

Abstract
T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive malignancy originating from T-cell precursors. The genetic landscape of T-ALL has been largely characterized by next-generation sequencing. Yet, the transcriptome of miRNAs (miRNome) of T-ALL has been less extensively studied. Using small RNA sequencing, we characterized the miRNome of 34 pediatric T-ALL samples, including the expression of isomiRs and the identification of candidate novel miRNAs (not previously annotated in miRBase). For the first time, we show that immunophenotypic subtypes of T-ALL present different miRNA expression profiles. To extend miRNome characteristics in T-ALL (to 82 T-ALL cases), we combined our small RNA-seq results with data available in Gene Expression Omnibus. We report on miRNAs most abundantly expressed in pediatric T-ALL and miRNAs differentially expressed in T-ALL versus normal mature T-lymphocytes and thymocytes, representing candidate oncogenic and tumor suppressor miRNAs. Using eight target prediction algorithms and pathway enrichment analysis, we identified differentially expressed miRNAs and their predicted targets implicated in processes (defined in Gene Ontology and Kyoto Encyclopedia of Genes and Genomes) of potential importance in pathogenesis of T-ALL, including interleukin-6–mediated signaling, mTOR signaling, and regulation of apoptosis. We finally focused on hsa-mir-106a-363 cluster and functionally validated direct interactions of hsa-miR-20b-5p and hsa-miR-363-3p with 3′ untranslated regions of their predicted targets (PTEN, SOS1, LATS2), overrepresented in regulation of apoptosis. hsa-mir-106a-363 is a paralogue of prototypic oncogenic hsa-mir-17-92 cluster with yet unestablished role in the pathogenesis of T-ALL. Our study provides a firm basis and data resource for functional analyses on the role of miRNA-mRNA interactions in T-ALL.
Abbreviations: ALL, acute lymphoblastic leukemia; EGIL, European Group for Immunological Classification of Leukemias; GEO, Gene Expression Omnibus; GO, Gene Ontology; isomiR, isoform of miRNA; KEGG, Kyoto Encyclopedia of Genes and Genomes; miRNome, transcriptome of miRNAs; MRE, miRNA response element; OR, Odds ratio; RT-qPCR, quantitative reverse transcription polymerase chain reaction; small RNA-seq, next-generation sequencing of small RNAs; T-ALL, T-cell acute lymphoblastic leukemia; 3′UTR, 3′ untranslated region
Introduction
miRNAs are small, nonprotein coding RNAs exerting negative regulatory control over their mRNA targets. miRNAs induce translational repression and enhance mRNA degradation upon specific binding of the seed sequence of miRNA to the miRNA response element (MRE), typically in the 3′ untranslated region (3′UTR) of mRNA [1], [2]. Thus, miRNA-mRNA interactions result in the silencing of mRNA expression and diminished level or lacking protein expression [3].
miRNAs are important regulators of gene expression and are involved in a multitude of biological processes, e.g., cell differentiation, including normal hematopoiesis, cell proliferation, apoptosis, cellular stress response, and many others. Aberrantly expressed miRNAs are implicated in the pathology of diseases, including malignancies, and are attractive candidate biomarkers and potential targets for therapy [4], [5], [6]. In neoplasms, upregulated miRNAs might serve as oncogenes by silencing the expression of mRNAs encoding tumor suppressor proteins. Downregulated tumor suppressor miRNAs contribute to oncogenesis by insufficient silencing of oncogenic mRNAs [7], [8]. The regulatory effect of a single miRNA might be subtle, and phenotypic effects of miRNAs' expression result from their involvement in intricate regulatory networks [9]. Thus, next-generation sequencing, enabling miRNA expression profiling in the whole-transcriptomic scale, importantly improves the possibilities to study miRNAs expression and to explore their biological implications.
In this study, we applied small RNA sequencing to investigate miRNA transcriptome of T-cell acute lymphoblastic leukemia (T-ALL) and to search for novel candidate oncogenic and tumor suppressor miRNAs and their targets.
T-ALL is an aggressive and heterogeneous malignancy originating from T-cell precursors (thymocytes). It accounts for approximately 15% of all acute lymphoblastic leukemia (ALL) cases in children and for approximately 25% in adults [10], [11]. With the advancement of high-throughput technologies, particularly next-generation sequencing, the molecular landscape of this leukemia has been largely characterized. The main focus, so far, has been on the characterization of the protein coding part of the genome and on the gene expression patterns specific for the subtypes of T-ALL. The genomic landscape of T-ALL has been widely characterized through whole exome sequencing and RNA sequencing [12], [13], [14], [15]. Yet, the transcriptome of miRNAs (miRNome) in T-ALL has been much less extensively studied thus far [16].
Here we present the results of small RNA sequencing performed in 34 pediatric T-ALL patients and 5 normal controls. In addition to broad characteristics of the miRNome of T-ALL, we also aimed to gain insights into the potential engagement of differentially expressed miRNAs in biological processes that might be of significance for T-ALL pathobiology. For this reason, we performed comprehensive target prediction and pathway enrichment analysis. We finally focused on selected miRNAs belonging to hsa-mir-106a-363 cluster, and we functionally validated direct interactions of hsa-miR-20b-5p and hsa-miR-363-3p with their targets predicted to be implicated in the positive regulation of apoptosis, namely, PTEN, SOS1, and LATS2. Importantly, hsa-mir-106a-363 cluster is a paralogue of the prototypic oncogenic hsa-mir-17-92 cluster, but its role in the pathogenesis of T-ALL is yet to be established.
Thus, we present a broad investigation of the landscape of miRNA expression in pediatric T-ALL and its potential implications for the biology of this disease. Such an approach, including miRNA-seq followed by target prediction, pathway enrichment analysis, and validation of miRNA-mRNA interactions, has not been applied in the study of pediatric T-ALL thus far. Our results form a firm basis and data resource for extended functional analyses on the role of miRNA-mRNA interactions in this disease.
Material and Methods
Patients' and Control Samples
Bone marrow samples were obtained from 34 patients with pediatric T-ALL at the time of diagnosis and from 5 healthy unrelated bone marrow donors aged <18 years. Samples were collected at the centers of Polish Pediatric Leukemia and Lymphoma Study Group, with the informed consent of the patients/legal guards, in accordance with Declaration of Helsinki. The study was approved by the Ethics Committee of the Medical University of Silesia (KNW/0022/KB1/145/I/11/12). Detailed immunophenotyping of T-ALL was performed using multicolor flow cytometry according to EuroFlow protocols [17], [18]. Characteristics of T-ALL patients from the study group and from the validation cohort are presented in supplementary Tables S1 and S2.
Bone marrow samples were subjected to isolation of mononuclear cells using density gradient centrifugation on Gradisol L (Aqua-Med), followed by immunomagnetic separation, with use of Human T Lymphocyte Enrichment Set-DM (Becton Dickinson) to obtain T-ALL cells and normal mature T lymphocytes (controls) for RNA isolation. We used negative immunomagnetic selection to avoid potential changes to the miRNome upon interaction of the cells with the antibodies, which might be the case in the positive selection approach. T-ALL cells and normal T lymphocytes were preserved from RNA degradation with use of Lysis Solution (miRCURY RNA Isolation Kit, Exiqon) and stored in −80°C until RNA isolation.
RNA Isolation and Quality Control
miRCURY RNA Isolation Kit Cell & Plant (Exiqon) was used for the extraction of total RNA, with the recovery of small RNA fraction. RNA isolates were DNase treated and purified with use of RNA Clean and Concentrator Kit (Zymo Research). RNA concentration was measured with Quantus Fluorometer (Promega) using Qubit HS RNA Assay Kit (Thermo Fisher Scientific). RNA integrity was determined with 4200 Tapestation using High Sensitivity RNA ScreenTape (Agilent Technologies) and 2100 Bioanalyzer using RNA 6000 Nano Assay (Agilent Technologies).
Small RNA Sequencing and Bioinformatics Analyses
Next-generation sequencing of small RNAs was performed by NGS Service Exiqon (Exiqon, Denmark). Libraries were generated with NEBNext Multiplex Small RNA Library Prep Set for Illumina (New England Biolabs). The quality of libraries was assesses based on size distribution and concentration using 2100 Bioanalyzer with DNA 1000 chip (Agilent Technologies). Small RNA-seq was performed using NextSeq500 Illumina and standard settings: 10 million reads/per sample, read length: 51 bp single-end. Small RNA sequencing data from this study is available in the ArrayExpress database (http://www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-7446.
Quality control of reads was conducted using FastQC ver. 0.11.5 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc), FastQ Screen ver. 0.5.1 [19], and a set of custom data processing and visualization scripts. Raw sequencing reads were adapter-trimmed using Cutadapt [20] (ver. 1.11) and aligned with Bowtie [21] (ver. 1.2.2) to a modified version of miRBase (ver. 21) created according to the miRge specifications [22]. We used an iterative alignment of reads: first reads were aligned to mature miRNA sequences (miRBase21); unaligned reads were sequentially matched against hairpin miRNA (miRBase21), noncoding RNAs, Ensembl cDNA database, and again to mature miRNA sequences (miRBase21) using less stringent criteria, as previously described [22]. Detection of candidate novel miRNAs was based on miRge2 for each individual sample. The results were later combined between samples based on genomic coordinates of identified miRNAs (partial overlap was considered sufficient).
Both known and candidate novel miRNAs, to which at least two reads were aligned in a single sample, were selected for further study. Read normalization, as well as identification of differentially expressed miRNAs, was conducted using edgeR [23], with Benjamini and Hochberg correction for multiple testing and .05 significance level. Read counts used in the tests were normalized in edgeR using Trimmed Mean of Mvalues (TMM) algorithm. The detection of differentially expressed miRNAs was conducted using various sample classification methods based on disease state, immunophenotype, maturation stage, European Group for Immunological Classification of Leukemias (EGIL) classification, etc. If more than two groups existed in a specific classification method, we conducted pairwise comparisons between them for each unique combination and additionally performed an ANOVA test independently for each miRNA with Benjamini and Hochberg correction for multiple testing.
To conduct a joint analysis with the Wallaert et al. dataset [16], we downloaded raw reads from the Sequence Read Archive database under accession SRP093752 (GEO accession number GSE89978) and processed them using identical workflow based on miRge [22]. Detection of differentially expressed miRNAs was conducted again using edgeR [23], accounting for batch effect by specifying an appropriate model matrix. ANOVA was conducted on edgeR normalized data after batch effect correction based on ComBat [24]. Results of all analyses were presented as heatmaps of Z-score normalized miRNA expression levels with dendrograms based on complete-linkage hierarchical clusterization and Euclidean distances.
