

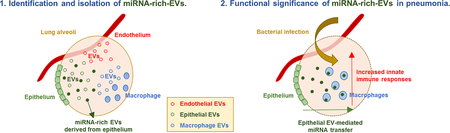
Abstract
Despite emerging interest in the role of extracellular vesicle (EV)-containing microRNAs (EV-miRNAs), the existence of functional EV-miRNAs under patho-physiological conditions has been viewed with skepticism. Due to the heterogenicity of EVs, several barriers related to EV-miRNA research are to be explored before the in vivo function of EV-miRNAs can be thoroughly delineated. For example, it has been reported that far less than one copy of a given miRNA can be detected per exosome. In this study, we demonstrated that miRNA-rich-EVs exist and can be consistently isolated using differential centrifugation & density-gradient fractionation from bronchoalveolar lavage fluid (BALF) in vivo. The absolute number of this ‘miRNA-rich’-EV population is only about 7.05 × 109 per mouse (6 % of total EVs). However, the RNA amount detected in this population of EVs represents approximately 39 % of the total EV RNAs in the BALF. In contrast, the remaining populations of BALF EVs (76 % of total EVs) contain extremely low concentrations of RNAs and miRNAs. The miRNA-rich-EVs in BALF are likely derived from alveolar epithelial type-I cells (ATIs). Notably, caveolin-1, a lipid raft protein, is exclusively detected in the miRNA-rich-EVs, suggesting the lipid raft protein as a biomarker of EV-miRNA enrichment. We further demonstrated that miRNAs contained in the ATI-EVs are actively delivered into alveolar macrophages, subsequently promoting inflammasome activation, neutrophil recruitment, and M1-macrophage polarization in response to P. aeruginosa pneumonia in vitro and in vivo. Collectively, we are the first to identify and characterize the miRNA-rich-EVs in BALF. These miRNA-rich EVs endorse pro-inflammatory responses in bacterial lung infection. Our study provides a novel insight into the development of biomarkers, therapeutic strategies and underlying mechanisms for lung pathology.
Keywords: extracellular vesicles, microRNA, acute lung injury, bacterial pneumonia, lung inflammation
Graphical Abstract
Introduction
Cells have been known for a long time to release vesicles to the extracellular compartment during cell apoptosis, and the fact that healthy or activated cells also release extracellular vesicles (EVs) has only been recently recognized [1]. So far, EVs have been isolated from most cell types and biological fluids including bronchial-alveolar lavage fluid (BALF) [2, 3]. The discovery of EVs in BALF offers a novel insight into lung physiology, pathology, and homeostasis [1].
Based on the recent statement from the International Society for Extracellular Vesicles (ISEV), EVs are generally classified into exosomes, microvesicles (MVs) and apoptotic bodies (ABs) according to their sizes, surface markers, and cellular generation mechanism [4, 5]. ABs are formed during the process of cell death and have the largest sizes among the three subgroups of EVs, roughly at 500 – 2,000 nm [6, 7]. Exosomes are nanometer-sized vesicles (50 – 150nm) of endocytic origin that form by inward budding of the multivesicular bodies (MVBs). Exosomes are released into the extracellular compartment when the MVEs fuse with the plasma membrane [8]. MVs are generated by direct budding of the plasma membrane and their sizes vary between 100 nm to 500 nm [9]. Although there is an attempt to define theses vesicles using specific protein markers, such as selectins, integrins and the CD40 ligand [10], exosomes and MVs seem to share most EV markers, such as CD63, CD81, CD9, ACTB, flotillin, and caveolin [11–13].
The amount, size and composition of EVs are heterogeneous and largely based on their cellular sources and biological environment. Despite accumulating evidence suggesting the critical functions of EV-containing miRNAs, challenges and skepticism exist on the concept that EV-miRNAs exert functional roles in vivo, particularly on the actual concentration of EV-miRNAs. Chevillet et al. reported that the average copy number of miRNAs in each EV is actually very low, even less than one copy per exosome [14]. These concerns prompted us to investigate whether there is a population of EVs that contain a high concentration of miRNAs and whether this population of “miRNA-rich EVs” can be isolated in a practical manner. This question is crucial for the development of EV-miRNA based biomarkers and EV-based therapeutics.
In this study, we identified the subpopulation of miRNA-rich EVs from mouse BALF using a novel method combining the differential ultracentrifugation and density gradient fractionation. The most widely used method for isolation of EVs is a differential ultracentrifugation. However, this method cannot completely separate each type of EVs due to the highly-present heterogeneity. Although the density gradient centrifugation has been used previously for detection of EVs, we are the first to use this method to separate EV sub-populations in body fluids. We attempted a dedicated method to obtain miRNA-rich EVs from BALF and characterized the features of this subgroup of EVs, including their cellular origin, uptake by recipient cells and functional roles in vivo. To the best of our knowledge, this is the first study identifying miRNA-enriched EV subpopulations in BALF and more importantly, provides a novel protocol to isolate the miRNA-rich EVs. This work potentially provides more advanced knowledge for the development of novel therapeutics and diagnostic biomarkers for human lung diseases.
Methods
Materials.
Anti-PDPN, anti-CD68, anti-SP-B, anti-Flot1, anti-CD9 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-caveolin-1 and anti-ACTB antibodies were purchased from Thermo Fisher (Waltham, MA) while Anti-Dicer-1 (#5362) and anti-agonate-2 (#2897) antibodies were purchased from Cell Signaling (Danvers, MA). Lipofectamine™ LTX Reagent with PLUS™ Reagent (15338030) and Lipofectamine™ MessengerMAX™ Transfection Reagent (LMRNA008, Thermo) were purchased from Thermo Fisher (Waltham, MA).
Animals.
Wild type and cav-1 knock out (Cavtm1Mls) C57BL/6 mice (6 to 8 weeks of age) were obtained from Jackson Laboratory (Bar Harbor, ME). All the protocols involving animals in this study were approved by the institutional animal care and use committee (IACUC). All experimental protocols and methods were approved by Boston University and were carried out in accordance with the approved guidelines.
EV isolation from BALF and cell-cultured medium.
