

SUMMARY
Matrix metalloproteinases (MMPs) are associated with decreased patient prognosis, but have failed as anti-invasive drug targets despite promoting cancer cell invasion. Through time-lapse imaging, optical highlighting, and combined genetic removal of the five MMPs expressed during anchor cell (AC) invasion in C. elegans, we find that MMPs hasten invasion by degrading basement membrane (BM). Though irregular and delayed, AC invasion persists in MMP- animals via adaptive enrichment of the Arp2/3 complex at the invasive cell membrane, which drives formation of an F-actin-rich protrusion that physically breaches and displaces BM. Using a large-scale RNAi synergistic screen and a genetically encoded ATP FRET sensor, we discover that mitochondria enrich within the protrusion and provide localized ATP that fuels F-actin network growth. Thus, without MMPs an invasive cell can alter its BM breaching tactics, suggesting that targeting adaptive mechanisms will be necessary to mitigate BM invasion in human pathologies.
Keywords: Invasion, basement membrane, matrix metalloproteinase, ATP transport, mitochondria, live imaging, actin dynamics
Graphical Abstract
eTOC Blurb
A requirement for MMPs in basement membrane invasion has been difficult to study. Kelley et al. uses C. elegans to genetically remove MMPs and demonstrate that cell invasion is delayed, but still persists through an adaptive cell response requiring increased F-actin protrusive force and localized ATP production by mitochondria.
INTRODUCTION
Basement membrane (BM) is a thin, dense, extracellular matrix (ECM) barrier that surrounds tissues (Yurchenco, 2011). BM composition and structure is highly conserved across species and formation of BM requires the initial deposition of non-cross-linked laminin networks that are subsequently strengthened by the addition of cross-linked collagen IV and other components (Pozzi et al., 2017). The resulting dense meshwork contains openings or pores smaller than 100nm in diameter, which is less than the diameter of smallest protrusions, and twenty-fold smaller than the 1–2 μm pore size known to limit migrations in vitro (Gaiko-Shcherbak et al., 2015; Inoué et al., 1983; Yurchenco et al., 1992). Despite its formidable barrier properties, specialized cells cross BMs to carry out many essential physiological processes including trophoblast invasion during embryo implantation, neural crest and muscle cell BM transmigrations during embryogenesis, and leukocytes crossing BMs during immune surveillance (Kelley et al., 2014; Madsen and Sahai, 2010). Invasion through BM is also misregulated in numerous immune disorders and is the defining step in metastasis, accounting for most cancer lethality (Hanahan and Weinberg, 2011; Menezes et al., 2016). Due to its importance, there has been great interest in understanding the mechanisms that promote BM invasion to design therapies that modify or block this cellular behavior, which has thus far remained elusive (Te Boekhorst and Friedl, 2016).
Matrix metalloproteinases (MMPs) are a family of zinc-dependent endopeptidases that cleave ECM proteins including collagen and laminin, and signaling proteins such as growth factors, chemokines, and cytokines (Overall and Kleifeld, 2006). This protease family is highly conserved, and each contains a signal peptide, a propeptide that maintains latency, and a catalytic domain (Cieplak and Strongin, 2017). MMPs are expressed during tissue remodeling events in development, wound healing, inflammation, and angiogenesis, and MMP overexpression is strongly associated with cancer cell invasion in human patients, mouse models, and in vitro assays (Deryugina and Quigley, 2006; Kumar et al., 2016). Within invading cells, several MMPs localize to invadopodia, invasive F-actin rich subcellular punctate structures transiently formed to facilitate invasion; invadopodial MMPs are hypothesized to enzymatically digest BM barriers, clearing a path for invasion (Castro-Castro et al., 2016; Sabeh et al., 2009). Cultured cells displaying a mesenchymal (MMP-dependent) phenotype can employ a compensatory amoeboid (MMP-independent) invasion mode when treated with MMP inhibitors in artificial matrices (Te Boekhorst and Friedl, 2016; Wolf et al., 2013). These studies, along with the failure of MMP inhibitors to show benefit in human clinical trials, suggests that cells in vivo might use, but not require, MMPs to cross endogenous BM (Cathcart et al., 2015; Coussens et al., 2002). Depending on cell type, amoeboid migration can propel cells through loose interstitial matrix barriers when pore sizes are in the range of 2.0–7.0 μm2 (Paul et al., 2017; Wolf et al., 2013), however, this “migration limit” is 70-times larger than the predicted gap sizes in the denser BM. Therefore, in the absence of MMPs, cells must employ additional unknown mechanisms for BM breaching.
Despite extensive study, the requirement for MMPs during cell invasion through BM remains controversial (Sabeh et al., 2009; Wolf et al., 2003), and the mechanistic link between MMPs and BM remodeling in vivo is unknown. This is due to the inability of in vitro models to fully recapitulate BM ultrastructure (density, cross-linking, and composition), and the complex signaling events that occur within a functioning tissue (Kelley et al., 2014). BM invasion in genetic mouse models is difficult to experimentally examine in vivo because cells often invade sporadically in visually inaccessible tissues. In addition, mice contain 24 MMPs, for which knockouts display minimal phenotypes most likely due to redundant roles in substrate specificity (Fanjul-Fernández et al., 2010; Page-McCaw, 2008).
AC invasion into the vulval epithelium during C. elegans development is a genetically and visually tractable in vivo model of cell invasion through BM (Sherwood and Sternberg, 2003; Sherwood et al., 2005). The AC is a specialized uterine cell that invades through the underlying BM to initiate uterine-vulval attachment. AC invasion is highly stereotyped and allows real-time visual analysis of invasion at the cell-BM interface. Further, the genetic pathways directing AC invasion are conserved with metastatic cells assayed in vitro. Genes regulating the actin cytoskeleton, BM composition, and protease secretion are upregulated prior to BM breaching (Hagedorn et al., 2013; Lohmer et al., 2016; Matus et al., 2014). Further, like cultured human cancer cells, the AC harbors BM breaching invadopodia (Hagedorn et al., 2013; Lohmer et al., 2014). The C. elegans genome encodes only six MMP genes, named zmp-1–6 (Zinc Metalloproteinase) (Altincicek et al., 2010). The simple tissue structure, visual accessibility, and genetic strengths make AC invasion an ideal model to elucidate the role of MMPs in BM invasion.
We genetically removed the five C. elegans MMPs (MMP- animals) that are expressed in or near the AC during BM invasion. Using live cell imaging, we found that AC invasion still occurs in MMP- animals through an increase in the F-actin nucleator Arp2/3 and F-actin growth at the invasive cell membrane, forming a large protrusion that physically tears and then displaces BM. Further, through a synergistic interaction screen, we discovered that mitochondria also respond to the loss of MMP’s and enrich at the site of BM breaching, where they provide localized ATP that fuels invasive F-actin network expansion. Together, these results demonstrate that invasive cells can adjust their BM invasion strategy after loss of MMPs, and suggest that therapeutic targeting of adaptive mechanisms might be required to block BM invasion in diseases such as cancer.
RESULTS
Five MMP genes are expressed within and near the invading AC
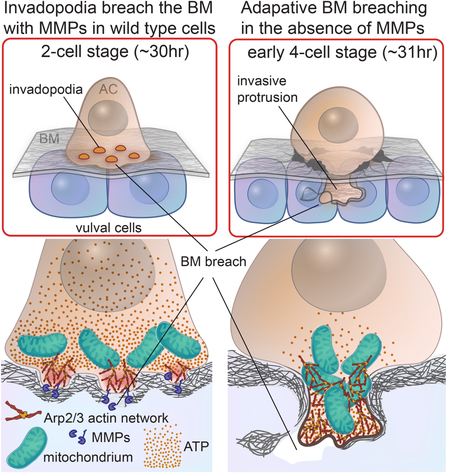
AC invasion is a highly stereotyped BM transmigration event that occurs in synchrony with the divisions of the underlying 1°fated P6.p vulval precursor cells and development of uterine tissue (Sherwood et al., 2005). AC invasion initiates at the late P6.p 2-cell stage and is completed by the mid P6.p 4-cell stage (a ~75 minute period; Figure 1A and 1B) (Sherwood and Sternberg, 2003). Of the six known MMP genes (Figure S1A, S2), three MMPs, zmp-1 (zmp-1>GFP), zmp-3 (zmp-3>ZMP-3::GFP) and zmp-6 (zmp-6>GFP), are expressed in the AC during invasion (Figure 1C), in keeping with previous studies (Matus et al., 2015; Sherwood et al., 2005). On the other hand, zmp-4 (zmp-4>GFP) is expressed in the body wall muscle (Figure S1B) and its protein (zmp-4>ZMP-4::GFP) accumulates in the juxtaposed epidermal and gonadal BM through which the AC invades (Figure 1C), while zmp-5 (zmp-5>GFP) is expressed in the dorsal uterine cells above the AC (Figure 1C, (Wang et al., 2014)). zmp-2 (zmp-2>ZMP-2::GFP) is undetectable near the AC, but is expressed in cells in the head and tail of the worm, consistent with previously published work (Figure S1C, (Altincicek et al., 2010)). We conclude that five of the six MMP genes encoded in the C. elegans genome are expressed at or near the site of AC invasion.
Figure 1. MMPs are located within and around the AC during BM invasion.

(A) The AC (cdft-3>mCherry::moeABD; green) before and after invasion through BM (laminin::GFP, magenta). (B) Timeline for AC invasion after hatching at 20°C is shown. During the P6.p 2-cell stage of the 1°vulval precursor ce lls (1°VPCs, blue), the AC (orange) breaches the BM (grey) with invadodia that transform into an invasive protrusion. At the P6.p 4-cell stage, the invasive protrusion grows and expands the BM opening before retracting. (C) Reporters for zmp expression (left) show that zmp-1, −3, and −6 are expressed in the AC (right, overlay in green on DIC image) during invasion. ZMP-4 protein localizes to the BM (arrows) and the zmp-5 gene is expressed in the dorsal uterine cells above the AC. Zmp-2 is not detectable at the invasion site (see Figure S1C). (D) Expression of zmp-1, −3, and −6 in the AC (left, fluorescence overlay on DIC) decreased after fos-1 RNAi-mediated knockdown (right). Normalized reduction in zmp mean fluorescence levels are shown in white (mean ± SD, p ≤ 0.001, n ≥ 10 for each treatment). (E) CRISPR-Cas9-mediated zmp-1 translational GFP knock-in (left panels, green in merged image) and invadopodia (middle top, marked with F-actin marker mCherry::moeABD, arrows) and the invasive protrusion (middle bottom, arrows). Overlay reveals colocalization with invadopodia and protrusion (right). Pearson’s correlation coefficient (r) values on merged images are representative of 10 animals examined. Scale bars, 5μm.
AC-expressed MMPs are regulated by Fos and ZMP-1 localizes to invadopodia
We next determined how the AC-expressed MMPs are controlled and where they localize during invasion. The bZIP Fos family of transcription factors are major regulators of MMP expression in vertebrates (Ganguly et al., 2013). The C. elegans Fos gene ortholog fos-1a is expressed in the AC during invasion, promotes AC invasion, and is required for zmp-1 expression (Sherwood et al., 2005), suggesting Fos regulation of MMPs may be conserved. Supporting this notion, we found that RNAi mediated loss of fos-1a markedly decreased expression of all AC expressed MMPs ((zmp-1, zmp-3, and zmp-6, (Figure 1D)). Thus, similar to vertebrates, C. elegans MMP transcription is controlled by Fos.
ZMP-1 is the only membrane-bound MMP in the worm, which is a class of MMPs strongly associated with invasion in vertebrates (Altincicek et al. 2010; Castro-Castro et al. 2016). By using CRISPR-Cas9 genome editing, we created a functional ZMP-1::GFP knock-in and found ZMP-1 colocalized with F-actin-rich invadopodia at the invasive cell membrane prior to and during BM breaching (Figure 1E). ZMP-1 localization is similar to vertebrate MT1-MMP, a membrane-bound MMP, that localizes to invadopodia in cultured tumor cells (Castro-Castro et al., 2016). ZMP-1 was also enriched and colocalized with F-actin within the large invasive protrusion that expands the hole in the BM (Figure 1E). These observations indicate that the transcriptional regulation and localization of AC expressed MMPs are similar to vertebrate IMPs.
