

Abstract
Chemical-stimuli-responsive nanotherapeutics have gained great interest in drug delivery and diagnosis applications. These nanotherapeutics are designed to respond to specific internal stimuli including pH, ionic strength, redox, reactive oxygen species, glucose, enzymes, ATP and hypoxia for site-specific and responsive or triggered release of payloads and/or biomarker detections. This review systematically and comprehensively addresses up-to-date technological and design strategies, and challenges nanomaterials to be used for triggered release and sensing in response to chemical stimuli.
Keywords: Nanotherapeutics, chemical stimuli, pH, ionic strength, redox potential, glucose, enzyme, reactive oxygen species, responsive release, targeting
Introduction
Although many therapeutics have been developed for the diagnoses and treatments of cancer, neurological disorders, infection, immunological diseases and other human diseases, the use of these therapeutics are still hampered by the lack of effective delivery systems [1,2]. The reason is because these therapeutics have very short half-lives, do not cross some biological barriers and are easily metabolized at other tissue sites [3]. Because nanoparticles are small in size with a large surface area and can be made of many types of materials with multifunctional surface groups, they hold significant advantages in transporting drugs across biological barriers [4], reducing drug clearance and improving drug stability and bioavailability at the targets [5–8]. These nanomaterials include inorganic and polymeric nanoparticles, polymeric micelles, liposomes, dendrimers, nanocapsules, nanotubes and nanogels, among others. They can control release of drugs in different manners and release rates owing to their different architectures. Nanoparticle is a general term for particles that are nano-sized and can have any shape. Polymeric micelles have a core–shell structure made of amphiphilic block copolymers [9]. Liposomes have a single or multiple lipid bilayer structure with their inside core and surface being hydrophilic [10]. Dendrimers have a highly branched treelike structure with many internal cavities and a central core that is hydrophobic [11]. Nanocapsules have a hollow core covered by a polymeric membrane, and can have any shape [12]. If the shape is a sphere the nanocapsule is called a hollow sphere. If the shape is cylindrical the nanocapsule is called a nanotube [13]. Nanogels have a physically or chemically crosslinked network structure [14].
When the nanomaterials contain stimuli-responsive moieties, they can release drugs in response to endogenous or exogenous stimuli. These stimuli could be of physical or chemical origin. The physical stimuli include temperature [15], electric fields, magnetic fields, ultrasound, light and X-rays [16]. They have the advantage of being easy to be controlled with reduced variability. However, they have disadvantages of being external, which can be costly and also not fully tolerated by the body. By contrast, many chemical stimuli are internal coming from microenvironmental changes in various organs, tissues or cells associated with tumors, inflammation, infection and other disease conditions. These chemical stimuli are: pH, ionic strength, redox, reactive oxygen species (ROS; an oxygen-containing compound that is particularly reactive), glucose, enzymes, ATP and hypoxia [17]. They can trigger selective drug release at the target site without the need of an external device. Specifically, these chemical stimuli are: variations of pH values in the gastrointestinal (GI) tract (stomach pH ~1–3, duodenum and ileum pH ~6.6–7.5), vaginal tract (pH ~4.2–5), subendosomal/lysosomal organelles (pH ~4.5–6.8) and tumor tissues (pH ~6.5–7.2); ionic strength; glutathione levels in cytoplasm and nuclei; ROS in mitochondria [18–20]; dysregulated enzyme levels in lysosomes; intracellular and extracellular levels of ATP; and hypoxia. Figure 1 schematically illustrates these chemical stimuli. In this review, we will discuss strategies to design chemical-stimuli-responsive nanotherapeutics and summarize their up-to-date biomedical applications.
Figure 1.

Schematic illustration of the common chemical stimuli present in various organs, cancerous tissues and cells. These chemical stimuli include: high ionic strength and dysregulated enzymes in the eye; variations in pH in the mouth and along the gastrointestinal tract; high blood glucose levels in patients with diabetes; higher levels of acidity and dysregulated enzymes in microenvironments of tumorous tissues; and high levels of acidity, reactive oxygen species (ROS), dysregulated enzymes and glutathione tripeptide (GSH) in cancerous cells.
pH-responsive nanotherapeutics
The pH of normal extracellular organelles and the bloodstream is ~7.4, whereas the pH is 1–3 in the stomach and 6.6–7.5 in the duodenum and ileum of the GI tract. The pH values of normal intracellular subendosomal and lysosomal organelles are 5.5–6.8 and 4.5–5.5, respectively [21]. In the diseased tissues, as a result of cancer, inflammation and infection, for example, the pH is usually decreased owing to dysregulated metabolism or irregular angiogenesis, which cause the rapid shortage of oxygen and nutrients resulting in a shift toward glycolytic metabolism [22]. The variations of the pH values in different organs, tissues, intracellular compartments and diseased tissues can be utilized to design nanomaterials that can control the release of therapeutics at the target site in response to the pH changes. Generally, pH-responsive nanomaterials contain either ionizable groups or blocks such as poly(acrylic acid) (pKa ~4.52), poly(methacrylic acid) (pKa ~5.5), poly(glutamic acid) (pKa ~4.9), poly(vinyl pyridine) (pKa ~4), chitosan (pKa ~6.2) and aminoalkyl methacrylate copolymer (pKa ~10) [23,24] (Figure 2a); or acid-labile bonds such as hydrazone orthoesters, benzoic imine and acetal [25–29] (Figure 2b). The ionizable groups experience solubility or conformational changes upon exposure to variations in pH, whereas acid-labile bonds can be cleaved in the acidic environment, resulting in the release of the drugs loaded in the nanomaterials at the target site.
Figure 2.

Chemical structures of commonly used (a) pH-responsive polymers that undergo pH-dependent conformational changes and (b) acid-labile linkages and their pHresponsive cleavage mechanism.
pH-sensitive compounds including glycocholate, Eudragit® polymer [Eudragit® L100 (MW 135 000 Da)], stearic acid, poly(acrylic acid) and poly(methacrylic acid) have been used to either coat or be chemically incorporated into nanoparticles made of liposomes, poly(lactic-co-glycolic acid) (PLGA) and silica to protect insulin [30], incretin hormone glucagon-like peptide-1 [31] or calcitonin [32], from the hostile acid environment in the stomach after oral administration, increasing the absorption of these therapeutics in the intestine, and thus increasing the bioavailability of these therapeutics. Because these nanocarriers are acidic, they are in their collapsed and hydrophobic state in the stomach as a result of the protonation of their carboxylic acid moieties at pH 1–3. After gastric passage, pH increases to 7.5 in the ileum, leading to the ionization of carboxylic acids and breakage of H-bonds. Consequently, nanoparticles swell and release the drug payload [33]. Peppas and colleagues showed that pH-sensitive nanospheres made of crosslinked poly(ethylene glycol) (PEG), grafted methacrylic acid or acrylic acid increased their hydrodynamic sizes from 200–350 nm to 600–2900 nm, ~tenfold, when pH was increased from 2 to 6 [34]. Both types of nanospheres released over 90% of the loaded insulin within 1.5 h at pH 7, mimicking physiological conditions. However, at low pH (i.e., pH 3), mimicking the stomach conditions, they both reduced or prevented the release of insulin and, furthermore, the nanospheres made of acrylic acid released significantly less insulin than those made of methacrylic acid, 75% vs ~10% in 1 h. Holst reported that silica nanocarriers coated with pH-responsive Eudragit® [poly(methacrylic acid methyl ester)] improved the oral bioavailability of incretin hormone glucagon-like peptide-1 (GLP-1) [35], a hormone secreted by the endocrine L cells of the intestinal mucosa that stimulates insulin secretion in response to meals and has antidiabetic potential [31]. Their results showed that the nanoparticles released <30% of the loaded GLP-1 at pH 1 but >80% at pH 7.4 during a 12 h period. The nanoparticles increased the oral hypoglycemic efficacy of GLP-1 by ~1.5 times and the intestinal mucosa permeability of GLP-1 by about five times in adult male Sprague–Dawley (SD) rats.
The pH variations in various intracellular compartments of cancer cells have been widely utilized to design nanoparticles for controlled release of anticancer drugs inside cancer cells to kill them. The mechanism-of-action is described here. After being internalized through endocytosis, drug-loaded pH-responsive nanomaterials containing acid-labile groups are entrapped in endocytic vesicles (endosomes). At early stages of development, endosomes are at a pH of ~6 but are acidified to pH 5.5 at later stages of development (late endosomes) owing to a proton-pump enzyme, resulting in partial drug release from the nanomaterials. After endosomal escape, the nanotherapeutics are taken up by more-acidified vesicles which are lysosomes with a pH of ~4.5. The highly acidic hostile environment within the lysosome, along with certain degradative enzymes, leads to the cleavage of acid-labile groups or breakage of nanocarriers to trigger the complete release of the encapsulated drugs [36] (Figure 3). For example, Deng et al. designed chitosan– silica hollow nanospheres to release tumor necrosis factor (TNF)α in MCF-7 cells in response to the acidic pH of the tumor microenvironment [37]. The chitosan–silica hollow nanospheres were prepared by reacting the silanol groups present on the surface of SiO2 nanoparticles with 3-glycidyloxypropyl-trimethoxysilane and then with chitosan to achieve crosslinking on the surface of SiO2 nanoparticles. The surface of the nanospheres was subsequently conjugated with ErbB2 monoclonal antibody to facilitate the uptake of the nanotherapeutics in tumor cells. BSA was used as a model protein drug to assess the release profile in PBS under various pH conditions. The nanospheres showed pH-dependent release of BSA with very little drug release <15% at pH 7.4 but a significantly higher amount (90%) at pH 4 in 100 h. The in vitro studies of antibody-conjugated and TNFα-loaded chitosan–silica hollow nanospheres showed a concentration-dependent killing effect in MCF-7 cells, with 90% cell viability at 50 μg/ml and <50% cell viability at 250 μg/ml. The in vivo experiments were conducted using athymic nude mouse models of HER-2-positive breast cancer where formulations were intraperitoneally administered to the mice once every 2 days. The weights of tumors treated with TNFα loaded on the CS–SiO2 nanospheres decreased onefold (45% of the control) – more than those treated with free-TNFα (~90% of the control) after 2 weeks of treatment. These results indicated that TNFα-loaded CS–SiO2 nanospheres were more effective in inhibiting tumor growth than free TNFα.
Figure 3.

Schematic mechanism of triggered drug release from pH-responsive nanocarriers in response to the acidic environment within the cancer cell. After being internalized through endocytosis, the nanocarriers are entrapped in endosomes. At early stages of development, endosomes are ~pH 6, but are acidified to pH 5.5 at later stages of development, resulting in partial drug release of the payload. After endosomal escape, the nanocarriers are taken up by lysosomes with a pH of ~4.5. The highly acidic hostile environment within the lysosome along with certain degradative enzymes lead to the full cleavage of acid-labile groups of nanocarriers to trigger the complete release of the drug encapsulated in the nanocarriers.
