

Abstract
Sublethal doses of γ-rays promote cancer cell invasion by stimulating a signaling pathway that sequentially involves p53, sulfatase 2 (SULF2), β-catenin, interleukin-6 (IL-6), signal transducer and activator of transcription 3 (STAT3), and Bcl-XL. Given that Bcl-XL can increase O2•− production by stimulating respiratory complex I, the possible role of mitochondrial reactive oxygen species (ROS) in γ-irradiation-induced cell invasion was investigated. Indeed, γ-irradiation promoted cell invasion by increasing mitochondrial ROS levels, which was prevented by metformin, an inhibitor of complex I. γ-Irradiation-stimulated STAT3 increased the expression of superoxide dismutase 2 (SOD2), a mitochondrial enzyme that catalyzes the conversion of O2•− to hydrogen peroxide (H2O2). In contrast to O2•−, H2O2 functions as a signaling molecule. γ-Irradiation consistently stimulated the Src-dependent invasion pathway in a manner dependent on both complex I and SOD2. SOD2 was also essential for the invasion of un-irradiated cancer cells induced by upregulation of Bcl-XL, an intracellular oncogene, or extracellular factors, such as SULF2 and IL-6. Overall, these data suggested that SOD2 is critical for the malignant effects of radiotherapy and tumor progression through diverse endogenous factors.
Subject terms: Oncogenes, Cell invasion
Cancer treatment: Stopping the spread after radiotherapy
A drug usually used to treat type 2 diabetes may also help to prevent cancer relapse following radiotherapy, which is commonly used to kill cancer cells. However, any tumor cells that survive radiation are highly invasive, sometimes causing tumors to spread. Hong-Duck Um and co-workers at the Korea Institute of Radiological & Medical Sciences in Seoul, South Korea, noticed that the surviving cells often showed higher levels of a key enzyme, superoxide dismutase 2 (SOD2), which is involved in energy production in the cellular powerhouse, the mitochondria. Artificially increasing levels of SOD2, without radiation, made cells more invasive. Treatment with metformin, which prevents production of the molecule that SOD2 acts on, prevented cells from becoming invasive. SOD2 has been implicated in many cancers, and is therefore a very promising therapeutic target.
Introduction
Ionizing radiation (IR), such as γ-irradiation, is a major therapeutic modality for treating cancer. In most patients, IR offers a significant survival benefit, but in some patients, local recurrence or distal metastasis following radiotherapy is a major therapeutic challenge. These undesirable consequences may reflect the regrowth or spread of cancer cells that survived radiotherapy. Studies using cultured cells and animal models have shown that sublethal doses of IR increase the mobility, invasiveness, and metastatic potential of cancer cells1,2, suggesting that IR promotes malignant behavior in postradiation tumors. Therefore, the cellular components involved in the malignant effects of IR should be defined to develop new strategies for improving the therapeutic effects of IR.
Mitochondria have emerged as central regulators of cancer cell invasion and metastasis, and reactive oxygen species (ROS) produced via the mitochondrial respiratory chain have been implicated as stimulators of various cellular pathways leading to cell migration and invasion3. The production of mitochondrial ROS is regulated by Bcl-2 family proteins4. Although they were originally identified as key regulators of cell death5, certain Bcl-2 family members also regulate cell migration, invasion, and cancer metastasis4. A well-characterized example is the group of pro-survival Bcl-2 family members, including Bcl-XL, Bcl-2, and Bcl-w, which stimulate complex I, a major source of ROS in the mitochondrial respiratory chain, to produce additional ROS. The ROS produced the following overexpression of Bcl-w, or Bcl-XL promote cell invasion by stimulating Src and its downstream signaling components6.
We have previously shown that sublethal doses of IR increase sulfatase 2 (SULF2) expression via the p53 transcription factor7. SULF2 is an extracellular sulfatase that modulates the signaling activities of diverse cell surface receptors8, and IR-induced SULF2 mediates the pro-invasive activity of IR by stimulating the signaling pathway that sequentially involves β-catenin, interleukin-6 (IL-6), and signal transducer and activator of transcription 3 (STAT3)7. STAT3 is a transcription factor that induces Bcl-XL expression9. Consistently, sublethal doses of IR increase the messenger RNA (mRNA) and protein levels of Bcl-XL in several cancer cell types, and Bcl-XL knockdown abolishes the pro-invasive activity of IR7,10, suggesting a role for Bcl-XL in IR-induced cell invasion. These results suggest the involvement of mitochondrial ROS in IR-induced cancer cell invasion. However, this possibility has not been directly addressed.