Previously reported protocols and techniques were applied to isolate EVs. The obtained mouse BALFs or cell-cultured medium were centrifuged at 300 g for 5 min to eliminate the inflammatory or dead cells. The supernatant was then collected and centrifuged at 1,200 g for 10 min to pellet cell debris. Finally, the resulting supernatant was ultracentrifuged at 100,000 g for 1 h to pellet total EVs. For isolating three subpopulations of EVs including apoptotic bodies (ABs), microvesicles (MVs), exosomes (Exos) [2, 6, 15, 16], the cell-depleted supernatant was centrifuged at 3,000 g for 10 min to pellet ABs [6, 7]. To isolate MVs, the AB-depleted supernatant was centrifuged at 12,000 g for 40 min [15, 17, 18]. Finally, the resulting supernatant was ultracentrifuged at 100,000 g for 1 h to pellet Exos [16, 19]. Isolated vesicles were re-suspended with cold PBS and analyzed using dynamic light scattering (DLS) instrument (Brookhaven 90plus Nano-particle Sizer), NanoSight (Malvern), and transmission electron microscopy (TEM).
Sucrose density gradient fractionation.
The multiple-step gradient layers including 2, 1.3, 1.16, 0.8, 0.5, and 0.25M density gradient layers were generated using sucrose (D(+)-Saccharose) reagent (S0389, Sigma-Aldrich, St. Louis, MO), which formed a density separation range between 1.01 – 1.299 g/ml after ultracentrifugation. Purified EVs were then loaded on the top of the step gradient sucrose layers, followed by 120,000 × g ultracentrifugation for 2 h (Optima LE-80K Ultracentrifuge, Beckman Coulter, Brea, CA). The fractions with different densities were sequentially collected from the top of the sample tube and their densities (g/ml) were measured using an electronic microbalance (OHAUS, USA). The density-fractionated EVs were stored at - 80° C and subjected to further experiments.
Bacterial lung infection.
Pseudomonas aeruginosa (P. aeruginosa) PA103 was cultured overnight in Luria-Bertani medium at 37 °C in a rotator at 250 RPM. 106 colony forming unit (CFU) was then intratracheally instilled into the mouse lung. At the designated time points after administration, mice were euthanized, and BALFs were collected from the mouse lungs, as described previously [20].
Isolation of primary macrophages.
BALFs were collected from mouse lung, followed by centrifugation of the collected samples at 300 g for 5 min. The obtained BALF inflammatory cells were then incubated in cell culture plates for 10 min to allow adhesion of alveolar macrophages (AMs) [21]. The adhered macrophages were then subjected to further experiments. For bacterial infection of the isolated AMs, 106 CFU/ml of P. aeruginosa was incubated with the AMs for the designated time points.
Isolation of cav-1−/− alveolar epithelial cells and immortalization.
Alveolar epithelial cells were isolated from mouse lung tissue as described previously [22]. WT and cav-1−/− mouse lungs were perfused with sterile PBS, followed by infusion of 2 ml dispase and 0.5 ml 1% agarose. Lung tissue was then dissociated in DMEM with 25 mM HEPES and 200 U/ml DNase. Isolated cells were stringed using sequential cell stringers (100μm, 40μm, and 20μm), and incubated on plates pre-coated with CD45 and CD16/32 antibodies for 2 h to remove myeloid lineage cells, followed by transferring of the suspended cells to non-coated plates to remove fibroblasts. After another 2 h incubation, suspended epithelial cells were cultured in DMEM containing 10% fetal bovine serum (FBS). Immortalization of the isolated primary cells was conducted using the simian virus 40 (SV40) T antigen, as described previously [23]. The established WT and cav-1−/− epithelial cells were verified with gene expressions of typical lung epithelial markers using qPCR (data not shown).
Cell culture.
Lung epithelial E10 cells [24], alveolar MH-S macrophages (ATCC® CRL-2019™), primary AMs and lung epithelial cells isolated from mouse lung were maintained in Dulbecco’s modified Eagle’s medium (DMEM) or RPMI-1640 with 10% FBS and 1% penicillin/streptomycin.
Deletion of Dicer1 in lung epithelial cells using CRISPR/Cas9.
pX330-sgRNA_Dicer_1 plasmid (#68807, Addgene) was co-transfected with EX-NEG-M56 plasmid (GeneCopoeia) expressing mCherry into lung epithelial E10 cells using lipofectamine 3000. The transfected cells were then selected using Geneticin selective antibiotic (100 μg/ml) or mCherry-positive cell sorting (MoFlo™ Astrios EQ Sorter, Beckman). The successful transfection and deletion of dicer1 were verified using fluorescence microscopy, qPCR and western blotting with specific primers and an antibody against dircer1. The established cells were subjected to further experiments.
Nanoparticle Tracking Analysis.
Nanoparticle Tracking Analysis was performed to determine the size and concentration of EVs at Nanomedicines Characterization Core Facility (The University of North Carolina at Chapel Hill, Chapel Hill, NC). Isolated EV samples were water-bath sonicated to help dispel aggregates and diluted to a concentration between 1×108 - 5×108 particles/mL in filtered PBS. The samples were then run on a NanoSight NS500 (NanoSight, Malvern Instruments, UK) to capture particles moving by way of Brownian motion. The hydrodynamic diameters were calculated using the Stokes-Einstein equation. The 100 nm standard particles and the diluent PBS alone were used for reference.
Electron microscopy and Dynamic Light Scattering analysis.
Purified EVs were fixed on the formvar-carbon coated mesh 400 grid according to the manufacturer’s recommendation (101Bio, Palo Alto, CA). EV structural images were generated using transmission electron microscopy (TEM) at the Experimental Pathology Laboratory Core, Boston University School of Medicine. Sizes of EVs were measured using dynamic light scattering (DLS) instrument (Brookhaven 90plus Nano-particle Sizer, Biomedical Engineering core, Boston University).
Quantitative real-time PCR.
Total RNAs were purified from the fractioned EVs or isolated AMs using MiRNeasy Mini Kits (Qiagen). Purified RNA concentration was measured using the NanoDrop Lite Spectrophotometer (Thermo Scientific), followed by reverse transcription to generate cDNAs. SYBR green-based real-time quantitative PCR (qPCR) was performed to detect specific mRNAs. For relative expression levels of mRNAs and miRNAs in cells, β-actin and U6 were used as reference housekeeping genes, respectively. For relative expression levels of miRNAs in EVs, vesicle number was used as a normalization control. The sequences of primers were shown in suppl. table 1.
EV uptake analysis.
An equal amount of isolated EVs was labeled with PKH67 (sigma), acridine orange (Thermo Fisher), or carboxyfluorescein succinimidyl ester (CFSE), according to the manufacturer’s recommendation. PKH67 was centrifuged at 16,000×g for 5 min to remove the PKH67 aggregates prior to EV labeling (suppl. fig. 1). Primary AMs were exposed to the labeled EVs to allow EV internalization into the macrophages overnight. Surface bounded-EVs were then extensively washed using an acidic buffer (pH 2.2), followed by cell fixation and Hoechst staining. The internalized EVs were analyzed using confocal microscopy and ImageJ software (NIH).