Min et al. fabricated pH-responsive polymeric micelles comprising a block copolymer of PEG and poly(β-amino ester), and loaded camptothecin into the micelles. The PEG block was hydrophilic and stayed on the surface of the micelles, whereas the poly(β-amino ester) block was hydrophobic, hydrolytically degradable and pH sensitive and stayed in the core of the micelles. The micelles were stable at neutral pH but underwent demicellization at pH 6.4 to release camptothecin [38]. Lee et al. also formulated pH-responsive polymeric micelles using a triblock copolymer [poly(L-lactic acid)-b-poly(ethylene glycol)-b-poly(L-histidine)] (PLA-PEG-PLH) [39]. Their rationale for designing the micelles were: (i) the PEG component of the triblock copolymer would enable the prolonged circulation of the micelles in the bloodstream; (ii) the PLA block would serve as a container to encapsulate hydrophobic drugs; and (iii) the PLH block would work as a pH-sensitive component because of the imidazole ring that had an electron lone pair on the unsaturated nitrogen that would provide PLH with an amphoteric property by a protonation–deprotonation equilibrium. To increase the cell membrane penetration of the micelles, the authors grafted the micelles with transactivator of transcription (TAT). TAT is an important cell-penetrating peptide derived from human immunodeficiency viruses (i.e., HIV-1 and HIV-2) and has been reported to facilitate the transport of nanoparticles, genes or proteins across cell membranes [40,41]. The authors further reported that TAT was hidden by the micelles during blood circulation but was exposed to the surface of the micelles at acidic pH in the tumor tissue to enhance the uptake of the micelles by MCF-7 cells. Once the micelles entered early endosomes of the cells, the acidic environment caused the degradation of PLA and consequently rapid release of doxorubicin loaded in the micelles. In a similar approach, pH-sensitive PEGylated long-circulating liposomes (HSPC:cholesterol and Doxil®), functionalized with TAT peptide and cancer-specific monoclonal antibody (mAb 2C5), were developed in such a way that TAT was grafted onto a small-chain PEG, whereas an acid-labile hydrazone group was introduced into a long-chain PEG [42]. At physiological pH, TAT molecules remained hidden owing to the shielding effect of the pH-responsive long PEG chains. However, once in contact with an acidic environment, the TAT molecule was exposed to the outlier of the liposomes owing to the degradation of the hydrazone bond and cleavage of the long-chain PEG. TAT-modified liposomes showed sixfold greater cellular uptake by MCF-7 human breast cancer cells and B16-F10 skin melanoma cells, compared with plain liposomes, and around a fourfold greater cancer-cell-killing effect of Doxil® grafted on the TAT-modified liposomes in B16-F10, HeLa and MCF-7 cells, compared with free doxorubicin at pH 5.
Besides the use for cancer treatments discussed above, pH-responsive nanotherapeutics have also been designed to treat infections. Radovic-Moreno et al. [43] formed micelles made of PLGA-b-polyhistidine-b-PEG triblock copolymer for controlled release of vancomycin, a glycopeptide-based antibiotic indicated to treat bacterial infections, specifically for drug-resistant strains of Staphylococcus aureus, at the infection site. Figure 4 illustrates that the vancomycin-containing micelles were stable in the bloodstream owing to the stealth property of PEG on the micelle surface and would not be endocytosed by nontarget cells owing to their slightly negative charge at physiological pH. At the site of infection, the bacterial cell wall was acidic and caused the polyhistidine in the micelles to be ionized and bound to the bacterial cell wall, and subsequently the release of the antibiotic vancomycin from the micelles to kill the bacterium.
Figure 4.

Schematic illustration of the targeted release of an antibiotic from pHresponsive nanocarriers into the interstices of a bacterium. The nanosystems were vancomycin-loaded micelles made of poly(lactic-co-glycolic acid)-b-polyhistidine-b- polyethylene glycol (PLGA-PLH-PEG) triblock copolymer. PLGA, PLH and PEG contributed to the core, interlayer and surface of the micelles, respectively. Vancomycin was loaded inside the hydrophobic core (PLGA block) of the micelles owing to its hydrophobicity. In the bloodstream, the pH was 7.4 and the micelles were stable at this physiological pH owing to the stealth property of hydrophilic PEG block on the micelle surface. The micelles would not be endocytosed by non-target cells owing to the slightly negative charge of PLH at physiological pH. At the site of infection, the pH was low and the high acidity ionized the polyhistidine component to render positive charges to the micelles. The positively charged micelles bound to the negatively charged bacterial cell wall, and subsequently released the loaded vancomycin antibiotic to kill bacteria. Adapted, with permission, from [43].
Ionic-strength-responsive nanotherapeutics
Various body fluids such as tears, blood, GI fluid and interstitial fluid contain cation and/or anion forms of ionizable groups. These ionizable groups can be utilized as internal-ionic-strength-stimuli for the controlled release of therapeutic molecules or diagnostic applications. The nanomaterials that respond to the variations in ionic strength undergo phase transition from hydrophobic to hydrophilic forms resulting in the increase of their particle size and solubility (or vice versa) to release drugs accordingly [44]. The common ionic-strength-responsive nanomaterials are fabricated in the form of ion-exchange nanoresins or nanofibers, metal-organic frameworks and self-assembled micelles [45–51]. Table 1 shows the chemical structures of the common ion-responsive resins and polymers that have been used for fabricating ionic-strength-responsive nanomaterials. The ionic-strength-responsive nanomaterials have demonstrated many advantages over nonresponsive nanomaterials in terms of better bioavailability, pharmacokinetics and sustained drug-release profiles after administration via nasal, oral, transdermal and/or ocular routes [52].
Table 1.
Chemical structures of common ion-responsive materials
| Name | Chemical structure | Applications | Refs |
|---|---|---|---|
| Ion-exchange resins |  |
Ionic strength sensors for environmental monitoring and healthcare screening | [45] |
 |
Taste masking and ion-responsive nasal drug delivery | [46] | |
| Poly(ethylacrylate-methylmethacrylate - trimethylammonioethyl methacrylate chloride) copolymers (Eudragit® RS/RL) | 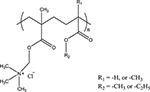 |
pH- and ionic- strength- responsive release of diltiazem HCl | [47] |
| Low methoxy pectin |  |
Nasal delivery of fentanyl | [48,49] |
| Poly(3-((2-((2,2-dimethylbutanoyl)oxy)ethyl)disulfaneyl)propanoic acid) and poly(2,2-dimethylbutanoic acid) |  |
Ionic-strength- and pH- responsive micelles for drug delivery | [50] |
| Polyelectrolyte block copolymer |  |
lonic-strength- and pH- responsive biomaterials | [51] |
Mercury ions are heavy metal ions that are detrimental to human health, and research on controlled drug release and biosensing in response to mercury ion concentration becomes increasingly important. Nanomaterials that are mercury-ion-responsive usually comprise oligonucleotides [53], DNA [54], organic chromophores [55], quantum dots [56] and conjugated polymers [57]. Zhang et al. developed DNA-tagged mesoporous silica nanoparticles to control the release of a model drug: Rhodamine 6G dye, in response to Hg2+ strength change in aqueous media [58]. Figure 5 illustrates the scheme for the formation and Rhodamine 6G release of the Hg2+ ionic-strength-responsive mesoporous silica nanoparticles [58]. The mesoporous silica nanoparticles were first surface-modified with an isocyanate group (-NCO) by reacting the nanoparticles with 3-(triethoxysilyl)propyl isocyanate. Rhodamine 6G was then loaded into the -NCO-modified nanoparticles. Amino-modified two-arm-DNA strands were then conjugated on the nanoparticles through the reaction of the amino groups of the DNA strands with the NCO groups on mesoporous silica nanoparticles. The arm-DNA strands were further crosslinked through a linker DNA by a hybridization reaction to cap the dye-loaded mesoporous silica nanoparticles. Upon exposure to Hg2+ ions the two DNA strands selectively bound to Hg2+ ions underwent dehybridization, leading the nanoparticles to be uncapped and subsequently release the loaded dye.
Figure 5.

Schematic illustration of mercuric-ion-triggered drug release from mesoporous silica nanoparticles (MSNs) that were capped by dsDNA. The hydroxy functional groups on the surfaces of the MSNs were first conjugated to the isocyanate groups (-NCO) of 3(triethoxysilyl)propyl isocyanate (MSN-NCO). After model drug rhodamine 6G dye was loaded into the MSNs (MSN-R6G), the -NCO groups were conjugated with aminomodified two-arm-DNA strands [MSN (uncapped)]. The arm-DNA strands were further hybridized by a linker DNA to cap the dye-loaded MSNs [MSN (capped)]. Upon exposure to Hg2+ ions, the DNA strands underwent dehybridization and released the loaded dye [MSN (after cleavage of linker DNA)]. Adapted, with permission, from [58].
In another strategy, low-methoxy pectin was used to control the release of fentanyl citrate in response to calcium ions existing in the nasal mucosa [59]. The low-methoxy pectin contains <50% methylated C-6 carboxyl groups and is capable of forming a gel in the presence of cations owing to the onset of intermolecular junction zones between homo-galacturonic smooth regions of different chains. The structure of such a junction zone is usually attributed to the so-called ‘egg box’ binding method, which initially combines two polymers to form a dimer followed by the development of weak inter-dimer aggregation, mainly directed by electrostatic interactions [60]. Such fentanyl-containing low-methoxy pectin was formulated into a nasal spray and used to conduct Phase I single-dose trials in 17 healthy adult volunteers [59]. The results showed that the nasal spray increased the maximum plasma concentration Cmax of fentanyl citrate 2.3-fold in comparison with orally administered fentanyl. Ion-strength-responsive controlled drug release can also be designed using nano ion-exchange resins, composed of polymers with ion-active sidechains of carboxylic acid, sulfonic acid, quaternary ammonium or tertiary amines [61]. Narisawa et al. loaded negatively charged theophylline or acetaminophen drug in ionexchange nanoresin made of positively charged poly(ethylacrylate-methylmethacrylate-trimethylammonioethyl methacrylate chloride) through electrostatic interactions [62]. Upon exposure to sodium, chloride or potassium counter-ions existing in saliva or GI fluids, the drug was released into the saliva or GI fluid by ionic exchange between the drug and the counter-ion on the nanoresins.
Besides their applications in controlled drug release, ionic-strength-responsive nanoparticles can also be used for biomedical diagnosis and detection. Zhang et al. synthesized a copolymer of oligo(ethylene glycol) methyl ether methacrylate and 2,2,2trifluoroethyl acrylate that underwent size shrinkage when 0.5 M NaCl salt was added into the polymer aqueous solution [63]. The shrinkage was due to partial dehydration of the copolymer by a salting-out effect through association of Na+ ions with the ketone oxygen of oligo(ethylene glycol) methyl ether methacrylate. The authors further utilized the ionic-strength-responsive conformational change of the copolymer as a potential noninvasive probe to detect cancer cells. The conformational change could be measured by 19F NMR which showed that T2 relaxation time was significantly lower in MCF-7 cancer cells (82 ms) than in normal cells (124 ms).
Redox-reduction-responsive nanotherapeutics
Redox-responsive nanocarriers are special chemically responsive systems that can control release of drugs in response to either reduction or oxidation conditions. The redox conditions usually occur inside the cell, triggering drug release in the cytosol and subsequently the cell nucleus [64]. The intracellular reducing conditions include the increase of the concentration of glutathione tripeptide (γ-glutamyl-cysteinyl-glycine) (GSH) from ~2–20 μmol in the extracellular milieu to 2–10 mmol in the cytosol [65–67]; and the increase of reducing potential by ~100–1000 times inside certain cancerous cells in human breast, prostate, colon and pancreatic cancers [68] compared with the extracellular milieu and bloodstream [69,70]. The oxidation conditions include ROS generated under oxidative stress. In this section, we will focus on redox-reduction-responsive nanotherapeutics. We will discuss ROS-responsive nanotherapeutics in the following section.
