing difference Fourier maps (Fo – Fc) of the apo-structure and the structure with the bound substrate analog revealed electron density for 6 from Gln3 to Gly16 (Fig. 2c), with the reactive Lys19 invisible due to disorder. Only one of the two symmetric binding sites of the homodimer is occupied by ligand because of occlusion of the other site by residues from the N terminus of a different protomer. Recognition of the ligand is mediated by residues from α-helix 1 of one polypeptide and α-helices 4–6 of the second monomer. Surprisingly, the ligand bound to the periphery of DurN and Ala6 (corresponding to the Dha6 involved in Lal formation) is far removed from any residues of the enzyme (Fig. 2c).
The structure of DurN with duramycin
The observed binding pose of 6 does not suggest an obvious mechanism of catalysis. We therefore determined the 1.66 Å resolution structure of DurN bound to duramycin. This structure revealed additional electron density in both binding sites of the DurN dimer, enabling modeling of the entire duramycin molecule, including the Lal linkage (Fig. 2d,e). Globally, the two ligand-bound structures are highly similar with an RMSD of 0.45 Å over 1,400 atoms (including ligands) (Fig. 2f). The duramycin-bound structure reaffirms that DurN does not provide any residues that could catalyze the Michael-type addition and suggests that DurN may mediate Lal formation by serving as a molecular scaffold to bring Dha6 and Lys19 in close proximity. Notably, a bidentate hydrogen-bond is observed between the guanidine of Arg17, the Hya15 hydroxyl group, and an amide backbone oxygen of Cys14 of duramycin (Fig. 2g). The Gln20 and Gln89 side chains of DurN further extend the hydrogen-bond network, and an electrostatic interaction is also observed between the carboxylate of Hya15 and Lys66 (Fig. 2g). These interactions position one of the carboxylate oxygens of Hya15 at 3.3 Å from the Lal secondary amine nitrogen. Thus, it appears that DurN holds the Hya15 carboxylate of the substrate in a conformation that is poised to activate Lys19 (Fig. 3). The other Hya15 carboxylate oxygen is 4.1 Å from the α-carbon of Ala6. This suggests that it would shield the Si face of the reactive Dha6, thus promoting protonation by solvent of the enolate formed upon the addition of Lys19 from the more accessible Re face, resulting in the correct (2S,9S)-Lal diastereomer (Fig. 3). The structures do not provide any evidence for activation of the carbonyl oxygen of Dha6. In fact, the Ψ angle in Ala6 in duramycin is close to zero, very likely due to electrostatic repulsion between the carboxylate of Hya15 and the amide carbonyl of Ala6, which is directed towards the solvent. This strongly suggests that the reacting Dha6 is fixed in an s-cis conformation prior to cyclization, which has been found to be the more reactive form in other Michael-type additions occurring in lanthipeptide biosynthesis20.
Figure 3: Proposed substrate-assisted Lal formation catalyzed by DurN.

Features in blue and green correspond to residues coming from the two monomers of DurN (see Fig. 2) and those in black correspond to its substrate/product.
Substrate-assisted Lal formation
The conformation of duramycin in the co-crystal structure is more constrained than the solution structure of its close analog cinnamycin bound to lysophosphatidylethanolamine (PDB 2DDE)9. Indeed, molecular dynamics simulations demonstrate that the conformation of duramycin relaxes upon removal of DurN (Supplementary Fig. 6). Notably, in the absence of DurN, Hya15 moves away from the Si face of Dha6. Given the apparent importance of the hydroxy group of Hya15 for positioning its carboxylate, we also performed MD calculations on the complex in which this functionality was removed (i.e. Hya15 replaced by Asp). In the absence of the β-hydroxy group, the carboxylate reorients to interact with Arg17 and Lys66, and thus the MD simulations predict it would no longer be available to act as a base (Fig. 4).
Figure 4. MD simulations indicating conformational changes of the Hya15Asp mutant compared to duramycin.

The complex between DurN and duramycin was investigated through 1.0 μs MD simulations. Duramycin backbone and sidechains are shown as dark and light blue lines, respectively. Lysinoalanine (Lal), Hya/Asp15, Arg17 and Lys66 are shown as red, blue, yellow and grey sticks, respectively. DurN is shown as grey (chain A) and brown (chain B) transparent cartoons. Nonpolar hydrogens have been omitted for clarity. a, The hydrogen bond network between Arg17, Lys66 (both located in the enzyme DurN), Hya15 and Ala6 (both located in the substrate) observed in the crystallographic structures is maintained throughout the trajectory, as reflected by the conserved χ dihedral angle of Hya15 (carboxylate side chain) of ca. +60°. b, However, removing the β-hydroxy group in the Hya15Asp mutant disrupts this hydrogen bond network and compromises the integrity of the active site; the carboxylate side chain undergoes a 120° conformational twist and engages in electrostatic interactions with Arg17 and Lys66, trapping the catalytic base in a non-catalytic conformation as revealed by its χ dihedral angle. This computational observation rationalizes the lack of catalytic activity of DurN with 9, the Hya15Asp mutant of 3. c,d, Conformational ensembles for each complex show five superimposed representative snapshots derived from the trajectories and reveal a more rigid complex when native duramycin is bound to DurN (c), unlike the more flexible arrangement with the Hya15Asp mutant (d).
To probe their roles, MBP-DurN variants of conserved residues (Supplementary Fig. 7) were constructed and their activities determined with substrate 3. Ala substitutions at positions involved in substrate binding (Arg17, Gln20, Gln89, and Lys66) resulted in inactive proteins, and the same outcome was observed for Arg18, Trp68, and Glu106 that are at the dimerization interface (Supplementary Figs. 8 and 9). To investigate the effect on affinity upon mutation of substrate binding residues, fluorescently labeled duramycin (Dur-FL, 7; Supplementary Fig. 3) was used for fluorescence polarization (FP) studies. Dur-FL bound to MBP-DurN with a Kd of 417 ± 6 nM (Supplementary Fig. 10a). Unlabeled duramycin and substrate 3 were then used in competition FP experiments providing inhibition constants (Ki) of 2.72 ± 0.01 μM and 16.8 ± 0.5 μM, respectively (Supplementary Fig. 10b). MBP-tagged DurN variants R17A, Q20A, and Q89A did not bind Dur-FL, R17K bound very weakly, and K66A provided a Ki of 7.43 ± 0.08 μM (Supplementary Fig. 10b). Collectively, the mutational data suggest that the binding modes observed in the co-crystal structures are functionally important because variants of residues that engage the substrate in the crystal are compromised with regards to both catalysis and substrate binding9.