Isolation of small/micro- and large-RNAs from EVs.
Small/miRNAs and large RNAs were separately purified from the miRNA-rich EVs using total exosome RNA isolation kit (cat n. 4478545, Invitrogen), according to the manufacturer’s instruction.
ELISA.
Conditioned medium of AMs which were pre-treated with ATP (1 mM) or mouse BALFs was collected and centrifuged at 300 g for 5 min to eliminate inflammatory cells. IL1β and MIP2 cytokine levels in the supernatants were then analyzed using DuoSet® ELISA Development Systems (R&D system), according to the manufacturer’s recommendation.
Caspase-1 enzymatic activity.
AMs were lysed with passive lysis buffer (Promega). The enzymatic activity of caspase-1 was then determined using Caspase-Glo® 1 Inflammasome Assay kit (Promega), according to the manufacturer’s recommendation.
Differential inflammatory cell count.
BALF inflammatory cells were cytocentrifuged at 300 × g for 5 min using a Shandon Cytospin 4 (Thermo Scientific, Rockford, IL). Slides were air-dried and stained with Hema 3™ Fixative and Solutions (PROTOCOL™). Differential cell (AMs and neutrophils) counts were evaluated under a light microscope, as described previously [15].
Statistical analysis.
For all experiments, the exact n values and statistical significances were shown in the corresponding figure and figure legends. Represented data of the identical results were shown in the presented figures. Statistical analyses were performed using two-tailed Student’s T-test and one-way ANOVA. Values of p < 0.05 were considered statistically significant (* P < 0.05, ** P < 0.01, *** P < 0.001).
Results
Isolation and characterization of BALF EVs after sucrose-density gradient fractionation.
In this study, we first developed a novel protocol to identify and isolate the miRNA-rich EVs from BALF using the sucrose-density-gradient. Initially, we isolated total EVs from mouse BALF using sequential centrifugation and confirmed that the BALF EVs are highly heterogeneous using transmission electronic microscopy (fig. 1A). We next established six differential density layers using 0.25 M, 0.5 M, 0.8 M, 1.16 M, 1.3 M, and 2 M sucrose and conducted a density-dependent separation and fractionation of the isolated BALF EVs, as illustrated in figure 1B. We measured the density of each fraction and confirmed that a successful density gradient was created (fig. 1B). The existence of EVs in each fractioned group was verified using nanoparticle-tracking analysis (NTA) (fig. 1C). The size range of fractioned EVs was between 50 – 500 nm. No significant differences in size were observed among each group of fractioned EVs (fig. 1C).
Figure 1. Characterization of BALF EVs after sucrose-density gradient fractionation.

(A-C) Total EVs were isolated from mouse BALF using sequential centrifugation and their morphology and heterogeneity were verified using transmission electronic microscopy (A). Six differential density layers were established using 0.25 M, 0.5 M, 0.8 M, 1.16 M, 1.3 M, and 2 M sucrose. The purified heterogeneous EVs were then further separated using the density gradient fractionation. The density of each fraction was shown (B). The fractioned EVs were identified using nanoparticle tracking analysis (NTA) (C) (n = 4 per group).
Identification of miRNA-rich EV subpopulations.
We next measured the number, size, protein amount, and RNA amount in each sub-group of fractioned EVs. The majority of EVs were detected in fraction 1–3 (F1–3). Fraction 2 (F2) possessed the highest counts of EVs (fig. 2A). However, when we calculated the average amount of proteins and RNAs in each EV, the EVs in F2 group had extremely low concentrations of proteins and RNAs compared with EVs obtained from the other fractions (fig. 2A). Interestingly, the RNA amount per EV was highest in the subpopulation of EVs obtained from F6–F8 (fig. 2A). The EVs in F6–F8 only represented approximate 6 percent (7.05 × 109 EVs) of total EVs obtained from BALF. However, the RNA amount in F6–F8 groups represented approximately 39% of the total BALF EV-RNAs (fig. 2B), suggesting that F6–F8 subpopulations are composed of RNA-rich EVs.
Figure 2. Identification of microRNA-enriched EV population.

(A-B) BALF EVs were separated using sucrose density gradient fractionation, followed by measuring total vesicle numbers, average vesicle size, total protein amount, and total RNA amount in each fraction (A, upper and middle panels). The proteins and RNAs were normalized to total vesicle number (A lower panels). The pie charts showed the proportion of vesicle number, protein amount, and RNA amount in the indicated frac tions (B) (mean ± SD, n = 4 per group). (C) Copy number of indicated inflammatory miRNAs in each fractioned EV was analyzed using quantitative real-time PCR. The copy number was normalized to vesicle number in each fraction (mean ± SD, n = 3 per group). (D) Soluble miRNAs were exposed to RNase at 37 °C for 10 min. The remaining intact miRNAs were then analyzed by qPCR (n = 3 per group). (E) The density-gradient fractioned EVs (F1–3 vs. F6–8) were exposed to RNase, followed by measuring miRNA expression in the fractioned EVs using qPCR. The expression levels were then normalized to vesicle number in each fraction (heatmap, n = 3 per group). * P < 0.05, ** P < 0.01, and *** P < 0.001 between the groups indicated.
Given the growing interests on EV-containing miRNAs as potential molecules involving the cell-cell cross-talk [25, 26], we next analyzed the absolute copy number of several well-recognized pro-inflammatory miRNAs [2, 15, 27–31] in the fractioned EVs. As shown in figure 2C, most EVs from F1–3 were miRNA deficient. Interestingly, we found that the miRNA copy numbers were dramatically elevated in EVs obtained from F6–8 (fig. 2C). To confirm that the detected miRNAs were contained in the EVs rather than “adherent” to the EVs or in “free” soluble form, EV-containing miRNAs were further analyzed from the fractioned EVs treated with RNase. RNase rapidly degrades RNAs outside the EVs, but not those encapsulated in the EVs [32, 33]. We confirmed that soluble miRNAs are rapidly degraded after RNase exposure (fig. 2D). As shown in figure 2E, miRNA expression including the above mentioned pro-inflammatory miRNAs was abundant in F6–8 EVs, even after RNase exposure, indicating that these miRNAs were enriched in F6–8 EVs obtained from BALF.