The nanocarriers that respond to reducing conditions usually contain disulfide bonds that are stable in the normal extracellular environment but undergo reduction in reducing conditions [71–73]. The disulfide bonds can be incorporated into the backbones, crosslinkers or side groups of the polymers that form the nanocarriers [66,71]. The polymers containing disulfide bonds can be formed into shell-sheddable, disassembled, shell-crosslinked [74,75] or core-crosslinked micelles [76], depending on the position of the crosslinking compound. One example of the shell-sheddable micelles is micelles made of a hydrophilic PEG shell and lipophilic poly(ε-benzyloxycarbonyl-L-lysine) core with intervening disulfide bonds placed in between by nanoprecipitation. The formed micelles showed better hemocompatibilities (<2% hemolysis) and cytocompatibilities (>98% cell viability at 0.1 mg/ml) in HeLa and HepG2 cells compared with positive control polyethylenimine, which showed <40% cell viability. They could load doxorubicin with ~30%wt loading efficiency, and rapidly released doxorubicin to greatly inhibit cell proliferation in HeLa and HepG2 cells pretreated with 10.0 mM GSH, but not in untreated cells [77]. Shell-crosslinked and core-crosslinked redox-responsive micelles can offer better stability along with higher drug loading than shell-sheddable and disassembled micelles because they can avoid the issue of the easy dissociation and aggregation of shell-uncrosslinked, shell-sheddable and disassembled micelles upon dilution and under the high shearing force in the circulation system. However, shell-crosslinked micelles have difficulty in being scaled up because they require highly diluted conditions for preparation to prevent undesired inter-nanoparticles crosslinking that might cause agglomeration of the nanocarriers [72,78]. Furthermore, crosslinking of the shell of micelles can decrease the surface hydrophilicity of the micelles causing instability and a short blood circulation time of the micelles [78]. By contrast, core-crosslinked micelles have a more stable and less aggregative architecture than shell-sheddable, disassembled and shell-crosslinked micelles because they maintain good surface hydrophilicity and do not disassociate easily in physiological conditions [79]. Under redox stimuli, if a disulfide bond is placed on the crosslinker of core-crosslinked micelles, it is cleaved and then the micelles are disassembled to release drugs. However, if the crosslinkers of core-crosslinked micelles are nondegradable and the linkage between the drug and micelles is a disulfide one, the micelles still release the drug in response to redox stimuli as a result of the cleavage of the disulfide bond. However, they do not disassemble but swell a little bit instead owing to the conversion of the disulfide bond into a thiol group [78]. Corecrosslinked redox-responsive micelles have less tendency to aggregate. These corecrosslinked micelles are usually designed from amphiphilic block copolymers that contain functional moieties: carboxylic acid, hydrazide, lipoyl, dithiopyridine, thiol and alkynyl in the hydrophobic block as pendant or end-capped groups for crosslinking [80,81]. For instance, poly(methacrylic acid) grafted with hydrazine was used as a hydrophobic block and PEG was used as a hydrophilic block in a copolymer to form micelles, and dithiodiethanoic acid was used as a crosslinker to react with hydrazide groups in saturated sodium bicarbonate solution, forming core-crosslinked dual redox- and pH-responsive micelles [82]. The core-crosslinked micelles had an average hydrodynamic diameter of ~56 nm smaller than that (~70 nm) of corresponding non-crosslinked micelles. They released >60% of the antitumor drug adriamycin under acidic and reductive conditions [pH 4, 15 mM dithiothreitol (DTT)], but <5% at physiological conditions (pH 7.5, 0 mM DTT) within 100 h. Although disulfide-containing nanomaterials are advantageous for controlling drug release in response to reducing conditions, they also have some limitations. The limitations include the fact that the disulfide bonds can be oxidized before reduction occurs, resulting in unwanted early and/or off-target drug release causing waste of the drug and side effects. Furthermore, the higher degree of crosslink core/shell redox-responsive nanoparticles could cause undesired slow response, whereas low crosslinking could lead to premature drug release. Therefore, the degree of crosslinking in core/shell-crosslinked redox-responsive nanoparticles must be optimized to achieve the desired therapeutic effects.
Nanomaterials containing diselenide have recently emerged as another type of redox-responsive nanotherapeutic. For example, Ma et al. synthesized a diselenide-containing biodegradable polyphosphoester block copolymer by using reduction-responsive di(1-hydroxylundecyl) diselenide as an initiator [83]. In a subsequent step, they functionalized doxorubicin with an azide group and conjugated it on the side chain of the diselenide-containing polyphosphoester via a click reaction which formed nanoparticles by self-assembly in aqueous medium. Their nanoparticles showed an average hydrodynamic diameter of 138 nm in PBS (pH 7.4) which swelled to larger aggregates when exposed to 10 mM GSH. These nanoparticles released <20% doxorubicin in PBS (pH 7.4); however, ~35% doxorubicin was released from nanoparticles when incubated with 10 mM GSH for 108 h. Diselenide-containing nanomaterials are even more sensitive to reducing conditions for site-specific drug release than the nanomaterials containing disulfide because the bond energy of Se–Se is lower than that of S–S [84]. However, they have drawbacks of poor aqueous solubility and difficulty in incorporating the diselenide bond into polymers [84], which are both factors that must be overcome before their future development into biomedical applications.
ROS-responsive nanotherapeutics
ROS are generated in several physiological reactions in the human body and play an important part in maintaining the normal functions of cells by regulating oxygen homeostasis. ROS also take part in several cell-signaling pathways, cell growth, migration, apoptosis, inflammation and extracellular matrix protein production [85]. General ROS in biological systems include hydrogen peroxide, hydroxyl radicals (OH·), superoxides (O2−) and peroxynitrites (ONOO−). Although moderate levels of ROS are essential for normal cell functions, abnormal levels or overproduction of ROS are concomitant with various diseases such as cancer, degenerative ailments, diabetes, cardiovascular disease, infection and inflammation [86–89]. From tenfold to 100-fold higher levels of ROS have been reported in the gastric mucosa of patients suffering from colon cancer, Helicobacter pylori infection, ulcerative colitis and Crohn’s disease in comparison with healthy people [90,91]. Subsequently, overexpressed ROS has been exploited as an internal chemical stimulus for designing smart nanotherapeutics for controlled drug delivery and other biomedical applications. Figure 6 illustrates the structures of the polymers that have been reported in the literature to be formed into ROSresponsive nanoparticles, as well as the oxidation–reduction (redox) reactions of these polymers in the presence of H2O2 or O2−.
Figure 6.

Schematic illustrations of the oxidation and cleavage mechanisms of common reactive oxygen species (ROS)-responsive polymers. The oxidation of (a) the sulfide group to sulfone, (b) the arylboronic ester group to phenylboronic acid and phenol, (c) the thioketal/thioether group to thiol, (d) the oligoproline peptides to pyrrolidin-2-one, peptide and carbon dioxide, (e) the selenide group to selenoxides/seleninic acids and selenones, and (f) the trisilane group to trisiloxane and silanetriol. These oxidations cause the polymers to undergo a phase change from hydrophobic to hydrophilic forms and/or degrade to enable controlled drug release.
Because sulfide can be oxidized to sulfone in an oxidative environment and sulfide-containing polymers change from the hydrophobic to hydrophilic phase upon oxidation [92], sulfide-containing polymers have been exploited as ROS-responsive nanotherapeutics. For example, Yu et al. designed sulfide-containing ROS-sensitive micelles using methoxy PEG-b-poly(diethyl sulfide) [93]. They labeled the micelles with Cy5.5 fluorescent dye, and found the labeled micelles accumulated significantly more in HCT116 colon cancer cells than in L929 mouse fibroblast cells owing to the higher level of H2O2 in the cancer cells than in the fibroblast cells. The micelles retained α-tocopheryl succinate in their hydrophobic core at physiological conditions. However, they rapidly released the drug in HCT116 colon cancer cells and killed >90% of the cancer cells but <50% of L929 mouse fibroblast cells at 100 μM after 48 h treatment. Cheng et al. developed phenyl-sulfide-containing mesoporous silica nanoparticles to control the release of doxorubicin in response to ROS stimuli [94]. Their results showed that the nanoparticles alone were not toxic (>95% cell viability at 100 μg/ml) to MCF-7 breast cancer cells and human umbilical vein endothelial cells. However, the anticancer drug doxorubicin released from the nanoparticles killed >60% MCF-7 cells and <30% of the endothelial cells at 100 μg/ml. These results indicated that doxorubicin-loaded phenylsulfide-containing mesoporous silica nanoparticles could selectively inhibit the growth of MCF-7 cells in response to the overexpressed ROS in MCF-7 cells.
Selenide-containing nanomaterials are the second class of ROS-sensitive nanomaterials. The selenide groups are converted into selenoxides or seleninic acids and selenones in the presence of ROS causing the selenide-containing nanomaterials to undergo a phase change from hydrophobic to hydrophilic forms. In addition, when selenide is in the diselenide form, it can be oxidized or reduced to seleninic acid resulting in the breakage of the diselenide bonds in the nanomaterials, and thus the nanomaterials become not only ROS-responsive but also redox-responsive [95]. For example, Ma et al. formulated dual redox- and ROS-responsive micelles using PEG-polyurethane-diselenide-PEG block copolymers [96]. Cryo-TEM images demonstrated that the micelles changed their shape from spherical to irregular structures when treated with 0.1% hydrogen peroxide solution for 2 h owing to the oxidation of Se–Se bonds in the micelles. The micelles rapidly released 90% rhodamine-B in 0.1% hydrogen peroxide medium (oxidative environment) within 4 h.
Poly(thioether ketal)-containing nanomaterials are the third class of ROS-responsive nanomaterials. The thioether groups are oxidized to thiol in the presence of ROS causing the nanomaterials to undergo a phase change from the hydrophobic to hydrophilic form. In addition, the ketal groups are sensitive to pH and can be cleaved by acid [97]. Therefore, poly(thioether ketal)-containing nanomaterials have dual ROS- and pH-responsive properties [98,99]. Wilson et al. prepared thioketal-containing nanoparticles composed of poly(1,4-phenyleneacetone dimethylene thioketal) for the oral co-delivery of siRNA and TNFα to treat intestinal inflammation caused by ulcerative colitis [99]. They tagged the siRNA with Cy3 dye and loaded it into the nanoparticles and analyzed the release of dye in 1 mM potassium superoxide or hydrogen peroxide solution. They found that <2% Cy3-siRNA was released in PBS (pH 7.4) after 12 h, whereas >5% and 8% Cy3siRNA was detected in the release medium containing potassium superoxide and hydrogen peroxide after 4 h and 12 h, respectively. Their in vivo biodistribution studies showed around threefold higher accumulation of the nanoparticles in dextran-sodium-sulfate-induced colitis mice than in healthy mice. Their in vivo bioeffect studies showed that the siRNA and TNFα-co-loaded nanoparticles decreased the colonic mRNA level of proinflammatory cytokines by fivefold and the myeloperoxidase activity by twofold in colitis mice when compared with PBS (pH 7.4), or siRNA and TNFα-co-loaded β-glucan particles as a control. These results indicated that the thioketal-containing nanoparticles were able to protect the siRNA and TNFα from the harsh environment of the GI tract, and thereby reduced inflammation and increased antimicrobial activity in the mouse model of ulcerative colitis.
Arylboronic-acid- or arylboronic-ester-containing nanomaterials are the fourth class of ROS-responsive nanomaterials. The arylboronic ester functional groups present in the core or on the surface of nanoparticles are oxidized to phenylboronic acid and phenol upon exposure to ROS, causing the nanomaterials to undergo phase change from the hydrophobic to hydrophilic form. Broaders et al. synthesized phenylboronic-ester-containing ROS-responsive micelles by using dextran conjugated with imidazoyl-carbamate-activated phenylboronic esters [100]. The micelles had hydrodynamic diameters of 100–200 nm in simulated physiological buffer but dissociated rapidly when 1 mmol H2O2 was added to the buffer, owing to the phase change governed by the oxidation of the arylboronic esters. When the micelles were in a simulated physiological buffer, they retained chicken egg albumin (CEA) inside them but released >90% CEA after 1% H2O2 was added to the buffer for 5 h. The CEA released from the micelles complexed with DC 2.4 murine dendritic cells to form CEA-derived CD8+ T cell epitope 27-fold more than nonresponsive PLGA nanoparticles after 6 h treatment.
Amino-acid- or peptide-containing nanomaterials are the fifth class of ROS-responsive nanomaterials. In particular, proline, aspartic acid and glutamic acid are susceptible to cleavage of their amide bonds by ROS, causing the nanomaterials to be degraded and subsequently provide controlled drug release [98,101]. For instance, Sung et al. synthesized oligoproline-derived PEGylated nanocarriers with an average size of 120 nm that were prone to degradation under ROS. They loaded plasmid DNA into the oligoproline-based micelles and achieved a 2.5-fold higher gene transfection efficiency in the in vitro model of pathogenic human coronary artery smooth muscle cells as compared with the nonresponsive control.