We also tested variants of substrate 3. To maintain the hydroxyl group of Hya but not its carboxylate, a variant of 3 containing Ser in place of Hya15 (8, Supplementary Fig. 3c) was prepared in similar fashion as 3 except for using DurA-D15S as a precursor peptide. 8 was a poor substrate for MBP-DurN (Supplementary Fig. 11), even though competition FP experiments show that this variant peptide inhibits Dur-FL binding with a Ki of 10.2 ± 0.3 μM (Supplementary Fig. 10b). Another variant of 3, with Asp in place of Hya15 (9), was prepared by removing DurX from the E. coli co-expression system (Supplementary Fig. 3f). This variant was not processed by MBP-DurN in vitro (Supplementary Fig. 11), in agreement with the predictions of the MD simulations described above. These data suggest that both functional groups on the Hya15 side chain (i.e. β-hydroxyl and γ-carboxylate) must be present for substrate to be processed by DurN.
Stereochemistry of enzymatic and non-enzymatic reactions
The observation that 9 was not a substrate for DurN in vitro was surprising because several studies have shown that deoxyduramycin/deoxycinnamycin can be obtained by co-expression of DurA/CinA with DurM/CinM and DurN/CinN21, 22. Thus, it appears that in bacteria, the absence of the hydroxy group on Asp15 does not prevent Lal formation. To further investigate this apparent contradiction, we obtained deoxyduramycin (10) by co-expressing DurA with DurM and DurN in E. coli (Supplementary Fig. 3g). The product was treated with Glu-C to remove most of the leader peptide, and the three residues remaining from the leader peptide were removed using aminopeptidase. To probe whether duramycin and deoxyduramycin contained Lal, each peptide was hydrolyzed under acidic conditions to their constituent amino acids, which were derivatized as described previously (see Methods)15. GC-MS analysis indicated that authentic duramycin isolated from the producer organism contained predominantly the natural ll-Lal isomer (Fig. 5a), with a small amount of epimer that is likely formed during acid hydrolysis6, 15. However, deoxyduramycin formed in E. coli contained about equal amounts of two diastereomers. Peptide 3 was then exposed to alkaline conditions that were previously used to form Lal non-enzymatically but for which the stereochemistry was not determined15. After removal of the leader peptide from the product peptide (11), hydrolysis and analysis by GC-MS, two diastereomers of Lal were detected in roughly equal amounts, which was also observed when using a commercial mixture of (2R,9S)-Lal and (2S,9S)-Lal standards (Fig. 5a). In contrast, product 4 obtained in vitro from DurN-catalyzed Lal formation again consisted predominantly of the (2S,9S)-Lal isomer. Collectively, these findings therefore suggest that Lal formation in E. coli when DurN is absent is non-stereoselective and may be non-enzymatic.
Figure 5: GC-MS analysis of derivatized lysinoalanine (Lal) and antimicrobial assay of duramycin and its analogs.

a, commercial Lal standard that is a mixture of diastereomers (top trace), Lal obtained by hydrolysis of authentic duramycin (second trace), Lal from enzymatically formed 4 (third trace), Lal from non-enzymatically formed 11 (fourth trace), and Lal from hydrolyzed deoxyduramycin made in E. coli (bottom trace). b, Agar diffusion growth inhibition assay for duramycin generated by DurN in vitro (I), chemically generated 11 (II), and duramycin from the native producer (III). Concentrations are in μM, and the indicator strain is Bacillus subtilis ATCC 6633. Ctrl, negative control containing buffer and the proteases used to remove the leader peptide. Experiment was performed once.
To explore the ramifications of the stereochemical information, we determined the antimicrobial activity of authentic duramycin, DurN-generated duramycin, and chemically generated duramycin by an agar diffusion assay against Bacillus subtilis ATCC 6633. These compounds would all contain the Hya15 and would only differ in the Lal. As expected based on the stereochemical analysis (Fig. 5a), the product of DurN catalysis showed a similar zone of growth inhibition as authentic duramycin, whereas non-enzymatically produced 11 showed a zone of inhibition roughly half the size (Fig. 5b).
Discussion
In this study, we provide the first in vitro demonstration that DurN catalyzes Lal formation, a step that had been recalcitrant in previous studies6, 15. By comparing enzymatic and non-enzymatic Lal generation, our data suggest that DurN greatly facilitates the reaction and controls its stereoselectivity. Furthermore, our data shows that the enzyme does not require the leader peptide for catalysis. DurN has a unique fold and dimeric architecture, and in co-crystal structures, the Lal is entirely solvent exposed and the Hya15 of the substrate is positioned to potentially act as a base during catalysis to deprotonate Lys19 to initiate the Michael addition. Additionally, Hya15 appears to shield the Si face of the enolate intermediate, such that protonation will only occur from the Re face, resulting in the correct (2S,9S)-Lal product. Mutational studies show that substrates that lack either the hydroxyl or carboxylate groups of Hya15 are not processed by DurN, despite maintaining binding affinity, suggesting both of these groups on the substrate are indispensable for catalysis. Merging the biochemical findings with structural and computational insights points to a mechanism whereby the substrate employs its own Hya15 as a catalytic base to facilitate a Michael addition and stereospecific enolate protonation, resulting in Lal formation between Lys19 and Dha6.
The biosynthetic gene clusters of lysinoalanine-containing natural products such as duramycin and cinnamycin are very similar (Supplementary Fig. 12a) and all encode a DurN-like protein. This relatively small protein family is comprised of similar proteins as shown by a tight sequence similarity network (SSN) of members identified by a BLAST search with DurN as query (Supplementary Fig. 12b)23. All identified homologs belong to duramycin-like lanthipeptide biosynthesis clusters that contain a type II lanthionine synthetase-like protein, a duramycin-like LanA protein, and a DurX-like protein. The natural products for some of these clusters have been characterized, and some of the LanN proteins (CinN, MarN) have been demonstrated to be important for Lal installation in vivo4, 6, 14, 15, 24. Since all of the LanA precursor peptides have a conserved Asp and a Lys close to the C terminus, it is likely that the substrate-assisted mechanism reported here is general for Lal formation in the biosynthesis of duramycin-like lanthipeptides.