The miRNA-rich EVs are caveolin-1 (cav-1) positive and most likely derived from lung epithelial cells.
EVs often carry the same markers as their “mother” cells [34]. We found that CD31, an endothelial marker, CD68, a macrophage marker, and SP-B, an alveolar epithelial cell type-II (ATII) marker were predominantly detectable in F1–4 (fig. 3A). On the other hand, PDPN, an alveolar epithelial cell type-I (ATI) maker, was broadly expressed over the EV fractions and covered the F6–8 EV fractions (fig. 3A), suggesting that F6–8 EVs were derived from lung epithelium. The most common EV proteins, such as cav-1, flotillin-1 (Flot1), and ACTB [11–13] were all detectable in the EV fractions (fig. 3A). Notably, we found that the expression of cav-1 was exclusively localized in F6–8 (fig. 3A). Cav-1 is predominately expressed in lung ATI cells and thought to be an ATI marker protein [24]. Therefore, we isolated EVs from the cell medium of ATI cells, followed by density-gradient fractionation. As shown in figure 3B, we confirmed the expression of PDPN and cav-1 in the fractioned ATI EVs, suggesting that the F6–8 BALF EVs are generated from cav-1-positive lung epithelium.
Figure 3. The microRNA-enriched EVs are exclusively cav-1 positive and most likely derived from ATI cells.

(A-B) Purified BALF EVs (A) and EVs from alveolar epithelial type-I cells (ATI) (B) were separated using sucrose density gradient fractionation, followed by western blot analysis using indicated antibodies. (C-D) ABs, MVs, and Exos were separately isolated from BALF using sequential ultracentrifugation. Size distributions of isolated vesicles were determined using dynamic light scattering (DLS) (C, upper panel) and NTA (C, lower panels). Expressions of indicated proteins in each EVs were examined using western blotting (D). (E) MVs were purified from BALF and separated using sucrose density gradient fractionation, followed by western blot analysis using indicated antibodies. (F) EVs were purified from WT or cav-1−/− ATI. The purified EVs were then separated using sucrose density gradient fractionation, followed by measuring protein amount (F, upper panel) and western blot analysis with indicated antibodies (F, low panel) (mean ± SD, n = 3 per group). (G-H) MiRNA expression was compared between fractioned EVs (F1–3 vs. F6–8 from WT ATIs) (G) or between WT EVs and cav-1−/− EVs from F6–8 (H) using qPCR analysis. (heatmap, n = 3–4 per group). *** P < 0.001 between the groups indicated.
We next isolated three types of BALF EVs, including apoptotic bodies (ABs), microvesicles (MVs), and exosomes (Exos) using sequential ultracentrifugation, as described previously [20], and verified their size distribution using dynamic light scattering (DLS) and Nanosight tracking analysis (NTA) (fig. 3C). As shown in figure 3D, we found that both cav-1 and PDPN were predominantly detectable in MVs, rather than ABs or Exos. We next fractioned the BALF MVs using the above sucrose-gradient. PDPN was still broadly detectable over the fractioned MVs and cav −1 was again abundant in F6–8 MVs (fig. 3E), indicating that cav-1-positive (miRNA-rich) EVs are mainly derived from BALF MVs, but the sequential ultracentrifugation alone is not sufficient to purify the miRNA-rich EVs due to the heterogeneity of BALF MVs. Meanwhile, CD9, which is another EV marker protein [11], was only detectable in low-density fractions (fig. 3E).
We next isolated EVs from WT and cav-1−/− ATIs. EVs were then fractioned using the sucrose density-gradient. ATI EVs were broadly detected over the fractions, and there were no significant differences between WT and cav-1−/− epithelial EVs (fig. 3F). We confirmed that cav-1 (fig. 3F) and various miRNAs are highly concentrated in the F6–8 EVs (fig. 3G). Deletion of cav-1 significantly up- or down-regulated the expressions of miRNAs in the miRNA-rich EVs (F6–8 EVs) (fig. 3H). Collectively, the data suggest that cav-1 is a central component and potential biomarker of miRNA-rich EVs.
The miRNA-rich EVs efficiently deliver their RNA compositions into alveolar macrophages (AMs).
We obtained 5×105 of AMs, and approximately 6×1010 of miRNA-deficient EVs (F2 EVs) and 5×109 of miRNA-enriched EVs (F7–8 EVs). The EV uptake can be saturated in the recipient cell [35, 36]. Thus, we first tested the saturation kinetics of EV uptake by AMs. The surface of EVs was labeled with PKH67 (green fluoresce) and primary AMs (105) were exposed to the 10-fold serial diluted PKH67-EVs (from 104 to 1010 EVs derived from F2 or F7/8 fraction). As shown in figure 4B, both EVs were effectively taken up by AMs with no significant difference. Notably, the EV uptake was almost saturated by 109 EVs exposure and was not significantly increased after exposure to 1010 EVs (fig. 4C). This result suggests that miRNA-rich EVs (109 EVs) are probably sufficient to deliver their contents into recipient AMs.
Figure 4. The microRNA-enriched EVs efficiently deliver their RNAs into AMs.

(A-C) Equal amounts of EVs from density gradient-fractioned BALF EVs (F2, F7, or F8) were labeled with PKH67 (staining for EV surface lipids) or OA (staining for EV-containing RNAs) as described in (A). Primary AMs (105 cells) were then exposed to the labeled EVs for 16 h to allow EV internalization, followed by acidic washing to remove the surface-bounded EVs. The saturation kinetics of EV uptake by the alveolar macrophage (105 cells) were shown (B-C) (mean ± SD, n = 3 per group). RNA transferring capacity of each fractioned EVs (109 EVs) into AMs (105 cells) was evaluated using confocal microscopy (D-E) (box and whisker plot, n = 4–6). * P < 0.05, ** P < 0.01, and *** P < 0.001 between the groups indicated.
Next, we evaluated the delivery efficiency of EV-RNAs. The same number of vesicles (109 EVs) of the fractioned EVs (F2, F7, and F8) were used for labeling their RNAs with Acridine Orange (AO) (red fluoresce) (fig. 4A). We demonstrated that F7 and F8 EVs efficiently deliver their RNA contents into primary AMs (105), compared to F2 EVs (fig. 4D and 4E).
MiRNAs in the miRNA-rich EVs promote innate immune responses in bacterial lung infection.