MMPs are not essential for AC invasion through BM
To determine if any of the five MMPs found in or near the AC are required for invasion, we examined AC invasion in mutant worms with deletion alleles that remove the catalytic domains of each MMP gene and are putative nulls (Figure S1A, S2, and S3A). Invasion was assessed in the late P6.p 4-cell stage (~45 min after wild-type animals complete invasion) by DIC imaging. BM breaching was determined by the presence (BM intact, blocked invasion) or absence (BM breached, invasion occurred) of the phase dense line of BM under the AC (Sherwood and Sternberg, 2003). Individual loss of any of the five MMPs located at or near the AC did not cause observable invasion defects at the late P6.p 4-cell stage (Figure S3B and Table 1). To account for the possibility that multiple MMPs may be functioning redundantly, animals were created with MMP deletion combinations including animals with all five MMP deletion alleles (quintuple-zmp mutant, called MMP-; see Figure S3B for combinations). Importantly, vulva induction and morphogenesis, which are required for AC invasion (Sherwood and Sternberg, 2003), were normal in MMP- animals (n=20/20). AC invasion occurred by the late P6.p 4-cell stage in all MMP mutant combinations, including the MMP- animals (Figure S3B and Table 1). Lastly, to ensure that zmp-2 does not compensate for the loss of zmp-1, −2, −4, −5 and −6, we confirmed that invasion proceeds when MMP- animals undergo zmp-2 RNAi treatment (see Table S1). In addition, we verified that GFP reporter expression in the AC and mRNA transcripts in whole worms were not increased in MMP- animals (Figure S3C, D). AC invasion was also resistant to commonly used MMP inhibitors (GM-6001 and BB-94, see Table S1 and Star Methods). We conclude that MMP proteases are not absolutely required for AC invasion, but do not rule out more subtle phenotypes not visible by DIC imaging.
Table 1.
Genetic analysis of the role of MMPs during AC invasion
| Genotype | RNAi Tx | Developmental P6.p Stage | Invasion Complete | n | |
|---|---|---|---|---|---|
| Screen of C. elegans MMP genes | |||||
| wild type | n/a | late four-cell | 100% | 50 | |
| zmp-1 (cg115) | n/a | late four-cell | 100% | 29 | |
| zmp-2 (tm3529)/+ | n/a | late four-cell | 100% | 22 | |
| zmp-3 (tm3482) | n/a | late four-cell | 100% | 50 | |
| zmp-4 (tm3078) | n/a | late four-cell | 100% | 59 | |
| zmp-4 (tm3484) | n/a | late four-cell | 100% | 26 | |
| zmp-5 (tm3209) | n/a | late four-cell | 100% | 15 | |
| zmp-6 (tm3073) | n/a | late four-cell | 100% | 50 | |
| zmp-6 (tm3385) | n/a | late four-cell | 100% | 23 | |
| zmp-1 (cg115); zmp-3 (tm3482) | n/a | late four-cell | 100% | 29 | |
| zmp-3 (tm3482); zmp-6 (tm3073) | n/a | late four-cell | 100% | 50 | |
| zmp-3 (tm3482); zmp-4 (tm3484) | n/a | late four-cell | 100% | 50 | |
| zmp-1 (cg115); zmp-6 (tm3073) | n/a | late four-cell | 100% | 50 | |
| zmp-1 (cg115); zmp-3 (tm3482); zmp-6 (tm3073) | n/a | late four-cell | 100% | 50 | |
| zmp-1 (cg115); zmp-3 (tm3482); zmp-4 (tm3484); zmp-6 (tm3073) | n/a | late four-cell | 100% | 50 | |
| MMP- | n/a | late four-cell | 100% | 50 | |
| Screen of MMP genes with AC membrane and BM markers | |||||
| wild type | n/a | early four-cell | 100% | 92 | |
| MMP- | n/a | early four-cell | 16% | 82 | |
| zmp-1 (cg115); zmp-6 (tm3073) | n/a | early four-cell | 27% | 54 | |
| zmp-3 (tm3482); zmp-4 (tm3484); zmp-5 (tm3209) | n/a | early four-cell | 89% | 59 | |
| RNAi genetic interaction experiments with AC membrane and BM markers | |||||
| wild type | ctl | early four-cell | 91% | 71 | |
| mid four-cell | 100% | ||||
| late four-cell | 100% | ||||
| MMP- | ctl | early four-cell | 33% | 71 | |
| mid four-cell | 79% | ||||
| late four-cell | 100% | ||||
| wild type | arx-2 | early four-cell | 31% | 97 | |
| mid four-cell | 59% | ||||
| MMP- | arx-2 | early four-cell | 8% | 100 | |
| mid four-cell | 15% | ||||
| MMP- | gon-1 | late four-cell | 100% | 20 | |
| MMP- | nas-37 | late four-cell | 100% | 20 | |
| MMP- | ant-1.1 | late four-cell | 30% | 66 | |
| Wild type | ant-1.1 | late four-cell | 88% | 40 | |
| MMP- | eif-1.A | late four-cell | 43% | 72 | |
| Wild type | eif-1.A | late four-cell | 97% | 29 | |
| MMP- | T04A8.6 | late four-cell | 51% | 74 | |
| Wild type | T04A8.6 | late four-cell | 95% | 21 | |
See Table S1 for additional scoring data, statistical information, and numbers of animals analyzed. Tx= Treatment; MMP-= zmp-1 (cg115); zmp-3 (tm3482); zmp-4 (tm3484); zmp-5 (tm3209); zmp-6 (tm3073); AC membrane marker = cdh-3>mCherry::PLCΔPH; BM marker = laminin::GFP
Additional zinc proteases such as ADAM, ADAMTS, cysteine, and serine proteases are implicated in cell invasion through ECM (Sevenich and Joyce, 2014). To determine if other proteases function with or compensate for MMP loss, we conducted an RNAi screen encompassing 87% (262/299) of the C. elegans genes with a putative protease or protease inhibitor domain in MMP- worms (Ihara et al., 2011) (Table S2). Failure of AC invasion disrupts uterine-vulval attachment and results in a protruding vulva (Pvl) phenotype (Sherwood and Sternberg, 2003). RNAi targeting nas-37 (Astracin-class metalloproteinase) and gon-1 (an ADAMTS9 ortholog) proteases resulted in significant numbers of Pvls (Table S2). Since defects in vulval muscle, neuronal innervation and uterine development can also cause Pvls, we scored AC invasion and found that neither gon-1 nor nas-37 enhanced the MMP- invasion defect (Table 1). These results suggest that protease-independent invasion may be occurring in MMP- animals.
MMPs accelerate AC invasion by removing BM
To determine if more subtle defects in AC invasion could be detected in MMP- worms, we examined the AC prior to, during, and after the normal time of invasion—P6.p 2-cell stage through the P6.p six-cell stage—by live-cell imaging of the BM component laminin (laminin::GFP). AC invasion was first viewed laterally and scored by examining the time of breach and subsequent BM gap enlargement (see Methods for timing of invasion, Table 1). Although AC invasion still occurred by the late P6.p 4-cell stage, breaching was delayed by approximately one hour in MMP- mutants compared to wild type animals (Figure 2A). Analysis of worms with two or more deletion alleles combined with laminin::GFP revealed that the combined loss of zmp-1 and zmp-6 (Table S1) phenocopied MMP- animals, suggesting that zmp-1 and zmp-6 are the dominant MMPs required for BM breaching. However, to ensure complete MMP loss, we continued to use the MMP- (quintuple mutant) animals for most analyses. To quantify BM removal rate, we examined invasion from a ventral perspective. Consistent with the delay in invasion, BM removal occurred more slowly in MMP- mutants than n wild-type animals (Figure 2B, Movie S1).
Figure 2. MMP loss delays BM breaching time and decreases ECM removal.

(A) Wild-type (left) and MMP- animals expressing laminin::GFP were scored for precise BM breaching time. Confocal imaging (ventral and lateral views of the same animal) at the early P6.p 4-cell stage show MMP- worm delayed in BM breaching. Arrows point to the BM breach (black area). The graph shows the percentage of AC invasion at each developmental time point (N ≥ 200 animals for each group, Table S1). Time after hatching at 20°C is shown. (B) 90 minute time-lapse of wild-type (top) and MMP- (bottom) animals shows BM removal after breaching is delayed in MMP- worms. Circular dotted lines show region of BM clearance at t=0. The graph shows quantification of the BM removal over time. Box plots show the average rates of BM clearing in wild-type animals (0.28 ± 0.08 μm2/min) and in MMP-mutants (0.09 ± 0.09 μm2/min, mean ± SD, p ≤ 0.01, Student’s t-test, n= 7 animals for wild-type: n=10 for MMP−). (C) Confocal sum projections of laminin::GFP (left panels) and collagen::mCh (right images) during AC invasion in wild type animals (upper) and in MMP- (lower) mutants show the progression of BM clearance by the AC. The fluorescence intensity of the BM at the perimeter of the cleared area (arrows) is increased in the absence of MMPs (~1.8 for laminin (1116 ± 352 vs 620 ± 75) and ~3.4 fold for collagen (1123 ± 426 vs 329 ± 279, mean ± SD, p≤0.01, Student’s t-test, wild-type, n=5 and 6; MMP-, n=9 and 5 for laminin::GFP and collagen::mCh, respectively)). (D) Schematic showing photoconversion of BM under the AC (green to red) before invasion. The amount of red BM physically displaced by the AC was calculated with post invasion images using regions 1–3 (see Methods). Grayscale (bottom, converted region is between red dotted lines) and spectral representation (top) of fluorescence intensity of optically converted laminin::Dendra show increased BM displacement in MMP- (right) animals (21 ± 0.09% in wild- type worms (left) versus 62 ± 35% in MMP- mutants, p≤0.05, mean ± SD, Student’s t-test, n=7 each group). Scale bars, 5μm.
MMPs have been proposed to aid invasion by enzymatically cleaving and degrading the ECM components of the BM (Cieplak and Strongin, 2017; Jacob and Prekeris, 2015). Previous work quantifying BM removal during AC invasion indicated that BM is both degraded and pushed aside (Hagedorn et al., 2013). We hypothesized that in the absence of MMPs, BM breaching and subsequent clearing might rely more on physical displacement. This is consistent with the density of the ring of laminin we see under the AC, suggesting significant BM displacement prior to the initial breach in MMP- animals (Figure 2A). We measured the mean ntensity of laminin::GFP bordering the site of invasion and found that it was significantly ncreased in MMP- animals throughout invasion (Figure 2C). A similar buildup of type IV collagen (EMB-9::mCherry) was also observed in MMP- worms (Figure 2C). To more directly assess BM displacement, we examined photoconvertible laminin::Dendra (Ihara et al., 2011). We photoconverted the laminin::Dendra in the area around the AC before invasion and then estimated the amount of BM displaced under the AC during invasion (see Methods, Figure 2D). Consistent with previous reports, we found that ~20% of the laminin was physically displaced in wild-type animals (Figure 2D) (Hagedorn et al., 2013). In contrast, ~60% of the laminin was displaced in MMP- animals, suggesting that BM opening proceeds predominantly through displacement, rather than degradation. Importantly, our analysis of BM removal did not capture aminin that moved beyond the edge of the gap or that which was torn off in MMP- animals (see below), thus our measurements likely underestimate displacement. Taken together, these data support the idea that MMPs accelerate invasion by removing and likely weakening the BM to increase the speed of BM breaching and displacement.