Analysis of isomiRs was based on the statistics provided by miRge [22], including the sequence variability of a single miRNA, occurrence in each sample, and entropy values calculated for each individual miRNA and sample. We used t test with Benjamini and Hochberg correction for multiple testing to compare the entropy values between T-ALL samples and controls in order to identify miRNAs that differ in the isomiR variability.
RT-qPCR Validation of Differentially Expressed miRNAs
Differentially expressed miRNAs were validated by RT-qPCR using miRNAs as endogenous controls, as previously described [25]. Briefly, RNA samples were reverse transcribed with TaqMan Advanced miRNA cDNA Synthesis Kit (Thermo Fisher Scientific) according to the manufacturer's protocol. TaqMan Fast Advanced Master Mix, predesigned TaqMan Advanced miRNA assays (Thermo Fisher Scientific), and 7900HT Fast Real-Time PCR System (Applied Biosystems) were used. Three endogenous control miRNAs were selected using a strategy based on a comprehensive assessment of expression stability in our small RNA-seq data and in RT-qPCR, as previously described [25]. Comparative delta CT method (ΔΔ CT) and Data Assist Software v. 3.01 (Thermo Fisher Scientific) were used for relative quantification of expression [26]. Two-tailed Student's t test was used to test for the significance of differences in expression between T-ALL samples and controls, with P < .05 for statistical significance.
miRNA Targets Prediction and Pathway Analysis
Target genes for differentially expressed miRNAs were identified using eight target prediction tools: DIANA-microT [27], ElMMo [28], MicroCosm [29], miRanda [30], miRDB [31], PicTar [32], PITA [33], and TargetScan [34], gathered trough the multiMiR Bioconductor library [35]. Target mRNAs were assumed to be regulated by a particular miRNA if they were predicted by more than five out of eight methods used. Target genes for differentially expressed miRNAs were tested for overrepresentation among biological processes and pathways, represented by relevant terms in Kyoto Encyclopedia of Genes and Genomes (KEGG), Reactome and Panther pathways, as well as Gene Ontology (GO). Overrepresentation of pathway associated genes among those regulated by differentially expressed miRNAs was tested using Fisher's exact test for pathways originating from KEGG, Reactome, and Panther databases. Gene Ontology processes were selected using conditional hypergeometric test implemented in the Bioconductor GOstats package (ver. 2.46). In all cases, Benjamini and Hochberg multiple testing correction was applied with a significance threshold of .05. The magnitude of overrepresentation was assessed based on odds ratio (OR) values, calculated using the following formula:
where q is the number of genes (predicted targets of differentially expressed miRNAs) involved in a given biological process, k is the total number of differentially expressed genes, m is the number of genes in a given process/pathway, and t is the total number of target genes reported in a given database.
Additionally, using the multiMiR Bioconductor library [35], we retrieved data from three databases of experimentally validated miRNA-mRNA interactions: miRecords [36], miRTarBase [37], and DIANA-TarBase [38]; two databases of diseases-related miRNAs: miR2Disease [39] and PhenomiR [40], and PharmacomiR [41], the miRNA Pharmacogenomics Database, enabling exploration of miRNAs and their drug-associated target genes.
Dual Luciferase Reporter Assays
Selected predicted miRNA-mRNA interactions were validated with Dual-Glo Luciferase Reporter Assay (Promega). HEK293T cells were cultured under standard conditions in Dulbecco's modified Eagle's medium (Gibco, Thermo Fisher Scientific) with 10% fetal bovine serum (Gibco, Thermo Fisher Scientific) and 1% penicillin/streptomycin solution (Sigma Aldrich). Twenty-four hours before transfection, cells were seeded on 24-well culture plate. Cells were subjected to transfection at 60% to 80% confluency using JetPrime DNA/siRNA Transfection Kit (Polyplus Transfection) to enable co-transfection with relevant miRNA mimics or negative (scrambled) control mimics (MirVana, Thermo Fisher Scientific) and pmirGLO plasmids (Promega), containing 3′UTR fragments of the selected target genes. miRNA mimics in final concentration of 50 μM and 125 ng of plasmid were used per well. 3′UTR fragments cloned into pmirGLO plasmid contained the predicted 6-8 nt long miRNA response element (MRE), flanked with 30 nt long regions on both sides. In Dual Luciferase rescue experiments, four point mutations were introduced to MRE region during oligonucleotide synthesis step to abolish the miRNA-mRNA interaction. Luciferase activity was measured with GloMax-Multi+ Detection System (Promega) after 48 hours from transfection. All experiments were performed in three replicates. Significant decrease in luciferase activity relative to control (scrambled miRNA) was indicative of direct interaction between seed sequence of miRNA (defined as the nucleotides at position 2-7 of the mature miRNA sequences) and MRE in 3′UTR of target mRNA. Statistical significance of results was calculated with unpaired two-tailed t test.
Results
Overview of miRNA-seq Performance
To investigate the spectrum of miRNAs expressed in pediatric T-ALL and to identify new candidate oncogenic and tumor suppressor miRNAs, we performed small RNA sequencing in 34 pediatric T-ALL samples and 5 normal controls. miRNA sequencing generated 908 million raw sequencing reads; on average, there were 23.3 million raw reads/sample. Prefiltering and filtering steps retained 22% (~4.3 million) of initial raw reads/sample. In total, 169 million reads were mapped to known mature miRNA sequences in miRBase21. The Phred quality scores for all reads, both T-ALL and control samples, were above 30% (>99.9% accuracy of base calling), representing high and uniform quality of reads. This refers to the mean Phred scores per read (per sequence quality) (Supplementary Figure S1) and to Phred scores along all reads (per base quality) (Supplementary Figure S2). The majority of reads were of approximate length of 22 bases, which represent the average length of miRNAs, as shown by sequence length distribution (Supplementary Figure S3). These data indicate high technical performance of miRNA sequencing and good quality of the samples.
Landscape of miRNA Transcriptome in T-ALL
Known miRNAs
The number of reads mapped to known miRNAs per T-ALL sample ranged from 0.7 to 9.9 million; on average, there were 4.35 million/T-ALL sample (Supplementary Figure S4). We identified 1462 miRNAs expresse in T-ALL samples (at least 2 reads/ sample), ranging from 409 to 934 miRNAs expressed per T-ALL sample. Out of these 1462 miRNAs, 452 were identified exclusively in T-ALL and not in the controls. The top 10 highly expressed miRNAs in T-ALL samples include hsa-miR-92a-3p; hsa-miR-128-3p; hsa-let-7a-5p/7c-5p; hsa-let-7f-5p; hsa-miR-148a-3p; hsa-let-7i-5p; hsa-let-7 g-5p; hsa-miR-181a-5p; hsa-miR-26a-5p; and hsa-miR-21-5p (Supplementary Figure S5).
To further characterize the landscape of miRNA expression in T-ALL patients, we analyzed the composition of the expression pattern of miRNA isoforms (miRNA variants deviating by a few nucleotides in length and sequence from the reference miRNA sequence, called isomiRs) [42], [43]. We also characterized the miRNome of T-ALL in terms of the identification of a spectrum of candidate novel miRNAs. We present the results of these analyses in the Supplementary materials (Expression of isomiRs; Candidate novel miRNAs).
miRNAs Differentially Expressed Between T-ALL Samples and Controls
To identify known miRNAs differentially expressed in T-ALL samples as compared to normal mature T lymphocytes, we applied edgeR [23] with Benjamini and Hochberg correction for multiple testing. Among 1505 known miRNAs expressed in this study, we identified 61 differentially expressed miRNAs: 23 miRNAs upregulated in T-ALL vs. controls, and 38 miRNAs downregulated (Table 1 and Figure 1). These differentially expressed miRNAs represent known and potential novel oncogenic and tumor suppressor miRNAs, respectively. Among overexpressed miRNAs, we identified miRNAs with already reported role as oncogenes in T-ALL, namely, hsa-miR-128-3p [44], [45], hsa-miR-181a, and hsa-miR-181b [46], [47], [48]. Remarkably, these miRNAs are highly expressed in our cohort of T-ALL patients (Figure S5 and Table 1). These miRNAs were previously reported to be highly expressed in T-ALL also in the studies using less sensitive, RT-qPCR–based approaches [44], [46], [47], [49]. Additionally, among miRNAs overexpressed in our T-ALL patients, we identified hsa-miR-153-3p and hsa-miR-6500-3p, which were also found to be overexpressed in the small RNA-seq study by Wallaert et al. [16]. These two miRNAs had relatively low expression levels in our T-ALL cohort, with mean read counts below 100 reads (Table 1).
Table 1.