The sixth class of ROS-responsive nanomaterials that has recently gained interest for biomedical applications are nanomaterials made of ferrocene-containing polymers [102–104]. Ferrocene is neutral and hydrophobic at physiological conditions, but changes to a hydrophilic and charged form (ferrocenium cation) to release payload upon oxidation by ROS [104]. Ferrocene-containing polymers have advantages of good stability and lower oxidation potential for ROS-responsive drug release. For example, Xu et al. synthesized ferrocene-containing amphiphilic block copolymer by atom-transfer radical-polymerization, using poly(N-acryloylmorpholine) and poly(2-acryloyloxyethyl ferrocenecarboxylate) as its hydrophilic and hydrophobic blocks, respectively [105]. The micelles showed average sizes of 61.7 nm in physiological conditions but swelled to 92.6 nm when exposed to oxidizing conditions. They further used the block copolymer micelles to encapsulate paclitaxel inside the hydrophobic core by dialysis with a loading efficiency of ~60%. The micelles released ~50% more (18.5% vs 12.2%) paclitaxel in medium containing 0.15% H2O2 than physiological medium at pH 7.4 at 60 h. Under acidic conditions (pH 5.8) and with a higher amount (1.8%) of H2O2 the micelles released ~60% paclitaxel at 60 h. Further studies of the ROS-responsive release under disease conditions are needed.
Although many ROS-sensitive nanotherapeutics have been developed for biomedical applications, several challenges need to be addressed before their successful translation into the clinic. The safety of the materials including linkers used for the design of ROS-responsive nanotherapeutics is the first challenge and must be investigated thoroughly. In adverse conditions, non-biocompatible components of the nanotherapeutics can cause undesirable inflammatory reactions, lead to overproduction of ROS and then consequently release the cargos in unwanted tissues. The second challenge in the clinical translation of ROS-responsive nanotherapeutics is that the levels of ROS in different patients and malignancies are very different. Therefore, the ROS-responsive nanotherapeutics should be designed based on an individual patient’s conditions and needs in the future.
Glucose-responsive nanotherapeutics
Glucose-responsive nanotherapeutics can release insulin in a programmable way and have been increasingly attractive for the management of diabetes [106–108]. They can be designed by the different strategies illustrated in Figure 7. The first strategy is to use phenylboronic acid (PBA)-based polymers [109,110]. The PBA molecule possesses two structural forms in the aqueous environment: an uncharged or hydrophobic form and a charged or hydrophilic form [111]. Upon glucose exposure in aqueous milieu, charged PBA forms a stable complex with the glucose through H-bonding, shifting the equilibrium to the direction of producing more hydrophilic forms of PBA (Figure 7a) [112,113]. When PBA is incorporated into nanoparticles loaded with insulin, insulin can be released in response to glucose concentration – the higher the glucose concentration the more hydrophilic PBA is produced and thus more insulin is released (Figure 7a). For example, a block copolymer containing poly(D-gluconamidoethyl-methacrylate) block and 3acrylamidophenylboronic-acid) was self-assembled into spherical nanoparticles in aqueous solution and could encapsulate insulin with 63% encapsulation efficiency and 11% loading capacity [114]. The nanoparticles swelled from 129 nm to 160 nm and released 40% more insulin in 48 h when glucose concentration in the release medium was increased from 0 to 3 mg/ml.
Figure 7.

Design strategies for glucose-responsive nanotherapeutics and mechanism of sensing and/or drug release. (a) Phenylboronic acid (PBA)-triggered glucose sensing and insulin release. Upon glucose exposure in aqueous milieu, the charged form of PBA attached to nanoparticles forms complexation with glucose by hydrogen binding, shifting the equilibrium to the direction of producing more hydrophilic forms of PBA to form complexation with glucose. Owing to the complexation, the nanoparticles undergo volumetric and phase change from hydrophillic to hydrophobic to release the loaded inuslin. The complexation between the PBA and glucose can also be preformed by conjuating them on different polymer chains of the nanoparticles before exposure to high glucose conditions. Under hyperglycemic conditions, the environmental glucose competes with the glucose conjugated on the nanoparticle to bind with the PBA causing the dissociation the polymer chains conjugated with glucose from the polymer chains conjugated with PBA, and subsquently the release of the loaded insulin. The PBA in the nanoparticles can be replaced with concanavalin A (ConA) which binds glucose with high specificity and affinity (b), or pH-sensitive polymers that become highly charged at low pH caused by gluconic acid which is generated by enzymatic degradation of glucose by glucose oxidase enzyme (c). Graphs were adapted, with permission, from [112,115].
The second strategy for designing glucose-responsive nanoparticles is to use polymers conjugated with glucose-binding protein such as concanavalin A (ConA) (Figure 7b). The ConA-conjugated polymers have amphiphilic characteristics and can self-assemble into supramolecular or micelle-like structures at lower glucose concentrations; however, they dissociate or swell at higher glucose concentrations ranging from 50.0 μM to 20.0 mM [115,116]. For example, Hurkat et al. coupled the carboxylic acid group of biodegradable poly(lactic-co-glycolic acid) (PLGA) with the amine group of ConA and assembled the obtained amphiphilic polymer into nanoparticles for oral insulin delivery [117]. Their in vivo studies showed that the ConA-functionalized PLGA nanoparticles were efficiently taken up by the intestine in Wistar rats, and effectively decreased the blood glucose level to 62.17 mg/dl within 4 h in the streptozotocin-induced diabetic rat model.
The third strategy for designing glucose-responsive nanoparticles is to conjugate glucose oxidase (a glucose-sensitive enzyme) to pH-responsive polymers [115]. Upon exposure to glucose, glucose oxidase enzymatically converts glucose into gluconic acid, and generates a decrease in pH in the microenvironment [118]. The change of pH causes the nanoparticles to disassemble, swell or degrade to release the loaded insulin (Figure 7c). The nanoparticles designed through this third strategy can be used as a sensor to detect or sense glucose levels in biological systems, and also glucose-responsive delivery systems to control the release of insulin. For example, Gu et al. developed such nanoparticles by using physically crosslinked microgels containing human recombinant insulin, and covalently bound glucose oxidase and catalase [119]. The role of the enzyme glucose oxidase was to generate a pH change in response to glucose. The role of the catalase was to regenerate oxygen to assist glucose oxidase catalysis and consume undesired hydrogen produced by glucose oxidation. The microgels released 20, 25 or 140 μg/ml insulin in the release media containing 0, 100 or 400 mg/dl glucose at 37°C, respectively, in 4 h. In the streptozotocin-induced type 1 diabetic mouse model, the enzyme-containing microgels continuously released insulin for 96 h to maintain normoglycemic levels in the mouse blood for 12 h. However, insulin alone without the microgels had only a 2 h effect on the glucose level control and the enzymes alone had no effect on the glucose level.
Glucose-responsive nanotherapeutics have clear advantages over conventional insulin formulations, including controlled insulin release in response to glucose level, decreased frequency of insulin injection, reduced adverse effects and better patient convenience. However, they are still at the development stage and face some limitations and challenges that need to be overcome before clinical translation and entry into the pharmaceutical market. The main challenge is how to construct nanotherapeutics to possess high sensitivity toward clinically relevant blood glucose levels, and reversibility to prevent excessive insulin release under hypoglycemic circumstances. If the high glucose sensitivity and reversibility functions can be achieved, the nanotherapeutics will mimic a natural pancreas, with the ability to control the release of insulin to maintain physiological blood glucose levels in the bloodstream.
Enzyme-responsive nanotherapeutics
Enzymes play a crucial part in cell regulation by accelerating the rate of various chemical reactions in the cells. Dysfunctional enzymes are correlated to numerous diseases, and thus are important targets for drug development and therapeutics [120]. Accordingly, nanodrug delivery systems can be designed to respond to the changes in enzyme levels in the body to diagnose and/or treat diseases. These enzyme-responsive nanotherapeutics are specifically programmed to respond to oxidoreductases, phospholipase and protease enzymes [121]. These enzymes can cause the condensation, hydrolysis, swelling, phosphorylation, dephosphorylation and other structural changes in nanotherapeutics leading to drug release at the target site (Figure 8a). The enzyme-responsive nanotherapeutics can be formulated by conjugating enzyme substrates to the polymers, peptides or other materials that are used to form the nanotherapeutics. For example, Lee et al. developed carrier–drug conjugates connected by linkers that were only cleaved by bacterial penicillin G amidase expressed in cells infected with Escherichia coli [122]. Specifically, they conjugated the antibiotic drug phenyl acetic acid with 4-hydroxymandelic acid through a peptide containing a YGRKKRRQRRRCNH2 sequence – a substrate of bacterial penicillin G amidase enzyme. In the cells infected with E. coli, the peptide was cleaved by the penicillin G amidase enzyme and, subsequently, phenylacetic acid was released to kill the infected cells. Insua et al. conjugated an anionic peptide (Ac-C-E-GLA-E-C-OH), a substrate of elastase from pathogenic Pseudomonas aeruginosa, with poly(ethylene imine) that had antimicrobial properties, and then formed the conjugates into polyion complex nanoparticles [123]. They adjusted the hydrodynamic diameters of the nanoparticles to be between 100 and 600 nm by changing the nitrogen:carboxylic-acid ratio. Upon exposure to a medium containing LasB elastase, a virulence enzyme produced by P. aeruginosa, the nanoparticles broke apart owing to the cleavage of the Ac-C-E-GLA-E-C-OH peptide by the elastase (Figure 8b). When the nanoparticles were exposed to HLE enzyme, an elastase produced by human leucocytes, minimal or no degradation of the nanoparticles was detected. These results suggested that the enzymatic degradation of the nanoparticles was specific to LasB elastase and thus P. aeruginosa, without affecting nonpathogenic strains.
Figure 8.

The chemistry involved in designing enzyme-responsive nanomaterials. (a) Common enzyme-derived reactions, including condensation, hydrolysis, phosphorylation and dephosphorylation, involved in designing enzyme-responsive nanomaterials. Adapted, with permission, from [121]. (b) Assembly and oxidative crosslinking of polyion complex nanoparticles from P1SH (Ac-C-E-GLA-E-C-OH) and antimicrobial branched poly(ethylene imine) (PEI). Degradation of polyion complex nanoparticles by LasB enzyme enabled payload release. Adapted, with permission, from [123]. (c) Graphical illustration of quantum dot nanoprobes for multiplexed detection of endonucleases. In the duplex, the fluorescence of the two quantum dots with different emissions is effectively quenched by inter- and intra-molecular quenchers. The target endonuclease disrupts the DNA duplex and results in the fluorescence emission. Adapted, with permission, from [126].
In another study, matrix metalloproteinase (MMP)-2-enzyme-responsive gelatin nanoparticles were developed to shrink from 100 to 10 nm upon exposure to MMP-2 overexpressed in a tumor microenvironment [124]. The shrunken nanoparticles also had a long circulation time and effectively penetrated through 300 μm from the injection site into the interstitial tumor pores of a mouse model of human fibrosarcoma after intratumoral injection, whereas corresponding non-enzyme-responsive silica nanoparticles showed little or no penetration. In addition, MMP-responsive and PEGylated lipids were also developed to modify adenoviral vectors to increase their tumor cell transduction and thereby reduce their immunogenicity [125]. in vitro infection experiments showed that the modified adenoviral vectors increased the gene expression in a HT1080 fibrosarcoma cell line threefold and 1.5-fold higher than naked adenoviral-5 and noncleavable PEG-lipid-modified adenoviral vectors, respectively. The modified adenoviral vectors also decreased the liver cytotoxicity by 1.2-fold and lowered the immune response as mediated by the production of cytokines from splenocytes by 1.6fold when compared with the naked adenovirus.