Large protrusions, rather than invadopodia, breach the BM in MMP- animals
AC invasion occurs via small dynamic invadopodia that initiate BM breaching, followed by the formation of a large protrusion that clears a wide gap in the BM (Hagedorn et al., 2013). To determine if the AC modifies its invasion mechanism in MMP- worms, we examined the AC with a probe for phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2; cdft-3>mCherry::PLCΔPH). PI(4,5)P2 localizes to the AC plasma membrane and concentrates at invadopodia (Hagedorn et al., 2013). Using live-cell imaging, we found that prior to BM breaching there were no differences in invadopodia number or diameter (Figure 3A, Movie S2). Thus, invadopodia appear to form normally in MMP- animals prior to BM breach.
Figure 3. Large protrusions breach the BM in the absence of MMPs.

(A) Ventral view 3D time series of dynamic invadopodia enriched with PtdIns(4,5)P2 (cdh-3>mCherry::PLCΔPH) in MMP- (bottom row) and wild-type (top row) animals at the early P6.p 2-cell stage. Colored spots are overlaid on invadopodia as identified and tracked by Imaris 3D software. No differences were detected in invadopodia number (left boxplot; 12.00 ± 4.82, n = 420 in MMP- versus 11.40 ± 3.46, mean ± SD, n = 298 wild type, p=0.06) or invadopodia diameter (right boxplot; 0.93 ± 0.37, N= 4608 MMP- versus 1.03 ± 0.39, N= 3429 wild-type, p<.0001, Student’s t-test). (B) Time series of the invasive protrusion (arrows, visualized with mCherry::PLCΔPH) that clears BM (purple arrowhead indicates BM breaching time) in wild type animals and initiates the BM breach in MMP- mutants. (C) At the time of BM breaching, an invadopodium (arrow; imaged with mCherry::PLCΔPH) occupies the BM gap (arrowhead; visualized with laminin::GFP) in wild type animals (upper panels). A large protrusion is associated with the BM breach in MMP- animals (lower panels). Isosurface renderings (magenta, dotted line represents the BM) were used to measure the volume of the AC’s protrusion that breached the BM and data from pooled animals is shown in the boxplot (17.3 ± μm3 MMP- versus 3.6 ± 1.9 μm3 wild-type, mean ± SD, p<0.01, Student’s t-test, N=5 each group). (D) Dorsal and ventral isosurface renderings (grayscale) of the BM breach in wild-type (top panels) and MMP- animals (bottom panels). Purple arrowheads point to the BM breach sites, and the cyan arrow points to a BM tear in the MMP- animal. Scale bars, 5μm.
To assess how the AC breaches the BM in MMP- animals, we performed time-lapse analysis from a lateral perspective. In wild type animals, one to four invadopodia breach the BM and a then single large protrusion grows from one of the breach sites ((Hagedorn et al., 2013), Figure 3B). MMP- animals breached the BM in a delayed manner with a single large protrusion that first deformed the BM. Small projections then emerged from these protrusions that broke through the BM (5/5 time-lapsed animals, Figure 3B). Single images captured at distinct time points during invasion confirmed that compared to wild type animals, where invadopodia made small BM breaches, in MMP- animals, a large protrusion distended the underlying BM (Figure 3C). Volumetric measurements revealed that a ~5-fold larger protrusion is required for BM penetration MMP- animals (Figure 3C). Dorsal and ventral isosurface renderings of BM from time-lapse imaging (Movie S3, ventral view unrendered time-lapse; Movie S4, rendered time point) revealed that in MMP- animals gross deformations in the BM were apparent in the dorsal tears (n=12/20 tears, Figure 3D, white arrow). These formed on the ventral side, often with tears (n=12/20 tears, Figure 3D, white arrow). These data suggest that in the absence of MMPs, the AC breaches the BM with physical force.
Loss of MMP function sensitizes invasion to Arp2/3 complex activity
We next investigated the source of the physical forces that allow MMP- AC invasion. Branched actin networks, driven by Arp2/3 complex actin nucleation, produce migratory and invasive membrane protrusions and has recently been implicated as playing a role in AC invasion (Cáceres et al., 2018; Swaney and Li, 2016). To determine if the Arp2/3 complex has a role in breaching the BM in MMP- animals, we first examined the Arp2/3 expression. The Arp2/3 complex in C. elegans is encoded by seven subunits (arx-1 through arx-7) (Sawa et al., 2003). A transcriptional reporter for arx-7 (arx-7>GFP) is expressed in the AC prior to and during invasion (Figure 4A). An ARX-2::GFP knock-in (Zhu et al., 2016) is also present in the AC and localizes to the invasive membrane (Figure 4C). To determine if Arp2/3 complex activity provides the protrusive force to drive BM invasion in MMP- animals, we treated wild-type and MMP- animals with RNAi targeting arx-2. Reduction of arx-2 resulted in significant invasion defects in wild-type animals (~40% invasion defect at the mid P6.p 4-cell stage, Figure 4B; Table 1). In MMP- worms, the invasion defect was enhanced from 20% to nearly 90% of RNAi treated animals (Figure 4B; Table 1). The enhancement of the invasion defect in MMP- animals and known role of the Arp2/3 branched actin networks in generating forces indicate that the Arp2/3 complex is crucial to BM breaching in the absence of MMPs.
Figure 4. An increase in Arp2/3 and F-actin support MMP-independent invasion.

(A) The Arp2/3 subunit arx-7 (arx-7>GFP) is expressed in the AC (arrow) during invasion. (B) Loss of MMPs sensitize animals to RNAi-mediated knockdown of the arx-7 (see Table S1) or arx-2 subunits. The percent of animals that display blocked, partial or normal invasion in wild type and MMP- animals is shown in the graph. (C) Wild type (left) and MMP- (right) ARX-2::GFP knock-in animals are shown in grayscale (top) and DIC overlayed by spectral imaging (bottom). The mean fluorescence of ARX-2::GFP at the invasive membrane (arrowheads) of the AC (arrow) is increased in MMP- animals (right boxplots; 7586 ± 1332 versus 5607 ± 991; mean ± SD, N= 10, each genotype; p<.01). (D) Representative 3D images of AC specific expression of F-actin (mCherry::moeABD) in grayscale (top). Isosurface rendering of F-actin intensity (magenta) in wild type versus MMP- animals. Graph shows quantification (1.58 ± 0.19 μm3 wild type vs 6.77 ± 3.54 μm3 MMP-, mean ± SD, p<.0001, Student’s t-test, n=15 each group). Scale bars, 5μm.
An adaptive response to MMP loss: the Arp2/3 complex and F-actin further enrich at the invasive front
In vitro atomic force microscopy studies of purified protein assemblies (Bieling et al., 2016) and manipulation of membrane tension with micropipettes in fish keratocytes (Mueller et al., 2017) have found that F-actin networks respond to mechanical loading by increasing in density, which enhances force production. Whether similar adaptive responses occur in vivo is not known (Plastino and Blanchoin, 2017). We hypothesized that the greater resistance exerted by the intact BM on the invading AC in MMP- worms may feedback on the F-actin network, causing the F-actin to respond by increasing its density. Consistent with this notion, mean ARX-2::GFP was 35% higher at the invasive membrane in MMP- animals (Figure 4C) and the volume of F-actin was ~40% compared to wild-type counterparts. Thus, in the absence of MMPs there is an adaptive increase in the Arp2/3 complex-driven invasive F-actin network, which likely enhances the force the AC generates to breach the BM.
Actomyosin contractility plays a minimal role in BM invasion
Chemical inhibition of MMPs in cultured cells leads to a switch from a mesenchymal to bleb-based amoeboid mode of migration dependent on Rho Kinase (ROCK) mediated actomyosin contractility (Te Boekhorst and Friedl, 2016). While examining AC morphology we detected spherical membrane structures that wild type animals never displayed (n > 100). ACs in MMP- animals often (~30%, n > 100) formed bleb-like structures that are attached to the cell body as well as detached tethered cell fragments ((Mayer et al., 2004; Wolf et al., 2003), Figure S3E). These structures never formed at the AC’s invasive front, were usually not in contact with the BM, and were never observed penetrating BM (n=50 animals observed, Figure S3F). Further, RNAi targeting of the C. elegans homologs of the actomyosin machinery—let-502 (ROCK), nmy-2 (non-muscle myosin heavy chain), mel-11 (myosin-associated serine/threonine protein phosphatase), mrck-1 (myosin light chain kinase), and mlc-4 (regulatory non-muscle myosin light chain)—in the presence or absence of MMPs produced minimal invasion defects (≥83% normal invasion, Table S1). Taken together, these data suggest that ROCK-mediated actomyosin-based contractility plays a minimal role in invasion in either the presence or absence of MMPs.
A genome-wide synergistic screen identifies genes that function with MMPs to promote invasion
In light of clinical MMP inhibitor trial failures (Cathcart et al., 2015; Te Boekhorst and Friedl, 2016), our results suggest that Arp2/3/F-actin and possibly other cell biological processes might compensate for lack of MMPs during tumor invasion through BMs. To identify these other mechanisms, we conducted a genome-wide synergistic interaction screen with MMP- mutant animals. Synergistic screens often reveal genes within separate pathways that work together to carry out a shared cell biological activity and are an underutilized strategy for anticancer therapeutics (Boone et al., 2007; O’Neil et al., 2017). To carry out this screen we took advantage of the invasion delay in MMP- animals, which does not result in a Pvl phenotype. We reasoned that knockdown of genes that synergistically interact with MMPs would fully block AC invasion in the MMP- strain and lead to a Pvl phenotype. We screened genes for which knockdown did not cause a highly penetrant Pvl phenotype in wild type worms, nor result in lethality or larval arrest. Three genes were recovered that met these criteria (Table S3; Movie S5): the mitochondrial adenine nucleotide transporter, ant-1.1 (Farina et al., 2008), the eukaryotic translation initiation factor, eif-1.A (Rhoads et al., 2006), and an ortholog of the yeast ribosome biogenesis promoting protein Nop15, T04A8.6 (Voutev et al., 2006). RNAi- mediated loss of these genes led to mild invasion defects on their own (ranging from 3–12%), however, RNAi-mediated loss in MMP- animals resulted in dramatic defects in AC invasion 46%−70%) (Figure S4A and Table 1). Translational reporters revealed that all three proteins are present at increased levels within the AC (Figure S4B–D), consistent with direct roles in promoting invasion.
Localized mitochondria and ATP production facilitates increased F-actin generation to breach BM in the absence of MMPs
We next wanted to understand the interplay of MMP loss and ant-1.1 reduction, as ant-1.1 knockdown in MMP- animals resulted in the greatest invasion defect (Table 1). The ant-1.1 gene encodes one of four C. elegans orthologs of the adenine nucleotide translocator family, an inner mitochondrial membrane protein that mediates exchange of cytosolic ADP for mitochondrial ATP, ensuring the intracellular supply of ATP produced by oxidative phosphorylation (Kunji et al., 2016). To independently test whether there is a greater requirement for ATP supply in the sensitized MMP- background, we used RNAi to knockdown genes encoding components of the electron transport chain, including cytochrome C (cyc-2.1), cytochrome C oxidase (cco-1), and cytochrome C reductase (cyc-1). All genes displayed synergistic defects for AC invasion in MMP- vs. wild-type animals, respectively (Table S4), supporting the notion of a greater dependency on ATP in MMP- animals.
To further understand how ANT-1.1 and mitochondria act in the absence of MMP’s, we examined the localization of ANT-1.1 using an in-frame translational GFP reporter (ant-1.1>ANT-1.1::GFP) (Farina et al., 2008). We found that ANT-1.1 polarized to the basal invasive membrane of the AC, but was not enriched basally in neighboring uterine cells (Figure 5C). A similar polarized pattern was observed for mitochondria (Mito Tracker Red CMXRos, Figure 5D). Strikingly, mitochondrial density was enriched more than 50% relative to wild-type at the invasive membrane in the MMP- animals, suggesting that mitochondria also adaptively increase at the AC’s invasive front. To determine if ATP production is also enriched here, we generated animals expressing a cytoplasmically localized fluorescence resonance energy transfer (FRET)-based ATP biosensor in the AC (cdh-3>ATeam). The sensitized emission (FRET/CFP ratios) in MMP- animals revealed that the FRET intensity was strongly polarized to the invasive membrane, indicating high levels of ATP at the invasive front (Figure 5E). Together, these results suggest that mitochondria polarize and further enrich in MMP- animals to deliver ATP to the site of invasion to allow BM breaching.