Online Methods
General methods
Positive numbers are used for amino acids in the core peptide counting towards the C-terminus from the leader peptide cleavage site. Negative numbers are used counting backwards from the cleavage site. Polymerase chain reactions (PCRs) were carried out on an automated thermocycler (C1000™, Bio-Rad). Gibson Assembly reaction solutions were made as reported25. DNA sequencing was performed by ACGT, Inc (Wheeling, IL). Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) analyses were carried out on an UltrafleXtreme mass spectrometer (Bruker Daltonics). Samples were desalted using ZipTipC18 (Millipore) and spotted onto a MALDI target plate with a matrix solution containing 35 mg/mL 2,5-dihydroxybenzoic acid (DHB) in 3:2 MeCN/H2O with 0.1% trifluoroacetic acid (TFA). Peptides were desalted by C18 solid-phase extraction (SPE) and purified by reversed-phase high performance liquid chromatography (RP-HPLC) on a Shimadzu LC-20AP equipped with a Phenomenex Luna® column (10 μm C18(2), 100 Å, 250 × 10 mm; part number: 00G-4253-N0) at a flow rate of 8 mL/min. An Agilent 1260 Infinity Quaternary LC System was employed for analytical HPLC with an Phenomenex Luna column (10 μm C18(2), 100 Å, 250 × 4.6 mm; part number: 00G-4253-E0) at a flow rate of 1 mL/min. Solvent A was 50 mM ammonium formate in H2O and solvent B was acetonitrile. A gradient of 5% solvent B to 60% solvent B over 30 min was used unless specified otherwise. Liquid chromatography electrospray ionization tandem mass spectrometry (LC/ESI-Q/TOF-MS/MS) was carried out using a Synapt ESI quadrupole TOF Mass Spectrometry System (Waters) equipped with an Acquity Ultra Performance Liquid Chromatography (UPLC) column (1.7 μm BEH C8, 130 Å, 1.0 × 100 mm, part number: 186002876). For all LC-MS or LC-MS/MS analysis, solvent A was water with 0.1% formic acid and solvent B was acetonitrile with 0.1% formic acid. An elution gradient of 15% solvent B to 60% solvent B over 20 min was used unless specified otherwise. For size-exclusion chromatography (SEC) studies, a HiLoad 16/60 column packed with Superdex 200 Prep Grade was used on an ÄKTA purifier eluting at 1.5 mL/min at 4 °C. GC-MS analysis was performed at the Roy J. Carver Metabolomics Center (UIUC) using an Agilent 7890 gas chromatograph with a ZB-1MS (25 m x 0.25 mm x 0.25 μm) column (Phenomenex).
Materials
Oligonucleotides were obtained from Integrated DNA Technologies. Restriction endonucleases, DNA polymerases, and T4 DNA ligase were purchased from New England Biolabs. Media components were obtained from Difco Laboratories. Lysinoalanine 2HCl was purchased from Bachem. Other chemicals were obtained from Sigma Aldrich or Fisher Scientific.
Strains and plasmids
E. coli DH5α was used as host for cloning and plasmid propagation and E. coli BL21 (DE3) as host for expression of proteins/peptides. pRSFDuet-1, pACYCDuet-1, and pETDuet-1 were obtained from Novagen. The pET His6 small ubiquitin-like modifier (Sumo) tobacco etch virus (TEV) protease LIC cloning vector (2S-T) was a gift from Scott Gradia (Addgene plasmid #29711). Procedures for construction of all plasmids and preparation of all proteins can be found in Supplementary Information 1.
Construction of pET28a/MBP-DurN and pMAL_2cx/DurN
pRSFDuet-1/DurN/DurX was reported previously14. pET28a/MBP was generously provided by Dr. Doug Mitchell26. durN was amplified from pRSFDuet-1/DurN/DurX and cloned into pET28a/MBP and pMAL_2cx C-terminal as fusion to the MBP sequence by Gibson Assembly to generate pET28a/MBP-DurN and pMAL_2cx/DurN, respectively. durN mutants were generated by site-directed mutagenesis. Primer sequences are listed in Supplementary Table 3.
Construction of pET/His6-Sumo-DurN, pACYCDuet/DurX, pRSFDuet-1/His6-DurA(S6A)/DurM, and pRSFDuet-1/His6-DurA(D15S)/DurM
Primers terminating with the ligation-independent cloning (LIC) v1 tags were designed for in-frame insertion of durN amplicons into the 2S-T vector to prepare pET/His6-Sumo-DurN (Supplementary Table 1). Amplicons were incubated with SspI-treated vector and assembled into a plasmid using the NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs) under the manufacturer’s recommended conditions. pACYCDuet-1/DurX was prepared by PCR of durX as well as the pACYCDuet-1 backbone from pACYCDurt-1/DurN(MCSI)/DurX(MCSII). Genes encoding DurA-S6A and DurA-D15S were prepared by site-directed mutagenesis of the template pRSFDuet-1/DurA/DurM using PCR and cloned into the MCSI of pRSFDuet-1/DurM by Gibson Assembly. Primer sequences are listed in Supplementary Table 3.
Production and purification of MBP-DurN and its mutants
E. coli BL21 (DE3) cells were transformed with pET28a/MBP-DurN (or mutants). Single colonies of chemically competent E. coli BL21(DE3) were grown in Luria-Broth (LB) media supplemented with kanamycin (50 μg/mL). A 5 mL starter culture was grown overnight and used to inoculate 1 L of LB media supplemented with kanamycin. Liquid cultures were grown at 37 °C with shaking (200 rpm) and induced with 0.15 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) when the OD600 reached 0.75 followed by a 20 min ice-bath incubation and further shaking for 20 h at 18 °C and 200 rpm. Cells were harvested by centrifugation at 4 °C, followed by suspension of the pellet in ~30 mL of binding buffer (50 mM Tris pH 7.5, 0.5 M NaCl, 2.5% (v/v) glycerol, 0.1% (v/v) Triton X-100), and lysis by sonication. The lysates were clarified by centrifugation at 4 °C. The supernatant was loaded onto a 5 mL amylose resin pre-equilibrated with binding buffer. The column was washed with 50 mL of wash buffer (50 mM Tris pH 7.5, 0.5 M NaCl, 2.5% (v/v) glycerol) and eluted with 20 mL of elution buffer (50 mM Tris pH 7.5, 0.15 M NaCl, 10 mM maltose, 2.5% (v/v) glycerol). Fractions containing the highest purity protein as judged by SDS-PAGE were pooled and further purified by size-exclusion chromatography (SEC) using storage buffer (50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 7.5, 50 mM NaCl, 2.5% (v/v) glycerol). The purified protein was concentrated using Amicon Ultra-4 centrifugal filters (30 kDa molecular weight cut-off (MWCO); Millipore Sigma) and frozen in liquid nitrogen for storage at −80 °C. Protein concentration was quantified by NanoDrop (Thermo Fisher Scientific) using an extinction coefficient of 83,070 M−1 cm−1 at 280 nm.