Besides the therapeutic effects discussed above, enzyme-responsive nanoparticles have also been designed for detecting enzymes in biological systems. For example, endonuclease, an enzyme that can cleave DNA and take part in DNA repair, replication and recombination, has been conventionally detected by using chromatography and gel electrophoresis techniques. However, these techniques are expensive, laborious, time-consuming and not very sensitive. To overcome these limitations, highly specific and sensitive nanoprobes have been synthesized for multiplexed detection of endonucleases by using quantum dots that were conjugated with endonuclease-cleavable DNA through succinimidyl-6-(β-maleimidopropionamido) hexanoate linkage [126]. The DNAconjugated quantum dots were also labeled with 4-[4-(dimethylaminophenylazo)]benzoic acid and BHQ-3 quenchers in sodium carbonate solution (Figure 8c). The obtained nanoprobes considerably decreased the background fluorescence in normal conditions owing to the quenchers. However, when the nanoprobes were exposed to endonucleases, fluorescence was increased owing to the cleavage of quencher-labeled DNA from the surface of quantum dots by endonucleases. The increase in fluorescence intensity is linearly proportional to the concentration of endonucleases so that the activity of endonucleases can be rapidly quantified. This quantum dot nanoprobe method is at least 100-times more sensitive than conventional methods.
Despite the emerging progress in enzyme-responsive drug delivery and diagnosis research, there are still many challenges that need to be addressed for future clinical use of enzyme-responsive technologies. The first challenge is that there is tremendous variety in enzyme dysregulation activities in different diseases, even at different stages of one disease. Further fundamental understanding of the spatial and temporal pattern of enzyme-responsive drug delivery systems is important for designing more-effective and -precise delivery vehicles. The second challenge is that there are many overlapping substrates between closely related enzyme families. More-specific designs of substrates that only respond to a specific enzyme should be carried out for enhanced delivery efficacy. Furthermore, comprehensive toxicology evaluations of the enzyme-responsive nanotherapeutics are essential for the safe clinical translation of this class of nanotherapeutics.
ATP-responsive nanotherapeutics
ATP is a multifunctional organic nucleotide that stores and transfers energy in cells. It is often referred as the ‘molecular unit of currency’ in living cells, and is composed of adenine, ribose and three phosphate groups that mediate important roles in many biological processes (e.g., cell division, DNA synthesis, triphosphoric acid cycle, membrane transport, neurotransmission, ion channels, muscle contraction and metabolism) [127,128]. An ATP level of 10–24 μM/million cells is vital for normal physiological functions, and higher levels of ATP have been associated with different disease conditions such as multidrug resistance, tumors and uncontrolled synaptic transmission in neurons [129,130]. During the past 10 years, overexpressed ATP at disease sites has become an attractive chemical stimulus in designing smart nanosystems for controlled drug delivery. Incorporation of functional molecules or moieties capable of distinguishing ATP from other cytosolic compounds is a common strategy to develop ATP-sensitive nanotherapeutics [131]. ssDNA aptamers that specifically bind ATP [128], enzymes that undergo conformational changes upon recognition by an ATP molecule [132] and phenylboronic-acid-grafted materials [133] that experience solubility changes after binding with ATP have been used as functional ATPsensitive modules. For example, He et al. conjugated an ATP-responsive aptamer on mesoporous silica nanocarriers (MSNs) for controlled release of Ru(bipy)32+ dye as a model drug [128]. The ATP aptamer was first hybridized with two ssDNA molecules to obtain a sandwich-type DNA complex. The two ssDNA molecules were then conjugated onto the MSN surface by a copper-catalyzed azide alkyne cycloaddition reaction as a cap to block the pores on the MSN surface so that Ru(bipy)32+ model drug loaded inside the MSNs would not come out. Upon exposure to 20 mM ATP, the aptamer formed stronger complexation with ATP than the ssDNA molecules leading to the following cascade event: departure of the aptamer from the MSNs, flexible movement of the ssDNA molecules, the exposure of the pores on the surface of the MSNs and 83.2% release of the loaded Ru(bipy)32+ from the pores. Similarly, Mo et al. also used the ATP-sensitive ssDNA aptamer strategy to control release of anticancer drug doxorubicin from nanogels in response to ATP-level change [134]. The unique thing about their nanogel systems was that the doxorubicin-loaded aptamer–DNA complex was embedded inside a crosslinked hyaluronic acid shell (Figure 9). The hyaluronic acid did not only protect the complex from degradation in the blood circulation but also enhanced the accumulation of the nanogels at the tumor site as it was an antigen that bound to the overexpressed CD44 receptor on the cancer cell membrane. At the tumor site, upregulated hyaluronidase hydrolyzed the hyaluronic acid on the nanogel surface leading to the exposure of the aptamer–DNA complex, and then overexpressed ATP broke down the aptamer–DNA complex leading to the release of doxorubicin. The results showed that the nanogels containing ATP aptamer inhibited tumor growth about 2- and 3-fold more efficiently than non-ATP-responsive and free-doxorubicin control groups, respectively, in an MDA-MB-231 tumor-bearing nude mouse model.
Figure 9.

Schematic illustrations of the cellular uptake and anticancer drug release of ATP-responsive nanotherapeutics in the tumor. (a) ATP-responsive nanocarrier consisting of a crosslinked hyaluronic acid shell and a core of doxorubicin–DNA motif along with protamine (a cationic molecule that can complex with negatively charged DNA, enhancing cell penetration and nuclear targeting). (b) Mechanism of ATP-triggered doxorubicin release from ATP-responsive DNA motif. Under overexpressed ATP conditions, DOX intercalated in the guanine–cytosine (GC)-rich pair and aptamer was released owing to the conversion of the aptamer from the duplex to tertiary structure. (c) The process of cellular uptake and ATP-responsive doxorubicin release from the nanogels: (i) accumulation of the nanogels at the tumor site by the process of passive and active targeting; (ii) specific binding of smart doxorubicin nanogels to overexpressed CD44 receptors on the tumor cells and degradation of hyaluronic acid shell by hyaluronase-rich tumor extracellular matrix; (iii) receptor-mediated endocytosis; (iv) endosomal/lysosomal escape of doxorubicin-loaded ATP-responsive DNA motif; (v) ATPtriggered doxorubicin release from the ATP-responsive DNA motif in the cytosol; and (vi) accumulation of doxorubicin in the cell nucleus. Adapted, with permission, from [134].
ATP-responsive biomaterials can also be designed by incorporating enzymes that undergo conformational changes upon recognition by an ATP molecule. Yuan et al. utilized adenylate kinase as a crosslinker to synthesize N-(2-hydroxypropyl)methacrylamide hydrogels, which shrunk when exposed to 8 mM ATP solution [132]. Although their system was composed of hydrogels, this strategy can be exploited to design ATP-responsive nanogels. In another study, Lai et al. used zincdipicolylamine analog (TDPA-Zn2+) to develop mesoporous silica nanoparticles coated with branched polypeptide [poly(aspartate-lysine)-b-aspartate] for the triggered release of fluorescein in response to ATP [129]. Their in vitro release studies showed >80% dye release in a medium containing 10 mM ATP for 4 h, whereas negligible dye release (<5%) was observed in the PBS buffer (pH 7.4, without ATP). This triggered release of dye was attributed to the competitive cleavage of the surface-coated polypeptide by the ATP molecule, because TDPA-Zn2+ has a higher binding affinity to ATP compared with the oligo-polypeptide. Another strategy to design ATP-responsive nanotherapeutics is the incorporation of phenylboronic acid into the nanoparticles, which has the tendency to form reversible covalent linkages with 1,2-diols present on the ribose ring of the ATP molecule [135]. For instance, Naito et al. designed 4-carboxy-3fluorophenylboronic-acid-conjugated polyion complex micelles composed of PEG-blockpoly(L-lysine) for the ATP-responsive delivery of cholesterol-modified siRNA [136]. The polyion complex micelles showed good stability in a medium containing 2 mM ATP, whereas triggered release of siRNA was detected in a medium with 10 mM ATP.
Although ATP-responsive nanotherapeutics are an interesting as well as emerging concept, they are still at an early stage of development and much more design and in vitro and in vivo evaluation and validation are needed for clinical translation. One of the major concerns of aptamer-based ATP-responsive nanocarriers is that aptamers are highly susceptible to nuclease-mediated degradation so that premature drug release at nontarget sites can occur and cause nonspecific cytotoxicity. ATP aptamers also have low specificity to their target molecule ATP (and derivatives) leading to ineffective targeting. Furthermore, because ATP aptamers are essentially ssDNA or RNA, possible immunogenic effects should be addressed before their clinical application. Moreover, above, we discussed that phenylboronic acid undergoes phase transition in the presence of high glucose levels. Therefore, phenylboronic-acid-conjugated ATP-responsive nanocarriers lack specificity to ATP and can release encapsulated drugs in the blood besides overexpressed ATP sites under hyperglycemic conditions. More-robust and specific ATP-responsive molecules or moieties need to be developed and incorporated into the nanocarriers.
Hypoxia-responsive nanotherapeutics
Hypoxia, a pathological disorder in which a tissue lacks the supply of sufficient oxygen, is an indication of various complex ailments such as cancer, cardiopathy, ischemia, rheumatoid arthritis and vascular diseases [137]. Clinical studies have demonstrated that tissue partial pressure of oxygen in patients with ischemic stroke or cancer is near zero mmHg, which is significantly lower than that in healthy tissues (30 mmHg) [138]. In addition, hypoxia causes production of lactic acid via anaerobic respiration leading to decreased pH and a reductive microenvironment at tumor sites. Therefore, nanomaterials containing pH- and/or redox-responsive hypoxia can be theoretically developed for controlled drug delivery in response to hypoxia [139]. However, these nanomaterials must have a redox potential of −200 to −400 mV to be responsive to hypoxic conditions [140]. The functional groups that meet the redox potential requirement such as quinones, aromatic nitro groups, aromatic and aliphatic N-oxides and cobalt complexes have been used to design hypoxia-responsive nanotherapeutics [141]. In particular, 2nitroimidazoles are the most commonly used molecules in the design of nanotherapeutics owing to their robust responsive behavior to hypoxia [142]. These molecules undergo structural changes from hydrophobic (2-nitroimidazoles) to hydrophilic (2aminoimidazoles) forms to release drugs under hypoxic conditions. For example, Thambi et al. grafted a 2-nitroimidazole derivative to the polymer backbone of carboxymethyl dextran to form self-assembled nanoparticles where 2-nitroimidazole was used as a self-assembly inducer as well as a hypoxia-responsive moiety [143]. They found that doxorubicin loaded in the nanoparticles was completely released out in 100 μM NADPH-containing PBS (pH 7.4), a hypoxic condition; whereas <50% of the drug was released in PBS (pH 7.4) without hypoxia over 12 h. They further reported that threefold shrinkage in the tumor volume was observed when doxorubicin-loaded hypoxia-responsive nanocarriers were administered in SCC7-tumor-bearing mice as compared with free doxorubicin. In another study, Liu et al. introduced hypoxia-sensitive nitroimidazole functional groups into malate dehydrogenase lipid molecules to form liposomes for controlled delivery of polo-like kinase 1 small interfering RNA (siRNA) to treat glioma [144]. Besides 2-nitroimidazoles, azobenzene, a hypoxia-degradable compound, is also used as a hypoxia-responsive functional group for the design of nanotherapeutics. For example, Xie et al. designed azobenzene-containing polyamidoamine dendrimers for co-delivery of doxorubicin and siRNA into tumors [145]. The doxorubicin was encapsulated in the hydrophobic core of the dendrimers by nanoprecipitation and the siRNA was electrostatically complexed with cationic primary amine on the surface of the dendrimers. The dendrimers were spherical with an average diameter of 197 nm under physiological conditions, but their average size decreased to 5.4 nm when treated with a reducing agent sodium thiosulfate. The azobenzene-containing dendrimers increased the cellular uptake of doxorubicin and siRNA by MCF-7 cells by ~2–3-fold when the environment was changed from physiological to hypoxic conditions.
Although hypoxia-responsive nanotherapeutics have emerged as promising smart systems for controlled and targeted drug delivery, their development is hampered by many limitations. One important limitation was that the nanotherapeutics cannot reach to the deep tumor tissues, even though these tissues have the highest level of hypoxia, because the blood that carries the nanotherapeutics cannot access the deep tumor tissues sufficiently. Therefore, design of nanotherapeutics with high permeability along with safe and sensitive characteristics is needed for the future clinical translation of hypoxia-responsive nanotherapeutics.