Figure 5. MMP- synergistic screen identifies a mitochondrial ADP/ATP translocase.

(A) RNAi clones targeting 11,511 genes were fed to newly hatched MMP- L1 animals. Adult worms with a Protruding vulval (Pvl) phenotype were scored for AC invasion defects if the gene did not cause Pvls in wild-type worms. (B) Representative images of AC (arrow) invasion in animals treated with ant-1.1 RNAi shows the blocked invasion in MMP- animals (arrowheads). (C) Grayscale (left) and spectral (right) imaging showing ANT-1.1 (ant-1.1>ANT-1.1::GFP) levels enriched in the AC (1.6 ± 0.06; n= 10; dotted outline). Within the AC, ANT-1.1 is enriched (1.5 ± 0.2; n=5; arrowhead) to the invasive membrane compared to neighboring uterine cells ((0.85 ± 0.2; n=5; mean ± SD, p=0.02) marked by asterisks, see Methods). (D) MitoTracker staining (top, and merged with DIC on bottom) shows a similar enrichment pattern to ANT-1.1 in the AC (arrow) compared to neighboring cells (AC/UC 1.8 ± 0.3, n=10; left bar graph and AC polarity 2.2 ± 0.9; n=11). Fluorescence intensity of MitoTracker Red is higher at the invasive membrane (arrowheads) in MMP- animals (boxplots MMP−= 7297 ± 1751, N=11; wild type=4436 ± 180, mean ± SD, N=10, p0.0001). (E) Representative images of sensitized emission (FRET/CFP ratios) spectra of the ATP biosensor in the AC (cdh-3>ATeam). ATP levels in the AC are highest at the invasive cell membrane (arrows) in MMP- animals (line graph of mean gray value plotted for 8 animals along the apical to basal (invasive) axis of the AC; bar graph = 2.5 ± 0.04, n=8). Scale bars, 5μm.
Active vesicle trafficking and cytoskeleton dynamics required for BM invasion are thought to be energy intensive (Altieri, 2017; Cunniff et al., 2016; Naegeli et al., 2017). We thus wanted to examine how mitochondria regulate BM invasion in MMP- animals, where there may be a greater energy requirement to breach the non-proteolyzed BM. In particular, we investigated the relationship of mitochondria to the F-actin network, as our results indicated that Arp2/3 complex-mediated F-actin polymerization was required for BM breaching in MMP- animals. Co-localization of mitochondria and F-actin during protrusion growth revealed that there was a tight association of mitochondria and F-actin, with mitochondria present at the base of the invasive F-actin protrusion prior to protrusion formation and then infiltrating into the protrusive F-actin network (n=6/6; Figure 6A and Movie S6). This association also occurs in wild type animals (Figure S4E). These observations suggest that the mitochondrial network provides a localized source of ATP to facilitate robust F-actin formation during BM invasion. Consistent with this notion, reduction of ant-1.1 in the MMP- animals resulted in a dramatic loss of the F-actin network at the invasive membrane: actin enrichment at the invasive membrane compared to the apical membrane fell from 3.6 to 1.7-fold and the total volume of F-actin decreased by 70% (Figure 6B). Taken together these results suggest a mechanism for the synergistic interaction between the loss of MMP activity and ANT-1.1—a reduction of localized ATP inhibits the adaptive increase in F-actin generation required to breach BM in MMP- animals (Figure 6C).
Figure 6. Mitochondria are tightly juxtaposed to the invasive F-actin networks.

(A) Mitochondria initially are juxtaposed to F-actin the invasive membrane before occupying the invasive protrusion alongside the F-actin network. Mitochondria staining is shown alone (DIOC6(3)); white, or merged with F-actin (mCherry::moeABD; magenta). Masked images and isosurface labeling of F-actin (magenta) and mitochondria (green) during early (left images), mid (center images) and late (right images) stages of AC invasion. (B) Actin enrichment decreased from 3.6 to 1.7 fold (p=0.0001) and the volume of the F-actin decreased by 70% (4.48 ± 2.34 to 1.31 ± 0.04 μm3; mean ± SD, p=0.006, n=8 each group) after treatment with ant-1.1 RNAi in MMP- animals. (C) Schematic diagram showing the time course of adaptive MMP- invasion. Invasion is delayed and is propelled by increased Arp2/3-mediated F-actin networks and enrichment of mitochondria/ATP (via ANT-1.1 ADP/ATP translocase), which helps form a large protrusion that breaches and displaces BM through physical forces. VDAC is an outer mitochondrial membrane pore that facilitates diffusion of small hydrophilic molecules such as ATP & ADP. Scale bars, 5 μm.
DISCUSSION
MMPs have long been associated with breaking down BM matrices during cell invasion in development and cancer (Kessenbrock et al., 2015; Shuman Moss et al., 2012). Supporting a function for MMPs, they are upregulated in carcinomas and invasive cell migrations in development (Brown and Murray, 2015; Rozario and DeSimone, 2010), localized to BM breaching invadopodial protrusions (Castro-Castro et al., 2016), and promote BM invasion in both in vitro and in xenograft assays (Brown and Murray, 2015; Lodillinsky et al., 2016). Our results establish that C. elegans MMPs promote BM cell invasion in vivo, but that they are not an obligatory component for BM breaching.
We found that three of the six encoded C. elegans MMPs are expressed in the AC during invasion (and two in neighboring tissues), the membrane-tethered MMP ZMP-1 localizes to AC-invadopodia, and optical highlighting of BM revealed that the C. elegans MMPs decrease BM levels at the site of invasion. The combined genetic loss of MMPs, however, only modestly delayed BM penetration. Importantly, extensive screening revealed that no other proteases enhanced the invasion defect in MMP- animals. Further, observations of the major BM components of laminin and type IV collagen along with optical highlighting experiments ndicated that the BM was largely displaced during MMP- invasion. Together these results strongly support the idea that the BM is crossed primarily through a physical route in MMP- animals. The BM in C. elegans is highly conserved with vertebrates, and type IV collagen, the main structural component, is present and cross-linked (Clay and Sherwood, 2015; Fidler et al., 2017). Further, the AC traverses two linked BMs during invasion—the neighboring vulval and uterine BMs (Morrissey et al., 2014). Thus, the AC can pierce a double BM barrier without MMPs, indicating the robustness of the invasive processes and providing a possible explanation for the failure of MMP inhibitor trials in cancer patients (Te Boekhorst and Friedl, 2016).
In response to MMP inhibition, cancer cells can switch from mesenchymal to amoeboid migration, allowing cells to squeeze through gaps in loose interstitial matrices (Wolf et al., 2013). Amoeboid migration is facilitated by Rho kinase (ROCK) mediated actomyosin contractility (Te Boekhorst and Friedl, 2016). Loss of actomyosin components did not enhance the MMP- AC invasion defect, suggesting that actomyosin contractility is not used as an adaptive mechanism to breach the BM. Recent in vitro and reconstitution studies found that branched F-actin networks formed by the Arp2/3 complex are inherently mechanosensitive and increase in network density and force generation in response to high loading (Plastino and Blanchoin, 2017). Whether Arp2/3 complex-based F-actin networks adaptively respond to altered loads in native tissue settings, however, is unclear. We discovered that in MMP- animals, Arp2/3 complex recruitment and F-actin formation increased along the AC invasive cell membrane, suggesting that the non-degraded BM may generate more load on the AC’s F-actin network. The expanded F-actin network that forms a large and dynamic protrusion likely generates more force, allowing the AC to breach the BM through physical means. Consistent with this idea, lamellipodia protrusions increase the force they exert when confronted with flexible glass fibers (Heinemann et al., 2011). Further, reduction of Arp2/3 complex activity dramatically enhanced the AC MMP- invasion defect, indicating a crucial role for the Arp2/3 complex in driving MMP- invasion. Recent studies have suggested that F-actin mediated forces play a role in breaching BM during AC invasion, but how these forces coordinate with MMP activity is unclear (Cáceres et al., 2018). Our results suggest that Arp2/3-branched F-actin generated forces and MMP-mediated BM degradation act together to drive BM invasion, and in the absence of MMPs the branched network increases and generates greater forces that breach the BM.
We performed a large-scale RNAi synergistic interaction screen to find additional genes that function with MMPs to promote BM invasion. We identified the mitochondrial ATP/ADP translocase ANT-1.1, which shuttles ATP out of the inner matrix, as a synergistic regulator of invasion (Farina et al., 2008; Kunji et al., 2016). Synergistic screens often detect genes that function in separate pathways that promote a common cellular function. Consistent with this idea, our results suggest that ANT-1.1 and mitochondria support BM breaching. We found that the ANT-1.1 protein and mitochondria polarize to the invasive cell membrane prior to and during invasion and that mitochondrial enrichment increases in the absence of MMPs. Using an ATP biosensor, we discovered that polarized mitochondria generate localized ATP along the AC’s invasive cell membrane. Loss of ANT-1.1 led to a dramatic reduction in Arp2/3 generated F- actin at the invasive membrane. Thus, polarized mitochondria appear to deliver ATP to fuel energy-dependent processes required for BM breaching--F-actin formation/turnover and possibly other energy consuming processes such as membrane trafficking (Altieri, 2017; Hagedorn et al., 2009; Naegeli et al., 2017).
The genetic interaction between loss of ant-1.1 and MMPs suggests that in the absence of MMPs, mitochondria become more essential, which is consistent with invasion driven primarily by highly energy-intensive physical means. ATP is not stored or kept at high levels in cells and its diffusion in the cytosol is thought to be limited by the dense and highly structured intracellular space (Zala et al., 2017; Zanotelli et al., 2018). Our results suggest that localized production is required to channel ATP to the highly focused and energy intensive process of BM breaching. The behavior of mitochondria is reminiscent of their activity in neurons, where mitochondria pool at energy-needy synapses and alter their localization in response to changing energy demands (Mironov, 2007; Schwarz, 2013). Mitochondria also polarize to the leading edge of several cancer cell lines in cell culture and localize to invasive cellular processes (Altieri, 2017; Arismendi-Morillo et al., 2012; Caino et al., 2015). Work in migrating ovarian cancer cells has suggested that mitochondrial movement is driven by high ATP utilization (Cunniff et al., 2016). The further enrichment of mitochondria in the AC of MMP- animals, may thus be in response to increased ATP utilization by the enlarged Arp2/3 F-actin network that breaches the BM, tying these adaptive processes together.