AMs are the first line of defense in lung alveoli [37, 38] and the most efficient cell type to uptake EVs in lung [20, 39]. To investigate the functional significance of the miRNA-rich EVs on AM function, we separately isolated the small/miRNAs and large RNAs from the fractioned EVs (F6–8) as shown in figure 5A–B. The small/miRNAs were more concentrated than large RNAs in the F6–8 EVs (fig. 5C). As illustrated in figure 5D, we delivered the isolated RNAs into AMs followed by infection of the AMs with pseudomonas aeruginosa (P. aeruginosa) for the indicated time points. The P. aeruginosa infection dramatically increased the pro-inflammatory TNFα, IL1β, and MIP2 gene in AMs (fig. 5E). Interestingly, the small/miRNA-delivered AMs were more reactive to increase IL1β and MIP2 gene during P. aeruginosa infection (fig. 5E). On the contrary, the delivery of large RNAs did not significantly alter the inflammatory responses of AMs (fig. 5E and suppl. fig. 2A). The pre-delivery of small/miRNAs also induced NLRP3 (fig. 5F) and enzymatic activity of caspase-1 (fig. 5G) in response to P. aeruginosa infection, indicating the inflammasome activation by small/miRNAs in the miRNA-rich EVs. Meanwhile, caspase-1 gene expression was not changed by the small/miRNA delivery (fig. 5F). We also confirmed that the small/miRNA-delivered AMs increased the production of IL1β and MIP2 in response to P. aeruginosa infection (fig. 5H). Additionally, we compared the effects of the miRNA-rich EVs (F6–8 EVs) with the miRNA-deficient EVs (F1–3 EVs). As shown in suppl. fig. 2B, pro-inflammatory genes (IL1β, IL6, MIP2, and NLRP3) in AMs treated with F6–8 EVs were significantly higher than those in AMs treated with F1–3 EVs under the P. aeruginosa infection.
Figure 5. EV-containing small/microRNAs stimulate inflammasome and MIP2 production in AMs after P. aeruginosa infection.

(A-C) Small/microRNAs and large RNAs were separately isolated from miRNA-rich EVs (F6–8 EVs) as illustrated in (A). The successful RNA isolation was verified using RNA electrophoresis (B) and the concentration of small/microRNAs and large RNAs were measured (C). (D-F) The purified RNAs were then directly transferred into AMs (105 MH-S cells) using liposomal transfection system (Lipofectamine LTX), followed by P. aeruginosa infection of the AMs for the indicated time points (D). Gene expression of inflammatory cytokines (E) and imflammasome components (F) were determined using qPCR (mean ± SD, n = 4). (G-H) AMs transfected with the small/miRNAs from F6–8 EVs were infected with P. aeruginosa for 3h. Capspase-1 enzymatic activity (G) and IL1β and MIP2 cytokine production (H) were then evaluated using luciferase assay and ELISA, respectively (mean ± SD or box and whisker plots, n = 4). * P < 0.05, ** P < 0.01 and *** P < 0.001 between the groups indicated.
Next, given that miRNA-rich EVs (F6–8 EVs) were derived from ATIs in vivo (fig. 3), we generated the miRNA-depleted ATIs using CRISPR/Cas9-mediated deletion of Dicer1. Dicer is required to produce matured miRNAs [40]. We confirmed the successful deletion of Dicer1 in ATIs using qPCR (fig. 6A) and western blot analysis (fig. 6B). Consistent with the previous report [41, 42], we also found a significant reduction of argonaute-2 (fig. 6B), which is a miRNA-interacting protein involved in the sorting of miRNAs into EVs [42, 43]. We next isolated EVs from ATIs (fig. 6C–D). Both WT and dicer1−/− ATIs actively produced EVs in the size range of 50–300 nm diameter (fig. 6C). MiRNA expression in both ATIs and ATI-derived EVs was suppressed after deletion of dicer1 (fig. 6D), indicating successful generation of miRNA-depleted ATI EVs. We next delivered the ATI EVs (109 EVs) into mouse lung using intratracheal instillation, followed by lung infection with P. aeruginosa for 3h, as illustrated in figure 6E. We initially confirmed that EVs were effectively taken up by AMs in vivo (fig. 6F). Consistent with our in vitro results, P. aeruginosa infection robustly induced IL1β and MIP2 production (fig. 6G) and recruitment of neutrophils into lung in vivo (fig. 6H–I). MIP2 is known as a key neutrophil chemoattractant [44]. Importantly, WT EV-instilled mice were more responsive to the P. aeruginosa infection, compared to the mice instilled with miRNA-depleted EVs (fig. 6G–I). We next isolated AMs from the bacteria-infected mouse lung BALF, and confirmed that caspase-1 enzymatic activity (fig. 6J), gene expressions of NLRP3, but not caspase-1 (fig. 6K), and the M1 macrophage-related markers (fig. 6L) were significantly upregulated in AMs from WT EV-instilled mice than those in AMs from the mice instilled with miRNA-depleted EVs. Taken together, our results show that ATI EV-miRNAs endorse the innate immune responses involving inflammasome activation, neutrophil recruitment and M1 macrophage polarization in bacterial lung infection.
Figure 6. MicroRNAs in ATI-derived EVs promote innate immune responses during P. aeruginosa infection in vivo.

(A-B) Plasmid containing Dicer1 gRNA and cas-9 was stably transfected into E10 ATIs. Deletion of Dicer1 in ATIs was confirmed using qPCR (mRNA region: 1017–1258 bp or 2645–2865 bp, which contain gRNA binding site) (A) and western blotting (B) (mean ± SD, n = 3). (C-D) EVs were isolated from the WT and Dicer1−/− ATIs, and the isolated EVs were verified using NTA (C). Global miRNA depletion in the dicer1−/− ATI and their EVs were evaluated using qPCR (D) (heatmap, n = 3). (E-L) EVs isolated from ATIs were intratracheally delivered into mouse lung (109 EVs per mouse), followed by P. aeruginosa infection of the lung. After 3h, the analyses of the BALFs were illustrated in (E). Frist, AMs were isolated from BALF. The uptake of exogenous EVs which were labeled with CFSE was visualized using confocal microscopy (F). Productions of IL1β and MIP2 in BALFs were measured using ELISA (G) (box and whisker plots, n = 4). Inflammatory cells in BALFs were cytospinned and H&E stained (H). The AMs and neutrophils were then counted under light microscope (I) (mean ± SD, n = 4). Caspase-1 enzymatic activities (J), gene expressions of NLRP3 and caspase-1 (K), and M1/M2-related gene expressions (L) in the isolated AMs were determined using luciferase reporter assay or qPCR (n = 4 per group).