miRNAs Differentially Expressed in 34 T-ALL Samples and Controls
| miRNA_ID | Average Read Counts |
log FC | P adj | |||
|---|---|---|---|---|---|---|
| All Samples |
Control |
T-ALL |
||||
| Raw | Normalized | Normalized | Normalized | |||
| miRNAs overexpressed in T-ALL vs. controls | ||||||
| hsa-miR-548a-3p | 21.2 | 20.1 | 0.0 | 23.0 | 7.674 | 8.27E-05 |
| hsa-miR-128-3p | 457,447.9 | 382,618.6 | 61,129.6 | 429,896.4 | 2.814 | 9.22E-05 |
| hsa-miR-181b-5p | 51,636.7 | 45,642.4 | 6860.0 | 51,345.7 | 2.904 | 1.28E-04 |
| hsa-miR-20b-5p | 3524.9 | 3005.6 | 296.2 | 3404.1 | 3.522 | 4.28E-04 |
| hsa-miR-6500-3p | 61.2 | 53.2 | 1.5 | 60.7 | 5.220 | .002 |
| hsa-miR-331-5p | 160.5 | 134.1 | 34.7 | 148.7 | 2.100 | .004 |
| hsa-miR-363-3p | 41,814.5 | 35,610.7 | 5759.5 | 40,000.6 | 2.796 | .008 |
| hsa-miR-153-3p | 71.8 | 51.4 | 0.0 | 59.0 | 9.031 | .008 |
| hsa-miR-466 | 127.4 | 112.5 | 0.0 | 129.1 | 10.157 | .009 |
| hsa-miR-130a-3p | 524.9 | 419.1 | 106.2 | 465.1 | 2.130 | .009 |
| hsa-miR-20b-3p | 65.1 | 58.7 | 7.9 | 66.2 | 3.053 | .010 |
| hsa-miR-210-3p | 508.4 | 460.4 | 75.8 | 517.0 | 2.766 | .010 |
| hsa-miR-181a-3p | 5371.8 | 4663.6 | 851.3 | 5224.2 | 2.617 | .010 |
| hsa-miR-4421 | 27.9 | 23.9 | 0.0 | 27.4 | 7.928 | .011 |
| hsa-miR-18b-5p | 15.8 | 11.9 | 0.0 | 13.6 | 6.937 | .012 |
| hsa-miR-181a-2-3p | 7392.8 | 6055.4 | 1753.8 | 6687.9 | 1.931 | .015 |
| hsa-miR-181a-5p | 159,408.7 | 146,775.2 | 31,833.1 | 163,678.4 | 2.362 | .015 |
| hsa-miR-625-3p | 3238.1 | 2702.5 | 638.3 | 3006.1 | 2.236 | .018 |
| hsa-miR-130b-3p | 381.5 | 337.0 | 109.9 | 370.4 | 1.753 | .025 |
| hsa-miR- 4687-5p | 26.9 | 24.8 | 1.6 | 28.2 | 4.070 | .035 |
| hsa-miR-4437 | 98.4 | 90.7 | 15.7 | 101.8 | 2.686 | .039 |
| hsa-miR-625-5p | 912.2 | 765.9 | 214.4 | 847.0 | 1.982 | .039 |
| hsa-miR-3609 | 993.4 | 1843.8 | 79.5 | 2103.2 | 4.723 | .041 |
| miRNAs underexpressed in T-ALL vs. controls | ||||||
| hsa-miR-574-5p | 652.8 | 542.1 | 3554.4 | 99.1 | −5.164 | 2.88E-19 |
| hsa-miR-10a-5p | 5025.9 | 4115.7 | 22,247.8 | 1449.2 | −3.940 | 5.11E-13 |
| hsa-miR-582-3p | 493.6 | 406.7 | 2490.7 | 100.3 | −4.633 | 4.16E-12 |
| hsa-miR-143-3p | 7581.1 | 6131.8 | 33,787.1 | 2064.9 | −4.032 | 1.51E-11 |
| hsa-miR-941 | 10,845.5 | 9357.4 | 34,576.7 | 5648.6 | −2.614 | 2.00E-09 |
| hsa-miR-145-5p | 75.5 | 63.8 | 364.7 | 19.5 | −4.212 | 2.87E-07 |
| hsa-miR- 27a-5p | 1526.9 | 1226.4 | 4706.0 | 714.6 | −2.719 | 1.04E-06 |
| hsa-miR-618 | 107.4 | 95.3 | 575.4 | 24.6 | −4.543 | 1.11E-06 |
| hsa-miR-24- 3p | 4801.6 | 4038.7 | 12,326.8 | 2819.9 | −2.128 | 2.39E-06 |
| hsa-miR-574- 3p | 75.1 | 65.3 | 325.6 | 27.0 | −3.587 | 2.43E-06 |
| hsa-miR-24-2-5p | 191.9 | 160.0 | 484.9 | 112.2 | −2.109 | 3.00E-06 |
| hsa-miR-145-3p | 76.8 | 66.7 | 330.4 | 27.9 | −3.568 | 5.64E-06 |
| hsa-miR-504-5p | 48.6 | 41.1 | 238.1 | 12.2 | −4.278 | 8.27E-05 |
| hsa-miR- 3690 | 50.4 | 46.9 | 223.2 | 21.0 | −3.409 | 2.45E-04 |
| hsa-miR- 223-5p | 6511.1 | 5472.8 | 18,853.6 | 3505.1 | −2.427 | 2.50E-04 |
| hsa-miR-199b-5p | 1728.1 | 1309.1 | 5169.0 | 741.4 | −2.801 | .001 |
| hsa-miR-550a-5p | 132.9 | 111.0 | 411.6 | 66.7 | −2.621 | .001 |
| hsa-miR-4695-3p | 3.6 | 3.3 | 25.6 | 0.0 | −7.824 | .001 |
| hsa-miR-30a-5p | 412.6 | 346.1 | 808.7 | 278.1 | −1.539 | .001 |
| hsa-miR-3909 | 176.9 | 149.9 | 365.8 | 118.2 | −1.625 | .002 |
| hsa-miR-2115-3p | 28.5 | 22.9 | 128.9 | 7.3 | −4.103 | .002 |
| hsa-miR-582-5p | 25.4 | 20.3 | 122.3 | 5.3 | −4.495 | .002 |
| hsa-miR-504-3p | 1.6 | 1.2 | 9.5 | 0.0 | −6.412 | .002 |
| hsa-miR- 23a-5p | 28.9 | 24.9 | 98.1 | 14.1 | −2.784 | .003 |
| hsa-miR-10b-5p | 211.4 | 146.8 | 629.1 | 75.9 | −3.047 | .006 |
| hsa-miR-4494 | 1.1 | 0.8 | 6.3 | 0.0 | −5.825 | .007 |
| hsa-miR-151a-3p | 2930.2 | 1886.3 | 6696.9 | 1178.8 | −2.506 | .009 |
| hsa-miR- 223-3p | 3083.8 | 2647.2 | 7802.3 | 1889.1 | −2.046 | .011 |
| hsa-miR-6865-3p | 0.7 | 0.6 | 4.5 | 0.0 | −5.331 | .012 |
| hsa-miR-7849-3p | 2.0 | 1.6 | 12.2 | 0.0 | −6.764 | .012 |
| hsa-miR- 1275 | 505.7 | 440.2 | 1198.6 | 328.6 | −1.866 | .012 |
| hsa-miR-338-5p | 38.7 | 32.3 | 128.7 | 18.1 | −2.821 | .013 |
| hsa-miR-3150b-5p | 1.1 | 0.9 | 6.3 | 0.1 | −4.945 | .016 |
| hsa-miR-3154 | 74.1 | 62.4 | 242.6 | 35.9 | −2.752 | .017 |
| hsa-miR-6823-5p | 0.6 | 0.4 | 3.4 | 0.0 | −4.956 | .017 |
| hsa-miR-10a- 3p | 12.8 | 10.5 | 48.4 | 4.9 | −3.273 | .024 |
| hsa-miR-143-5p | 16.6 | 13.1 | 75.6 | 3.9 | −4.234 | .036 |
| hsa-miR-4745-3p | 1.1 | 1.0 | 7.8 | 0.0 | −6.125 | .036 |
Controls, normal mature T lymphocytes of bone marrow donors aged <18 years; Average Read Counts Normalized, average TMM normalized reads; FC, fold change; P adj, Benjamini and Hochberg correction for multiple testing and .05 significance level. miRNAs are sorted based on increasing P adj values and decreasing log FC values.
Figure 1.

miRNA expression levels in T-ALL vs. controls.
Diagonal plot showing the expression levels of miRNAs in T-ALL samples vs. controls (mature T lymphocytes of bone marrow). Dots represent miRNAs that are: significantly overexpressed in T-ALL (red), significantly underexpressed in T-ALL (blue), not differentially expressed (gray). The top 10 upregulated and downregulated miRNAs, according to log FC values, are depicted with miRNA IDs.
Among downregulated miRNAs, we found hsa-miR-30a-5p with already suggested role as a tumor suppressor in T-ALL [50], as well as hsa-miR-3909 and hsa-miR-1275, reported as downregulated in the study by Wallaert et al. [16]. Mean read counts in T-ALL samples for these 3 miRNAs were below 350 reads (Table 1). Thus, these miRNAs are not only downregulated as compared to normal controls but additionally belong to miRNAs with relative low expression in our cohort of T-ALL patients. Interestingly, among miRNAs downregulated in T-ALL vs. normal mature T lymphocytes, we identified hsa-miR-223-5p and hsa-miR-223-3p. hsa-miR-223 has been previously reported to be among the top 10 most highly expressed miRNAs in the cohort of 50 T-ALL patients analyzed with RT-qPCR by Mavrakis et al. [51]. The group further showed oncogenic potential of this miRNA in a mouse model [51]. Yet, in our cohort of 34 T-ALL patients, hsa-miR-223-5p and hsa-miR-223-3p were less abundantly expressed, ranked at position 67 and 87 of most highly expressed miRNAs. As compared to normal mature T lymphocytes, we found hsa-miR-223-5p and miR-223-3p downregulated.
Using unsupervised hierarchical clusterization based on Euclidean distances and complete-linkage method (agglomerative), we showed that the expression of 61 differentially expressed miRNAs allows to clearly segregate T-ALL samples from normal mature T lymphocytes (Figure 2).
Figure 2.

Differentially expressed miRNAs in T-ALL samples and controls.
Heatmap and dendrograms of Z-score normalized miRNA expression levels created for miRNAs that differentiate T-ALL samples from normal controls (mature T lymphocytes of bone marrow). Rows represent miRNAs; columns represent samples. Dendrograms are based on complete-linkage hierarchical clusterization and Euclidean distances. The fold changes and the abundance of miRNAs (both in log scale) are shown on the left side of the plot.
Landscape of miRNAs Transcriptome in T-ALL: a Broader Perspective
Having characterized the landscape of miRNA expression in 34 T-ALL samples in relation to 5 samples of normal mature T lymphocytes as controls, we aimed to broaden the perspective of the investigation of miRNome in pediatric T-ALL. To this aim, we performed a combined analysis of our miRNA-seq data and the results of small RNA sequencing of 48 samples of pediatric T-ALL and 4 samples of normal thymocytes [16]. The work by Wallaert et al. [16] represents thus far the only available miRNA-seq dataset obtained in a cohort of pediatric T-ALL patients. We used uniform bioinformatics pipeline to integrate and analyze our miRNA-seq data and the dataset publically available in GEO database (accession number GSE89978) [16]. Before the integration of the two data sets, we used ComBat [24] for the correction for batch effects. Thus, we decreased the potentially nonbiological variation between the two data sets, associated with technical differences in the studies, as illustrated by principal component analysis plots for the two datasets, before and after the correction (Supplementary Figure S6).
The combined analysis of the 2 data sets enabled to characterize the landscape of miRNA expression in a total of 82 pediatric T-ALL patients, the largest group analyzed by small RNA-sequencing thus far. We identified 1628 expressed miRNAs in T-ALL samples; out of these, 415 were expressed exclusively in T-ALL and not in the controls. The top 10 highly expressed miRNAs in the combined dataset included: hsa-miR-92a-3p; hsa-miR-181a-5p; hsa-let-7f-5p; hsa-miR-26a-5p; hsa-let-7a-5p/7c-5p; hsa-miR-128-3p; hsa-miR-191-5p; hsa-miR-148a-3p; hsa-miR-21-5p; and hsa-miR-30d-5p (Supplementary Figure S7).