Cell invasion through BM barriers defines the initiation of metastatic cancer, however, there are no effective therapies targeting this lethal aspect of cancer progression (Te Boekhorst and Friedl, 2016). In yeast, synergistic screens have revealed that redundant and overlapping gene networks regulate cellular processes such as cell division, polarity, and DNA repair (O’Neil et al., 2017). Our genetic results indicate that cell invasion through BM is also highly robust and has built-in feedback mechanisms that allow for vigorous adaptation to ensure invasion completion. Synergistic screens for cell invasion through BM in vivo have not been previously possible, as they require large-scale screening in multicellular animals to find rare interactions. Our synergistic interaction screen during C. elegans AC invasion provides a framework for uncovering these robust networks, which will likely be crucial in developing more effective and more specific therapies to modulate cell invasion through BM barriers.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David R. Sherwood (david.sherwood@duke.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Worm handling and strains
Caenorhabditis elegans were reared under standard conditions at 15-to-25°C (Brenner, 1974). N2 Bristol strain was used as wild-type. Strains were viewed at room temperature. All animals scored were hermaphrodites during the L3 stage when the anchor cell (AC) invades. AC invasion was precisely staged in reference to vulval precursor cell (VPC) divisions and gonad development as previously described (Sherwood et al., 2005). Briefly, the AC is positioned over the central P6.p VPC cell prior to invasion in the early L3 larval stage. During the mid L3 stage the P6.p cell divides once (P6.p 2-cell stage). At the late P6.p 2-cell stage the AC initiates BM breach (near the time with the distal tip cells of the gonad arm begin migrating dorsally). At the P6.p 2-to-4-cell transition (when the P6.p daughters divide) the AC protrusion forms and clears an opening in the basement membrane and extends around and between the P6.p vulval precursor cell descendants. In the text and figures, we use a ‘>‘ symbol for linkages to a promoter and a ‘::’ symbol for linkages that fuse open reading frames (Ziel et al., 2009). The following alleles and transgenes were used: qyIs187 [zmp-2>GFP], qyIs201 [zmp-3>zmp-3::GFP], qyIs190 [zmp-4>zmp-4::GFP], qyIs245 [zmp-5>GFP], qyEx200 [zmp-6>GFP], qyIs192 [zmp-6>GFP], fos-1>rde-1, sEx16163 [T048.6>T048.6::GFP], sEx14510 [eif-1.A>EIF-1.A::GFP], pmcIs1 [ant-1.1>ANT-1.1::GFP], sbEx133 [AW20>mel-11::GFP], axIs1943 [FM050> mCherry::mlc-4], zhEx401 [let-502::nls::gfp, Plin-48::gfp], qyEx182[arx-7>arx-7::gfp]; LGI LP132 nmy-2(cp7[nmy-2::gfp]), qyIs57 [cdh-3 > mCherry::moeABD]; LGII qyIs17 [zmp-1>mCherry], qyIs23 [cdh-3>PH], rrf-3 (pl426), rol-6 (su1006); emb-9> emb-9::mCherry (qyIs45) LGIII zmp-1 (cg115), zmp-6 (tm3073), zmp-6 (tm3385), rde-1 (ne219); zmp-1(qy17[zmp-1::gfp +loxP]) LGIV qyIs108 [laminin::dendra]; LGV zmp-3 (tm3482), zmp-4 (tm3484), zmp-4 (tm3078), qyIs127 [laminin::mCh], cas607 [arx-2::gfp knock-in]; LGX zmp-5 (tm3209), qyIs166 [cdh-3>GFP::CAAX]
METHODS DETAILS
Generation of quintuple-mutant animals
zmp-1 and zmp-6 reside in close proximity (0.66 centimorgans apart) on chromosome V, and these mutations were linked by using gene mutations flanking zmp-1 that display obvious plate level phenotypes (Dpy and Unc). Specifically, hermaphrodite worms carrying mutations in unc-119, zmp-1, and dpy-1 (unc-119 zmp-1 dpy-1) on chromosome V were crossed with males harboring the zmp-6 mutant allele. We recovered worms that appeared Dpy (not Unc) in the F2 generation. PCR was then used for genotyping to check for the presence of the zmp-6 deletion (worms were zmp-1 zmp-6 dpy-1). zmp-3 and zmp-4 both reside on chromosome III. These genes are 22 centimorgans apart. Since the chance of recombination is high, we linked these genes by screening for and recovering recombinants through genotyping 200 F2 progeny from zmp-3 hermaphrodite worms crossed with male zmp-4 mutant worms. Quadruple mutants (zmp-1 zmp-6 (III); zmp-3 zmp-4 (V)) were created through the use of a strain that contained visual markers on chromosomes III M3 (egl-17>YFP) and V M5 (cdh-3>CFP). Males harboring both M3 and M5 markers (M3, M5) were crossed to zmp-1 zmp-6 (III) hermaphrodites. Male cross progeny were then mated to zmp-3 zmp-4 (V) hermaphrodites. Hermaphrodite progeny were recovered (1 per plate) that lacked M3 and contained M5. Self-progeny that did not contain M5 (these are homozygous zmp-3 zmp-4 (V) animals) were genotyped for the presence of the zmp-1 zmp-6 deletions (25% and 50% of the animals are homozygous or heterozygous for the mutation, respectively). Quintuple mutant animals were derived by crossing M3, M5 males to zmp-5 (X) hermaphrodites. Male progeny were then crossed to zmp-1 zmp-6 (III); zmp-3 zmp-4 (V) hermaphrodites. Hermaphrodite cross progeny were recovered that contained M3 and M5 and self-progeny were isolated that lacked both M3 and M5 and were subsequently genotyped for zmp-5. A similar strategy was used for crossing in cell biological markers. Cell biological marker strains were crossed with M3, M5 males, and then F1 males were crossed into quintuple hermaphrodites. M3, M5 animals were then excluded in the following generation, while the biological markers were retained and animals were genotyped for the five zmp deletions in the self-progeny to identify homozygous individuals. Primers used for genotyping are listed below.
| Target | Primer type | Primer sequence | Amplicon |
|---|---|---|---|
| zmp-1 | Forward | CCAGACCTAACCCAACATCC | Forward + Reverse = 2692 in wild type worms (no band); 386 in mutants; Internal Forward + Reverse = 452 in wild type worms |
| Reverse | CCAAAGATTGTGCCCAACAG | ||
| Internal Forward | CCGATTGTTGTGCAGTATCG | ||
| zmp-2 | Forward | AAACCTTGCAGTTTGACGGAGC | Forward + Reverse = 846 in wild type worms, 556 in mutants; Internal Forward + Reverse = 611 in wild type worms |
| Reverse | ATAATTCAGTGCCCGAGAGC | ||
| Internal Forward | GAAGGCCGACGCTCTAATG | ||
| zmp-3 | Forward | GGTCTTCCGATTTGTCAAAGG | Forward + Reverse = 495 in wild type worms, 197 in mutants; Internal Forward + Reverse primers = 364 in wild type |
| Reverse | GCTTCCGGCTCCATAAATCG | ||
| Internal Forward | CAGCACTTCGGACATCAAAA | ||
| zmp-4 | Forward | TCGCTTCCAATCTGCATTCC | Forward + Reverse = 697 in wild-type worms, 259 in mutants; Forward + Internal Reverse= 240 in wild type worms |
| Reverse | AAAGAACTAGAGGAGCCCTCG | ||
| Internal Reverse | CTTGTGACTTGTATGCCCATTCC | ||
| zmp-5 | Forward | TACAAACAGTACACGGGAGC | Forward + Reverse = 840 in wild-type worms, 687 in mutants; Internal Forward + Reverse = 700 in wild type worms |
| Reverse | AGACTTGCACTCCCTATTGG | ||
| Internal forward | GCGTCAGAACATCACATTTGG | ||
| zmp-6 | Forward | CATCTTCTGCCATGCCATCC | Forward + Reverse = 991 in wild type worms, 600 in mutants; Internal Reverse + Forward = 320 in wild type worms, no band in worms with the deletion |
| Reverse | AGAACCTACACTCAAAGAGAGC | ||
| Internal Reverse | ACAGGGGGAGTTGGGTTAAT |
Construction of fusion proteins
In text, figures, and methods, we use “>” to denote linkage to a promoter and to denote linkages of open reading frames. zmp-3>::zmp-3::GFP and zmp-6>zmp-6 reporters were described previously (Matus et al., 2015). A fragment containing the entire zmp-3 (5.4 kb) genomic region prior to its stop codon, or the zmp-6 promoter (7.8 kb), was amplified from N2 genomic DNA and then linked to GFP amplified from pPD95.75 by PCR fusion. zmp-4>zmp-4::GFP, and zmp-4>GFP were constructed by PCR fusion of the zmp-4 promoter (0.5 kb) region with (4.3 kb) or without the genomic region to GFP. zmp-2>zmp-2::GFP was construction by amplifying two fragments (3.3 kb flanked by Sma-1 and Nhe-1, and 4.2 kb flanked with Nhe-1 and Kpn-1 restriction sites) from N2 genomic DNA and cloned into pPD95.75. zmp-5>GFP was generated from amplifying the promoter region (7 kb) from genomic N2 DNA and then linked to GFP amplified from pPD95.75 by PCR fusion. For arx-7>GFP a 755 bp A fragment containing the arx-7 (755 bp) promoter region was amplified from N2 genomic DNA and then linked to GFP amplified from pPD95.75 by PCR fusion. Constructs were coinjected with 50 ng/μl unc-119 rescue DNA, ~50ng/pl pBsSK, and 25 ng/pl EcoRI cut salmon sperm DNA into unc-119(ed4) hermaphrodites. Multiple extrachromosomal lines with high transmission frequency were established and selected lines were integrated by gamma irradiation. Integrated lines were evaluated that produced 100% unc-119-rescued progeny, and selected lines were outcrossed to remove any background mutations.
Construction of the zmp-1 GFP knock-in strain
Since, ZMP-1 is a GPI membrane anchored protein with a N-terminal signal sequence, the GFP tag was inserted between the end of the hemopexin domain and the beginning of the GPI sequence, similar to prior approaches tagging transmembrane MMPs in mammalian systems (Radichev et al., 2010). The endogenous zmp-1 locus was tagged with GFP using CRIPSR- Cas9 mediated homologous recombination (qy17[zmp-1::gfp +loxP], (Dickinson and Goldstein, 2016)). Briefly, an sgRNA with the sequence 5’-GAACAAGCTTCTCATTGCGA-3’ targeted Cas9 to induce a double strand break in the stalk region of zmp-1. The double stranded break was repaired from a construct containing 1.5–2 KB of 5’ and 3’ genomic regions a mNeonGreen tag inserted between Cys-492 and Pro-493 of zmp-1. The homologous repair template and Cas9- sgRNA plasmids were coinjected into the gonad of young adult N2 worms. Animals that were recombinant were identified in the F3 offspring of injected animals based on the presence of selectable markers (dominant-negative sqt-1 rol phenotype and hygromycin resistance). Following strain isolation, the selectable markers were removed from the genome through Cre- Lox recombination and proper genome editing was confirmed by amplification and sequencing of the edited region.
Microscopy, image acquisition, processing, and analysis
Confocal images were acquired using a camera (EM-CCD; Hamamatsu Photonics) and a spinning disk confocal microscope (CSU-10; Yokogawa) mounted on a microscope (Axiolmager; Carl Zeiss) with a Plan-APOCHROMAT 100×/1.4 oil differential interference contrast objective and controlled by by iVision software (Biovision Technologies) or microManager (Edelstein et al., 2010). All images within each experiment were acquired using identical settings. Acquired images were processed using ImageJ 1.46r and Photoshop (CS6 Extended; Adobe). 3D reconstructions were built from confocal z-stacks, analyzed, and exported as .mov files using IMARIS 9.0.2 (Bitplane, Inc.). Figures were constructed using Illustrator (CS6 Extended; Adobe), and graphs were exported from JMP Pro (SAS). Movies were annotated using Photoshop, compressed with Handbrake and combined to run alongside other movies using Pic Stitch. Quantitative analyses of AC-invadopodia, invasive protrusion, or BM breach formation was done using either ImageJ, Imaris, or both. For time-lapse microscopy, worms were anesthetized in 0.2% tricaine and 0.02% levamosile in M9 and then transferred to 5% noble agar pads, sealed with VALAP, and imaged at 23°C. See Kelley et. al. for detailed worm staging, microscopy and data handling protocols and video tutorials.