ROS include free radicals, such as superoxide anion (O2•−) and hydroxyl radical (HO•), as well as nonradical molecules, such as hydrogen peroxide (H2O2). Among these free radicals, H2O2 has a relatively long half-life and can freely diffuse to induce signaling11. Therefore, it is thought that H2O2 is the effector ROS that modulates activities of signaling molecules. However, the mitochondrial respiratory chain produces O2•− that needs to be converted to H2O2 to modulate signaling. Superoxide dismutase (SOD) is a metalloenzyme that catalyzes the conversion of O2•− to H2O2. In mammals, there are three distinct types of SOD as follows: Cu/ZnSOD (SOD1), MnSOD (SOD2), and extracellular Cu/ZnSOD (SOD3)12. SOD1 is the major intracellular form of SOD, and it is localized primarily in the cytosol. In contrast, SOD2 is exclusively localized to the mitochondrial matrix. This feature of SOD2 suggests that it may be involved in the conversion of mitochondrial O2•− to H2O2, thus contributing to cell invasion. Hence, the present study investigated the potential role of SOD2 in IR-induced cell invasion. The possibility was indeed supported by our data. The present findings demonstrated that SOD2 also mediates the invasion of un-irradiated cancer cells induced by upregulation of diverse oncogenic components, supporting the role of SOD2 in tumor progression. Therefore, SOD2 is a potential target for preventing cancer cell invasion following radiotherapy and suppressing tumor progression under diverse conditions.
Materials and methods
Antibodies and recombinant proteins
The following antibodies were used in the present study: anti-SOD2 from Enzo Life Sciences (Farmingdale, NY, USA); anti-IL-6 and anti-β-catenin from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti-Bcl-XL, anti-STAT3, anti-Src, and anti-phospho-Src from Cell Signaling Technology (Danvers, MA, USA); and anti-β-actin from Sigma-Aldrich (St. Louis, MO, USA). Recombinant human IL-6 was purchased from Millipore (Darmstadt, Germany).
siRNA and shRNAs
Small interfering RNA (siRNAs) targeting IL-6 (S7312) and Bcl-XL (120717) were purchased from Ambion (Austin, TX, USA). siRNAs targeting SOD2 (sc-41655), β-catenin (sc-44275), and STAT3 (sc-29209) as well as lentiviruses expressing small hairpin RNAs (shRNAs) targeting SOD2 (sc-41655-V), Bcl-XL (sc-43630-V), and SULF2 (sc-63088-V) were obtained from Santa Cruz Biotechnology.
Cell culture, transfection, infection, and treatments
All cell lines used in this study, except for HCT116/p53wt and HCT116/p53(−/−) colon cancer cells (a generous gift from Dr. Bert Vogelstein), were obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in RPMI-1640 (A549, H1299, and H460 lung cancer cells) or DMED (MCF-7 breast cancer and HCT116 cells) medium supplemented with 10% heat-inactivated fetal bovine serum (FBS). Expression constructs for SOD2, SULF2, and Bcl-XL were prepared using the pcDNA3 vector (Invitrogen, Carlsbad, CA, USA). Expression constructs and siRNAs were introduced into cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. The transfectants were used for experiments after 48 h of recovery. For long-term gene silencing, cells were infected with lentiviruses containing shRNAs targeting SOD2 in the presence of polybrene (5 μg/mL) according to the manufacturer’s protocols. Infected cells were selected with puromycin (2 μg/mL). For irradiation, cells were exposed to the specified doses of γ-rays from a 137Cs γ-ray source (Atomic Energy of Canada, Chalk River, Canada) at a dose rate of 3 Gy/min. Alternatively, cells were treated with the indicated concentrations of IL-6.
Western blotting
Cell lysates were prepared using previously described methods13. Proteins in the lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, electrotransferred onto nitrocellulose membranes (Millipore), and analyzed using the specified antibodies and an ECL detection system (Bio-Rad, Hercules, CA, USA).
RT-PCR and quantitative real-time PCR
Reverse transcription-PCR (RT-PCR) was performed by amplifying complementary DNA (cDNA) in Premix PCR solution (Takara, Shiga, Japan) with SOD2 primers (5′-GGA-AGC-CAT-CAA-ACG-TGA-CTT-3′ and 5′-GTG-CTC-CCA-CAC-ATC-AAT-CC-3′). Quantitative real-time PCR was performed using the SYBR Fast Universal qPCR Kit (Kapa Biosystems, Wilmington, MA, USA) and SOD2 primers (5′-GGC-CTA-CGT-GAA-CAA-CCT-GAA-3′ and 5′-CTG-TAA-CAT-CTC-CCT-TGG-CCA-3′). GAPDH was amplified in both PCR assays with the following primers as an internal control for normalization: 5′-CAT-CTC-TGC-CCC-CTC-TGC-TGA-3′ and 5′-GGA-TGA-CCT-TGC-CCA-CAG-CCT-3′. The RT-PCR and real-time PCR results were analyzed by agarose gel electrophoresis and an IQ-5 Real-Time System (Bio-Rad), respectively.
Invasion assay
As described previously14, cells in serum-free medium were seeded onto the upper surfaces of Matrigel-coated Transwell chambers (BD Biosciences, San Jose, CA, USA). The lower compartments of the chambers were filled with medium supplemented with 10% heat-inactivated FBS. After 16 h of incubation, cells that invaded the lower surface of the filter were stained with the Diff-Quick Kit (Fisher Scientific, Waltham, MA, USA) and counted under a microscope.