This set of miRNAs show much overlap with the top 10 highly expressed miRNAs in our original miRNA-seq data obtained in 34 T-ALL patients (Supplementary Figures S5, S7, S8). miRNAs not overlapping between these 2 lists of “top 10 highly expressed miRNAs,” namely, hsa-miR-30d-5p and hsa-miR-191-5p, were also highly expressed in our 34 T-ALL patients, ranked at positions 13 and 19, respectively, while hsa-let-7i-5p and hsa-let-7g-5p were ranked at positions 13 and 15 of the highly expressed miRNAs in the combined dataset. Of note, hsa-miR-92 and hsa-miR-26a were previously identified by Mavrakis et al. [51] to be among the top 10 highly expressed miRNAs in a cohort of 50 T-ALL patients in the RT-qPCR–based study (Supplementary Figure S8).
What is more, hsa-let-7a-5p/7c-5p (among the top 10 highly expressed miRNAs in our original miRNA-seq data and in the combined dataset) was also identified among the most stably expressed miRNAs across 34 T-ALL samples, 6 T-ALL cell lines, normal mature T lymphocytes, and CD34+ and CD4 + CD8 + CD3+ thymocytes, as we previously reported [25]. Of note, in our analysis of isomiRs' expression, we found that hsa-let-7a-5p/7c-5p is expressed in T-ALL samples and in normal mature T lymphocytes exclusively in its canonical form (no isomiRs). Thus, we used this miRNA as one of endogenous control miRNAs in our RT-qPCR validation of miRNA-seq results.
In the combined dataset, we also aimed to identify miRNAs differentially expressed in T-ALL as compared to control normal mature T lymphocytes and thymocytes. Out of 1729 miRNAs expressed in the analyzed samples, 103 miRNAs were differentially expressed: 11 miRNAs were overexpressed in T-ALL vs. controls, and 92 miRNA were underexpressed in T-ALL as compared to controls (Supplementary Table 3 and Figure 3).
Figure 3.

Differentially expressed miRNAs in combined analysis of 82 T-ALL samples, normal mature T lymphocytes, and thymocytes.
Heatmap and dendrograms of Z-score normalized miRNA expression levels created for miRNAs that differentiate T-ALL samples from normal controls (mature T lymphocytes and thymocytes). Rows represent miRNAs; columns represent samples. Dendrograms are based on complete-linkage hierarchical clusterization and Euclidean distances. The fold changes and the abundance of miRNAs (both in log scale) are shown on the left side of the plot. IHG, Institute of Human Genetics; UG, Ghent University.
Interestingly, the analysis in the combined datasets revealed a set of miRNAs which we found differentially expressed in our original analysis of 34 T-ALL patients. This included 4 upregulated miRNAs, hsa-miR-20b-5p, hsa-miR-153-3p, hsa-miR-625-5p, and hsa-miR-466, and a set of 24 downregulated miRNAs, including hsa-miR-3909 and hsa-miR-1275. We also checked the convergence of the results of the combined data sets analysis with differentially expressed miRNAs reported by Wallaert et al. [16]. This analysis revealed 4 overlapping upregulated miRNAs, hsa-miR-374b-3p, hsa-miR-3913-5p, hsa-miR-181d-5p, and hsa-miR-153-3p; and 13 overlapping downregulated miRNAs, including hsa-miR-3909 and hsa-miR-1275. Of note, the latter two miRNAs and hsa-miR-153-3p were overlapping between our original data set and the results by Wallaert et al. [16].
miRNA Expression Profile In Relation to Immunophenotypic Stages of T-ALL Classification
We also aimed to verify whether miRNA expression profile can separate T-ALL samples representing different immunophenotypic subtypes, according to the EGIL [52]. Three of the 4 immunophenotypic subtypes were represented in our group of 34 T-ALL patients (Supplementary Table S1). Since data on EGIL classification were not available for the T-ALL samples from the UG dataset, we subtracted them from the analysis. We retained the CD34+ and CD4 + CD8 + CD3+ thymocyte samples in the analysis since they represent very early immature precursors and more mature precursors of normal T lymphocytes. Using ANOVA test (independently for each miRNA with Benjamini and Hochberg correction for multiple testing) and unsupervised hierarchical clusterization, we showed that expression profile of 28 miRNAs separate T-ALL samples from 3 different EGIL subtypes into 3 major clusters (Figure 4). Nearly all immature T-ALL samples (EGIL II), 8/9 (89%), group together with the 2 samples of early thymocytes (CD34+). Nearly all cortical T-ALL samples (EGIL III), 8/9 (89%), and the majority (14/16, 88%) of mature T-ALL samples group in the second cluster with the 2 samples of CD4 + CD8 + CD3+ thymocytes. Of note, these two thymic samples group in between the two parts of the cluster, one with the predominance of the cortical T-ALL samples (EGIL III) and the second cluster containing almost exclusively mature T-ALL cases (EGIL IV). Normal mature T lymphocytes group separately. Thus, we show that distinct immunophenotypic subtypes of T-ALL present differing miRNA expression profiles.
Figure 4.

Differentially expressed miRNAs between T-ALL samples representing different immunophenotypic subtypes of T-ALL.
Heatmap and dendrograms of Z-score normalized miRNA expression levels created for miRNAs that differentiate T-ALL samples representing immunophenotypic subtypes of T-ALL (according to EGIL classification) and from normal controls (mature T lymphocytes and thymocytes). Differentially expressed miRNAs were identified using ANOVA test independently for each miRNA, with Benjamini and Hochberg correction for multiple testing. Rows represent miRNAs; columns represent samples. Dendrograms are based on complete-linkage hierarchical clusterization and Euclidean distances. The fold changes and the abundance of miRNAs (both in log scale) are shown on the left side of the plot.
Target Prediction and Pathway Enrichment Analysis: miRNA-mRNA Interactions of Potential Importance for Biology of T-ALL
To explore the potential implications of aberrant expression of miRNAs for the biology of T-ALL, we performed a comprehensive in silico analysis aimed at the prediction of mRNA targets, regulated by the 61 miRNAs we identified as differentially expressed between T-ALL and normal mature T lymphocytes. We took a comprehensive approach using multiple target prediction tools and retrieving data from several databases, as described in detail in Materials and Methods.
In total, this analysis resulted in 117,203 miRNA-mRNA interactions, predicted by any of the eight target prediction algorithms; each interaction of miRNA seed sequence with MRE in a target gene was counted as 1 record. Thus, the total number of predicted target genes was much lower, 7609 predicted targets. Additionally, the analysis resulted in 17,143 records of miRNA-mRNA interactions validated by different experimental approaches. The total number of records retrieved from pharmacogenomics database representing miRNA-mRNA associations with drugs was 33. Additionally, the analysis resulted in 1466 records retrieved from databases of miRNAs related to diseases, including malignancies. This enabled us to gain a comprehensive overview of our 61 differentially expressed miRNAs and their mRNA targets, with additional view on their involvement in diseases and drugs response.
But to further explore the implications of aberrant miRNA expression in T-ALL and to identify novel miRNA-mRNA interactions with oncogenic and tumor suppressor potential in this diseases, we focused on target mRNAs, uniformly predicted by five out of eight target prediction algorithms. This reduced the number of mRNA targets of interest from 7609 to 225 (Figure 5).
Figure 5.
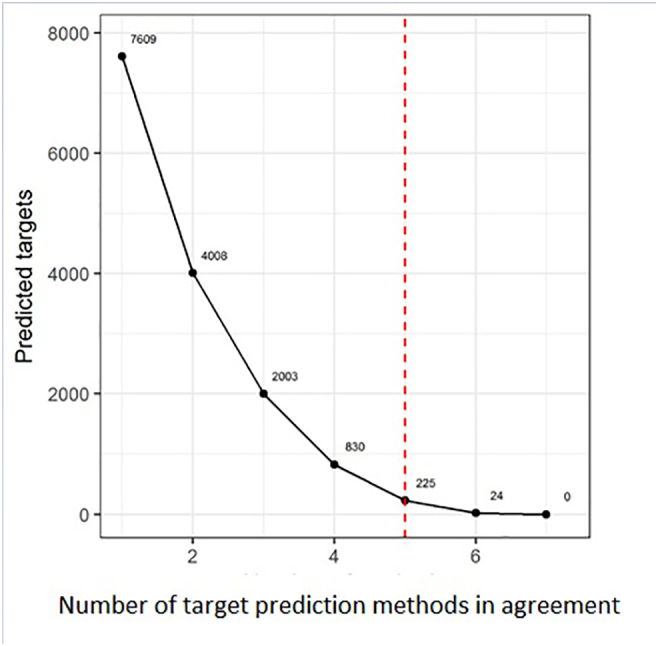
Number of mRNA targets for miRNAs differentially expressed in T-ALL vs. controls related to the number of prediction algorithms in agreement.
To get insights into the potential pathogenic role of differentially expressed miRNAs and their 225 selected target mRNAs, we tested these target genes for overrepresentation in biological processes and pathways, defined by terms in GO, KEGG, Reactome, and Panther databases. The targets for our differentially expressed miRNAs were significantly enriched in 42 pathways from GO and in 66 terms from KEGG database (Supplementary Table S2). No significant results were retrieved from Reactome and Panther. Since the vast majority of KEGG terms did not seem of significance for T-ALL, we further focused on GO terms (Figure 6). Importantly, among the top 10 pathways with the highest odds ratio, we identified several processes of significance for T-ALL pathogenesis, namely, signal transduction by trans-phosphorylation, interleukin-6–mediated signaling pathway, negative regulation of TOR signaling, positive regulation of apoptotic process, regulation of growth, and regulation of phosphorylation. We then focused on the positive regulation of apoptosis due to high position in the OR rank (OR = 2.68, P = .018) and clear importance for the oncogenic process.
Figure 6.

Pathway enrichment analysis for targets of miRNAs aberrantly expressed in T-ALL.
Odds ratio for the selected GO terms, identified using conditional hypergeometric test, with Benjamini and Hochberg correction for multiple testing and .05 significance level. The size of the dots (gene count) represents the number of genes (predicted targets of differentially expressed miRNAs) involved in a given biological process; the color of the dots represents P value. The terms are sorted alphabetically. Processes with the highest OR values (from the top 10) are depicted with *.
Overexpressed miRNAs from mir-106a-363 Cluster: Potentially Implicated in Deregulation of Apoptosis in T-ALL
Using the results of our comprehensive analysis with multiMiR R package, we searched through the differentially expressed miRNAs and their targets, associated with the positive regulation of apoptosis, with the aim to identify potential novel miRNA-mRNA interactions with the oncogenic role in T-ALL.