RNA Interference
RNAi was delivered by feeding worms E. coli feeding strain HT115(DE3) expressing double stranded RNA (Fire et al., 1998). Bacteria harboring an empty RNAi vector (L4440) was used as a negative control for all RNAi experiments. Transcription of RNAi vector expression was induced with 1mM Isopropyl b-D-1-thiogalactopyranoside (IPTG) and cultures were plated on plates containing NGM and topical application of 5μL each of 30mg/mL carbenicillin and 1M IPTG. We generated a set of 261 RNAi clones targeting an overlapping set of genes predicted to have protease activity from the two commercially available genome-wide RNAi libraries, the C. elegans RNAi library (Source BioScience) (Kamath et al., 2003) and the C. elegans ORF-RNAi library (Source BioScience) (Rual et al., 2004). Our combined protease library targeted 88% (262) of the 299 genes with predicted protease or protease inhibitor domains in the C. elegans genome (see Table S2, (Ihara et al., 2011)). The whole genome RNAi screen included all the clones within the GE Healthcare RNAi library containing (11,511 clones, 55% of genome, (Rual et al., 2004)). In both screens, RNAi feeding was performed following L1 synchronization by hypochlorite treatment. MMP- L1 animals expressing a BM marker (laminin::dendra (qyIs108); zmp-1 (cg115); zmp-3 (tm3482); zmp-4 (tm3484); zmp-5 (tm3209); zmp-6 (tm3073)) were fed on bacterial lawns of Escherichia coli expressing double stranded RNA for ~51 hours, in six-well plates, and screened for the presence of a Protruding vulval (Pvl) phenotype using a dissecting microscope. 50–100 animals were examined per well and the number of Pvl animals recorded. The empty RNAi vector, L4440, was used as a negative control and an RNAi clone encoding fos-1, a transcription factors known to produce Pvl phenotypes following RNAi knockdown (Matus et al., 2010; Sherwood et al., 2005), was used as a positive control. All RNAi clones that resulted in the presence of multiple Pvl animals (>10) and had no ‘protruding vulva’ annotation in Wormbase under ‘Phenotypes’ were re-screened alongside wild-type counterparts (see Table S3). Clones that had a high number of Pvl in MMP- animals compared to wild-type animals over multiple experiments were scored for AC invasion defects (see Table S1). Following the initial RNAi high-throughput screen, the RNAi vector encoding double stranded RNA targeting all putative hits were sequenced to verify the correct insert and these clones were then used in subsequent experiments and delivered by feeding to synchronized L1- arrested larvae. For experiments targeting fos-1a, arx-2, and Rho/Rock pathway genes the corresponding ORF-RNAi library clone (Kamath et al., 2003; Rual et al., 2004) was delivered by feeding using the same methods as described above.
Mitochondrial Dyes
MitoTracker Red CMXRos (Molecular Probes) was diluted in M9 to a final concentration of 10nM. DiOC6(3) (3,3’-Dihexyloxacarbocyanine Iodide; Thermo) was reconstituted in DMSO, and diluted in M9 to a final concentration of 0.4 μl/ml. Either dye was added (200 μl) to the OP50 bacterial lawn of a 5cm plate and allowed to dry before L1 synchronized larvae were added. Worms remained on plates during development and were imaged at the L3 stage.
MMP inhibitor treatment
Previous reports demonstrated that recombinant protein products derived from the cloning of zmp-1, zmp-2, and zmp-3 and were sensitive to BB-94 in vitro (Wada et al., 1998). Synchronized L1 C. elegans mutants (bus-5(e2688)) highly sensitized to chemical uptake were incubated in 1 mM batimastat (BB94), or ilomostat (GM-6001, both from Calbiochem, USA) dissolved in soybean oil. Late L3 animals were scored for AC invasion defects and adult worms were scored for Pvl phenotypes.
QUANTIFICATION AND STATISTICAL ANALYSIS
Intensities and Optical highlighting of BM components
Quantification of percent laminin displacement was done by photoconverting all of the laminin::Dendra within a 15-μm radius of the AC with a compound light microscope (Zeiss Imager.A1) using the DAPI filter at the early P6.p two-cell stage (prior to invasion). Worms were recovered to OP50 plates for 2–3 h, and then reimaged from the ventral perspective on the Yokogawa CSU-10 spinning disk confocal. Sum projections of confocal z-stacks were then analyzed using ImageJ 1.46g software. Mean BM intensity, area, and integrated densities were measured sum projections of confocal z-stacks (0.5 μm slices). Three measurements were taken with ImageJ. First, the freehand line tool was used to outline and measure the BM area cleared by the AC (breach, region 1 in Figure 2D). Second, a freehand 15-pixel-wide line was drawn around the BM directly adjacent to the cleared area to measure BM accumulated at the edge of the hole (boundary, region 2). Third, a 15-pixel-wide line measured the BM just beyond area 2 (further from the hole) which represents uninterrupted BM (control, region 3 in the schematic). The following formula (developed by Hagedorn et al., 2013) was used to calculate the percent BM displaced: [((Mean fluorescence intensity of region 2) × (Area of region 2) − (Mean fluorescence intensity of region 3) × (Area of region 2) / (Mean fluorescence intensity of region 3) × (Area of region 1)) × 100]
Colocalization Analysis
Colocalization analysis of ZMP-1::GFP and mCherry::moeABD was performed on confocal z- sections using the JACoP Plugin in ImageJ (https://imagej.nih.gov/ij/plugins/track/jacop.html). Pearson’s correlation coefficients (r) are reported and are representative of 10 animals imaged ventrally (invadopodia) or laterally (invasive protrusion).
Analysis of AC Polarity, Enrichment, and Invasive Membrane Enrichment
For analysis of Mitotracker and ant-1.1>ANT-1.1::GFP up-regulation in the AC, regions of interest were drawn in ImageJ around the AC and then a neighboring uterine cell in the same animal; mean fluorescence intensity of each cell was then determined. Up-regulation was calculated as the following ratio: [AC mean intensity - background]/[neighboring uterine cell mean intensity - background]. Polarity was determined using the ratio of the mean fluorescence intensity from a 5-pixel-wide line scan drawn along the invasive and apicolateral membranes of ACs and in some cases neighboring uterine cells. Polarity was calculated as the following ratio: [invasive membrane mean intensity - background]/[apicolateral membrane mean intensity - background]. Enrichment of ARX-2::GFP or MitoTracker at the invasive membrane was calculated from invasive membrane mean intensity - background using a 15-pixel wide line.
Scoring of F-actin volume, invadopodia dynamics and BM removal rates
See our published Nature Protocols manuscript (Kelley et al., 2017) for detailed methods incuding video tutorials specific to these methods. In brief, confocal Z-stacks were used to make 3D reconstructions of F-actin networks in ACs expressing the F-actin probe mCherry::moeABD using Imaris (Bitplane, Inc.). Isosurface renderings of mCherry::moeABD were created setting a threshold that outlined the dense F-actin network at the invasive membrane in wild-type ACs. Quantitative measurements were then made for the volume and amount of fluorescence intensity with these isosurface renderings. AC invadopodia dynamics were quantified using the “Spots” module within Imaris, which facilitated the tracking of cdh-3 > mCherry::PLCδPH at the invasive membrane of the AC over time in wild type and MMP- animals. Quantification of BM- hole expansion over time was measured by exporting a time-series montage showing a ventral view of BM removal. Binary thresholds were applied using ImageJ to fit the BM hole and the following equation was used: BM Removed (μm2) = Area value × (μm of error bar/pixels width of error bar)2.
ATeam Sensitized Emission
Images for FRET were obtained on a Zeiss 880 Airyscan Inverted Confocal with an 63x/1.4 Oil objective at a constant pixel size of 0.15um with 408 and 514 lasers. Acceptor photobleaching experiments were used to reveal a FRET efficiency of approximately 10%. For sensitized emission, excitation and emission parameters and laser power were kept constant throughout imaging and were matched across channels. Lines that genetically encode the fluorescent ATP biosensor (ATeam, (Imamura et al., 2009; Yoshida et al., 2017)) optimized for use in C. elegans (AT1.03L, (Tsuyama et al., 2013)) were created by PCR fusion to an AC specific promoter (cdh-3>ATeam). The ATeam reporter is composed of the epsilon subunit of the bacterial F0F1-ATP synthase located between Venus and Cyan Fluorescent Protein. ATP binding to the epsilon subunit brings the two fluorescent proteins close to each other and increases FRET efficiency. Images were captured in the following manner: CFP (458 nm excitation and 463–517 nm emission); YFP (514nm excitation and 516–589 nm emission); and FRET (458 nm excitation and 516–589 nm emission). Donor and acceptor bleed through was calculated using “Fret and Colocalization Analyzer” ImageJ plug-in with images of animals containing cdh-3 > CFP and cdh-3 > YFP alone (n=5 each). Non-normalized FRET index was subsequently calculated using the same plug-in. Areas of false FRET were eliminated from non-colocalized areas of donor and acceptor fluorophores.
RNAi isolation and RT-qPCR
Wild type and MMP- animals (~500) were collected in M9, washed and re-suspended in 500 ul TRI Reagent (Molecular Research Center, USA), and then vortexed at room temperature for 20–30 minutes before 50 ul of BCP- Phase Separation Reagent was added (Molecular Research Center, USA). Samples were mixed by inversion and incubated at room temperature for 5 minutes before centrifugation at 13,000g @ −4° for 10 minutes. The aqueous phase containing RNAi was recovered and precipitated with an equal volume of isopropanol. Finally, the RNAi was washed with 70% ethanol before resuspension in 100ul of DPEC water. RNA samples where then treated with Turbo DNAse (Ambion by Life Technologies, USA) and concentrated with RNeasy MinElute Clean-Up Kit (Qiagen, USA). The quantity and quality of RNA extracted was determined by PCR and Agilent Bioanalyzer by the Microbiome Core within the Duke Center for Genomic and Computational Biology. Two-hundred nanograms of each extracted RNA sample was retrotranscribed to cDNA and quantified using the iTaq Universal SYBR Green One Step Kit (BioRad, Hercules, CA, USA). Real-time qPCR reactions were performed in an Eppendorf Mastercycler ep RealPlex thermocycler for 40 cycles with the following parameters: 95°C for 5 min to activate Taq polymerase, denaturation for 10 s at 95°C, annealing/extension for 30 s at 55°C. One final extension step was added for 1 min at 55°C. Cycle threshold (CT) was determined automatically by the Eppendorf RealPlex software. Primers were designed for intron spanning (exon junction) and off-target annealing checked by BLAST. Amplification efficiencies for each pair of primers targeting zmp-2 as well as the reference control, Y45F10D.4 (Zhang et al., 2012) were determined by qRT-PCR on three 10-fold serial dilutions of the same sample. These efficiencies were used to calculate RNA enrichment relative to the reference gene (Y45F10D.4). A melting curve analysis was performed after each run to ensure the specificity of products. Each experiment consisted of three biological replicate samples, each with three technical replicates.
Blinding and Unbiasing of Data
For polarity and fluorophore intensity measurements, data sets were randomized using an ImageJ macro (courtesy of Martin Hoehne) to blind analysis. For samples in which blind analysis was not possible, randomly selected samples were chosen for re-analysis to confirm precision of measurements.
Statistical analysis
For quantification of invasion, fluorescence levels and localization sample size was validated a posteriori for variance and statistical significance. The variance was similar within each experimental data set, as assessed for normality using a Shapiro-Wilk’s normality test for each data series. Statistical analyses were performed using either a two-tailed unpaired Student’s t-test or a Fisher’s exact probability test using JMP Pro 13 software (SAS). Figure legends specify when each test was used and the number of animals examined.
Supplementary Material
Movie S1. BM hole opening is delayed and irregular in MMP- (quintuple mutant) animals, Related to Figure 2B. Ventral view time-lapses (3 animals per genotype) shows the BM (laminin::GFP) gap opening in wild-type (left) and MMP- animals (right). The time points were acquired every 60 s over 90 minutes. The movies were constructed from projections of confocal z-section (step size of 0.5 μm).
Table S2. Protease screen, related to STAR Methods.
Table S3. Synthetic whole-genome screen, Related to Figure 5.
Table S4. AC invasion screen of electron transport chain and ATP synthase genes, Related to Figure 5.
Movie S2. Invadopodia form normally in the absence of MMPs, Related to Figure 3A. Ventral view time-lapses display invadopodia formation at the AC-BM interface. Wild type (left) and MMP- animals (right) express the invadopodia membrane marker cdh-3 mCherry::PLCΔPH. Colored spots are overlaid on invadopodia as identified and tracked by Imaris 3D software. The time points were acquired every 30 s over 30 minutes. The movies were constructed from projections of confocal z-section (step size of 0.5 μm).