Analysis of mitochondrial ROS levels
Cells were exposed to 10 μM MitoSOX Red (Invitrogen) or 5 μM Peroxy Orange-1 (Tocris Bioscience, Bristol, UK) for 30 min, and cell-associated fluorescence was analyzed by flow cytometry.
Clonogenic assay
Various numbers of cells infected with the specified lentiviruses were seeded in triplicate into 60 mm dishes (100, 200, 400, and 800 cells/dish). After 24 h of incubation, cells were exposed to different doses of γ-rays (1, 3, 5, and 7 Gy). Irradiated and untreated control cells were cultured for 14 days. The number of colonies was counted with a colony counter (Imaging Products, Hollywood, CA, USA), and clonogenic survival was calculated as described previously15.
Statistical analysis
All experiments were performed at least three times to obtain means and standard deviations. Statistical significance was determined with one-way analysis of variance (GraphPad Software, La Jolla, CA, USA), and p values <0.05 were considered significant.
Results
Sublethal doses of IR increase SOD2 expression via the p53/SULF2/β-catenin/IL-6/STAT3 pathway
To investigate the potential involvement of SOD2 in IR-induced cell invasion, p53wt-expressing (H460 and A549 lung cancer cells as well as HCT116 colon cancer cells) and p53null cells (H1299 lung cancer cells) were irradiated with sublethal doses of γ-rays. Irradiation elevated protein levels of SOD2 in the p53wt-expressing cells but not in the p53null cells (Fig. 1a). Consistently, knockout of p53 in HCT116 cells abolished IR-induced SOD2 accumulation. It has been previously confirmed that p53 protein levels in p53wt-expressing cells are elevated upon γ-irradiation, but that p53 expression is not detected in p53null or p53-knockout cells even after γ-irradiation16–18. These findings suggested that the γ-irradiation mediated increase in SOD2 levels is p53 dependent.
Fig. 1. IR induces SOD2 expression via the p53/SULF2/β-catenin/IL-6/STAT3 pathway.

a–d Western blotting and RT-PCR were performed 48 h after γ-irradiation. a H460 and A549 lung cancer cells (p53wt) were infected with lentiviruses expressing control (nontargeting sequence) or SULF2-specific shRNA. These transfectants, along with H1299 lung cancer cells (p53null) and p53wt-expressing or p53-knockout HCT116 colon cancer cells, were irradiated with the indicated doses of γ-rays, and SOD2 levels were compared by western blot analysis using β-actin as a loading control. SULF2 expression was compared by RT-PCR using GAPDH as a loading control. b A549 and H460 cells were transfected with an empty or SULF2 expression vector, and SOD2 protein and SULF2 mRNA levels were compared. c H460 cells treated with a control or an siRNA targeting β-catenin, IL-6, or STAT3 were irradiated with 2 Gy of γ-rays, and the levels of the indicated proteins were compared. d H460 cells infected with the lentiviruses indicated in a were irradiated, and SOD2 mRNA levels were analyzed by RT-PCR. e H460 cells treated with a control or a STAT3-targeting siRNA were irradiated, and SOD2 mRNA levels were compared by quantitative real-time PCR at 24 and 48 h after irradiation
p53 mediates IR-induced cell invasion by stimulating cellular pathways sequentially involving SULF2, β-catenin, IL-6, and STAT37. To investigate the relationship between this pathway and SOD2 induction, SULF2 was knocked down in H460 and A549 cells using a specific shRNA, which abolished or attenuated IR-induced SOD2 accumulation (Fig. 1a). Consistently, SOD2 protein levels were increased following overexpression of SULF2 in un-irradiated cells (Fig. 1b), confirming that SULF2 increases SOD2 protein levels. Moreover, knockdown of β-catenin, IL-6, or STAT3 using specific siRNAs reduced IR-induced accumulation of SOD2 protein (Fig. 1c), suggesting that IR increases SOD2 levels via the p53/SULF2/β-catenin/IL-6/STAT3 pathway.
In addition, IR elevated mRNA levels of SOD2, and this increase was abolished by knockdown of SULF2 (Fig. 1d) or STAT3 (Fig. 1e), suggesting that IR induces SOD2 protein accumulation by increasing its mRNA levels via STAT3 stimulated by the SULF2 pathway. The ability of STAT3 to induce SOD2 gene expression has also been reported in a mouse model of ischemic brain injury19. Moreover, the promoter region of SOD2 has been shown to contain STAT3-binding sites19.
SOD2 mediates IR-induced cell invasion
To investigate the potential role of SOD2 in IR-induced cell invasion, SOD2 was overexpressed in H460 and A549 cells. The invasiveness of both cell lines was enhanced by SOD2 overexpression (Fig. 2a). Consistently, IR-induced cell invasion was reduced by knockdown of SOD2 (Fig. 2b), suggesting that SOD2 mediates IR-induced cell invasion. Consistent with the role of SOD2, its knockdown also abolished cell invasion induced by SULF2 overexpression (Fig. 2c) or IL-6 treatment (Fig. 2d).
Fig. 2. SOD2 mediates IR-induced cell invasion.