Tag removal of MBP-DurN and its mutants
Tobacco etch virus (TEV) protease was added to MBP-tagged protein at a 1:10 (w/w) ratio in digestion buffer (50 mM HEPES pH 7.5, 300 mM KCl, 50 mM NaCl, 2.5% (v/v) glycerol), and the reaction was left at 4 °C overnight. The sample was loaded onto 5 mL of HisTrap resin pre-equilibrated with digestion buffer and washed with 10 mL of the same buffer. Tag-free proteins were eluted with elution buffer (10 mM imidazole, 50 mM HEPES pH 7.5, 300 mM KCl, 50 mM NaCl, 2.5% (v/v) glycerol). Fractions containing the highest purity protein as judged by SDS-PAGE were pooled and concentrated using Amicon Ultra-4 centrifugal filters (10 kDa MWCO, Millipore Sigma) to ≤ 5 mL and injected onto a 120 mL Superdex 200 10/300 GL column (GE Healthcare) to purify by SEC using 300 mM KCl, 20 mM HEPES pH 7.5 as running buffer.
Production and purification of Sumo-DurN
Transformation of E. coli BL21 (DE3) and IPTG-mediated over-expression was carried out as described for MBP-DurN. Cell pellets were harvested from the cultures by centrifugation at 3,000 ×g followed by suspension of the pellet in ~30 mL of buffer (50 mM Tris pH 8, 500 mM NaCl, 10% (v/v) glycerol). Cells were lysed by sonication and the lysates were clarified by centrifugation at 4 °C. The supernatant was loaded onto 5 mL of HisTrap resin pre-equilibrated with binding buffer (50 mM Tris pH 8, 500 mM KCl, 10% (v/v) glycerol). The column was washed with 50 mL of wash buffer (50 mM Tris pH 8, 30 mM imidazole, 500 mM KCl, 10% (v/v) glycerol) and eluted with 20 mL of elution buffer (binding buffer with 500 mM imidazole). Fractions containing the highest purity protein, as judged by SDS-PAGE, were pooled and further purified by SEC in 50 mM HEPES pH 7.5, 2.5% glycerol, 300 mM KCl. The purified protein was concentrated using Amicon Ultra-4 centrifugal filters (10 kDa MWCO, Millipore Sigma) and frozen in liquid nitrogen for storage at −80 °C. Protein concentration was quantified by NanoDrop using an extinction coefficient of 20,970 M−1 cm−1 at 280 nm. Sumo-DurN was used for the data in Supplementary Figure 10.
Analytical size-exclusion chromatography
Following amylose-resin and size-exclusion chromatography, N-terminal MBP-tagged DurN variants were concentrated to ≥ 5 mg/mL prior to injecting 0.5 mL onto a 30 mL Superdex 200 10/300 GL column (GE Healthcare) equilibrated with 0.1 M KCl, 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 7.5. A similar running buffer was used to elute proteins at a flow rate of 1 mL/min. N-terminal Sumo-tagged DurN variants were analyzed similarly following their purification by Ni-affinity and size-exclusion chromatography. In addition, tag-free DurN was analyzed using a similar procedure. The retention times of DurN variants were compared to those of commercially available protein standards: carbonic anhydrase (bovine erythrocytes; 29 kDa), albumin (bovine serum; 66 kDa), and alcohol dehydrogenase (yeast; 150 kDa) purchased from Sigma-Aldrich and analyzed using a similar procedure. Overlaid chromatograms for each DurN variant suggest similar homodimeric states (Supplementary Figure 11).
Production and purification of compounds 1, 2, 3, 6, 8, 9, 4, and deoxyduramycin
E. coli BL21(DE3) cells were transformed with pRSFDuet/His6-DurA/DurM and pACYCDuet/DurX to prepare compound 1 or with pRSFDuet/His6-DurA/DurM and pACYCDuet/DurN/DurX to prepare compound 2. Treatment with endoproteinase GluC generated peptide 3 (from 1) and 4 (from 2). Cells were transformed with pRSFDuet/His6-DurA-Dha6Ala/DurM and pACYCDuet/DurX to prepare 6, with pRSFDuet/His6-DurA-Asp15Ser/DurM and pACYCDuet/DurX to prepare 8, or with pRSFDuet/His6-DurA/DurM to prepare 9. Transformants were plated on a LB agar plate containing 50 mg/L kanamycin and 50 mg/L chloramphenicol. E. coli BL21 (DE3) cells were transformed with pRSFDuet/His6-DurA/DurM, pMAL-DurN to prepare deoxyduramycin and the transformants were plated on a LB agar plate containing 50 mg/L kanamycin and 50 mg/mL ampicillin. The following procedures are the same for all preparations. A single colony was picked and grown in 5 mL of LB at 37 °C for 12 h containing antibiotics as described above, and the resulting culture was used to inoculate 1 L of LB containing the same antibiotics. When the OD600 reached 0.75, the cultures were cooled on ice for 30 min, and then IPTG was added to 1.0 mM. The cells were cultured at 18 °C for another 18 h before harvesting by centrifugation at 8,000 × g for 10 min. Cell pellets were resuspended at room temperature in LanA buffer 1 (6 M guanidine hydrochloride, 500 mM NaCl, 0.5 mM imidazole, 20 mM NaH2PO4, pH 7.5 at 25 °C) followed by sonication. The lysates were centrifuged at 18,000 ×g for 30 min and supernatants were passed through 0.45-μm syringe filters. His-tagged modified peptides were purified by immobilized metal affinity chromatography (IMAC) loaded with nickel as previously described14. The fractions with the desired peptide were desalted by preparative RP-HPLC using an Agilent Bond Elut C18 column (3 mL). The desalted peptides were lyophilized, dissolved in 50 mM Tris pH 7.5 buffer, and digested by endoproteinase GluC at 20:1 (w/w). The digested reaction mixtures were purified by reversed-phase prep-HPLC (see General Methods).