Discussion
In the past decade, growing research has focused on the potential of circulating miRNAs to serve as biomarkers of human diseases and disease progression, particularly in cancer and metastasis [45, 46]. However, the diagnostic specificity and reproducibility of the reported “marker miRNAs” remain a challenge. In human body fluid and blood, a complex mixture of miRNAs is derived from various cell and tissue types [45]. Therefore, to develop a disease-specific marker, miRNAs from specific EVs derived from the same group of cells are much more attractive to yield more consistent and reproductive results. MiRNAs secreted to the extracellular space are either incorporated into EVs or in a vesicle-free form bound to various protein and lipoprotein complexes [45]. Intraluminal RNA content is thought to be relatively stable and the EV membrane protects the RNA cargo from enzymatic degradation. Therefore, EV-containing miRNAs are thought to be a more consistent source of miRNA biomarkers. Furthermore, EVs not only serve as a transporter that exports the intracellular miRNA, but also deliver miRNA cargo to recipient cells [43, 47]. Emerging interests are growing on the “EV-mediated delivery” as a therapeutic strategy. However, despite the enthusiasm about EV-containing miRNAs, many challenges remain on EV-miRNA research.
The first challenge is that the category or nomenclature of EVs remains unsettled. For example, it is unclear whether an exosome or microvesicle (MV) can be defined by size, surface markers or biogenesis. Even if the exosome and MV are defined by all of the above, there is an overlap in size range between exosomes and MVs. Do MVs and exosomes that are similar in size contain similar miRNA profiles? Can MV- or exosome-containing miRNAs serve as biomarkers? Apparently, it has been reported that far less than one molecule of a given miRNA can be detected per EV [14], which raises skepticism on whether EV-containing miRNAs are biologically meaningful and whether EV-containing miRNAs can serve as biomarkers or simply as non-specific cell debris.
On the other hand, it can be argued that due to a high variation of EV isolation methods, certain steps of the EV isolation that the above investigators used results in a low miRNA yield and a low sensitivity. Our previous reports also show that a large amount (copy number) of miRNAs exist in MVs [2, 15] but not in exosomes, consistent with the previous reports. In this study, we found that most BALF-derived EVs have very low concentration of proteins, RNAs, and miRNAs (fig. 2A–E), probably due to the heterogenicity of EVs and the mixture of a large amount of non-RNA-containing EVs. The uncertainty of isolated EV populations (exosomes vs. MVs vs. ABs) makes EV-miRNAs more difficult to serve as biomarkers or therapeutic strategies. Therefore, we challenge the idea of using MV or exosome as a category to isolate the miRNA-rich EVs. In fact, no matter what the nomenclature is defined, EVs and their subpopulations will be useful for developing diagnostic and prognostic miRNAs only if they can be isolated with high purity reproducibly and can retain sufficient RNAs for analyses [45].
Using the conventional ultracentrifugation and sucrose-gradients, we newly identified a subpopulation of BALF EVs which are miRNA-rich (fig. 2C and E). More interestingly, this subpopulation of miRNA-rich EVs are mainly derived from type I alveolar epithelial cells (ATI) and are caveolin-1 (cav-1) – rich EVs. Cav-1 is a well-documented ATI cell marker, further confirming that this group of EVs is derived from ATI cells. Although the number of vesicles in this subpopulation is relatively low compared to the total amount of BALF EVs, they play an essential role in modulating the phenotype of the recipient alveolar macrophage (fig. 5 and fig. 6). Lung alveoli are compactly surrounded by lung epithelium (95 % of ATI) [48], and AMs are the main type of immune cells (above 90 %) in the alveoli [49, 50]. Recently, we and other groups have shown that BALF EVs are mainly derived from lung epithelial cells and AMs [2, 15, 51]. Interestingly, we can detect alveolar macrophage-derived EVs (CD68+) only in the low-density fractions (fig. 3A). Meanwhile, epithelial EVs (PDPN+) were broadly detected over most of the density fractions. The miRNA-rich EVs were exclusively localized in the high-density fractions, indicating that density-dependent EV separation would be useful to identify specific types of EV populations.
EVs are composed of lipids, proteins, and nucleic acids [52]. Those components are largely heterogeneous based on their cellular source and probably contribute to determining their EV density. Many factors contribute to the density of EVs, including but not limited to lipids (cholesterol, fatty acid, triglyceride, and phospholipids), membrane proteins and protein modifications (glycoprotein, integral protein, globular protein, peripheral protein and protein channel), carbohydrates, RNA length and structure [11]. Notably, we found that EV-containing lipid-raft proteins (flotillin and caveolin) are mainly localized in the high-density fractions (fig. 3). Lipid rafts are specialized glycolipoprotein microdomains at the cell membrane and are 3 to 5-fold enriched in cholesterol, sphingolipids such as sphingomyelin, and various receptor proteins [53, 54]. They play essential roles in the regulation of endo- and exocytosis of many intracellular components and mediating signaling pathways of important cellular activities [53, 54]. Interestingly, RNAs were also concentrated in the high-density fractioned EVs (fig. 2), suggesting the contribution of lipid rafts and RNAs to their EV density.
Cav-1 is highly expressed in ATI [24] and the primary structural component of caveolae, which is responsible for the trafficking of diverse cellular molecules [55]. Cav-1 has also been identified as a common component of EVs [12]. Here, we reported that cav-1 is exclusively expressed in the miRNA-rich EVs which are derived from lung epithelium (fig. 3), suggesting that cav-1 is a vital component and potential biomarker of miRNA-rich EVs. Lung alveolar epithelium is a pivotal regulator of the immune system in lung homeostasis and disease conditions [24]. Recently, we have reported the lung epithelial EVs as a novel regulator of the lung inflammation in acute lung injury (ALI) [2, 15]. Lung epithelial EVs are dramatically increased during the development of ALIs, and the EV-containing specific miRNAs are responsible for macrophage migration and activation [2, 15]. Moreover, it has been demonstrated that cav-1 involves the inflammatory lung responses after LPS challenge [56]. Here we show a novel insight that cav-1/miRNA-rich EVs promote AM-mediated inflammatory responses including inflammasome activation and M1-macrophage polarization after bacterial lung infection.