a H460 and A549 cells were transfected with control (empty vector) or SOD2 expression vector. After 48 h of incubation, a cell invasion assay was performed. Cells that invaded through Matrigel-coated polycarbonate filters were imaged, and the number of invaded cells was counted and plotted. SOD protein levels were compared by western blot analysis. b H460 cells infected with lentiviruses expressing a control or SOD2-targeting shRNA were irradiated, and invasiveness and protein levels were then analyzed 48 h after irradiation. c H460 cells were cotransfected with a SULF2 expression vector and SOD2 siRNA as per the indicated combinations, and cell invasiveness was then assessed. d H460 cells infected with lentiviruses expressing control RNA or SOD2-specific shRNA were incubated in the presence or absence of IL-6 (100 ng/mL) for 24 h. Cell invasiveness was then assessed
IR induces activation of the mitochondrial ROS/Src-dependent invasion pathway
In response to IR, STAT3 increases the mRNA and protein levels of Bcl-XL, which is essential for IR-induced cell invasion10. To eliminate the possibility of Bcl-XL influencing SOD2 expression, Bcl-XL was overexpressed in H460 cells. Bcl-XL overexpression did not significantly affect SOD2 mRNA and protein levels (Fig. 3a), confirming the irrelevance of Bcl-XL in SOD2 induction. These data suggested that STAT3 promotes the expression of Bcl-XL and SOD2 in response to γ-irradiation. Notably, these two proteins co-accumulated at 24 and/or 48 h postradiation (Fig. 3b). This phenomenon was observed in four different p53wt cancer cell lines, including lung (H460 and A549), colon (HCT116/p53wt), and breast cancer cells (MCF-7), and the post-irradiation peak accumulation time was cell type-dependent. The co-induction of Bcl-XL and SOD2 implied their cooperation in IR-induced cellular responses.
Fig. 3. IR induces the mitochondrial ROS/Src-dependent invasion pathway.

a H460 cells were transfected with a control or a Bcl-XL-expression vector. After 24 and 48 h of incubation, SOD2 mRNA levels were compared by quantitative real-time PCR (top), and the levels of the indicated proteins were compared by western blotting (bottom). b The indicated cells were irradiated with the specified doses of γ-rays. Western blotting for SOD2 and Bcl-XL was performed at 24 and 48 h after irradiation. c Irradiated (2 Gy, 48 h) and untreated control H460 cells were incubated in the presence or absence of NAC (5 mM) and metformin (5 mM) for 1 h, and mitochondrial ROS levels were compared by staining cells with MitoSOX Red probes and analyzing them by flow cytometry. d Irradiated and untreated control cells were analyzed for their invasiveness in the presence or absence of NAC or metformin. The levels of Src and phosphorylated Src in the cells were compared by western blotting
Bcl-XL overexpression increases the ability of complex I to produce ROS, which, in turn, stimulates the Src-dependent invasion pathway6. Given that IR upregulates Bcl-XL10, it may also stimulate the ROS/Src pathway. Because complex I produces O2•−, the levels of mitochondrial O2•− levels in control and irradiated cells were compared using MitoSox Red, a probe specific for mitochondrial O2•−. IR increased mitochondrial O2•− levels, which was prevented by N-acetylcysteine (NAC), an ROS scavenger, or metformin, an inhibitor of complex I20 (Fig. 3c), suggesting that complex I contributes to IR-induced O2•− production. IR also increased Src phosphorylation, and both IR-induced cell invasion and Src phosphorylation were abolished by NAC or metformin (Fig. 3d). Similar effects on IR-induced Src phosphorylation were observed following Bcl-XL knockdown (Fig. 4a, left). These results suggested that O2•− produced by the Bcl-XL/complex I pathway contributes to the ability of IR to promote the Src-dependent invasion pathway.
Fig. 4. Bcl-XL and SOD2 functionally cooperate to promote Src phosphorylation and cell invasion.

a H460 cells treated with a control, Bcl-XL-targeting, or SOD2-targeting siRNA were irradiated, and the levels of the indicated proteins were compared 24 h after irradiation. b H460 cells infected with lentiviruses expressing the specified shRNAs were transfected with a Bcl-XL or SOD2 expression vector in the indicated combinations. Cellular invasiveness and the levels of Bcl-XL and SOD2 were compared after 48 h of incubation. c H460 cells were transfected with a Bcl-XL or SOD2 expression vector in the indicated combinations. Cellular invasiveness and the levels of the indicated proteins were compared after 48 h of incubation. d H460 cells treated with a control or SOD2-targeting siRNA were irradiated (2 Gy). After 48 h of incubation, irradiated and untreated control cells were analyzed for their levels of O2•− and H2O2 using MitoSox Red and Peroxy Orange-1, respectively. e H460 cells were transfected with a SULF2 expression vector and SOD2-targeting siRNA in the indicated combinations (left). Alternatively, H460 cells infected with lentiviruses expressing a control or SOD2-targeting shRNA were incubated in the presence or absence of 100 ng/mL IL-6 (right). After treatment for 24 h, cellular H2O2 levels were compared using Peroxy Orange-1
SOD2 mediates IR-induced Src phosphorylation in a cooperative manner with Bcl-XL
While complex I produces O2•−, H2O2 is the ROS that stimulates Src phosphorylation21. Therefore, SOD2 may mediate IR-induced cell invasion by converting O2•− to H2O2. IR-induced Src phosphorylation was attenuated by SOD2 knockdown (Fig. 4a, right). SOD2 knockdown also abolished Bcl-XL-induced cell invasion (Fig. 4b, left), a result consistent with the view that SOD2 acts downstream of Bcl-XL to promote cell invasion. However, cell invasion induced by SOD2 overexpression was also prevented by Bcl-XL knockdown (Fig. 4b, right), suggesting that SOD2 requires Bcl-XL for cell invasion. The functional interdependence of Bcl-XL and SOD2 supports the view that both O2•− production (by Bcl-XL) and its conversion to H2O2 (by SOD2) are critical for cell invasion. The functional cooperation between Bcl-XL and SOD2 was further supported by the finding that overexpression of both Bcl-XL and SOD2 was superior to overexpression of either alone for inducing Src phosphorylation and cell invasion (Fig. 4c).