Nineteen genes, predicted targets of our differentially expressed miRNAs, were enriched in the positive regulation of apoptosis pathway. These included CDK19, FAP, FBXW7, FGD4, FNIP1, LATS2, MAP3K11, NCOA1, NET1, NEUROD1, NOX4, NSMAF, PPARG, PTEN, SERINC3, SOS1, SOS2, TXNIP, and UBC. These genes are predicted and/or validated targets of four miRNAs downregulated in our cohort of T-ALL patients, hsa-miR-30a-5p, hsa-miR-199b-5p, hsa-miR-145-5p, and hsa-miR-223-3p, and five upregulated miRNAs: hsa-miR-130a-3p, hsa-miR-130b-3p, hsa-miR-363-3p, hsa-miR-20b-5p, and hsa-miR-18b-5p (Supplementary Table S4).
We then focused on upregulated miRNAs since their overexpression, resulting in the silencing of their targets, may potentially lead to an antiapoptotic phenotype and enhanced proliferation of T-ALL cells. Importantly, three of these upregulated miRNAs, namely, hsa-miR-363-3p, hsa-miR-20b-5p, and hsa-miR-18b-5p, belong to one cluster of miRNAs. These three miRNAs are transcribed from one genomic locus (hsa-mir-106a-363 cluster; chrX: 134169378-chrX: 134170278) as a long noncoding transcript, further processed to mature miRNAs, which potentially cooperate in the regulation of genes and pathways [53]. Of note, hsa-mir-106a-363 cluster is a paralogue of the hsa-mir-106b-25 cluster and a prototypic oncogenic hsa-mir-17-92 cluster [54]. miRNAs within these clusters share high homology of their mature sequences within the miRNA gene families (Figure 7) and thus may regulate analogous targets and have overlapping roles [55].
Figure 7.

Paralogous miRNA clusters: mir-17-92, mir-106a-363, and mir-106b-25.
The colors indicate miRNAs belonging to one family and thus sharing high homology of mature miRNA sequence, including identity of the seed sequence.
Using RT-qPCR, we confirmed overexpression of two miRNAs from hsa-mir-106a-363 cluster, hsa-miR-20b-5p and hsa-miR-363-3p, in the study group and in the validation cohort of T-ALL patients using normal mature T lymphocytes and, in case of hsa-miR-20b-5p, also using thymocytes as controls, as previously described [25]. We also validated expression levels of hsa-miR-128-3p and hsa-miR-181a-5p, both among the top 10 most highly expressed in our miRNA-seq data and overexpressed as compared to normal T lymphocytes, and hsa-miR-1275, downregulated in our miRNA-seq data, in line with the downregulation in T-ALL patients in the study by Wallaert et al. [16]. The comparison of expression data from miRNA-seq and RT-qPCR documenting positive validation of our miRNA-seq results is presented in Supplementary Figure S9.
We then aimed at the validation of selected miRNA-mRNA interactions with a potential role in the pathogenesis of T-ALL. From the list of genes enriched in positive regulation of apoptosis term that were predicted by five of eight algorithms to be targeted by hsa-miR-20b-5p and hsa-miR-363-3p, we selected four miRNA-mRNA interactions not previously validated in functional studies and/or not reported in the context of T-ALL. These included SOS1 potentially targeted by hsa-miR-20b-5p, LATS2 targeted by hsa-miR-363-3p, and PTEN targeted by both hsa-miR-20b-5p and hsa-miR-363-3p.
Using Dual Luciferase Reporter Assay, we validated direct interactions of hsa-miR-20b-5p and hsa-miR-363-3p with the 3′UTR regions of the selected targets. Co-transfection of HEK293T cells with mimics of hsa-miR-20b-5p or hsa-miR-363-3p and pmirGLO vectors, containing relevant 3′UTR regions of selected genes, resulted in significant decrease in the activity of reporter luciferase relative to controls (co-transfection with scrambled miRNAs). This effect was not observed in case of pmiRGLO plasmids containing mutants of the relevant 3′UTR regions of target genes (Figure 8). Thus, we in vitro validated direct interactions of hsa-miR-20b-5p and hsa-miR-363-3p with 3′UTRs of their targets potentially implicated in the biology of T-ALL.
Figure 8.

Validation of interaction between studied miRNAs and their predicted target 3′UTRs in HEK293T cell line via Dual Luciferase Reporter Assay.
WT, wild-type sequence; MUT, sequence with mutations introduced within miRNA binding site in 3′UTR; *P < .05; **P < .01; ***P < .001; ns, not significant. The graphs present the decrease of relative luciferase activity in the presence of miRNA mimic in reference to scrambled control. Below each graph, the predicted interaction sites in miRNAs and mRNAs are shown, with indication of nucleotides in mRNAs mutated in rescue experiment. (A) Interaction between LATS2 3′UTR and hsa-miR-363-3p. (B) Interaction between PTEN 3′UTR and hsa-miR-363-3p. (C) Interaction between SOS1 3′UTR and hsa-miR-20b-5p. (D) Interaction between PTEN 3′UTR and hsa-miR-20b-5p.
Discussion
Landscape of miRNA Transcriptome in T-ALL
We present the results of small RNA sequencing performed in 34 samples of T-ALL and 5 samples of normal mature T lymphocytes as controls aimed at the description of miRNome in pediatric T-ALL. For a more comprehensive insight into the landscape of miRNA transcriptome in this leukemia, we broadened our analysis by combining our results with data of 48 T-ALL samples published by Wallaert et al. [16], thus far the only available dataset regarding miRNA expression profiling in T-ALL patients with small RNA-seq. The knowledge on miRNA expression in T-ALL provided by previous works was based on RT-qPCR approaches, which enabled the analysis of expression of preselected sets of miRNAs [51], [56], [57]. The only previous study using high-throughput sequencing was that by Schotte et al. [58]. Yet, the aim of this work was to define miRNA profiles differing between subtypes of acute lymphoblastic leukemia; patient samples, including 10 samples of T-ALL, were pooled to represent the subtypes. This precluded the analysis of the landscape of miRNA expression across individual T-ALL patients. Most recently, a study applying small RNA-seq to eight T-ALL samples was published by Dos Santos Almeida [59]. The study was aimed at the identification of miRNAs differing between T-ALL and B-ALL. Normal controls were not analyzed in this study, which precluded the identification of miRNAs differentially expressed between T-ALL cells and normal counterparts. Since small RNA-seq data were not made publically available, we could not include them in our analysis of combined data sets regarding T-ALL patients.
With our study, we provide an overview of the miRNome identified in 34 T-ALL patients, further combined with the data by Wallaert et al. [16] for a total number of 82 T-ALL cases. Thus, we report on miRNA expression in the largest cohort analyzed by small RNA-seq to date.
We observed that the sets of miRNAs of the highest expression in T-ALL samples are strongly overlapping between our original miRNA-seq data and in the broader analysis of 82 patients. Thus, we show that miRNAs most abundantly expressed in T-ALL samples include hsa-miR-92a-3p, hsa-miR-181a-5p, hsa-miR-128-3p, hsa-let-7f-5p, hsa-let-7a-5p/7c-5p, hsa-miR-26a-5p, hsa-miR-148a-3p, and hsa-miR-21-5p. Of note, 2 of these miRNAs, hsa-miR-92 and hsa-miR-26a, were previously reported to be among the top 10 highly expressed miRNAs in T-ALL samples in an RT-qPCR–based study [51]. Although the results of miRNA profiling with miRNA-seq and RT-qPCR cannot be directly compared due to the differing scope of miRNAs covered by both approaches, our results confirm previous findings from less accurate methods. With this small RNA-seq analysis, we extensively broaden the knowledge on miRNA expression in T-ALL and we aim to comprehensively characterize the miRNome of this disease.
We demonstrate that T-ALL samples present substantial diversity of expressed miRNA isoforms (isomiRs). The majority (80%) of miRNAs expressed in 34 T-ALL samples was represented by up to 100 isoforms/miRNA. With more stringent criteria focusing on miRNAs most abundantly expressed in T-ALL (at least 4 reads in more than 60% of T-ALL samples), we show that the diversity of isomiRs expression is still high, with nearly 70% of miRNAs represented by up to 10 isoforms. It is now recognized that different classes of isomiRs contribute to the intricacy of miRNA-dependent regulation of gene expression: 5′ isomiRs affect the seed sequence and thus the targeting of mRNAs [60]; other classes of isomiRs are postulated to change the activity and turnover of miRNAs [61]. The analysis of the composition of different classes of isomiRs was beyond the scope of our study. Yet, our results point to the complexity of miRNAs' involvement in the pathogenesis of T-ALL.
Additionally, we report for the first time that T-ALL samples originating from T-cell precursors at various stages of maturation, thus classified into separate immunophenotypic T-ALL subtypes, present with differing miRNA expression profiles. The inclusion of thymocytes in this analysis enabled to demonstrate that immature T-ALL samples (EGIL II) show similarity of miRNA expression with early precursors of T-cells (CD34+ thymocytes), while cortical (EGIL III) and mature (EGIL IV) T-ALL samples group with more mature precursors (CD4 + CD8 + CD3+). Thus, we demonstrate that small RNA-seq provides insights into immunophenotypic heterogeneity of T-ALL and holds potential for improved classification of this leukemia.
Novel Candidate Oncogenic and Tumor Suppressor miRNAs in T-ALL
We also aimed at the identification of miRNAs differentially expressed between T-ALL samples and controls, which represent candidate oncogenic and tumor suppressor miRNAs. With our results, we point to a set of miRNAs identified as differentially expressed in our original data (34 T-ALL samples), the combined data sets (82 T-ALL samples), and the results by Wallaert et al. (48 T-ALL cases) [16]. These miRNAs, overlapping between at least two of these datasets, include several overexpressed miRNAs: hsa-miR-20b-5p, hsa-miR-153-3p, hsa-miR-625-5p, hsa-miR-466, hsa-miR-374b-3p, hsa-miR-3913-5p, hsa-miR-181d-5p, and a set of downregulated miRNAs, with hsa-miR-3909 and hsa-miR-1275 identified uniformly in all three datasets. Interestingly, the isoforms of two of these miRNAs have been previously reported to act as oncogenes in T-ALL, namely, miR-20a [51], hsa-miR-181a, and hsa-miR-181b [46], [47], [48]. Yet the role of hsa-miR-20b-5p, hsa-miR-181d-5p, and the remaining differentially expressed miRNAs in the biology of T-ALL remains to be established.