Concluding remarks and future perspectives
Chemical-stimuli-responsive nanomaterials that can control drug release and detect biomarkers in response to chemical stimuli including pH, ionic strength, redox, ROS, glucose, enzymes, ATP and hypoxia hold great potential as smart nanotherapeutics for the treatments, diagnoses and management of a variety of diseases in the future. Many strategies have been explored to program them with the desired multifunctionality by incorporating chemical-stimuli-responsive bridges or groups on the surface, interface or cavities of a variety of nanomaterials. Recently, a few chemical-stimuli-responsive nanotherapeutics have advanced into early clinical trial stages, including Merck’s glucose-sensitive smart insulin delivery system which is in Phase I [146] and a pHresponsive camptothecin-conjugated cyclodextrin nanoparticle in Phase II for the treatment of tumors [147]. However, like the conventional nanotherapeutics, the number of chemical-stimuli-responsive nanocarriers that have advanced into late clinical trials is very limited, which can be attributed to various challenges. The first challenge is nonspecific uptake of nanocarriers by nontarget tissues. This is mainly associated with nonspecific adsorption of proteins from the biological milieu on the surfaces of nanomaterials leading to a protein corona being formed [148,149]. The absorbed proteins start to degrade leading to the aggregation and/or the phagocytosis of the nanomaterials and the accumulation of the nanomaterials in the nontarget organs [148,149]. In line with this fact, a recent meta-study of preclinical analysis of nanocarriers developed for the treatment of various tumors revealed that a median of ~0.7% of the administered nanocarriers reached the target sites [150].
The second major challenge is lack of clearance of nanomaterials from the body after delivering the drug to the desired site. Most of the nanotherapeutics investigated have sizes beyond the renal threshold and cannot be efficiently removed from the body via the kidney and they tend to accumulate in the body and cause toxicological concerns. This is particularly a major issue with nonbiodegradable nanomaterials. As for degradable nanomaterials, even though they can degrade to the size below to the renal threshold, their degraded fragments can be sequestered in lysosomal and other compartments of cells to cause toxic side-effects [148]. The third challenge for the clinical translation of nanotherapeutics is that – particularly for smart nanocarriers designed based on targeting moieties – it is rare to find receptors that are expressed exclusively by the diseased tissues. For example, folate receptor is overexpressed in a large number of malignancies, but it is also expressed in a moderate-to-high level in healthy organs such as the small intestine, placenta and kidneys. Besides, folate receptor expression by malignant cells is not homogeneous. As a result, there would not be specific and uniform distribution of nanotherapeutics at target sites. Therefore, successful clinical translation of nanotherapeutics demands overcoming those major hurdles and smart nanocarriers such as chemical-stimuli-responsive nanocarriers would have the edge over the conventional nanotherapeutics in this regard. Future research on chemical-stimuli-responsive nanotherapeutics should be focused on optimization of chemistry and architecture, efficient uptake by cells and tissues, effective permeability across biological barriers [142–146,148–151], no or minimal off-target delivery, ultimate safety, rapid response to chemical stimuli, predictable pharmacokinetics (ADME) and pharmacodynamics, desired therapeutic effects, product stability and scale-up manufacturing.
Teaser:
This review systematically addresses up-to-date technology, design strategies and challenges for chemical-stimuli-responsive nanotherapeutics for controlled drug delivery, diagnostics and other biomedical applications.
Highlights:
Various chemical stimuli existing in the biological systems are discussed
Strategies for designing chemical stimuli-responsive nanocarriers are summarized
Biomedical applications of chemical stimuli-responsive nanocarriers are reviewed
Limitations and future perspectives of current nanotherapeutics are addressed
Acknowledgments
This work was financially supported by NIH R01EY023853. We thank Kumar K. Niloy for his assistance in writing the ATP-responsive and hypoxia-responsive nanotherapeutics sections.
Biography
 Muhammad Gulfam
Muhammad Gulfam
Dr Muhammad Gulfam earned a BS from University of Agriculture, Faisalabad, Pakistan. In 2009, he was awarded MS-level training with the Korean University/Industry Fellowship, Higher Education Commission, Pakistan, to pursue his MS study in bionano engineering at Hanyang University, South Korea. He was awarded a prestigious fellowship for his PhD study in nanomedicine and pharmaceutical innovation through the European Commission, Education, Audiovisual and Cultural Executive Agency. After graduation with his PhD from the University of Nottingham, UK, Dr Gulfam joined Dr Lowe’s lab as a Postdoctoral Fellow. His research interests include development of smart polymeric nanomaterials for tissue engineering, drug delivery and controlled-release applications.
 Fitsum F. Sahle
Fitsum F. Sahle
Dr Fitsum Sahle earned a PhD in pharmaceutical technology and biopharmaceutics from Martin Luther University, Halle/Saale, Germany. After his PhD he worked for 2.5 years as an Assistant Professor at the School of Pharmacy, Addis Ababa University, Ethiopia. Later he joined the Freie Universität Berlin, Germany, as an Alexander von Humboldt/George-Forster Postdoctoral Fellow and worked for 3 years on areas of nanotechnology and transdermal and transfollicular drug delivery. Currently, Dr Sahle is a postdoctoral fellow in Dr Lowe’s lab and is working on development of smart polymeric nanomaterials for drug delivery and tissue engineering.
 Tao L. Lowe
Tao L. Lowe
Dr Tao Lowe is currently an Associate Professor of Pharmaceutical Sciences and Biomedical Engineering at the University of Tennessee Health Science Center. Dr Lowe’s research activities include design and development of multifunctional biomaterials for targeted and sustained drug and gene delivery, regenerative medicine, stem cell engineering and biosensing for the diagnoses and treatment of brain and eye diseases, cancers, bone fractures and cartilage damage, as well as contraception. She has authored many high-impact peerreviewed articles and US and international patents; and has lectured extensively throughout the global scientific community. Her research has been supported by NIH, DOD, Coulter Foundation and JDRF, among others.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and revisew of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Blanco E et al. (2015) Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol 33, 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Janagam DR et al. (2017) Nanoparticles for drug delivery to the anterior segment of the eye. Adv. Drug Deliv. Rev 122, 31–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu L et al. (2014) Overcoming the blood–brain barrier in chemotherapy treatment of pediatric brain tumors. Pharm. Res 31, 531–540 [DOI] [PubMed] [Google Scholar]
- 4.Gil ES et al. (2012) β-Cyclodextrin-poly(β-amino ester) nanoparticles for sustained drug delivery across the blood–brain barrier. Biomacromolecules 13, 3533–3541 [DOI] [PubMed] [Google Scholar]
- 5.Gao GH et al. (2012) The use of pH-sensitive positively charged polymeric micelles for protein delivery. Biomaterials 33, 9157–9164 [DOI] [PubMed] [Google Scholar]
- 6.Suma T et al. (2012) Smart multilayered assembly for biocompatible siRNA delivery featuring dissolvable silica, endosome-disrupting polycation, and detachable PEG. ACS Nano 6, 6693–6705 [DOI] [PubMed] [Google Scholar]
- 7.Ryu JH et al. (2010) Self-cross-linked polymer nanogels: a versatile nanoscopic drug delivery platform. J. Am. Chem. Soc 132, 17227–17235 [DOI] [PubMed] [Google Scholar]
- 8.Janagam DR et al. (2017) Nanoparticles for drug delivery to the anterior segment of the eye. Adv. Drug Deliv. Rev 122, 31–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yokoyama M (2014) Polymeric micelles as drug carriers: their lights and shadows. J. Drug Target. 22, 576–583 [DOI] [PubMed] [Google Scholar]
- 10.Pattni BS et al. (2015) New developments in liposomal drug delivery. Chem. Rev 115, 10938–10966 [DOI] [PubMed] [Google Scholar]
- 11.Wu L-P. et al. (2015) Dendrimers in medicine: therapeutic concepts and pharmaceutical challenges. Bioconj. Chem 26, 1198–1211 [DOI] [PubMed] [Google Scholar]
- 12.Kothamasu P et al. (2012) Nanocapsules: the weapons for novel drug delivery systems. BioImpacts 2, 71–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Filova E et al. (2015) The diameter of nanotubes formed on Ti-6Al-4V alloy controls the adhesion and differentiation of Saos-2 cells. Int. J. Nanomed 10, 7145–7163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neamtu I et al. (2017) Basic concepts and recent advances in nanogels as carriers for medical applications. Drug Deliv. 24, 539–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim YS et al. (2018) Thermoresponsive-co-biodegradable linear–dendritic nanoparticles for sustained release of nerve growth factor to promote neurite outgrowth. Mol. Pharm 15, 1467–1475 [DOI] [PubMed] [Google Scholar]
- 16.Sahle FF et al. (2018) Design strategies for physical-stimuli-responsive programmable nanotherapeutics. Drug Discov Today 23, 992–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pacardo DB et al. (2015) Programmable nanomedicine: synergistic and sequential drug delivery systems. Nanoscale 7, 3381–3391 [DOI] [PubMed] [Google Scholar]
- 18.Mura S et al. (2013) Stimuli-responsive nanocarriers for drug delivery. Nat. Mater 12, 991–1003 [DOI] [PubMed] [Google Scholar]
- 19.Yin Q et al. (2013) Reversal of multidrug resistance by stimuli-responsive drug delivery systems for therapy of tumor. Adv. Drug Deliv. Rev 65, 1699–1715 [DOI] [PubMed] [Google Scholar]
- 20.Jhaveri A et al. (2014) Stimuli-sensitive nanopreparations for combination cancer therapy. J. Control. Release 190, 352–370 [DOI] [PubMed] [Google Scholar]
- 21.Hu Y-B et al. (2015) The endosomal-lysosomal system: from acidification and cargo sorting to neurodegeneration. Transl. Neurodegen 4, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Webb BA et al. (2011) Dysregulated pH: a perfect storm for cancer progression. Nat. Rev. Cancer 11, 671. [DOI] [PubMed] [Google Scholar]
- 23.Biswas S et al. (2016) Recent advances in polymeric micelles for anti-cancer drug delivery. Eur. J. Pharm. Sci 83, 184–202 [DOI] [PubMed] [Google Scholar]
- 24.Knipe JM et al. (2016) Enzyme- and pH-responsive microencapsulated nanogels for oral delivery of siRNA to induce TNF-α knockdown in the intestine. Biomacromolecules 17, 788–797 [DOI] [PubMed] [Google Scholar]
- 25.Bae Y and Kataoka K (2009) Intelligent polymeric micelles from functional poly(ethylene glycol)-poly(amino acid) block copolymers. Adv. Drug Deliv. Rev 61, 768–784 [DOI] [PubMed] [Google Scholar]
- 26.Dong D-W. et al. (2013) Comparative studies of polyethylenimine–doxorubicin conjugates with pH-sensitive and pH-insensitive linkers. J. Biomed. Mater. Res. Part A 101, 1336–1344 [DOI] [PubMed] [Google Scholar]