To directly confirm the role of SOD2 in the conversion of IR-induced O2•− to H2O2, the levels of O2•− and H2O2 were determined using MitoSox Red and Peroxy Orange-122, respectively. SOD2 knockdown increased the amount of O2•− and decreased H2O2 levels in both control and irradiated cells (Fig. 4d). Consistently, SULF2 overexpression and IL-6 treatment increased H2O2 levels, and this increase was prevented by SOD2 knockdown (Fig. 4e). Therefore, these findings clearly demonstrated that SOD2 contributes to the conversion of O2•− generated via the IR-induced SULF2/IL-6 pathway to H2O2.
SOD2 promotes cell invasion without altering radiosensitivity
SOD2 contributes to radioresistance in certain but not all cell types23,24. Colony-forming assays revealed that SOD2 knockdown did not significantly influence the radiosensitivity of H460 and A549 cells (Fig. 5). However, IR-induced cell invasion was inhibited by SOD2 knockdown (Fig. 2b), suggesting that SOD2 mediates IR-induced cell invasion without altering cellular radiosensitivity.
Fig. 5. SOD2 does not influence radiosensitivity of H460 and A549 cells.
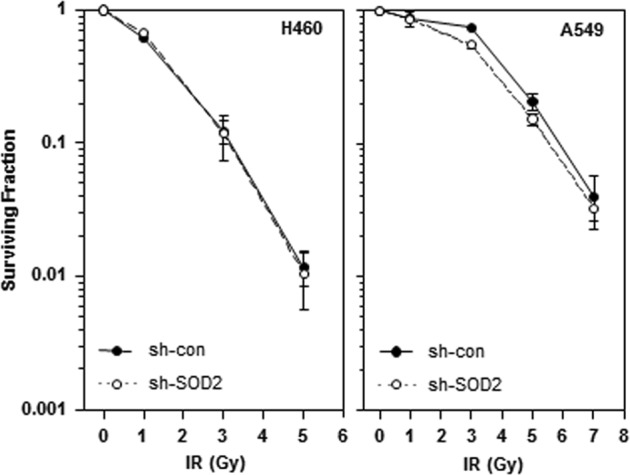
H460 and A549 cells were infected with lentiviruses expressing a control or SOD2-targeting shRNA and irradiated with the indicated doses of γ-rays, and cell survival was then analyzed by clonogenic assays
Discussion
In the present study, overexpression or knockdown SOD2 in lung cancer cells showed that SOD2 promotes cancer cell invasion. As similar results have been reported for other types of cancer, such as fibrosarcoma25, salivary26, and tongue carcinomas27,28, the pro-invasive activity of SOD2 appears to be a common feature of diverse cancers. This view is consistent with the report that SOD2 is upregulated in many cancer types, including lung, breast, colon, brain, thyroid, gastric, and salivary cancers, and especially late-stage metastatic cancers12. SOD2 upregulation in cancer cells has been correlated with distant metastasis, poorer prognosis, and lower overall and disease-free survival12,28. Therefore, SOD2 upregulation is critical for tumor progression.