Of note, in both data sets, regarding 34 T-ALL and 82 T-ALL patients, we observed that the number of miRNAs downregulated as compared to relative controls outnumbers that of the upregulated miRNAs. This is a common trait observed in cancer cells, regardless of the type of malignancy [62]. Due to established role of miRNAs in the regulation of differentiation processes, this global downregulation of miRNAs is hypothesized to reflect pathological condition of the cells, possessing higher potential to proliferate than to differentiate [63], which is one of the hallmarks of cancer. This massive downregulation of miRNAs is hypothesized to result in the global release of oncogenes from the negative control exerted by tumor suppressor miRNAs and thus contribute to oncogenesis. It is also possible that the increased number of downregulated miRNAs as compared to upregulated ones might be an artifact of the normalization procedure. The disproportion in the number of up- and downregulated miRNAs found in a particular experiment might be caused by a small number of miRNAs being highly expressed in one group of samples but not in the other. By consuming a large proportion of the library size, the remaining miRNAs might tend to be undersampled and may falsely appear as downregulated. Yet, in this work, we used Upper Quantile and TMM normalization methods designed with the aim to prevent such artifacts; they omit the most highly expressed features before calculating scaling factors.
What is more, in our miRNA-seq analysis of 34 T-ALL patients, we identified 254 candidate novel miRNAs (not previously reported in miRBase21). These include 10 miRNAs differentially expressed between T-ALL samples and controls, all 10 being underexpressed and representing novel candidate tumor suppressors miRNAs in this disease. Thus, our results point to a set of miRNAs, already annotated in miRBase and newly identified, which might prove of importance for functional studies on the pathogenesis of this disease.
miRNA-mRNA Interactions Potentially Contributing to T-ALL
Comprehensive target prediction followed by pathway enrichment analysis revealed biological processes potentially regulated by differentially expressed miRNAs. Among the top 10 highly enriched processes, we identified several of interest for the biology of this disease.
The term “signal transduction by trans-phosphorylation” was on top of the enriched processes. This enrichment is due to the involvement of two genes coding protein kinases, STK39 (serine/threonine kinase 39) and WNK1 (lysine deficient protein kinase 1), being predicted targets of miRNAs we found differentially expressed in our study, hsa-miR-30a-5p and hsa-miR-130a-3p, respectively. Thus far, there are no data on the engagement of these kinases in the pathogenesis of T-ALL. Overexpression of STK39 has recently been reported to have oncogenic potential in non–small cell lung cancer [64] and osteosarcoma [65]. STK39 was functionally demonstrated to increase cell proliferation when overexpressed [64] and to repress cell proliferation, migration, and invasion when knocked down [64], [65]. Thus, downregulation of hsa-miR-30a-5p predicted to target STK39 mRNA may potentially contribute to leukemia by upregulation of this kinase. WNK1 kinase is involved in MAPK (mitogen-activated protein kinase) signaling as an activator of ERK5 (extracellular-signal-regulated kinase 5), which stimulates cell proliferation but has also proapoptotic substrates such as BAD (Bcl-2-associated death promoter) [66], [67]. WNK1 mutations were reported in patients with breast, lung, and ovarian cancer [68] and more recently in chronic lymphocytic leukemia [69], but its role as either an oncogene or a tumor suppressor is yet not clear. Overexpression of hsa-miR-130a-3p predicted to target WNK1 might potentially contribute to T-ALL pathogenesis by disruption of MAPK/ERK5 signaling.
“Interleukin-6–mediated signaling pathway” is another term we found significantly enriched with targets of differentially expressed miRNAs. These include IL6ST (interleukin 6 signal transducer) predicted target of hsa-miR-223-3p, GAB1 (GRB2 associated binding protein 1), and STAT3 (signal transducer and activator of transcription 3), the latter two being predicted targets of hsa-miR-20b-5p. IL6ST (gp130) is a non–ligand-binding component of functional receptor for IL6. GAB1 is an adaptor protein recruited in many signaling cascades of receptor tyrosine kinases, including IL6 pathway [70]. Binding of IL6 to its receptor triggers JAK/STAT signaling, with the final activation of STAT3 transcription factor and subsequent expression of its target genes [71]. IL6 signaling pathway is primarily known for its involvement in inflammation, but it is also crucial for hematopoiesis, as it promotes T-cell proliferation by prevention from apoptosis [72]. Aberrant IL6/JAK/STAT signaling has been shown to contribute to several types of malignancies [73], [74], [75], [76]. Yet, our results indicating potential involvement of these two miRNAs and their targets enriched in IL6/JAK/STAT signaling seem inconsistent: hsa-miR-223-3p was downregulated in T-ALL samples in our study, thus potentially leading to the upregulation of IL6-mediated signaling, while we found hsa-miR-20b-5p to be upregulated, thus potentially resulting in GAB1 and STAT3 repression. These observations might seemingly be in conflict considering the involvement of miRNAs in intricate networks of the regulation of gene expression. Moreover, the nature of the pathway enrichment analysis is to generate hypotheses for functional verification.
In turn, our results on the enrichment of miRNA targets in the term “negative regulation of TOR signaling” are very consistent and generate promising hypothesis. All miRNAs (hsa-miR-130a-3p, hsa-miR-363-3p, hsa-miR-18b-5p, hsa-miR-130b-3p) which predicted targets (FNIP1, HIF1A, PRKAA1, SESN3, SH3BP4, TSC1) are enriched in this signaling pathway, are overexpressed in T-ALL samples in our study. This upregulation of miRNAs and the resulting downregulation of the genes involved in the negative control over mTOR signaling may potentially contribute to T-ALL by enhanced activity of this signaling cascade. mTOR is a central regulator of cell growth, proliferation, and survival in response to growth factors but also signaling from PI3K, MAPK, and AMPK cascades [77]. Despite existing data indicating the involvement of these genes: FNIP1 [78], HIF1A [79], [80], PRKAA1 [81], [82], SESN3 [83], SH3BP4 [84], and TSC1 [82], [85], [86], [87] in various malignancies, their role in aberrant mTOR signaling as a pathogenic mechanism in T-ALL is largely unknown. The activation of mTOR pathway in T-ALL might have practical consequences because pharmacological inhibition of the mTOR signaling with sirolimus or everolimus can be considered [88].
Overexpressed miRNAs from mir-106a-363 Cluster and Their Validated Targets: Potential Contribution to T-ALL by Dysregulation of Apoptosis
Importantly, we identified “positive regulation of apoptosis” among the top processes highly enriched in genes, predicted to be targeted by miRNAs differentially expressed in T-ALL. We further focused on upregulated miRNAs and their targets involved in this pathway since miRNA-mediated repression of these genes could potentially contribute to T-ALL by inducing resistance to apoptosis and thus enhancing proliferation of leukemic cells. Several of these genes, e.g., PTEN [89], FBXW7 [90], BCL2L11 [82], [91], and CDK19 [92], have already reported roles as tumor suppressors in various malignancies, including T-ALL. The most widely recognized mechanisms of the loss of their function are inactivating mutations and/or deletions, as well as hypermethylation of their promoter regions. Yet, the mechanisms causing posttranslational repression of these genes, including silencing by aberrantly overexpressed miRNAs, have been extensively studied recently.
Of note, three of the upregulated miRNAs predicted to target genes involved in the positive regulation of apoptosis, hsa-miR-363-3p, hsa-miR-20b-5p, and hsa-miR-18b-5p, belong to hsa-mir-106a-363 cluster, a paralogue of a prototypic oncogenic hsa-mir-17-92 cluster [54]. The oncogenic role of hsa-mir-106a-363 cluster has been much less extensively studied, specifically in the context of leukemia. There are few reports indicating an oncogenic potential of miRNAs from this cluster in human malignancies [93], [94], [95]. Interestingly, Dylla and Jedlicka demonstrated the importance of the cooperative oncogenic activity of miRNAs from this cluster in Ewing sarcoma cell lines; blockade of individual miRNAs had little or no inhibitory effect on growth of the cells in contrast to the blockade of the whole cluster [94]. Thus, simultaneous overexpression of hsa-miR-363-3p, hsa-miR-20b-5p, and hsa-miR-18b-5p we observed in T-ALL samples and significant enrichment of their targets in the positive regulation of apoptosis pointed our attention to these miRNAs. We further focused on hsa-miR-363-3p and hsa-miR-20b-5p since we confirmed their overexpression in the study group and in the validation cohort of T-ALL patients using RT-qPCR [25]. With the aim to point to novel candidate miRNA-mRNA interactions with potential importance for T-ALL, we validated four miRNA-mRNA interactions not yet validated and/or not reported to be implicated in this disease. Using Dual Luciferase Reporter Assay, we showed direct interactions of these two miRNAs with 3′UTRs of their predicted targets: hsa-miR-363-3p with LATS2 and PTEN; hsa-miR-20b-5p with SOS1 and PTEN.
PTEN (phosphatase and tensin homolog) is a tumor suppressor, acting as a negative regulator of PI3K/AKT/mTOR pathway. PTEN inactivation, due to mutations and deletions, has been reported to contribute to many malignancies, including T-ALL [96], [97], [98]. Interaction of miR-20b-5p with 3′UTR of PTEN has been reported in the context of oncogenic mechanisms in breast cancer [99] and colorectal cancer [100] yet not in the context of T-ALL pathogenesis. The interaction of PTEN with hsa-miR-363-3p and the interaction of LATS2 with hsa-miR-363-3p have not been functionally validated thus far. LATS2 (large tumor suppressor 2) is a tumor suppressor, acting as a positive regulator of p53 activity [101], shown to be downregulated in many types of cancer [102], [103], [104], including ALL [105]. The interaction of hsa-miR-20b-5p with SOS1 has been reported in thyroid cancer, though it has been shown to display tumor suppressor effects in thyroid carcinoma cells [106]. SOS1 (Son of Sevenless 1) is an activator of the RAS signaling pathway, crucial for normal thymocyte development. Knockout of Sos1 in mice was shown to cause a block in the differentiation of T-cell precursors [107], [108], [109]. Posttranscriptional silencing of these three genes by hsa-miR-20b-5p and hsa-miR-363-3p, overexpressed in T-ALL patients, may contribute to the pathogenesis of this disease. Thus, we in vitro validated direct miRNA-mRNA interactions of potential interest for functional studies in T-ALL.
Conclusions
We comprehensively investigated the miRNome of pediatric T-ALL with the aim at broad characterization of the landscape of miRNA expression in this disease. We specifically focused on miRNAs most abundantly expressed in T-ALL and miRNAs differentially expressed between T-ALL samples and controls. We also point to the complexity of miRNA expression in T-ALL by providing data on the expression of isomiRs, candidate novel miRNAs (not previously annotated in miRBase), and by the analysis of differential miRNA expression in immunophenotypic classes of T-ALL. By comprehensive target prediction and pathway enrichment analysis, we point to miRNAs and their target genes potentially implicated in the processes crucial for the biology of T-ALL, such as signaling cascades and regulation of apoptosis. We functionally validated several of these miRNA-mRNA interactions. With this study, we provide a firm basis and data resource for extended functional analyses on the role of miRNA-mRNA interactions in this disease.