To prepare deoxyduramycin, the peptide was digested with aminopeptidase (0.1 U per μg of peptide) at pH 8.5 at room temperature overnight and purified by analytical HPLC (General Methods).
Dithiothreitol (DTT) assay to detect uncyclized Dha
DTT was dissolved in water at 1 M then aliquoted and stored at −20 °C for up to two weeks. DTT solutions were added to the peptide solutions to give a final concentration of 250 mM, and pH was adjusted (if necessary) to 7.5. Reactions were conducted at 37 °C for 30 min before.
Preparation of duramycin and 11 (non-enzymatic reaction) in vitro
Compound 3 was incubated with MBP-DurN (5:1, peptide : enzyme) in 100 mM Tris-HCl pH 7.5, 300 mM KCl, and 10 mM MgCl2 at 37 °C for 12 h. No DTT addition to the peptide indicated full conversion of 3. The reaction mixture was purified by analytical HPLC (see General Methods) and fractions containing 3 (judged by MS) were lyophilized. The dried peptide was dissolved in 50 mM Tris pH 8.5, then aminopeptidase was added at 0.1 U/μg of peptide, and the reaction mixture was incubated at 37 °C for 12 h to remove the residual leader peptide residues. The reaction mixture was analyzed by analytical HPLC (General Methods), MALDI-TOF-MS, LC-MS, and LC/MSMS indicating production of duramycin. For the preparation of 11, compound 3 was incubated in 50 mM Tris-HCl pH 10 at r.t. for 12 h and the DTT assay indicated full consumption of 3. Purification and removal of residual leader peptide residues to yield 11 was carried out as described above. Peptide concentrations were estimated by comparing the peak area at 214 nm in the LC chromatogram to that of authentic duramycin.
Activity assays for DurN and its mutants with 3 and its variants
All assays were performed with 50 μM modified DurA or its variants with 10 μM or 5 μM MBP-DurN or its mutants in 100 mM Tris-HCl pH 7.5, 300 mM KCl, and 50 mM MgCl2. A positive control with MBP-DurN and compound 3 and a negative control with just MBP and compound 3 were included as well as a negative control where MBP-DurN (or its mutants) was omitted; control reactions were conducted in parallel to the reaction of interest under the same conditions. Reactions were incubated at 37 °C for 1 h then treated with DTT using the procedure described above. Samples were analyzed by LC-MS and/or LC-MS/MS and relative activities measured by end-point assays. The area of peaks of products with and without DTT adduct were determined, and reaction yield was calculated as [area of non-adduct]/([area of non-adduct]+[area of adduct]).
Protein crystallization
Pooled fractions from MBP tag removal and purification of MBP-DurN (see Supplementary Information 1) were concentrated to ≤ 5 mL and injected onto a 120 mL Superdex 200 10/300 GL column (GE Healthcare) to purify by SEC using 300 mM KCl, 20 mM HEPES pH 7.5 as running buffer.
DurN (apo): Following SEC, DurN was concentrated to 7 mg/mL and crystallized in 2 μL hanging drops at 16 °C whereby DurN solution was mixed 1:1 (v:v) with a reservoir solution containing 25% polyethylene glycol (PEG) 3350, 0.2 M magnesium chloride, and 0.1 M bis-tris pH 6.5. Apo DurN also produced similar diffraction quality crystals using a different reservoir solution containing 20% PEG 8000, 0.2 M magnesium acetate, and 0.1 M sodium cacodylate pH 6.0. In both cases, rod-shaped crystals grew to a maximum size within 48 h.
DurN + compound 6: Following SEC, DurN was concentrated to 9 mg/mL and incubated with 1.8 mM compound 6 for 30 min on ice. This solution was mixed 1:1 (v:v) with a reservoir solution containing 25% PEG 3350, 0.2 M MgCl2, and 0.1 M bis-tris pH 6.0 in 2 μL hanging drops incubated at 16 °C. Hexagonal shaped crystals grew to maximum size within 48 h.
DurN + duramycin: Following SEC, DurN was concentrated to 6 mg/mL and incubated with 1.5 mM duramycin for 30 min on ice. Insoluble particulates were pelleted by centrifugation and the supernatant was mixed 1:1 (v:v) with a reservoir solution containing 22.5% PEG 3350, and 0.1 M 2-(N-morpholino)ethanesulfonic acid (MES) pH 5.6 in 2 μL hanging drops at 16 °C. Thick rod-shaped crystals grew to a maximum size within 72 h.
Crystallographic data collection, structure solutions, and refinement
Data were collected at the Advanced Photon Source at Argonne National Lab using the Life-Science Collaborative Access Team 21-ID-D, 21-ID-F, and 21-ID-G beamlines. Before collecting data, crystals were cryoprotected by immersing the crystals in similar solutions supplemented with 20% ethylene glycol and then flash-freezing in liquid nitrogen. To obtain phases experimentally, co-crystals of DurN and compound 6 were soaked with 0.5 M potassium iodide for 24 h at 16 °C and used to collect diffraction data at 8 keV. Data were initially processed using autoPROC27 and further processed in autoSHARP to obtain phase information. The initial model was rebuilt using Buccaneer28 and refined using REFMAC529 in the CCP4 Software Suite. Diffraction datasets corresponding to apo DurN crystals and DurN-duramycin co-crystals were phased after similar processing in autoPROC followed by molecular replacement using Phaser MR30. Ligand parameters for compound 6 and duramycin were determined using Phenix eLBOW31 and modeled into their corresponding electron densities, followed by iterative rounds of automated REFMAC5 refinement and manual rebuilding in Coot32. Solvent molecules and potassium ions were first incorporated using Phenix Refine33 or REFMAC5 then manually curated in Coot.