An important finding of the present study was that SOD2 functions as a key mediator of IR-induced cancer cell invasion, which was initially indicated by the finding that SOD2 mRNA and protein levels were elevated in cancer cells that survived γ-irradiation. In addition, the ability of IR to increase SOD2 expression has been reported in human fibroblasts29 as well as in mouse brain and gut30. In the present study, the IR-induced SOD2 expression was mediated by the p53/SULF2/β-catenin/IL-6/STAT3 pathway, which has been previously shown to mediate IR-induced cancer cell invasion7. Moreover, prevention of IR-induced cancer cell invasion by SOD2 knockdown directly supports the role of SOD2 in the malignant effects of sublethal doses of IR. This finding and our previous findings7,10 demonstrate that IR-stimulated STAT3 promotes cancer cell invasion by inducing the expression of SOD2 and Bcl-XL. Based on the recent report that Bcl-XL upregulation increases the ability of complex I to produce ROS, which then stimulate the Src-dependent invasion pathway6, the ability of IR to utilize the mitochondrial pathway and promote cancer cell invasion was investigated. The findings demonstrated that IR increased mitochondrial ROS production and Src phosphorylation. Notably, ROS production, Src phosphorylation, and IR-induced cell invasion were prevented by treatment with an ROS scavenger (NAC) or a complex I inhibitor (metformin), thereby supporting the view that IR stimulates the Src-dependent invasion pathway by promoting complex I-dependent ROS production. Thus, SOD2 likely supports IR-induced cell invasion by converting O2•− generated by complex I to H2O2, a signaling molecule that stimulates Src phosphorylation21, which was supported by the finding that SOD2 knockdown abolished IR-induced Src phosphorylation. Similar effects were observed when Bcl-XL was knocked down, indicating that both Bcl-XL and SOD2 are required for IR-mediated induction of the Src-dependent invasion pathway. Thus, IR-induced Bcl-XL may increase the ability of complex I to produce O2•−, which is then converted to H2O2 by SOD2 (Fig. 6). The ability of SOD2 to convert IR-induced O2•− to H2O2 was further confirmed by determining levels of O2•− and H2O2 using specific dyes, namely, MitoSox Red and Peroxy Orange-1, respectively. According to the present model, SOD2 acts downstream of Bcl-XL in an IR-induced signaling pathway, leading to cell invasion. However, despite this hierarchy, the functional relationship of Bcl-XL and SOD2 was cooperative. Both Bcl-XL and SOD2 were required to promote cell invasion, and their co-expression induced Src phosphorylation and cell invasion much more effectively than either alone, suggesting that both O2•− production and its conversion to H2O2 are required for the pro-invasive activity of IR. The present data also suggested that IR fulfils these two requirements by co-inducing Bcl-XL and SOD2 via STAT3. This co-induction was verified in lung, colon, and breast cancer cells. Therefore, the model depicted in Fig. 6 is a general mechanism applicable to diverse cancer types.
Fig. 6. Schematic model for the role of mitochondria in IR-induced cell invasion.
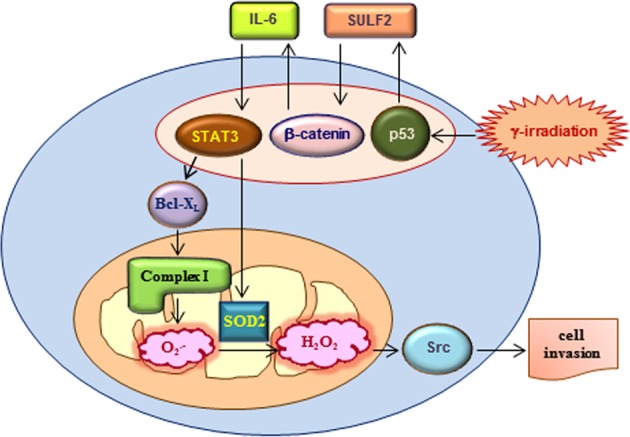
STAT3 stimulated by IR via the p53/SULF2/β-catenin/IL-6 pathway induces Bcl-XL and SOD2 expression. Bcl-XL increases the ability of complex I to produce O2•−, and SOD2 converts mitochondrial O2•− to H2O2, which, in turn, acts as a signaling molecule to promote cell invasion
The ability of p53 to induce SOD2 expression has been reported by other investigators31,32. Although the consensus p53-binding sequences have been identified in the promoter region of SOD231, they are not required for the transcriptional activation of SOD2 by p5332, suggesting that p53 induces SOD2 expression indirectly, which is consistent with the present model (Fig. 6).
Some investigators have reported that SOD2 protects cells from IR23, while others have reported that SOD2 is irrelevant in radioresistance24. While the reason for this discrepancy is still unclear, the present study demonstrated that SOD2 did not significantly influence the radiosensitivity of H460 and A549 cells. Considering that SOD2 plays an essential role in the IR-induced invasion of H460 cells, it is clear that SOD2 mediates IR-induced cell invasion without altering cellular radiosensitivity.
The present data also suggested that the pro-invasive role of SOD2 is not restricted to extrinsic treatments, such as radiotherapy, as it was also involved in intrinsic factor-induced cell invasion. In the present study, SOD2 was essential for cell invasion induced by Bcl-XL, an oncogene upregulated in many cancers4. In addition, SOD2 mediated cell invasion induced by extracellular factors, such as IL-6 and SULF2, suggesting that SOD2 plays an essential role in cell invasion induced in response to tumor microenvironment changes. Thus, SOD2 is critical for tumor progression under diverse conditions.