EVs derived from dicer−/− epithelial cells attenuated the macrophage activation and neutrophil recruitment into lungs after bacterial infection (fig. 6), suggesting that EV-miRNAs play more crucial roles than EV-containing proteins. Proteins which are encapsulated into EVs may have lost their critical structure and domains due to truncation or fragmentation. On the other hand, miRNAs, only approximately 22 nucleotide-long, are relatively stable under the shield of EVs [32, 33]. Pro-inflammatory function of EV-containing miRNA has been reported [2, 57]. For instance, miR-221 which is highly expressed in miRNA-rich EVs (fig. 2C) downregulates SOCS1, a suppressor of cytokine signaling and production [58]. Moreover, SOCS1 is known to inhibit inflammasome activation [59].
Although the amount of these miRNA-rich EVs are low (9% of the total BALF EVs), their uptake by macrophages potentially out-weighs other populations of EVs, given the abundant cav-1 in these EVs. Cav-1 has been well known for its regulatory roles in mediating endocytosis. It is very possible that macrophages engulf EVs not only via phagocytosis, but also via endocytosis mediated by lipid raft proteins. Future studies are required to compare the uptake of different subpopulations of EVs by different cells in order to further address the underlying functional roles of each EV population.
One pitfall of this study is that conventional methods using ultracentrifugation and sucrose gradients must be used to isolate EVs here. This process is time-consuming. Numerous commercial kits for isolation and purification of EVs have been developed, but whether these kits can be used to obtain miRNA-rich EVs remain unexplored.
In summary, despite many reports on the functional significance of the EV-containing miRNAs, the actual concentration of miRNAs in the heterogeneous EVs has been a challenge for biomarker development and functional studies. Our study contributes to a novel method to identify miRNA-rich EVs in body fluids and confirms their functional significance in vivo.
Supplementary Material
Highlights.
miRNA-enriched extracellular vesicles (EVs) exist in bronchoalveolar lavage fluids
miRNA-rich EVs can be isolated using density-gradient fractionation
miRNA-rich EVs are most likely derived from lung epithelial type-I cells
Caveolin-1, a lipid raft protein, is a potential biomarker of miRNA-rich EVs.
miRNA-rich EVs contribute to innate immune responses after bacterial lung infection
Acknowledgments
This work is support by NIH R21 AI121644 (Y.J.), NIH R33 AI121644R01 (Y.J.), NIH R01 GM127596–01 (Y.J.), NIH R01 GM111313 (Y.J.) and Wing Tat Lee Awards (Y.J.). The authors thank Matthew Haney at Center for Nanotechnology in Drug Delivery, UNC Eshelman School of Pharmacy, for his technical assistance with NTA analysis.
This work is support by NIH R21 AI121644 (Y.J.), NIH R33 AI121644R01 (Y.J.), NIH R01 GM127596–01 (Y.J.), NIH R01 GM111313 (Y.J.) and Wing Tat Lee Awards (Y.J.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tkach M and Thery C, Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell, 2016. 164(6): p. 1226–32. [DOI] [PubMed] [Google Scholar]
- 2.Lee H, et al. , Epithelial cell-derived microvesicles activate macrophages and promote inflammation via microvesicle-containing microRNAs. Sci Rep, 2016. 6: p. 35250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moldovan L, et al. , Analyzing the circulating microRNAs in exosomes/extracellular vesicles from serum or plasma by qRT-PCR. Methods Mol Biol, 2013. 1024: p. 129–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crescitelli R, et al. , Distinct RNA profiles in subpopulations of extracellular vesicles: apoptotic bodies, microvesicles and exosomes. J Extracell Vesicles, 2013. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turturici G, et al. , Extracellular membrane vesicles as a mechanism of cell-to-cell communication: advantages and disadvantages. Am J Physiol Cell Physiol, 2014. 306(7): p. C621–33. [DOI] [PubMed] [Google Scholar]
- 6.Atkin-Smith GK, et al. , Isolation of cell type-specific apoptotic bodies by fluorescence-activated cell sorting. Sci Rep, 2017. 7: p. 39846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu Z, et al. , Macrophage-derived apoptotic bodies promote the proliferation of the recipient cells via shuttling microRNA-221/222. J Leukoc Biol, 2017. 101(6): p. 1349–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnstone RM, et al. , Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J Biol Chem, 1987. 262(19): p. 9412–20. [PubMed] [Google Scholar]
- 9.Ng YH, et al. , Endometrial exosomes/microvesicles in the uterine microenvironment: a new paradigm for embryo-endometrial cross talk at implantation. PLoS One, 2013. 8(3): p. e58502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Todorova D, et al. , Extracellular Vesicles in Angiogenesis. Circ Res, 2017. 120(10): p. 1658–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keerthikumar S, et al. , ExoCarta: A Web-Based Compendium of Exosomal Cargo. J Mol Biol, 2016. 428(4): p. 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Logozzi M, et al. , High levels of exosomes expressing CD63 and caveolin-1 in plasma of melanoma patients. PLoS One, 2009. 4(4): p. e5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee Y, El Andaloussi S, and Wood MJ, Exosomes and microvesicles: extracellular vesicles for genetic information transfer and gene therapy. Hum Mol Genet, 2012. 21(R1): p. R125–34. [DOI] [PubMed] [Google Scholar]
- 14.Chevillet JR, et al. , Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc Natl Acad Sci U S A, 2014. 111(41): p. 14888–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee H, et al. , Lung Epithelial Cell-Derived Microvesicles Regulate Macrophage Migration via MicroRNA-17/221-Induced Integrin beta1 Recycling. J Immunol, 2017. 199(4): p. 1453–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee HD, et al. , Exosome release of ADAM15 and the functional implications of human macrophage-derived ADAM15 exosomes. FASEB J, 2012. 26(7): p. 3084–95. [DOI] [PubMed] [Google Scholar]
- 17.Han CZ, et al. , Macrophages redirect phagocytosis by non-professional phagocytes and influence inflammation. Nature, 2016. 539(7630): p. 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu R, et al. , Highly-purified exosomes and shed microvesicles isolated from the human colon cancer cell line LIM1863 by sequential centrifugal ultrafiltration are biochemically and functionally distinct. Methods, 2015. 87: p. 11–25. [DOI] [PubMed] [Google Scholar]
- 19.Lee HD, Kim YH, and Kim DS, Exosomes derived from human macrophages suppress endothelial cell migration by controlling integrin trafficking. Eur J Immunol, 2014. 44(4): p. 1156–69. [DOI] [PubMed] [Google Scholar]