Movie S3. MMP- invasion, Related to Figure 3B–D. Ventral view imaging of wild type (left) and MMP- (right) animal coexpressing BM (laminin::GFP, middle panels) and AC (cdh-3 > mCherry::PLCΔPH, lower; overlay top panels) markers shows delayed and irregular BM breaching in the MMP- animal. The time points were acquired every 60 s over 90 minutes. The movies were constructed from projections of confocal z-section (step size of 0.5 μm).
Movie S4. BM breaching in the absence of MMPs, Related to Figure 3D. 3-D Isosurface rendering of the BM (laminin::GFP) at the time of breach in MMP- and wild type animals (generated from Movie S3). Rotation is used to highlight the structure of the dorsal (on which the AC sits) and ventral (where the protrusion exits) sides of the BM. MMP- animals have excess BM built-up on the dorsal membrane and the ventral membrane displays a small tear where the protrusion has punched through.
Movie S5. Plate level phenotype of MMP- young adult worms after ant-1.1 knockdown, Related to Figure 5A, B. Movie shows MMP- animals fed control (left) or ant-1.1 (right) RNAi beginning at the L1 stage. Worms were imaged after development at 18°C for 52 hrs. RNAi mediated knockdown of ant-1.1 results in sterile animals with protruding-vulva (Pvl) phenotype, but otherwise appear healthy and do not show altered mobility. Worms were imaged at 21x and the 12 second Movie runs at 2x normal speed.
Movie S6. Mitochondrial infiltration into the invasive protrusion, Related to Figure 5A. Isosurface labeling of F-actin (magenta) and mitochondria (green, stained with DiOC6(3)) during early (left), mid (images), and late (images) stages of AC invasion in MMP- animals. Images are rotated to highlight the 3D structure of the actin-mitrochondrial interface.
Table S1: AC invasion scoring data, related to Table 1.
Highlights.
MMPs accelerate but are not essential for invasion through basement membrane (BM)
In MMP- animals a large protrusion breaches BM instead of invadopodia
Arp2/3-F-actin networks increase in density to allow BM breaching without MMPs
Mitochondria deliver more localized ATP for F-actin network growth without MMPs
ACKNOWLEDGEMENTS
We thank Sam Johnson and Lisa Cameron of the Duke University LMCF for imaging advice, K. Gordon for comments on the manuscript, and J. Hartman for advice with experiments. We thank Agnieszka Kawska (info@illuscientia) for her work on schematics. E.H. is supported by postdoctoral fellowship 129351-PF-16–024-01-CSM from the American Cancer Society. D.Q.M. was supported by the Leukemia and Lymphoma Society (3601–11/388–0036) and the National Cancer Institute (4R00CA154870–03); A.J.S. was supported by the American Cancer Society; and L.C.K. and D.R.S. are supported by the National Institutes of Health (F32GM103148, NIGMS R35, MIRA GM118049 and R21HD084290). J.P. was supported by Foundation Association pour la Recherche sur le Cancer (ARC) and the Idex ANR-10-IDEX- 0001–02 PSL.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATIONS OF INTEREST
The authors declare no competing interests.
REFERENCES
- Altieri DC (2017). Mitochondria on the move: emerging paradigms of organelle trafficking in tumour plasticity and metastasis. Br J Cancer 117, 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altincicek B, Fischer M, Fischer M, Lüersen K, Boll M, Wenzel U, and Vilcinskas A (2010). Role of matrix metalloproteinase ZMP-2 in pathogen resistance and development in Caenorhabditis elegans. Dev Comp Immunol 34, 1160–1169. [DOI] [PubMed] [Google Scholar]
- Arismendi-Morillo G, Hoa NT, Ge L, and Jadus MR (2012). Mitochondrial network in glioma’s invadopodia displays an activated state both in situ and in vitro: potential functional implications. Ultrastruct Pathol 36, 409–414. [DOI] [PubMed] [Google Scholar]
- Bieling P, Li T-D, Weichsel J, McGorty R, Jreij P, Huang B, Fletcher DA, and Mullins RD (2016). Force Feedback Controls Motor Activity and Mechanical Properties of Self-Assembling Branched Actin Networks. Cell 164, 115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone C, Bussey H, and Andrews BJ (2007). Exploring genetic interactions and networks with yeast. Nat Rev Genet 8, 437–449. [DOI] [PubMed] [Google Scholar]
- Brenner S (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GT, and Murray GI (2015). Current mechanistic insights into the roles of matrix metalloproteinases in tumour invasion and metastasis. J Pathol 237, 273–281. [DOI] [PubMed] [Google Scholar]
- elegans C Deletion Mutant Consortium (2012). large-scale screening for targeted knockouts in the Caenorhabditis elegans genome. G3 (Bethesda) 2, 1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cáceres R, Bojanala N, Kelley LC, Dreier J, Manzi J, Di Federico F, Chi Q, Risler T, Testa I, Sherwood DR, et al. (2018). Forces drive basement membrane invasion in Caenorhabditis elegans. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caino MC, Ghosh JC, Chae YC, Vaira V, Rivadeneira DB, Faversani A, Rampini P, Kossenkov AV, Aird KM, Zhang R, et al. (2015). PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc Natl Acad Sci U S A 112, 8638–8643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Castro A, Marchesin V, Monteiro P, Lodillinsky C, Rosse C, and Chavrier P (2016). Cellular and Molecular Mechanisms of MT1 -MMP-Dependent Cancer Cell Invasion. Annu Rev Cell Dev Biol 32, 555–576. [DOI] [PubMed] [Google Scholar]
- Cathcart J, Pulkoski-Gross A, and Cao J (2015). Targeting matrix metalloproteinases in cancer: bringing new life to old ideas. Genes Dis. 2, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieplak P, and Strongin AY (2017). Matrix metalloproteinases - From the cleavage data to the prediction tools and beyond. Biochim Biophys Acta. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay MR, and Sherwood DR (2015). Basement Membranes in the Worm: A Dynamic Scaffolding that Instructs Cellular Behaviors and Shapes Tissues. Curr Top Membr 76, 337–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Fingleton B, and Matrisian LM (2002). Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science 295, 2387–2392. [DOI] [PubMed] [Google Scholar]
- Cunniff B, McKenzie AJ, Heintz NH, and Howe AK (2016). AMPK activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Mol Biol Cell 27, 2662–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deryugina EI, and Quigley JP (2006). Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev 25, 9–34. [DOI] [PubMed] [Google Scholar]
- Dickinson DJ, and Goldstein B (2016). CRISPR-Based Methods for Caenorhabditis elegans Genome Engineering. Genetics 202, 885–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein A, Amodaj N, Hoover K, Vale R, and Stuurman N (2010). Computer control of microscopes using μManager. Curr Protoc Mol Biol Chapter 14, Unit14.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanjul-Fernández M, Folgueras AR, Cabrera S, and López-Otín C (2010). Matrix metalloproteinases: evolution, gene regulation and functional analysis in mouse models. Biochim Biophys Acta 1803, 3–19. [DOI] [PubMed] [Google Scholar]
- Farina F, Alberti A, Breuil N, Bolotin-Fukuhara M, Pinto M, and Culetto E (2008). Differential expression pattern of the four mitochondrial adenine nucleotide transporter ant genes and their roles during the development of Caenorhabditis elegans. Dev Dyn 237, 1668–1681. [DOI] [PubMed] [Google Scholar]
- Farooqui S, Pellegrino MW, Rimann I, Morf MK, Müller L, Fröhli E, and Hajnal A (2012). Coordinated lumen contraction and expansion during vulval tube morphogenesis in Caenorhabditis elegans. Dev Cell 23, 494–506. [DOI] [PubMed] [Google Scholar]
- Fidler AL, Darris CE, Chetyrkin SV, Pedchenko VK, Boudko SP, Brown KL, Gray Jerome W, Hudson JK, Rokas A, and Hudson BG (2017). Collagen IV and basement membrane at the evolutionary dawn of metazoan tissues. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, and Mello CC (1998). Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811. [DOI] [PubMed] [Google Scholar]
- Gaiko-Shcherbak A, Fabris G, Dreissen G, Merkel R, Hoffmann B, and Noetzel E (2015). The acinar cage: basement membranes determine molecule exchange and mechanical stability of human breast cell acini. PLoS ONE 10, e0145174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly K, Rejmak E, Mikosz M, Nikolaev E, Knapska E, and Kaczmarek L (2013). Matrix metalloproteinase (MMP) 9 transcription in mouse brain induced by fear learning. J Biol Chem 288, 20978–20991. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Hagedorn EJ, Yashiro H, Ziel JW, Ihara S, Wang Z, and Sherwood DR (2009). Integrin acts upstream of netrin signaling to regulate formation of the anchor cell’s invasive membrane in C. elegans. Dev Cell 17, 187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagedorn EJ, Ziel JW, Morrissey MA, Linden LM, Wang Z, Chi Q, Johnson SA, and Sherwood DR (2013). The netrin receptor DCC focuses invadopodia-driven basement membrane transmigration in vivo. J Cell Biol 201, 903–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Heinemann F, Doschke H, and Radmacher M (2011). Keratocyte lamellipodial protrusion is characterized by a concave force-velocity relation. Biophys J 100, 1420–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihara S, Hagedorn EJ, Morrissey MA, Chi Q, Motegi F, Kramer JM, and Sherwood DR (2011). Basement membrane sliding and targeted adhesion remodels tissue boundaries during uterine-vulval attachment in Caenorhabditis elegans. Nat Cell Biol 13, 641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura H, Nhat KPH, Togawa H, Saito K, Iino R, Kato-Yamada Y, Nagai T, and Noji H (2009). Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc Natl Acad Sci U S A 106, 15651–15656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoué S, Leblond CP, and Laurie GW (1983). Ultrastructure of Reichert’s membrane, a multilayered basement membrane in the parietal wall of the rat yolk sac. J Cell Biol 97, 1524–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob A, and Prekeris R (2015). The regulation of MMP targeting to invadopodia during cancer metastasis. Front. Cell Dev. Biol 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, et al. (2003). Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421, 231–237. [DOI] [PubMed] [Google Scholar]
- Kelley LC, Lohmer LL, Hagedorn EJ, and Sherwood DR (2014). Traversing the basement membrane in vivo: a diversity of strategies. J Cell Biol 204, 291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LC, Wang Z, Hagedorn EJ, Wang L, Shen W, Lei S, Johnson SA, and Sherwood DR (2017). Live-cell confocal microscopy and quantitative 4D image analysis of anchor-cell invasion through the basement membrane in Caenorhabditis elegans. Nat Protoc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessenbrock K, Wang C-Y, and Werb Z (2015). Matrix metalloproteinases in stem cell regulation and cancer. Matrix Biol 44-46, 184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Kapoor A, Desai S, Inamdar MM, and Sen S (2016). Proteolytic and non- proteolytic regulation of collective cell invasion: tuning by ECM density and organization. Sci Rep 6, 19905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunji ERS, Aleksandrova A, King MS, Majd H, Ashton VL, Cerson E, Springett R, Kibalchenko M, Tavoulari S, Crichton PG, et al. (2016). The transport mechanism of the mitochondrial ADP/ATP carrier. Biochim Biophys Acta 1863, 2379–2393. [DOI] [PubMed] [Google Scholar]
- Lodillinsky C, Infante E, Guichard A, Chaligné R, Fuhrmann L, Cyrta J, Irondelle M, Lagoutte E, Vacher S, Bonsang-Kitzis H, et al. (2016). p63/MT1-MMP axis is required for in situ to invasive transition in basal-like breast cancer. Oncogene 35, 344–357. [DOI] [PubMed] [Google Scholar]