Metformin is a first-line medication for patients with type 2 diabetes because it reduces hyperglycemia by suppressing hepatic gluconeogenesis33. However, accumulating evidence has supported the potential of metformin for cancer therapy. Metformin has been shown to inhibit the growth of many cancer types34, and among cancer patients with diabetes, metformin users show better survival than non-users35. Metformin also improves tumor responses to radiotherapy by acting as a radiosensitizer36. As metformin is an inhibitor of complex I20, the ability of this drug to disrupt the malignant actions of IR was investigated. This possibility was supported by the finding that metformin suppressed the ability of IR to increase mitochondrial ROS production, Src phosphorylation, and cellular invasiveness. The present findings supported the view that metformin may improve the therapeutic effects of IR not only by acting as a radiosensitizer but also by preventing malignant actions of IR.
In conclusion, the present study showed that SOD2 is a key mediator of IR-induced cancer cell invasion, thereby supporting the critical role of mitochondria in cancer cell invasion and metastasis4. Considering the short half-life of O2•− 11, mitochondrial SOD2 may have an advantage for converting mitochondrial O2•− to H2O2. Therefore, mitochondrial components may be potential therapeutic targets for overcoming the malignant effects of IR.
Acknowledgements
This work was supported by grants from the National Research Foundation of Korea (2017R1D1A1B03032395 and 2017R1C1B2006273) and the Korea Institute of Radiological and Medical Sciences. This work was also funded by the Ministry of Science and ICT (MSIT), Republic of Korea (50531-2018).
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Chan-Hun Jung, Eun Mi Kim
References
- 1.Fujita M, Yamada S, Imai T. Irradiation induces diverse changes in invasive potential in cancer lines. Semin. Cancer Biol. 2015;35:45–52. doi: 10.1016/j.semcancer.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 2.Canphausen K, et al. Radiation therapy to a primary tumor accelerates metastatic growth in mice. Cancer Res. 2001;61:2207–2211. [PubMed] [Google Scholar]
- 3.Wu WS. The signaling mechanism of ROS in tumor progression. Cancer Metastas. Rev. 2006;25:695–705. doi: 10.1007/s10555-006-9037-8. [DOI] [PubMed] [Google Scholar]
- 4.Um HD. Bcl-2 family proteins as regulators of cancer cell invasion and metastasis: a review focusing on mitochondrial respiration and reactive oxygen species. Oncotarget. 2016;7:5193–5203. doi: 10.18632/oncotarget.6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Delbridge AR, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer. 2016;16:99–109. doi: 10.1038/nrc.2015.17. [DOI] [PubMed] [Google Scholar]
- 6.Kim EM, Park JK, Hwang SG, Um HD. Src and epidermal growth factor receptor mediate the pro-invasive activity of Bcl-w. Tumour Biol. 2016;37:1245–1252. doi: 10.1007/s13277-015-3917-x. [DOI] [PubMed] [Google Scholar]
- 7.Jung CH, et al. Involvement of SULF2 in γ-irradiation-induced invasion and resistance of cancer cells by inducing IL-6 expression. Oncotarget. 2016;7:16090–16103. doi: 10.18632/oncotarget.7449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hammond E, Khurana A, Shridhar V, Dredge K. The role of heparanase and sulfatases in the modification of heparan sulfate proteoglycans within the tumor microenvironment and opportunities for novel cancer therapeutics. Front. Oncol. 2014;4:195. doi: 10.3389/fonc.2014.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiong A, Yang Z, Shen Y, Zhou J, Shen Q. Transcription factor STAT3 as a novel molecular target for cancer prevention. Cancers (Basel) 2014;6:926–957. doi: 10.3390/cancers6020926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ho JN, et al. Bcl-XL and STAT3 mediate malignant actions of gamma-irradiation in lung cancer cells. Cancer Sci. 2010;101:1417–1423. doi: 10.1111/j.1349-7006.2010.01552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rhee SG. Redox signaling: hydrogen peroxide as intracellular messenger. Exp. Mol. Med. 1999;31:53–59. doi: 10.1038/emm.1999.9. [DOI] [PubMed] [Google Scholar]
- 12.Che M, Wang R, Li X, Wang HY, Zheng XFS. Expanding roles of superoxide dismutases in cell regulation and cancer. Drug Discov. Today. 2016;21:143–149. doi: 10.1016/j.drudis.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jung CH, et al. Bmal1 suppresses cancer cell invasion by blocking the phosphoinositide 3-kinase-Akt-MMP-2 signaling pathway. Oncol. Rep. 2013;29:2109–2113. doi: 10.3892/or.2013.2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jung CH, et al. Mdm2 increases cellular invasiveness by binding to and stabilizing the Slug mRNA. Cancer Lett. 2013;335:270–277. doi: 10.1016/j.canlet.2013.02.035. [DOI] [PubMed] [Google Scholar]
- 15.Cho HJ, et al. Luteolin acts as a radiosensitizer in non-small cell lung cancer cells by enhancing apoptotic cell death through activation of a p38/ROS/caspase cascade. Int. J. Oncol. 2015;46:1149–1158. doi: 10.3892/ijo.2015.2831. [DOI] [PubMed] [Google Scholar]