Differential scanning fluorimetry (DSF)
Tag-free DurN purified by SEC was solvent exchanged into a buffer lacking potassium or magnesium ions by subjecting to additional SEC on a 30 mL Superdex 200 10/300 GL column (GE Healthcare) equilibrated with 0.25 M NaCl and 20 mM HEPES pH 7.5. Protein was eluted using a similar running buffer and pooled fractions were concentrated to 1 mg/mL. SYPRO orange dye was added to the protein at 8X final concentration and incubated at room temperature for 20 min. High-Profile 96-Well Semi-Skirted PCR plates (Bio-Rad Laboratories, Inc.) were used to serially titrate either KCl or MgCl2 into the protein-dye mixture from 300 mM to 150 μM or 200 mM to 100 μM, respectively, and incubated for 25 min at room temperature. Melting curves were obtained using a CFX Connect Real-Time PCR Detection System (Bio-Rad Laboratories, Inc.) where the sample was initially held at 25 °C for 3 min then incrementally ramped to 98 °C at 0.5 °C every 10 s with plate reading steps at every increment. The channel was set to “FRET” to satisfy the excitation and emission wavelengths of the SYPRO orange dye (λex 470 nm /λem 570 nm). Data corresponding to the melt curves and first derivatives of the melt curves were obtained in the Bio-Rad CFX Manager 3.1 Data Analysis tool and subsequently plotted in OriginPro 2016 (OriginLab).
Mutational studies of DurN
A number of residues make hydrophobic contacts at the dimeric interface (i.e. Leu22, Val24, Leu25, Leu28, Leu39, Leu43, Leu51, Ile54, Trp68, Leu72, Trp73, Leu85, Ile111, and Leu113). Additional electrostatic interactions are observed intermolecularly between Asp12-Lys103 and Arg18-Glu106, as well as intramolecularly between Lys34-Glu37 and Arg53-Glu82. Several MBP-tagged and site-directed DurN variants were constructed based on the co-crystal structures and sequence conservation between DurN homologs (Supplementary Fig. 12). Specifically, Ala substitutions were generated at Arg17, Arg18, Gln20, Cys26, Lys66, Trp68, Ser79, Gln89, and Glu101 in addition to a Lys substitution at Arg17, and an end-point substrate conversion assay using compound 3 was performed (see Methods). Aside from DurN variants C26A and S79A, a diminution in activity relative to the wild-type DurN was observed for all DurN variants. These activity data are rationalized by the structural data as residues Arg17, Gln20, and Lys66 are implicated in substrate binding, and residues Arg18, Trp68, and Glu101 are important for dimerization. Unsurprisingly, mutations at Cys26 and Ser79 exhibit no activity loss, as these residues are solvent exposed and distal to the ligand binding site in the crystal structures.
Preparation and purification of Dur-FL
Duramycin (1 mg) was dissolved in 50 mM HEPES pH 7.5. Carboxyfluorescein succinimidyl ester (2 mg) was dissolved in 10 μL of dimethyl sulfoxide and added (total reaction volume = 1 mL). The reaction was left at r.t. in the dark for 2 h, desalted and analyzed by MS indicating both doubly and singly acylated duramycin. The products were purified by analytical HPLC (see General Methods), lyophilized, and stored at −20 °C. Peptide concentration was estimated by comparing the peak area at 214 nm to that of duramycin and NHS-Fluorescence standard.
Fluorescence polarization (FP) binding studies
Serial dilutions (1:1) for MBP-DurN or its mutants were prepared with FP buffer (300 mM KCl, 20 mM HEPES, pH 7.5) in a 96-well PCR plate. Each well contained 45 μL solution. The highest MBP-DurN concentration was 15 μM. For each well, 45 μL Dur-FL of a 20 nM stock solution was added, to give a final concentration of 10 nM. Then, 80 μL from each well was transferred to a well in a black 384-well plate (i.e. each well contains 80 μL liquid, with 15 uM~500 pM MBP-DurN or its mutants, and 10 nM Dur-FL). FP analysis was performed on a BioTek Synergy H4 Hybrid reader. The detection method was set to “fluorescence” and read type was “endpoint”. Measurements were made by excitation at 485 nm (20 nm bandwidth) and emission at 528 nm (20 nm bandwidth). The Corning™ black 384-well plate (Corning™ 3575) was incubated in the plate reader for 5 min at 25 °C, shaken for 1 min, and followed by data collection. Polarization was calculated as Polarization = [I(parallel)-I(perpendicular)]/[I(parallel)-I(perpendicular)]. Dissociation constant (Kd) values were calculated using a dose-response curve (equation below) in Origin Pro 9.1 (OriginLab) with three independent titrations. Background fluorescence from the proteins alone was subtracted from the fluorescence polarization signal obtained with the fluorophore.
Where y is polarization, A1 is the minimum polarization, A2 is the maximum polarization, X0 is the IC50, p is the Hill coefficient, and x is the concentration of Dur-FL. A receptor depletion equation was also used to calculate the Kd for comparison and did not yield a significant difference.
Competition fluorescence polarization (FP) binding studies
The same procedures were applied for all competition FP studies unless specified otherwise. The inhibitor of interest was prepared at 200 μM in water, and serially diluted 1:1 with FP buffer (300 mM KCl, 20 mM HEPES, pH 7.5) to provide 16 wells with concentrations ranging from 100 μM to 0.003 μM in a 96-well PCR plate. A solution of MBP-DurN (2 μM) and Dur-FL (20 nM) was prepared in the same FP buffer and incubated at r.t. for 5 min before transferring 45 μL to each well, then 80 μL of the mixtures in each well were transferred to a black-bottomed 384-well plate (i.e. 80 μL wells containing 1 μM MBPDurN, 10 nM Dur-FL, and inhibitor concentration ranging from 100 μM to 3 nM). IC50 values were calculated from the 50% saturation point using a dose-response curve fit in Origin Pro 9.1 (OriginLab) with three independent titrations. All IC50 values were transferred to Ki values by applying the Munson-Rodbard equation (see equation below)34.
Where yo is the initial bound to free ratio for the labeled species before perturbation of equilibrium by the added inhibitor, and LT is the total amount of labeled species.