- Lohmer LL, Kelley LC, Hagedorn EJ, and Sherwood DR (2014). Invadopodia and basement membrane invasion in vivo. Cell Adh Migr 8, 246–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmer LL, Clay MR, Naegeli KM, Chi Q, Ziel JW, Hagedorn EJ, Park JE, Jayadev R, and Sherwood DR (2016). A Sensitized Screen for Genes Promoting Invadopodia Function In Vivo: CDC-42 and Rab GDI-1 Direct Distinct Aspects of Invadopodia Formation. PLoS Genet 12, e1005786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen CD, and Sahai E (2010). Cancer dissemination--lessons from leukocytes. Dev Cell 19, 13–26. [DOI] [PubMed] [Google Scholar]
- Matus DQ, Chang E, Makohon-Moore SC, Hagedorn MA, Chi Q, and Sherwood DR (2014). Cell division and targeted cell cycle arrest opens and stabilizes basement membrane gaps. Nat Commun 5, 4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matus DQ, Li X-Y, Durbin S, Agarwal D, Chi Q, Weiss SJ, and Sherwood DR (2010). In vivo identification of regulators of cell invasion across basement membranes. Sci Signal 3, ra35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matus DQ, Lohmer LL, Kelley LC, Schindler AJ, Kohrman AQ, Barkoulas M, Zhang W, Chi Q, and Sherwood DR (2015). Invasive Cell Fate Requires G1 Cell-Cycle Arrest and Histone Deacetylase-Mediated Changes in Gene Expression. Dev Cell 35, 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer C, Maaser K, Daryab N, Zänker KS, Bröcker EB, and Friedl P (2004). Release of cell fragments by invading melanoma cells. Eur J Cell Biol 83, 709–715. [DOI] [PubMed] [Google Scholar]
- Menezes ME, Das SK, Minn I, Emdad L, Wang XY, Sarkar D, Pomper MG, and Fisher PB (2016). Detecting tumor metastases: the road to therapy starts here. Adv Cancer Res 132, 1–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov SL (2007). ADP regulates movements of mitochondria in neurons. Biophys J 92, 2944–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissey MA, Keeley DP, Hagedorn EJ, McClatchey STH, Chi Q, Hall DH, and Sherwood DR (2014). B-LINK: a hemicentin, plakin, and integrin-dependent adhesion system that links tissues by connecting adjacent basement membranes. Dev Cell 31, 319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller J, Szep G, Nemethova M, de Vries I, Lieber AD, Winkler C, Kruse K, Small JV, Schmeiser C, Keren K, et al. (2017). Load adaptation of lamellipodial actin networks. Cell 171, 188–200.e16. [DOI] [PubMed] [Google Scholar]
- Naegeli KM, Hastie E, Garde A, Wang Z, Keeley DP, Gordon KL, Pani AM, Kelley LC, Morrissey MA, Chi Q, et al. (2017). Cell Invasion In Vivo via Rapid Exocytosis of a Transient Lysosome-Derived Membrane Domain. Dev Cell 43, 403–417.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neil NJ, Bailey ML, and Hieter P (2017). Synthetic lethality and cancer. Nat Rev Genet 18, 613–623. [DOI] [PubMed] [Google Scholar]
- Overall CM, and Kleifeld O (2006). Tumour microenvironment - opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer 6, 227–239. [DOI] [PubMed] [Google Scholar]
- Page-McCaw A (2008). Remodeling the model organism: matrix metalloproteinase functions in invertebrates. Semin Cell Dev Biol 19, 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul CD, Mistriotis P, and Konstantopoulos K (2017). Cancer cell motility: lessons from migration in confined spaces. Nat Rev Cancer 17, 131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plastino J, and Blanchoin L (2017). Adaptive Actin Networks. Dev Cell 42, 565–566. [DOI] [PubMed] [Google Scholar]
- Pozzi A, Yurchenco PD, and Iozzo RV (2017). The nature and biology of basement membranes. Matrix Biol 57–58, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radichev IA, Remacle AG, Shiryaev SA, Purves AN, Johnson SL, Pellecchia M, and Strongin AY (2010). Biochemical characterization of the cellular glycosylphosphatidylinositol- linked membrane type-6 matrix metalloproteinase. J Biol Chem 285, 16076–16086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads RE, Dinkova TD, and Korneeva NL (2006). Mechanism and regulation of translation in C. elegans. WormBook 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozario T, and DeSimone DW (2010). The extracellular matrix in development and morphogenesis: a dynamic view. Dev Biol 341, 126–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rual J-F, Ceron J, Koreth J, Hao T, Nicot A-S, Hirozane-Kishikawa T, Vandenhaute J, Orkin SH, Hill DE, van den Heuvel S, et al. (2004). Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res 14, 2162–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeh F, Shimizu-Hirota R, and Weiss SJ (2009). Protease-dependent versus - independent cancer cell invasion programs: three-dimensional amoeboid movement revisited. J Cell Biol 185, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawa M, Suetsugu S, Sugimoto A, Miki H, Yamamoto M, and Takenawa T (2003). Essential role of the C. elegans Arp2/3 complex in cell migration during ventral enclosure. J Cell Sci 116, 1505–1518. [DOI] [PubMed] [Google Scholar]
- Schwarz TL (2013). Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevenich L, and Joyce JA (2014). Pericellular proteolysis in cancer. Genes Dev 28, 2331–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood DR, and Sternberg PW (2003). Anchor cell invasion into the vulval epithelium in C. elegans. Dev Cell 5, 21–31. [DOI] [PubMed] [Google Scholar]
- Sherwood DR, Butler JA, Kramer JM, and Sternberg PW (2005). FOS-1 promotes basement-membrane removal during anchor-cell invasion in C. elegans. Cell 121, 951–962. [DOI] [PubMed] [Google Scholar]
- Shuman Moss LA, Jensen-Taubman S, and Stetler-Stevenson WG (2012). Matrix metalloproteinases: changing roles in tumor progression and metastasis. Am J Pathol 181, 1895–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney KF, and Li R (2016). Function and regulation of the Arp2/3 complex during cell migration in diverse environments. Curr Opin Cell Biol 42, 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Te Boekhorst V, and Friedl P (2016). Plasticity of Cancer Cell Invasion-Mechanisms and Implications for Therapy. Adv Cancer Res 132, 209–264. [DOI] [PubMed] [Google Scholar]
- Tsuyama T, Kishikawa J, Han Y-W, Harada Y, Tsubouchi A, Noji H, Kakizuka A, Yokoyama K, Uemura T, and Imamura H (2013). In vivo fluorescent adenosine 5’- triphosphate (ATP) imaging of Drosophila melanogaster and Caenorhabditis elegans by using a genetically encoded fluorescent ATP biosensor optimized for low temperatures. Anal Chem 85, 7889–7896. [DOI] [PubMed] [Google Scholar]
- Voutev R, Killian DJ, Ahn JH, and Hubbard EJA (2006). Alterations in ribosome biogenesis cause specific defects in C. elegans hermaphrodite gonadogenesis. Dev Biol 298, 45–58. [DOI] [PubMed] [Google Scholar]
- Wada K, Sato H, Kinoh H, Kajita M, Yamamoto H, and Seiki M (1998). Cloning of three Caenorhabditis elegans genes potentially encoding novel matrix metalloproteinases. Gene 211, 57–62. [DOI] [PubMed] [Google Scholar]
- Wang Z, Linden LM, Naegeli KM, Ziel JW, Chi Q, Hagedorn EJ, Savage NS, and Sherwood DR (2014). UNC-6 (netrin) stabilizes oscillatory clustering of the UNC-40 (DCC) receptor to orient polarity. J Cell Biol 206, 619–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, Deryugina EI, Strongin AY, Bröcker E-B, and Friedl P (2003). Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J Cell Biol 160, 267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, Te Lindert M, Krause M, Alexander S, Te Riet J, Willis AL, Hoffman RM, Figdor CG, Weiss SJ, and Friedl P (2013). Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol 201, 1069–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Alfaqaan S, Sasaoka N, and Imamura H (2017). Application of FRET-Based Biosensor “ATeam” for Visualization of ATP Levels in the Mitochondrial Matrix of Living Mammalian Cells. Methods Mol Biol 1567, 231–243. [DOI] [PubMed] [Google Scholar]
- Yurchenco PD (2011). Basement membranes: cell scaffoldings and signaling platforms. Cold Spring Harb Perspect Biol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurchenco PD, Cheng YS, and Colognato H (1992). Laminin forms an independent network in basement membranes. J Cell Biol 117, 1119–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zala D, Schlattner U, Desvignes T, Bobe J, Roux A, Chavrier P, and Boissan M (2017). The advantage of channeling nucleotides for very processive functions. [version 2; referees: 3 approved]. F1000Res 6, 724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotelli MR, Goldblatt ZE, Miller JP, Bordeleau F, Li J, VanderBurgh JA, Lampi MC, King MR, and Reinhart-King CA (2018). Regulation of ATP utilization during metastatic cell migration by collagen architecture. Mol Biol Cell 29, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziel JW, Hagedorn EJ, Audhya A, and Sherwood DR (2009). UNC-6 (netrin) orients the invasive membrane of the anchor cell in C. elegans. Nat Cell Biol 11, 183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen D, Smith MA, Zhang B, and Pan X (2012). Selection of reliable reference genes in Caenorhabditis elegans for analysis of nanotoxicity. PLoS ONE 7, e31849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Chai Y, Jiang Y, Li W, Hu H, Li W, Wu J-W, Wang Z-X, Huang S, and Ou G (2016). Functional coordination of WAVE and WASP in c. elegans neuroblast migration. Dev Cell 39, 224–238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. BM hole opening is delayed and irregular in MMP- (quintuple mutant) animals, Related to Figure 2B. Ventral view time-lapses (3 animals per genotype) shows the BM (laminin::GFP) gap opening in wild-type (left) and MMP- animals (right). The time points were acquired every 60 s over 90 minutes. The movies were constructed from projections of confocal z-section (step size of 0.5 μm).
Table S2. Protease screen, related to STAR Methods.
Table S3. Synthetic whole-genome screen, Related to Figure 5.
Table S4. AC invasion screen of electron transport chain and ATP synthase genes, Related to Figure 5.
Movie S2. Invadopodia form normally in the absence of MMPs, Related to Figure 3A. Ventral view time-lapses display invadopodia formation at the AC-BM interface. Wild type (left) and MMP- animals (right) express the invadopodia membrane marker cdh-3 mCherry::PLCΔPH. Colored spots are overlaid on invadopodia as identified and tracked by Imaris 3D software. The time points were acquired every 30 s over 30 minutes. The movies were constructed from projections of confocal z-section (step size of 0.5 μm).
Movie S3. MMP- invasion, Related to Figure 3B–D. Ventral view imaging of wild type (left) and MMP- (right) animal coexpressing BM (laminin::GFP, middle panels) and AC (cdh-3 > mCherry::PLCΔPH, lower; overlay top panels) markers shows delayed and irregular BM breaching in the MMP- animal. The time points were acquired every 60 s over 90 minutes. The movies were constructed from projections of confocal z-section (step size of 0.5 μm).
Movie S4. BM breaching in the absence of MMPs, Related to Figure 3D. 3-D Isosurface rendering of the BM (laminin::GFP) at the time of breach in MMP- and wild type animals (generated from Movie S3). Rotation is used to highlight the structure of the dorsal (on which the AC sits) and ventral (where the protrusion exits) sides of the BM. MMP- animals have excess BM built-up on the dorsal membrane and the ventral membrane displays a small tear where the protrusion has punched through.
Movie S5. Plate level phenotype of MMP- young adult worms after ant-1.1 knockdown, Related to Figure 5A, B. Movie shows MMP- animals fed control (left) or ant-1.1 (right) RNAi beginning at the L1 stage. Worms were imaged after development at 18°C for 52 hrs. RNAi mediated knockdown of ant-1.1 results in sterile animals with protruding-vulva (Pvl) phenotype, but otherwise appear healthy and do not show altered mobility. Worms were imaged at 21x and the 12 second Movie runs at 2x normal speed.
Movie S6. Mitochondrial infiltration into the invasive protrusion, Related to Figure 5A. Isosurface labeling of F-actin (magenta) and mitochondria (green, stained with DiOC6(3)) during early (left), mid (images), and late (images) stages of AC invasion in MMP- animals. Images are rotated to highlight the 3D structure of the actin-mitrochondrial interface.
Table S1: AC invasion scoring data, related to Table 1.