- 27.Chen W et al. (2009) pH-Responsive biodegradable micelles based on acid-labile polycarbonate hydrophobe: synthesis and triggered drug release. Biomacromolecules 10, 1727–1735 [DOI] [PubMed] [Google Scholar]
- 28.Masson C et al. (2004) pH-sensitive PEG lipids containing orthoester linkers: new potential tools for nonviral gene delivery. J. Control. Release 99, 423–434 [DOI] [PubMed] [Google Scholar]
- 29.Deng H et al. (2015) Balancing the stability and drug release of polymer micelles by the coordination of dual-sensitive cleavable bonds in cross-linked core. Acta Biomater. 11, 126–136 [DOI] [PubMed] [Google Scholar]
- 30.Cui M et al. (2015) Liposomes containing cholesterol analogues of botanical origin as drug delivery systems to enhance the oral absorption of insulin. Int. J. Pharm 489, 277–284 [DOI] [PubMed] [Google Scholar]
- 31.Qu W et al. (2012) A silica-based pH-sensitive nanomatrix system improves the oral absorption and efficacy of incretin hormone glucagon-like peptide-1. Int. J. Nanomed 7, 4983–4994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen C et al. (2013) Orally delivered salmon calcitonin-loaded solid lipid nanoparticles prepared by micelle-double emulsion method via the combined use of different solid lipids. Nanomedicine 8, 1085–1100 [DOI] [PubMed] [Google Scholar]
- 33.Colombo P et al. (2009) Novel platforms for oral drug delivery. Pharm. Res 26, 601–611 [DOI] [PubMed] [Google Scholar]
- 34.Foss AC et al. (2004) Development of acrylic-based copolymers for oral insulin delivery. Eur. J. Pharm. Biopharm 57, 163–169 [DOI] [PubMed] [Google Scholar]
- 35.Holst JJ (2007) The physiology of glucagon-like peptide 1. Physiol. Rev 87, 1409–1439 [DOI] [PubMed] [Google Scholar]
- 36.Selby LI et al. (2017) Nanoescapology: progress toward understanding the endosomal escape of polymeric nanoparticles. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology 9, e1452–n/a [DOI] [PubMed] [Google Scholar]
- 37.Deng Z et al. (2011) Hollow chitosan–silica nanospheres as pH-sensitive targeted delivery carriers in breast cancer therapy. Biomaterials 32, 4976–4986 [DOI] [PubMed] [Google Scholar]
- 38.Min KH et al. (2010) Tumoral acidic pH-responsive MPEG-poly(β-amino ester) polymeric micelles for cancer targeting therapy. J. Control. Release 144, 259–266 [DOI] [PubMed] [Google Scholar]
- 39.Lee ES et al. (2008) Super pH-sensitive multifunctional polymeric micelle for tumor pH(e) specific TAT exposure and multidrug resistance. J. Control. Release 129, 228–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ziegler A et al. (2005) The cationic cell-penetrating peptide CPP(TAT) derived from the HIV-1 protein TAT is rapidly transported into living fibroblasts: optical, biophysical, and metabolic evidence. Biochemistry 44, 138–148 [DOI] [PubMed] [Google Scholar]
- 41.Rudolph C et al. (2003) Oligomers of the arginine-rich motif of the HIV-1 TAT protein are capable of transferring plasmid DNA into cells. J. Biol. Chem 278, 11411–11418 [DOI] [PubMed] [Google Scholar]
- 42.Koren E et al. (2012) Multifunctional PEGylated 2C5-immunoliposomes containing pH-sensitive bonds and TAT peptide for enhanced tumor cell internalization and cytotoxicity. J. Control. Release 160, 264–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Radovic-Moreno AF et al. (2012) Surface charge-switching polymeric nanoparticles for bacterial cell wall-targeted delivery of antibiotics. ACS Nano 6, 4279–4287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kathmann EEL et al. (1997) Water-soluble polymers. Electrolyte- and pHresponsive zwitterionic copolymers of 4-[(2-acrylamido-2-methylpropyl)- dimethylammonio]butanoate with 3-[(2-acrylamido-2-methyl- propyl)dimethylammonio]propanesulfonate. Macromolecules 30, 5297–5304 [Google Scholar]
- 45.Nucara L et al. (2017) Ionic strength responsive sulfonated polystyrene opals. ACS Appl. Mater. Interfaces 9, 4818–4827 [DOI] [PubMed] [Google Scholar]
- 46.Guo X (2009) Ion-exchange resins as drug delivery carriers. J. Pharm. Sci 98, 3886–3902 [DOI] [PubMed] [Google Scholar]
- 47.Bodmeier R et al. (1996) The influence of buffer species and strength on diltiazem HCl release from beads coated with the aqueous cationic polymer dispersions, Eudragit RS, RL 30D. Pharm. Res 13, 52–56 [DOI] [PubMed] [Google Scholar]
- 48.Illum L (2012) Nasal drug delivery – recent developments and future prospects. J. Control. Release 161, 254–263 [DOI] [PubMed] [Google Scholar]
- 49.Watts P and Smith A (2009) PecSys: in situ gelling system for optimised nasal drug delivery. Expert Opin. Drug Deliv 6, 543–552 [DOI] [PubMed] [Google Scholar]
- 50.Ghosh S et al. (2009) Redox, ionic strength, and pH sensitive supramolecular polymer assemblies. J. Polym. Sci. Part A Polym. Chem 47, 1052–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stubenrauch K et al. (2009) pH and ionic strength responsive polyelectrolyte block copolymer micelles prepared by ring opening metathesis polymerization. J. Polym. Sci. Part A Polym. Chem 47, 1178–1191 [Google Scholar]
- 52.Takayuki Y et al. (2016) Ion-responsive drug delivery systems. Curr. Drug Target 17, 1–14 [Google Scholar]
- 53.Chiang C-K. et al. (2008) Oligonucleotide-based fluorescence probe for sensitive and selective detection of mercury(II) in aqueous solution. Anal. Chem 80, 3716–3721 [DOI] [PubMed] [Google Scholar]
- 54.Liu J and Lu Y (2007) Rational design of “turn-on” allosteric DNAzyme catalytic beacons for aqueous mercury ions with ultrahigh sensitivity and selectivity. Angew. Chem. Int. Ed. Engl 46, 7587–7590 [DOI] [PubMed] [Google Scholar]
- 55.Che Y et al. (2008) Ultraselective fluorescent sensing of Hg2+ through metal coordination-induced molecular aggregation. Chem. Commun 12, 1413–1415 [DOI] [PubMed] [Google Scholar]
- 56.Li H et al. (2007) Calixarene capped quantum dots as luminescent probes for Hg2+ ions. Mater. Lett 61, 1474–1477 [Google Scholar]
- 57.Liu X et al. (2007) Optical detection of mercury(II) in aqueous solutions by using conjugated polymers and label-free oligonucleotides. Adv. Mater 19, 1662 [Google Scholar]
- 58.Zhang Y et al. (2012) DNA-capped mesoporous silica nanoparticles as an ionresponsive release system to determine the presence of mercury in aqueous solutions. Anal. Chem 84, 1956–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fisher T et al. (2009) Fentanyl pectin nasal spray (FPNS) with PecSys® provides most favorable pharmacokinetic/tolerability profile compared with nasal chitosanbased fentanyl and oral transmucosal fentanyl citrate (OTFC). J. Pain 10, S45 [Google Scholar]
- 60.Capel F et al. (2006) Calcium and acid induced gelation of (amidated) low methoxyl pectin. Food Hydrocolloids 20, 901–907 [Google Scholar]
- 61.Yoshida T et al. (2013) pH- and ion-sensitive polymers for drug delivery. Expert Opin. Drug Deliv 10, 1497–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Narisawa S et al. (1994) An organic acid-induced sigmoidal release system for oral controlled-release preparations. Pharm. Res 11, 111–116 [DOI] [PubMed] [Google Scholar]
- 63.Zhang C et al. (2016) Ion-responsive 19F MRI contrast agents for the detection of cancer cells. ACS Sensors 1, 757–765 [Google Scholar]
- 64.Lili Y et al. (2016) Intracellular doxorubicin delivery of a core cross-linked, redoxresponsive polymeric micelles. Int. J. Pharm 498, 195–204 [DOI] [PubMed] [Google Scholar]
- 65.Schafer FQ and Buettner GR (2001) Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med 30, 1191–1212 [DOI] [PubMed] [Google Scholar]
- 66.Li J (2012) Redox-sensitive micelles self-assembled from amphiphilic hyaluronic acid-deoxycholic acid conjugates for targeted intracellular delivery of paclitaxel. Biomaterials 33, 2310–2320 [DOI] [PubMed] [Google Scholar]
- 67.Estrela JM et al. (2006) Glutathione in cancer biology and therapy. Crit. Rev. Clin. Lab. Sci 43, 143–181 [DOI] [PubMed] [Google Scholar]
- 68.Acharya A et al. (2010) Redox regulation in cancer: a double-edged sword with therapeutic potential. Oxidative Medicine Cellular Longevity 3, 23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Balendiran GK et al. (2004) The role of glutathione in cancer. Cell. Biochem. Funct 22, 343–352 [DOI] [PubMed] [Google Scholar]
- 70.Ballatori N et al. (2009) Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem 390, 191–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Roy D et al. (2010) Future perspectives and recent advances in stimuli-responsive materials. Prog. Polym. Sci 35, 278–301 [Google Scholar]
- 72.Fleige E et al. (2012) Stimuli-responsive polymeric nanocarriers for the controlled transport of active compounds: concepts and applications. Adv. Drug Deliv. Rev 64, 866–884 [DOI] [PubMed] [Google Scholar]
- 73.Wang Y-C et al. (2011) Redox-responsive nanoparticles from the single disulfide bond-bridged block copolymer as drug carriers for overcoming multidrug resistance in cancer cells. Bioconj. Chem 22, 1939–1945 [DOI] [PubMed] [Google Scholar]
- 74.Zhang K et al. (2009) Cationic shell-crosslinked knedel-like nanoparticles for highly efficient gene and oligonucleotide transfection of mammalian cells. Biomaterials 30, 968–977 [DOI] [PubMed] [Google Scholar]
- 75.Zhang K et al. (2009) Cationic shell-crosslinked knedel-like nanoparticles for highly efficient gene and oligonucleotide transfection of mammalian cells. Biomaterials 30, 968–977 [DOI] [PubMed] [Google Scholar]
- 76.Talelli M et al. (2015) Core-crosslinked polymeric micelles: principles, preparation, biomedical applications and clinical translation. Nano Today 10, 93–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ding J et al. (2013) Biocompatible reduction-responsive polypeptide micelles as nanocarriers for enhanced chemotherapy efficacy in vitro. J. Mater. Chem. B 1, 69–81 [DOI] [PubMed] [Google Scholar]
- 78.Wang H et al. (2013) Redox-responsive, core-cross-linked micelles capable of ondemand, concurrent drug release and structure disassembly. Biomacromolecules 14, 3706–3712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.van Nostrum CF (2011) Covalently cross-linked amphiphilic block copolymer micelles. Soft Matter 7, 3246–3259 [Google Scholar]
- 80.Sun H et al. (2014) Reduction-responsive polymeric micelles and vesicles for triggered intracellular drug release. Antioxid Redox Signal. 21, 755–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhao Y et al. (2015) Advanced stimuli-responsive polymer nanocapsules with enhanced capabilities for payloads delivery. Polym. Chem 6, 4197–4205 [Google Scholar]
- 82.Wei C et al. (2011) Dual stimuli-responsive polymeric micelles exhibiting “AND” logic gate for controlled release of adriamycin. Macromol. Rapid Commun 32, 451–455 [DOI] [PubMed] [Google Scholar]
- 83.Ma G et al. (2018) Dual-responsive polyphosphoester-doxorubicin prodrug containing a diselenide bond: synthesis, characterization, and drug delivery. ACS Biomater. Sci. Eng 4, 2443–2452 [DOI] [PubMed] [Google Scholar]
- 84.Huo M (2014) Redox-responsive polymers for drug delivery: from molecular design to applications. Polym. Chem 5, 1519–1528 [Google Scholar]
- 85.Touyz RM and Schiffrin EL (2004) Reactive oxygen species in vascular biology: implications in hypertension. Histochem. Cell Biol 122, 339–352 [DOI] [PubMed] [Google Scholar]
- 86.Braunersreuther V and Jaquet V (2012) Reactive oxygen species in myocardial reperfusion injury: from physiopathology to therapeutic approaches. Curr. Pharm. Biotechnol 13, 97–114 [DOI] [PubMed] [Google Scholar]
- 87.Bae S et al. (2016) Hydrogen peroxideresponsive nanoparticle reduces myocardial ischemia/reperfusion injury. J. Am. Heart Assoc. Cardiovasc. Cerebrovas. Dis 5, e003697 [Google Scholar]