Acknowledgments
Acknowledgements
The authors wish to acknowledge Prof. Pieter van Vlierberghe (Center for Medical Genetics Ghent, Ghent University, Belgium), who kindly provided RNA samples obtained from CD34+ and CD4 + CD8 + CD3+ normal thymocyte subsets. We are also grateful to Prof. Anton Langerak and Joyce Schilperoord-Vermeulen (Erasmus MC, Department of Immunology, Rotterdam, the Netherlands) for their support in obtaining RNA from normal thymocytes. The authors express their appreciation to all the clinicians of the Polish Pediatric Leukemia and Lymphoma Study Group centers for providing the samples and related data for this study.
Author Contributions
M. D. (M. Dawidowska) conceived and supervised the study, analyzed and interpreted the data, and wrote the manuscript; R. J. carried out bioinformatics and statistical analyses and provided relevant graphics; M. D. (M. Drobna), B. S.-Z., and M. K. carried out RT-qPCR miRNA profiling and dual luciferase reporter assays; the graphics for these analyses were provided by M. D. (M. Drobna); B. S.-Z. contributed to the conception of the study; Ł. S. and L. M. performed flow cytometry immunophenotyping of T-ALL samples at diagnosis; A. L. contributed to RT-qPCR miRNA profiling; M. L., M. U., and K. K. contributed to sample acquisition; J. R. K. coordinated patients' treatment and data collection at the centers of Polish Pediatric Leukemia and Lymphoma Study Group; T. S. coordinated acquisition of samples and related data; M. W. was in charge of overall direction and funding. All authors critically revised the manuscript and approved the submitted version.
Footnotes
Conflict of Interest: The authors declare no conflicts of interest.
Funding: This work was supported by the National Science Center, Poland [grant numbers: 2014/15/B/NZ2/03394 and 2017/25/N/NZ2/01132] and by the National Centre for Research and Development, Poland [grant number: STRATEGMED3/304586/5/NCBR/2017]. B. S.-Z. received financial resources as part of financing the doctoral scholarship from the National Science Center, Poland (ETIUDA scholarship number: 2017/24/T/NZ5/00359).
Supplementary data to this article can be found online at https://doi.org/10.1016/j.neo.2019.01.004.
Contributor Information
Małgorzata Dawidowska, Email: malgorzata.dawidowska@igcz.poznan.pl.
Roman Jaksik, Email: roman.jaksik@polsl.pl.
Monika Drobna, Email: monika.drobna@igcz.poznan.pl.
Bronisława Szarzyńska-Zawadzka, Email: bronislawa.szarzynska-zawadzka@igcz.poznan.pl.
Maria Kosmalska, Email: maria.kosmalska@igcz.poznan.pl.
Łukasz Sędek, Email: lsedek@sum.edu.pl.
Ludomiła Machowska, Email: ludomilamachowska@poczta.fm.
Anna Lalik, Email: anna.lalik@polsl.pl.
Monika Lejman, Email: monika.lejman@umlub.pl.
Marek Ussowicz, Email: ussowicz@tlen.pl.
Krzysztof Kałwak, Email: krzysztof.kalwak@gmail.com.
Jerzy R. Kowalczyk, Email: jkowalcz@dsk.lublin.pl.
Tomasz Szczepański, Email: szczep57@poczta.onet.pl.
Michał Witt, Email: michal.witt@igcz.poznan.pl.
Appendix A. Supplementary data
Supplementary Figures & Supplementary Data.
Supplementary Tables
References
- 1.Bartel DP. MicroRNAs: Target Recognition and Regulatory Functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang R, Su B. Small but influential: the role of microRNAs on gene regulatory network and 3'UTR evolution. J Genet Genomics. 2009;36:1–6. doi: 10.1016/S1673-8527(09)60001-1. [DOI] [PubMed] [Google Scholar]
- 3.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 4.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 5.Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med. 2014;20:460–469. doi: 10.1016/j.molmed.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 6.Drobna M, Szarzyńska-Zawadzka B, Dawidowska M. T-cell acute lymphoblastic leukemia from miRNA perspective: Basic concepts, experimental approaches, and potential biomarkers. Blood Rev. 2018;32(6):457–472. doi: 10.1016/j.blre.2018.04.003. [DOI] [PubMed] [Google Scholar]
- 7.Svoronos AA, Engelman DM, Slack FJ. OncomiR or Tumor Suppressor? The Duplicity of MicroRNAs in Cancer. Cancer Res. 2016;76:3666–3670. doi: 10.1158/0008-5472.CAN-16-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 9.Bracken CP, Scott HS, Goodall GJ. A network-biology perspective of microRNA function and dysfunction in cancer. Nat Rev Genet. 2016;17:719–732. doi: 10.1038/nrg.2016.134. [DOI] [PubMed] [Google Scholar]
- 10.Belver L, Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer. 2016;16:494–507. doi: 10.1038/nrc.2016.63. [DOI] [PubMed] [Google Scholar]
- 11.Hefazi M, Litzow MR. Recent Advances in the Biology and Treatment of T Cell Acute Lymphoblastic Leukemia. Curr Hematol Malig Rep. 2018;13:265–274. doi: 10.1007/s11899-018-0455-9. [DOI] [PubMed] [Google Scholar]
- 12.Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, McCastlain K, Edmonson M, Pounds SB, Shi L. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49:1211–1218. doi: 10.1038/ng.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen B, Jiang L, Zhong ML, Li JF, Li BS, Peng LJ, Dai YT, Cui BW, Yan TQ, Zhang WN. Identification of fusion genes and characterization of transcriptome features in T-cell acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2018;115:373–378. doi: 10.1073/pnas.1717125115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Keersmaecker K, Atak ZK, Li N, Vicente C, Patchett S, Girardi T, Gianfelici V, Geerdens E, Clappier E, Porcu M. Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat Genet. 2013;45:186–190. doi: 10.1038/ng.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Atak ZK, Gianfelici V, Hulselmans G, De Keersmaecker K, Devasia AG, Geerdens E, Mentens N, Chiaretti S, Durinck K, Uyttebroeck A. Comprehensive analysis of transcriptome variation uncovers known and novel driver events in T-cell acute lymphoblastic leukemia. PLoS Genet. 2013;9 doi: 10.1371/journal.pgen.1003997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wallaert A, Van Loocke W, Hernandez L, Taghon T, Speleman F, Van Vlierberghe P. Comprehensive miRNA expression profiling in human T-cell acute lymphoblastic leukemia by small RNA-sequencing. Sci Rep. 2017;7 doi: 10.1038/s41598-017-08148-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Dongen JJM, Lhermitte L, Boettcher S, Almeida J, van der Velden VHJ, Flores-Montero J, Rawstron A, Asnafi V, Lecrevisse Q, Lucio P. EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes. Leukemia. 2012;26:1908–1975. doi: 10.1038/leu.2012.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalina T, Flores-Montero J, van der Velden VHJ, Martin-Ayuso M, Boettcher S, Ritgen M, Almeida J, Lhermitte L, Asnafi V, Mendonca A. EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia. 2012;26:1986–2010. doi: 10.1038/leu.2012.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wingett SW, Andrews S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research. 2018;7:1338. doi: 10.12688/f1000research.15931.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2012;17:10–12. [Google Scholar]
- 21.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baras AS, Mitchell CJ, Myers JR, Gupta S, Weng LC, Ashton JM, Cornish TC, Pandey A, Halushka MK. miRge - A Multiplexed Method of Processing Small RNA-Seq Data to Determine MicroRNA Entropy. PLoS One. 2015;10 doi: 10.1371/journal.pone.0143066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. (Oxford, England) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 25.Drobna M, Szarzyńska-Zawadzka B, Daca-Roszak P, Kosmalska M, Jaksik R, Witt M, Dawidowska M. Identification of Endogenous Control miRNAs for RT-qPCR in T-Cell Acute Lymphoblastic Leukemia. Int J Mol Sci. 2018;19 doi: 10.3390/ijms19102858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 27.Maragkakis M, Reczko M, Simossis VA, Alexiou P, Papadopoulos GL, Dalamagas T, Giannopoulos G, Goumas G, Koukis E, Kourtis K. DIANA-microT web server: elucidating microRNA functions through target prediction. Nucleic Acids Res. 2009;37:W273–W276. doi: 10.1093/nar/gkp292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaidatzis D, van Nimwegen E, Hausser J, Zavolan M. Inference of miRNA targets using evolutionary conservation and pathway analysis. BMC Bioinformatics. 2007;8:69. doi: 10.1186/1471-2105-8-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004;2 doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43:D146–D152. doi: 10.1093/nar/gku1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 33.Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. 2007;39:1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- 34.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 35.Ru Y, Kechris KJ, Tabakoff B, Hoffman P, Radcliffe RA, Bowler R, Mahaffey S, Rossi S, Calin GA, Bemis L. The multiMiR R package and database: integration of microRNA-target interactions along with their disease and drug associations. Nucleic Acids Res. 2014;42:e133. doi: 10.1093/nar/gku631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiao FF, Zuo ZX, Cai GS, Kang SL, Gao XL, Li TB. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009;37:D105–D110. doi: 10.1093/nar/gkn851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chou CH, Chang NW, Shrestha S, Hsu SD, Lin YL, Lee WH, Yang CD, Hong HC, Wei TY, Tu SJ. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016;44:D239–D247. doi: 10.1093/nar/gkv1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vlachos IS, Paraskevopoulou MD, Karagkouni D, Georgakilas G, Vergoulis T, Kanellos I, Anastasopoulos IL, Maniou S, Karathanou K, Kalfakakou D. DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res. 2015;43:D153–D159. doi: 10.1093/nar/gku1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang Q, Wang Y, Hao Y, Juan L, Teng M, Zhang X, Li M, Wang G, Liu Y. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009;37:D98–104. doi: 10.1093/nar/gkn714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruepp A, Kowarsch A, Schmidl D, Buggenthin F, Brauner B, Dunger I, Fobo G, Frishman G, Montrone C, Theis FJ. PhenomiR: a knowledgebase for microRNA expression in diseases and biological processes. Genome Biol. 2010;11:R6. doi: 10.1186/gb-2010-11-1-r6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rukov JL, Wilentzik R, Jaffe I, Vinther J, Shomron N. Pharmaco-miR: linking microRNAs and drug effects. Brief Bioinform. 