Derivatization of lysinoalanine (Lal) standard
MeOD-d4 (3 mL) was cooled in a 50 mL pear-shaped glass flask immersed in an ice-water bath. Acetyl chloride (0.9 mL) was added dropwise. This solution was added to 2 mg Lal standard (LL and DL-lysinoalanine mixture, Bachem) and the reaction mixture was heated at 110 °C for 1 h under reflux, then allowed to cool down to r.t. in air. The solvent was removed by a stream of nitrogen gas. Dichloromethane (3 mL) and pentafluoropropionic anhydride (1 mL) were added to the flask cooled in an ice-water bath. The mixture was heated at 110 °C under reflux for 30 min, allowed to cool to r.t., and the solvent was removed under a stream of nitrogen gas for 2 h. For each sample, 800 μL of pyridine was used to dissolve all resulting compounds, and the solutions were directly analyzed by GC-MS.
Hydrolysis and derivatization of duramycin, compound 11, and deoxyduramycin
The same hydrolysis and derivatization procedures were applied for all Lal-containing samples. To 1 mg of peptide, 3 mL of 6 M deuterium chloride in D2O was added to the sample (~1 mg) in a glass pressure tube (Ace Glass). The reaction mixture was heated without stirring at 80 °C for 4 h. The solution was transferred to a 50 mL pear-shaped bottle, and the solvent was removed with a rotary evaporator. Then the same derivatizing procedures were used as described for the derivatization of Lal standard.
GC-MS analysis of lysinoalanine standard, duramycin, compound 11, and deoxyduramycin
The derivatized samples were analyzed by GC-MS using an Agilent 7890 gas chromatograph equipped with a ZB-1MS column (Phenomenex, 30 m x 0.25 mm x 0.25 μm). Samples were dissolved in 300 μL of pyridine and a small volume introduced to the instrument via a splitless injection at a flow rate of 1.6 mL/min helium gas. The front inlet was set to 230 °C for initial temperature, the purge flow was 20 mL/min, the purge time was 1 min, and the total flow was 24 mL/min. The flow rate in the column was 1.6 mL/min. The temperature method used for the oven was 100 °C for 2 min, followed by 100 °C to 215 °C at 2.5 °C /min, and 215 °C for 2 min. The mass spectrometer was operated in simultaneous scan/selected-ion monitoring (SIM) mode, monitoring at the characteristic fragment masses of 190 Da, 230 Da, 405 Da, 420 Da, 433 Da, 468 Da, 643 Da, and 705 Da, for Lal residues.
Structure preparation for computational studies
The crystallographic coordinates of duramycin bound to DurN homodimer were used to generate the starting structures for the simulations. The 11 unresolved resides at both N-termini (MKSAKEPTTIYQ) were modeled as random coils in some simulations; the presence or absence of this highly flexible region did not have a significant impact in the observed results. Different crystallographic ions (K+ and Mg2+) were tested in the simulations, the best results being obtained with K+. The uncyclized duramycin precursor was modeled from co-crystalized duramycin by simply breaking the Cβ(Dha6)–N(Lys19) bond, slightly separating the fragments and minimizing the resulting structure with molecular mechanics using the appropriate generated parameters. When necessary mutations such as Hya15Asp in duramycin were introduced manually using PyMOL 1.8 (PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC).
MD simulations
Unconstrained MD simulations in water were performed using the GPU accelerated code (pmemd)35–37 of the Amber 16 and AmberTools 17 package38. Parameters for non-natural dehydro amino acids, (methyl)lanthionine, hydroxylated aspartic acid (Hya) and lysinoalanine (Lal) were generated within the antechamber module using the upgraded version of the general AMBER force field (gaff2)38, with partial charges set to fit the electrostatic potential generated at the HF/6–31G(d) level by the RESP (restrained electrostatic potential) model39. The charges were calculated according to the Merz-Singh-Kollman scheme40, 41 using the Gaussian 16 package42. Each peptide was immersed in a pre-equilibrated cubic box with a 10 Å buffer of TIP3P14 water molecules using the leap module, resulting in the addition of around 15,000 solvent molecules. The systems were neutralized by addition of explicit counter ions (K+ and Cl–). All subsequent calculations were done using an evolved version of the Stony Brook modification of the Amber 99 force field (ff14SB)43. A two-stage geometry optimization approach was performed. The first stage minimizes the positions of solvent molecules and ions imposing positional restraints on solute by a harmonic potential with a force constant of 500 kcal mol−1 Å−2, and the second stage was a minimization of all the atoms in the simulation cell. The systems were gently heated under periodic-boundary conditions at a constant pressure of 1 atm. Water molecules were treated with the SHAKE algorithm such that the angle between the hydrogen atoms was kept fixed. Long-range electrostatic effects were modelled using the particle mesh Ewald method44. An 8 Å cutoff was applied to Lennard–Jones and electrostatic interactions. Harmonic restraints of 10 kcal/mol were applied to the solute, and the Andersen equilibration scheme was used to control and equalize the temperature. The time step was kept at 1 fs during the heating stage, allowing potential inhomogeneities to self-adjust for 100 ps. Each system was then equilibrated for 2 ns with a 2 fs time step at a constant volume and temperature of 300 K. Production trajectories were run for 1.0 μs under the same conditions.
Data availability
Atomic coordinates and structure factors for the reported crystal structure have been deposited in the Protein Data Bank under accession codes 6C0G, 6C0H, and 6C0Y.
Supplementary Material
Acknowledgements
The authors thank Dr. L. Huo for providing plasmids, Dr. A. Vladimirovich Ulanov (UIUC Metabolomics Center) for assistance with GC-MS analysis, and K. Brister and the staff at LS-CAT at the Advanced Photon Source (Argonne National Laboratory) for assistance with X-ray crystallography data acquisition. This work was supported by the National Institutes of Health (R37 GM 058822 to W.A.V. and R01 GM079038 to S.K.N.), and D.G.I. MINECO/FEDER (grants CTQ2015–70524-R and RYC-2013–14706 to G.J.O. and a predoctoral fellowship to C.D.N).