- 20.Lee H, et al. , Functional Evidence of Pulmonary Extracellular Vesicles in Infectious and Noninfectious Lung Inflammation. J Immunol, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang X, Goncalves R, and Mosser DM, The isolation and characterization of murine macrophages. Curr Protoc Immunol, 2008. Chapter 14: p. Unit 14 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang X, et al. , p62 sequestosome 1/light chain 3b complex confers cytoprotection on lung epithelial cells after hyperoxia. Am J Respir Cell Mol Biol, 2013. 48(4): p. 489–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kemp SJ, et al. , Immortalization of human alveolar epithelial cells to investigate nanoparticle uptake. Am J Respir Cell Mol Biol, 2008. 39(5): p. 591–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamamoto K, et al. , Type I alveolar epithelial cells mount innate immune responses during pneumococcal pneumonia. J Immunol, 2012. 189(5): p. 2450–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valadi H, et al. , Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol, 2007. 9(6): p. 654–9. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, et al. , Exosome and exosomal microRNA: trafficking, sorting, and function. Genomics Proteomics Bioinformatics, 2015. 13(1): p. 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paraskevi A, et al. , Circulating MicroRNA in inflammatory bowel disease. J Crohns Colitis, 2012. 6(9): p. 900–4. [DOI] [PubMed] [Google Scholar]
- 28.Zhu D, et al. , MicroRNA-17/20a/106a modulate macrophage inflammatory responses through targeting signal-regulatory protein alpha. J Allergy Clin Immunol, 2013. 132(2): p. 426–36 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casadei L, et al. , Exosome-Derived miR-25–3p and miR-92a-3p Stimulate Liposarcoma Progression. Cancer Res, 2017. 77(14): p. 3846–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou Y, et al. , miRNA-221–3p Enhances the Secretion of Interleukin-4 in Mast Cells through the Phosphatase and Tensin Homolog/p38/Nuclear Factor-kappaB Pathway. PLoS One, 2016. 11(2): p. e0148821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Armstrong DA, et al. , Pulmonary microRNA profiling: implications in upper lobe predominant lung disease. Clin Epigenetics, 2017. 9: p. 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng L, et al. , Exosomes provide a protective and enriched source of miRNA for biomarker profiling compared to intracellular and cell-free blood. J Extracell Vesicles, 2014. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koga Y, et al. , Exosome can prevent RNase from degrading microRNA in feces. J Gastrointest Oncol, 2011. 2(4): p. 215–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suchorska WM and Lach MS, The role of exosomes in tumor progression and metastasis (Review). Oncol Rep, 2016. 35(3): p. 1237–44. [DOI] [PubMed] [Google Scholar]
- 35.Harris DA, et al. , Exosomes released from breast cancer carcinomas stimulate cell movement. PLoS One, 2015. 10(3): p. e0117495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heusermann W, et al. , Exosomes surf on filopodia to enter cells at endocytic hot spots, traffic within endosomes, and are targeted to the ER. J Cell Biol, 2016. 213(2): p. 173–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guth AM, et al. , Lung environment determines unique phenotype of alveolar macrophages. Am J Physiol Lung Cell Mol Physiol, 2009. 296(6): p. L936–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karagianni AE, et al. , The equine alveolar macrophage: functional and phenotypic comparisons with peritoneal macrophages. Vet Immunol Immunopathol, 2013. 155(4): p. 219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang D, et al. , Exosome-Mediated Small RNA Delivery: A Novel Therapeutic Approach for Inflammatory Lung Responses. Mol Ther, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomou T, et al. , Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature, 2017. 542(7642): p. 450–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bodak M, et al. , Dicer, a new regulator of pluripotency exit and LINE-1 elements in mouse embryonic stem cells. FEBS Open Bio, 2017. 7(2): p. 204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martinez NJ and Gregory RI, Argonaute2 expression is post-transcriptionally coupled to microRNA abundance. RNA, 2013. 19(5): p. 605–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKenzie AJ, et al. , KRAS-MEK Signaling Controls Ago2 Sorting into Exosomes. Cell Rep, 2016. 15(5): p. 978–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feng L, et al. , Modulation of neutrophil influx in glomerulonephritis in the rat with anti-macrophage inflammatory protein-2 (MIP-2) antibody. J Clin Invest, 1995. 95(3): p. 1009–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Witwer KW, Circulating microRNA biomarker studies: pitfalls and potential solutions. Clin Chem, 2015. 61(1): p. 56–63. [DOI] [PubMed] [Google Scholar]
- 46.Endzelins E, et al. , Detection of circulating miRNAs: comparative analysis of extracellular vesicle-incorporated miRNAs and cell-free miRNAs in whole plasma of prostate cancer patients. BMC Cancer, 2017. 17(1): p. 730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Villarroya-Beltri C, et al. , Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun, 2013. 4: p. 2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barkauskas CE, et al. , Lung organoids: current uses and future promise. Development, 2017. 144(6): p. 986–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reynolds HY, Bronchoalveolar lavage. Am Rev Respir Dis, 1987. 135(1): p. 250–63. [DOI] [PubMed] [Google Scholar]
- 50.Vlahos R and Bozinovski S, Role of alveolar macrophages in chronic obstructive pulmonary disease. Front Immunol, 2014. 5: p. 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soni S, et al. , Alveolar macrophage-derived microvesicles mediate acute lung injury. Thorax, 2016. 71(11): p. 1020–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yanez-Mo M, et al. , Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles, 2015. 4: p. 27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lingwood D and Simons K, Lipid rafts as a membrane-organizing principle. Science, 2010. 327(5961): p. 46–50. [DOI] [PubMed] [Google Scholar]
- 54.Simons K and Sampaio JL, Membrane organization and lipid rafts. Cold Spring Harb Perspect Biol, 2011. 3(10): p. a004697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jin Y, et al. , Caveolin-1: a critical regulator of lung injury. Am J Physiol Lung Cell Mol Physiol, 2011. 300(2): p. L151–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garrean S, et al. , Caveolin-1 regulates NF-kappaB activation and lung inflammatory response to sepsis induced by lipopolysaccharide. J Immunol, 2006. 177(7): p. 4853–60. [DOI] [PubMed] [Google Scholar]
- 57.Alexander M, et al. , Exosome-delivered microRNAs modulate the inflammatory response to endotoxin. Nat Commun, 2015. 6: p. 7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kinjyo I, et al. , SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity, 2002. 17(5): p. 583–91. [DOI] [PubMed] [Google Scholar]
- 59.Zhang L, et al. , SOCS-1 Suppresses Inflammation Through Inhibition of NALP3 Inflammasome Formation in Smoke Inhalation-Induced Acute Lung Injury. Inflammation, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.