- 16.Kim EM, et al. Thep53/p21 complex regulates cancer cell invasion and apoptosis by targeting Bcl-2 family proteins. Cancer Res. 2017;77:3092–3100. doi: 10.1158/0008-5472.CAN-16-2098. [DOI] [PubMed] [Google Scholar]
- 17.Kim EM, Kim J, Um HD. Bcl-2 protein targeting by the p53/p21 complex-response. Cancer Res. 2018;78:2772–2774. doi: 10.1158/0008-5472.CAN-17-3919. [DOI] [PubMed] [Google Scholar]
- 18.Essmann F, et al. Irradiation-induced translocation of p53 to mitochondria in the absence of apoptosis. J. Biol. Chem. 2005;280:37169–37177. doi: 10.1074/jbc.M502052200. [DOI] [PubMed] [Google Scholar]
- 19.Jung JE, Kim GS, Narasimhan P, Song YS, Chan PH. Regulation of Mn-superoxide dismutase activity and neuroprotection by STAT3 in mice after cerebral ischemia. J. Neurosci. 2009;29:7003–7014. doi: 10.1523/JNEUROSCI.1110-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wheaton WW, et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife. 2014;3:e02242. doi: 10.7554/eLife.02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhuang S, Schnellmann RG. H2O2-induced transactivation of EGF receptor requires Src and mediates ERK1/2, but not Akt, activation in renal cells. Am. J. Physiol. Ren. Physiol. 2004;286:F858–F865. doi: 10.1152/ajprenal.00282.2003. [DOI] [PubMed] [Google Scholar]
- 22.Hong Y, Li L, Luan G, Drlica K, Zhao X. Contribution of reactive oxygen species to thymineless death in Escherichia coli. Nat. Microbiol. 2017;2:1667–1675. doi: 10.1038/s41564-017-0037-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Motoori S, et al. Overexpression of mitochondrial manganese superoxide dismutase protects against radiation-induced cell death in the human hepatocellular carcinoma cell line HLE. Cancer Res. 2001;61:5382–5388. [PubMed] [Google Scholar]
- 24.Veldwijk MR, et al. Recombinant adeno-associated virus 2-mediated transfer of the human superoxide-dismutase gene does not confer radioresistance on HeLa cervical carcinoma cells. Radiother. Oncol. 2004;72:341–350. doi: 10.1016/j.radonc.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 25.Connor KM, et al. Manganese superoxide dismutase enhances the invasive and migratory activity of tumor cells. Cancer Res. 2007;67:10260–10267. doi: 10.1158/0008-5472.CAN-07-1204. [DOI] [PubMed] [Google Scholar]
- 26.Chang B, et al. SOD2 deregulation enhances migration, invasion and has poor prognosis in salivary adenoid cystic carcinoma. Sci. Rep. 2016;6:25918. doi: 10.1038/srep25918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X, et al. Deregulation of manganese superoxide dismutase (SOD2) expression and lymph node metastasis in tongue squamous cell carcinoma. BMC Cancer. 2010;10:365. doi: 10.1186/1471-2407-10-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Z, et al. Manganese superoxide dismutase induces migration and invasion of tongue squamous cell carcinoma via H2O2-dependent Snail signaling. Free Radic. Biol. Med. 2012;53:44–50. doi: 10.1016/j.freeradbiomed.2012.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akashi M, et al. Irradiation increases manganese superoxide dismutase mRNA levels in human fibroblasts. Possible mechanisms for its accumulation. J. Biol. Chem. 1995;270:15864–15869. doi: 10.1074/jbc.270.26.15864. [DOI] [PubMed] [Google Scholar]
- 30.Veeraraghavan J, Natarajan M, Herman TS, Aravindan N. Low-dose γ-radiation-induced oxidative stress response in mouse brain and gut: regulation by NFκB-MnSOD cross-signaling. Mutat. Res. 2011;718:44–55. doi: 10.1016/j.mrgentox.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 31.Hussain SP, et al. p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer Res. 2004;64:2350–2356. doi: 10.1158/0008-5472.CAN-2287-2. [DOI] [PubMed] [Google Scholar]
- 32.Dhar SK, Xu Y, St. Clair DK. Nuclear factor kappaB- and specificity protein 1-dependent p53-mediated bi-directional regulation of the human manganese superoxide dismutase gene. J. Biol. Chem. 2010;285:9835–9846. doi: 10.1074/jbc.M109.060715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kirpichnikov D, McFarlane SI, Sowers JR. Metformin: an update. Ann. Intern. Med. 2002;137:25–33. doi: 10.7326/0003-4819-137-1-200207020-00009. [DOI] [PubMed] [Google Scholar]
- 34.Zi F, et al. Metformin and cancer: an existing drug for cancer prevention and therapy. Oncol. Lett. 2018;15:683–690. doi: 10.3892/ol.2017.7412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandulache VC, et al. Association between metformin use and improved survival in patients with laryngeal squamous cell carcinoma. Head Neck. 2014;36:1039–1043. doi: 10.1002/hed.23409. [DOI] [PubMed] [Google Scholar]
- 36.Koritzinsky M. Metformin: a novel biological modifier of tumor response to radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2015;93:454–464. doi: 10.1016/j.ijrobp.2015.06.003. [DOI] [PubMed] [Google Scholar]