References
- 1.Kessler H et al. The structure of the polycyclic nonadecapeptide Ro 09–0198. Helv. Chim. Acta 71, 1924–1929 (1988). [Google Scholar]
- 2.Hayashi F et al. The structure of PA48009: the revised structure of duramycin. J. Antibiot 43, 1421–1430 (1990). [DOI] [PubMed] [Google Scholar]
- 3.Fredenhagen A et al. Duramycins B and C, two new lanthionine containing antibiotics as inhibitors of phospholipase A2. Structural revision of duramycin and cinnamycin. J. Antibiot 43, 1403–1412 (1990). [DOI] [PubMed] [Google Scholar]
- 4.Chen E et al. Mathermycin, a Lantibiotic from the Marine Actinomycete Marinactinospora thermotolerans SCSIO 00652. Appl. Environ. Microbiol 83, e00926–00917 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kodani S, Komaki H, Ishimura S, Hemmi H & Ohnishi-Kameyama M Isolation and structure determination of a new lantibiotic cinnamycin B from Actinomadura atramentaria based on genome mining. J. Ind. Microbiol. Biotechnol 43, 1159–1165 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Smith TE et al. Accessing chemical diversity from the uncultivated symbionts of small marine animals. Nat. Chem. Biol 14, 179–185 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pomorski T, Hrafnsdóttir S, Devaux PF & van Meer G Lipid distribution and transport across cellular membranes. Semin. Cell Dev. Biol 12, 139–148 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Iwamoto K et al. Curvature-dependent recognition of ethanolamine phospholipids by duramycin and cinnamycin. Biophys. J 93, 1608–1619 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hosoda K et al. Structure determination of an immunopotentiator peptide, cinnamycin, complexed with lysophosphatidylethanolamine by 1H-NMR. J. Biochem 119, 226–230 (1996). [DOI] [PubMed] [Google Scholar]
- 10.Richard AS et al. Virion-associated phosphatidylethanolamine promotes TIM1-mediated infection by Ebola, dengue, and West Nile viruses. Proc. Natl. Acad. Sci. U.S.A 112, 14682–14687 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oliynyk I, Varelogianni G, Roomans GM & Johannesson M Effect of duramycin on chloride transport and intracellular calcium concentration in cystic fibrosis and non-cystic fibrosis epithelia. Apmis 118, 982–990 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Zhao M, Li Z & Bugenhagen S 99mTc-labeled duramycin as a novel phosphatidylethanolamine-binding molecular probe. J. Nucl. Med 49, 1345–1352 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Delvaeye T et al. Noninvasive whole-body imaging of phosphatidylethanolamine as a cell death marker using (99m)Tc-duramycin during TNF-induced SIRS. J. Nucl. Med 59, 1140–1145 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Huo L, Ökesli A, Zhao M & van der Donk WA Insights into the Biosynthesis of Duramycin. Appl. Environ. Microbiol 83, e02698–02616 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ökesli A, Cooper LE, Fogle EJ & van der Donk WA Nine post-translational modifications during the biosynthesis of cinnamycin. J. Am. Chem. Soc 133, 13753–13760 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen E et al. Mathermycin, a Lantibiotic from the Marine Actinomycete Marinactinospora thermotolerans SCSIO 00652. Appl. Environ. Microbiol 83, e00926–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burley SK et al. RCSB Protein Data Bank: Sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Sci 27, 316–330 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holm L & Rosenström P Dali server: conservation mapping in 3D. Nucleic Acids Res 38, W545 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vagin AA et al. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Biol. Crystallogr 60, 2184–2195 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Tang W, Jiménez-Osés G, Houk KN & van der Donk WA Substrate control in stereoselective lanthionine biosynthesis. Nat. Chem 7, 57–64 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lopatniuk M, Myronovskyi M & Luzhetskyy A Streptomyces albus: A new cell factory for non-canonical amino acids incorporation into ribosomally synthesized natural products. ACS Chem. Biol 12, 2362–2370 (2017). [DOI] [PubMed] [Google Scholar]
- 22.O’Rourke S, Widdick D & Bibb M A novel mechanism of immunity controls the onset of cinnamycin biosynthesis in Streptomyces cinnamoneus DSM 40646. J. Ind. Microbiol. Biotechnol 44, 563–572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerlt JA et al. Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta 1854, 1019–1037 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Widdick DA et al. Cloning and engineering of the cinnamycin biosynthetic gene cluster from Streptomyces cinnamoneus cinnamoneus DSM 40005. Proc. Natl. Acad. Sci. U.S.A 100, 4316–4321 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods References
- 25.Gibson DG et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Lee SW et al. Discovery of a widely distributed toxin biosynthetic gene cluster. Proc. Natl. Acad. Sci. U.S.A 15, 5879–5884 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vonrhein C et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr 67, 293–302 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cowtan K The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr 62, 1002–1011 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Murshudov G et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr 67, 355–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCoy AJ et al. Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moriarty NW, Grosse-Kunstleve RW & Adams PD electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr 65, 1074–1080 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Afonine PV et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr 68, 352–367 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Munson PJ & Rodbard D An Exact Correction to the “Cheng-Prusoff” Correction. J. Recept. Res 8, 533–546 (1988). [DOI] [PubMed] [Google Scholar]
- 35.Götz AW et al. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput 8, 1542–1555 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pierce LC, Salomon-Ferrer RF, de Oliveira CA, McCammon JA & Walker RC Routine access to millisecond time scale events with accelerated molecular dynamics. Theory Comput 8, 2997–3002 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le Grand S, Götz AW & Walker RC SPFP: Speed without compromise-A mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun 184, 374–380 (2013). [Google Scholar]
- 38.Wang J, Wolf RM, Caldwell JW, Kollman PA & Case DA Development and testing of a general amber force field. J. Comput. Chem 25, 1157–1174 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Bayly CI, Cieplak P, Cornell WD & Kollman PA A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem 97, 10269–10280 (1993). [Google Scholar]
- 40.Besler BH, Merz KM & Kollman PA Atomic charges derived from semiempirical methods. J. Comput. Chem 11, 431–439 (1990). [Google Scholar]
- 41.Singh UC & Kollman PA An approach to computing electrostatic charges for molecules. J. Comput. Chem 5, 129–145 (1984). [Google Scholar]
- 42.Frisch MJ et al. , Edn. 09, Revision A.1 (Gaussian, Inc., Wallingford CT; 2016). [Google Scholar]
- 43.Maier JA et al. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 11, 3696–3713 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Darden T, York D & Pedersen L Particle mesh Ewald: An N-log(N) method for Ewald sums in large systems. J. Chem. Phys 98, 10089–10092 (1993). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the reported crystal structure have been deposited in the Protein Data Bank under accession codes 6C0G, 6C0H, and 6C0Y.