- 88.Lee D et al. (2013) H2O2-responsive molecularly engineered polymer nanoparticles as ischemia/reperfusion-targeted nanotherapeutic agents. Sci. Rep 3, 2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Winterbourn CC (2008) Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol 4, 278. [DOI] [PubMed] [Google Scholar]
- 90.Peng YC et al. (2008) Chemiluminescence assay of mucosal reactive oxygen species in gastric cancer, ulcer and antral mucosa. Hepatogastroenterology 55, 770–773 [PubMed] [Google Scholar]
- 91.Kountouras J et al. (2001) Reactive oxygen metabolites and upper gastrointestinal diseases. Hepatogastroenterology 48, 743–751 [PubMed] [Google Scholar]
- 92.Lee SH et al. (2013) Current progress in reactive oxygen species (ROS)-responsive materials for biomedical applications. Adv. Healthcare Mater 2, 908–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yu L-Y et al. (2016) Specific cancer cytosolic drug delivery triggered by reactive oxygen species-responsive micelles. Biomacromolecules 17, 3040–3047 [DOI] [PubMed] [Google Scholar]
- 94.Cheng Y et al. (2017) Free-blockage mesoporous anticancer nanoparticles based on ROS-responsive wetting behavior of nanopores. Small 13, 1701942–n/a [DOI] [PubMed] [Google Scholar]
- 95.Han P et al. (2010) Oxidation-responsive micelles based on a selenium-containing polymeric superamphiphile. Langmuir 26, 14414–14418 [DOI] [PubMed] [Google Scholar]
- 96.Ma N et al. (2010) Dual redox responsive assemblies formed from diselenide block copolymers. J. Am. Chem. Soc 132, 442–443 [DOI] [PubMed] [Google Scholar]
- 97.Zhang Y et al. (2017) ROS-switchable polymeric nanoplatform with stimuliresponsive release for active targeted drug delivery to breast cancer. ACS Appl. Mater. Interfaces 9, 12227–12240 [DOI] [PubMed] [Google Scholar]
- 98.Saravanakumar G et al. (2017) Reactive-oxygen-species-responsive drug delivery systems: promises and challenges. Adv. Sci 4, 1600124–n/a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wilson DS et al. (2010) Orally delivered thioketal nanoparticles loaded with TNFalpha-siRNA target inflammation and inhibit gene expression in the intestines. Nat. Mater 9, 923–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Broaders KE et al. (2011) A biocompatible oxidation-triggered carrier polymer with potential in therapeutics. J. Am. Chem. Soc 133, 756–758 [DOI] [PubMed] [Google Scholar]
- 101.Wang M et al. (2014) Reactive oxygen species-responsive protein modification and its intracellular delivery for targeted cancer therapy. Angew. Chem. Int. Ed 53, 13444–13448 [DOI] [PubMed] [Google Scholar]
- 102.Staff RH et al. (2014) Hydrophobic nanocontainers for stimulus-selective release in aqueous environments. Macromolecules 47, 4876–4883 [Google Scholar]
- 103.Scheid D et al. (2016) Synthesis of breathing metallopolymer hollow spheres for redoxcontrolled release. Macromol. Rapid Commun 37, 1573–1580 [DOI] [PubMed] [Google Scholar]
- 104.Enrique L and Nicola T (2013) Oxidationresponsive polymers: which groups to use, how to make them, what to expect from them (biomedical applications). Macromol. Chem. Phys 214, 143–158 [Google Scholar]
- 105.Xu F et al. (2017) Redox-responsive self-assembly micelles from poly(Nacryloylmorpholine-block-2-acryloyloxyethyl ferrocenecarboxylate) amphiphilic block copolymers as drug release carriers. ACS Appl. Mater. Interfaces 9, 5181–5192 [DOI] [PubMed] [Google Scholar]
- 106.Peng Q et al. (2012) A rapid-acting, long-acting insulin formulation based on a phospholipid complex loaded PHBHHx nanoparticles. Biomaterials 33, 1583–1588 [DOI] [PubMed] [Google Scholar]
- 107.Zhao L et al. (2017) Glucose oxidase-based glucose-sensitive drug delivery for diabetes treatment. Polymers 9, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhao L et al. (2015) Competitive binding-accelerated insulin release from a polypeptide nanogel for potential therapy of diabetes. Polym. Chem 6, 3807–3815 [Google Scholar]
- 109.Matsumoto A et al. (2012) A synthetic approach toward a self-regulated insulin delivery system. Angew. Chem. Int. Ed 51, 2124–2128 [DOI] [PubMed] [Google Scholar]
- 110.Matsumoto A et al. (2010) A totally synthetic glucose responsive gel operating in physiological aqueous conditions. Chem. Commun 46, 2203–2205 [DOI] [PubMed] [Google Scholar]
- 111.Chou DH-C et al. (2015) Glucose-responsive insulin activity by covalent modification with aliphatic phenylboronic acid conjugates. Proc. Natl. Acad. Sci. U. S. A 112, 2401–2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Akira M et al. (2012) A synthetic approach toward a selfregulated insulin delivery system. Angew. Chem. Int. Ed 51, 2124–2128 [DOI] [PubMed] [Google Scholar]
- 113.Yang J and Cao Z (2017) Glucose-responsive insulin release: analysis of mechanisms, formulations, and evaluation criteria. J. Control. Release 263 (suppl. C), 231–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Guo Q et al. (2014) Phenylboronate-diol crosslinked glycopolymeric nanocarriers for insulin delivery at physiological pH. Soft Matter 10, 911–920 [DOI] [PubMed] [Google Scholar]
- 115.Veiseh O et al. (2015) Managing diabetes with nanomedicine: challenges and opportunities. Nat. Rev. Drug Discov. 14, 45–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ye T et al. (2014) Synthesis and volume phase transition of concanavalin Abased glucose-responsive nanogels. Polym. Chem 5, 186–194 [Google Scholar]
- 117.Hurkat P et al. (2012) Concanavalin A conjugated biodegradable nanoparticles for oral insulin delivery. J. Nanoparticle Res 14, 1219 [Google Scholar]
- 118.Gu Z et al. (2013) Glucose-responsive microgels integrated with enzyme nanocapsules for closed-loop insulin delivery. ACS Nano 7, 6758–6766 [DOI] [PubMed] [Google Scholar]
- 119.Gu Z et al. (2013) Glucose-responsive microgels integrated with enzyme nanocapsules for closed-loop insulin delivery. ACS Nano 7, 6758–6766 [DOI] [PubMed] [Google Scholar]
- 120.de la Rica R et al. (2012) Enzyme-responsive nanoparticles for drug release and diagnostics. Adv. Drug Deliv. Rev 64, 967–978 [DOI] [PubMed] [Google Scholar]
- 121.Zelzer M et al. (2013) Enzyme responsive materials: design strategies and future developments. Biomater. Sci 1, 11–39 [DOI] [PubMed] [Google Scholar]
- 122.Lee M-R. et al. (2004) Targeted enzyme-responsive drug carriers: studies on the delivery of a combination of drugs. Angew. Chem. Int. Ed 43, 1675–1678 [DOI] [PubMed] [Google Scholar]
- 123.Insua I et al. (2016) Enzyme-responsive polyion complex (PIC) nanoparticles for the targeted delivery of antimicrobial polymers. Polym. Chem 7, 2684–2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wong C et al. (2011) Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proc. Natl. Acad. Sci. U. S. A 108, 2426–2431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wan Y et al. (2013) Enzyme-responsive liposomes modified adenoviral vectors for enhanced tumor cell transduction and reduced immunogenicity. Biomaterials 34, 3020–3030 [DOI] [PubMed] [Google Scholar]
- 126.Huang Y et al. (2011) Intermolecular and intramolecular quencher based quantum dot nanoprobes for multiplexed detection of endonuclease activity and inhibition. Anal. Chem 83, 8913–8918 [DOI] [PubMed] [Google Scholar]
- 127.Khakh BS and Burnstock G (2009) The double life of ATP. Scientific American 301, 84–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.He X et al. (2012) ATP-responsive controlled release system using aptamerfunctionalized mesoporous silica nanoparticles. Langmuir 28, 12909–12915 [DOI] [PubMed] [Google Scholar]
- 129.Lai J et al. (2015) Real-time monitoring of ATP-responsive drug release using mesoporous-silica-coated multicolor upconversion nanoparticles. ACS Nano 9, 5234–5245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lu Y et al. (2016) Bioresponsive materials. Nat. Rev. Mater 2, 16075 [Google Scholar]
- 131.Sun W and Gu Z (2016) ATP-responsive drug delivery systems. Expert Opin. Drug Deliv 13, 311–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yuan W et al. (2008) Smart hydrogels containing adenylate kinase: translating substrate recognition into macroscopic motion. J. Am. Chem. Soc 130, 15760–15761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mitsuru N et al. (2012) A phenylboronatefunctionalized polyion complex micelle for ATPtriggered release of siRNA. Angew. Chem. Int. Ed 51, 10751–10755 [DOI] [PubMed] [Google Scholar]
- 134.Mo R et al. (2014) ATP-triggered anticancer drug delivery. Nat. Commun 5, 3364. [DOI] [PubMed] [Google Scholar]
- 135.Okuro K et al. (2016) Boronic acid-appended molecular glues for ATP-responsive activity modulation of enzymes. J. Am. Chem. Soc 138, 5527–5530 [DOI] [PubMed] [Google Scholar]
- 136.Mitsuru N et al. (2018) Enhanced intracellular delivery of siRNA by controlling ATPresponsivity of phenylboronic acidfunctionalized polyion complex micelles. Macromol. Biosci 18, 1700357. [DOI] [PubMed] [Google Scholar]
- 137.Harris AL (2002) Hypoxia--a key regulatory factor in tumour growth. Nat. Rev. Cancer 2, 38–47 [DOI] [PubMed] [Google Scholar]
- 138.Takasawa M et al. (2008) Applications of nitroimidazole in vivo hypoxia imaging in ischemic stroke. Stroke 39, 1629–1637 [DOI] [PubMed] [Google Scholar]
- 139.Mo R and Gu Z (2016) Tumor microenvironment and intracellular signalactivated nanomaterials for anticancer drug delivery. Materials Today 19, 274–283 [Google Scholar]
- 140.Hunter FW et al. (2016) Hypoxia-activated prodrugs: paths forward in the era of personalised medicine. Br. J. Cancer 114, 1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chenggen Q et al. (2016) Lightactivated hypoxiaresponsive nanocarriers for enhanced anticancer therapy. Adv. Mater 28, 3313–3320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Okuda K et al. (2012) 2-Nitroimidazole-tricarbocyanine conjugate as a nearinfrared fluorescent probe for in vivo imaging of tumor hypoxia. Bioconj. Chem 23, 324–329 [DOI] [PubMed] [Google Scholar]
- 143.Thambi T et al. (2014) Hypoxia-responsive polymeric nanoparticles for tumortargeted drug delivery. Biomaterials 35, 1735–1743 [DOI] [PubMed] [Google Scholar]
- 144.Liu HM et al. (2017) Hypoxia-responsive ionizable liposome delivery siRNA for glioma therapy. Int. J. Nanomedicine 12, 1065–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Xie Z et al. (2018) Targeting tumor hypoxia with stimulus-responsive nanocarriers in overcoming drug resistance and monitoring anticancer efficacy. Acta Biomater. 71, 351–362 [DOI] [PubMed] [Google Scholar]
- 146.Kaarsholm NC et al. (2018) Engineering glucose responsiveness into insulin. Diabetes 67, 299–308 [DOI] [PubMed] [Google Scholar]
- 147.Svenson S et al. (2011) Preclinical to clinical development of the novel camptothecin nanopharmaceutical CRLX101. J. Control. Release 153, 49–55 [DOI] [PubMed] [Google Scholar]
- 148.Soni KS et al. (2016) Nanogels: an overview of properties, biomedical applications and obstacles to clinical translation. J. Control. Release 240, 109–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bobo D et al. (2016) Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date. Pharm. Res 33, 2373–2387 [DOI] [PubMed] [Google Scholar]
- 150.Wilhelm S et al. (2016) Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater 1, 16014 [Google Scholar]
- 151.Sun W et al. (2017) Leveraging physiology for precision drug delivery. Physiol. Rev 97, 189–225 [Google Scholar]