2014;15:648–659. doi: 10.1093/bib/bbs082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morin RD, O'Connor MD, Griffith M, Kuchenbauer F, Delaney A, Prabhu AL, Zhao Y, McDonald H, Zeng T, Hirst M. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008;18:610–621. doi: 10.1101/gr.7179508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neilsen CT, Goodall GJ, Bracken CP. IsomiRs--the overlooked repertoire in the dynamic microRNAome. Trends Genet. 2012;28:544–549. doi: 10.1016/j.tig.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 44.Mets E, Van Peer G, Van der Meulen J, Boice M, Taghon T, Goossens S, Mestdagh P, Benoit Y, De Moerloose B, Van Roy N. MicroRNA-128-3p is a novel oncomiR targeting PHF6 in T-cell acute lymphoblastic leukemia. Haematologica. 2014;99:1326–1333. doi: 10.3324/haematol.2013.099515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamada N, Noguchi S, Kumazaki M, Shinohara H, Miki K, Naoe T, Akao Y. Epigenetic regulation of microRNA-128a expression contributes to the apoptosis-resistance of human T-cell leukaemia Jurkat cells by modulating expression of Fas-associated protein with death domain (FADD) Biochimica Et Biophysica Acta-Molecular Cell Research. 2014;1843:590–602. doi: 10.1016/j.bbamcr.2013.11.022. [DOI] [PubMed] [Google Scholar]
- 46.Yan ZX, Zheng Z, Xue W, Zhao MZ, Fei XC, Wu LL, Huang LM, Leboeuf C, Janin A, Wang L. MicroRNA181a Is Overexpressed in T-Cell Leukemia/Lymphoma and Related to Chemoresistance. Biomed Res Int. 2015;10 doi: 10.1155/2015/197241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Verduci L, Azzalin G, Gioiosa S, Carissimi C, Laudadio I, Fulci V, Macino G. microRNA-181a enhances cell proliferation in acute lymphoblastic leukemia by targeting EGR1. Leuk Res. 2015;39:479–485. doi: 10.1016/j.leukres.2015.01.010. [DOI] [PubMed] [Google Scholar]
- 48.Fragoso R, Mao T, Wang S, Schaffert S, Gong X, Yue SBA, Luong R, Min H, Yashiro-Ohtani Y, Davis M. Modulating the Strength and Threshold of NOTCH Oncogenic Signals by mir-181a-1/b-1. PLoS Genet. 2012;8 doi: 10.1371/journal.pgen.1002855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nemes K, Csoka M, Nagy N, Mark A, Varadi Z, Danko T, Kovacs G, Kopper L, Sebestyen A. Expression of Certain Leukemia/Lymphoma Related microRNAs and its Correlation with Prognosis in Childhood Acute Lymphoblastic Leukemia. Pathol Oncol Res. 2015;21:597–604. doi: 10.1007/s12253-014-9861-z. [DOI] [PubMed] [Google Scholar]
- 50.Ortega M, Bhatnagar H, Lin AP, Wang L, Aster JC, Sill H, Aguiar RCT. A microRNA-mediated regulatory loop modulates NOTCH and MYC oncogenic signals in B- and T-cell malignancies. Leukemia. 2015;29:968–976. doi: 10.1038/leu.2014.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mavrakis KJ, Van der Meulen J, Wolfe AL, Liu XP, Mets E, Taghon T, Khan AA, Setti M, Rondou P, Vandenberghe P. A cooperative microRNA-tumor suppressor gene network in acute T-cell lymphoblastic leukemia (T-ALL) Nat Genet. 2011;43 doi: 10.1038/ng.858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bene MC, Castoldi G, Knapp W, Ludwig WD, Matutes E, Orfao A, Vantveer MB. Proposals for the immunological classification of acute leukemias. Leukemia. 1783-1786;1995:9. [PubMed] [Google Scholar]
- 53.Yuan X, Liu C, Yang P, He S, Liao Q, Kang S, Zhao Y. Clustered microRNAs' coordination in regulating protein-protein interaction network. BMC Syst Biol. 2009;3:65. doi: 10.1186/1752-0509-3-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR. Targeted deletion reveals essential and overlapping functions of the miR-17 similar to 92 family of miRNA clusters. Cell. 2008;132:875–886. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khuu C, Utheim TP, Sehic A. The Three Paralogous MicroRNA Clusters in Development and Disease, miR-17-92, miR-106a-363, and miR-106b-25. Scientifica (Cairo) 2016;2016:1379643. doi: 10.1155/2016/1379643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schotte D, Chau JCK, Sylvester G, Liu G, Chen C, van der Velden VHJ, Broekhuis MJC, Peters T, Pieters R, den Boer ML. Identification of new microRNA genes and aberrant microRNA profiles in childhood acute lymphoblastic leukemia. Leukemia. 2009;23:313–322. doi: 10.1038/leu.2008.286. [DOI] [PubMed] [Google Scholar]
- 57.Schotte D, De Menezes RX, Moqadam FA, Khankahdani LM, Lange-Turenhout E, Chen CF, Pieters R, Den Boer ML. MicroRNA characterize genetic diversity and drug resistance in pediatric acute lymphoblastic leukemia. Haematologica-the Hematology Journal. 2011;96:703–711. doi: 10.3324/haematol.2010.026138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schotte D, Moqadam FA, Lange-Turenhout EAM, Chen C, van Ijcken WFJ, Pieters R, den Boer ML. Discovery of new microRNAs by small RNAome deep sequencing in childhood acute lymphoblastic leukemia. Leukemia. 2011;25:1389–1399. doi: 10.1038/leu.2011.105. [DOI] [PubMed] [Google Scholar]
- 59.Dos Santos Almeida R., Costa E Silva M., Coutinho L.L., Gomes R.G., Pedrosa F., Massaro J.D., Donadi E.A., Lucena-Silva N. MicroRNA expression profiles discriminate childhood T- from B-acute lymphoblastic leukemia. Hematol Oncol. 2018:1–10. doi: 10.1002/hon.2567. [DOI] [PubMed] [Google Scholar]
- 60.Tan GC, Chan E, Molnar A, Sarkar R, Alexieva D, Isa IM, Robinson S, Zhang S, Ellis P, Langford CF. 5' isomiR variation is of functional and evolutionary importance. Nucleic Acids Res. 2014;42:9424–9435. doi: 10.1093/nar/gku656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gebert LFR, MacRae IJ. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol. 2019;20:21–37. doi: 10.1038/s41580-018-0045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 63.Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T, Livingston DM, Rajewsky K. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 2005;19:489–501. doi: 10.1101/gad.1248505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li Z, Zhu W, Xiong L, Yu X, Chen X, Lin Q. Role of high expression levels of STK39 in the growth, migration and invasion of non-small cell type lung cancer cells. Oncotarget. 2016;7:61366–61377. doi: 10.18632/oncotarget.11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang T, Zhou Y, Cao Y, Tao J, Zhou ZH, Hang DH. STK39, overexpressed in osteosarcoma, regulates osteosarcoma cell invasion and proliferation. Oncol Lett. 2017;14:4599–4604. doi: 10.3892/ol.2017.6728. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 66.Ségaliny AI, Tellez-Gabriel M, Heymann MF, Heymann D. Receptor tyrosine kinases: Characterisation, mechanism of action and therapeutic interests for bone cancers. J Bone Oncol. 2015;4:1–12. doi: 10.1016/j.jbo.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McCormick JA, Ellison DH. The WNKs: atypical protein kinases with pleiotropic actions. Physiol Rev. 2011;91:177–219. doi: 10.1152/physrev.00017.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang L, Fan J, Francis JM, Georghiou G, Hergert S, Li S, Gambe R, Zhou CW, Yang C, Xiao S. Integrated single-cell genetic and transcriptional analysis suggests novel drivers of chronic lymphocytic leukemia. Genome Res. 2017;27:1300–1311. doi: 10.1101/gr.217331.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Takahashi-Tezuka M, Yoshida Y, Fukada T, Ohtani T, Yamanaka Y, Nishida K, Nakajima K, Hibi M, Hirano T. Gab1 acts as an adapter molecule linking the cytokine receptor gp130 to ERK mitogen-activated protein kinase. Mol Cell Biol. 1998;18:4109–4117. doi: 10.1128/mcb.18.7.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol. 1998;161:4652–4660. [PubMed] [Google Scholar]
- 73.Brooks GD, McLeod L, Alhayyani S, Miller A, Russell PA, Ferlin W, Rose-John S, Ruwanpura S, Jenkins BJ. IL6 Trans-signaling Promotes KRAS-Driven Lung Carcinogenesis. Cancer Res. 2016;76:866–876. doi: 10.1158/0008-5472.CAN-15-2388. [DOI] [PubMed] [Google Scholar]
- 74.Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Klöppel G, Yoshimura A, Reindl W, Sipos B, Akira S. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19:456–469. doi: 10.1016/j.ccr.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 76.Becker C, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A, Burg J, Strand S, Kiesslich R, Huber S. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004;21:491–501. doi: 10.1016/j.immuni.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 77.Pópulo H, Lopes JM, Soares P. The mTOR signalling pathway in human cancer. Int J Mol Sci. 2012;13:1886–1918. doi: 10.3390/ijms13021886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hasumi H, Baba M, Hasumi Y, Lang M, Huang Y, Oh HF, Matsuo M, Merino MJ, Yao M, Ito Y. Folliculin-interacting proteins Fnip1 and Fnip2 play critical roles in kidney tumor suppression in cooperation with Flcn. Proc Natl Acad Sci U S A. 2015;112:E1624–E1631. doi: 10.1073/pnas.1419502112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen M, Lu J, Wei W, Lv Y, Zhang X, Yao Y, Wang L, Ling T, Zou X. Effects of proton pump inhibitors on reversing multidrug resistance via downregulating V-ATPases/PI3K/Akt/mTOR/HIF-1α signaling pathway through TSC1/2 complex and Rheb in human gastric adenocarcinoma cells in vitro and in vivo. Onco Targets Ther. 2018;11:6705–6722. doi: 10.2147/OTT.S161198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen WM, Huang MD, Kong R, Xu TP, Zhang EB, Xia R, Sun M, De W, Shu YQ. Antisense Long Noncoding RNA HIF1A-AS2 Is Upregulated in Gastric Cancer and Associated with Poor Prognosis. Dig Dis Sci. 2015;60:1655–1662. doi: 10.1007/s10620-015-3524-0. [DOI] [PubMed] [Google Scholar]
- 81.Obba S, Hizir Z, Boyer L, Selimoglu-Buet D, Pfeifer A, Michel G, Hamouda MA, Gonçalvès D, Cerezo M, Marchetti S. The PRKAA1/AMPKα1 pathway triggers autophagy during CSF1-induced human monocyte differentiation and is a potential target in CMML. Autophagy. 2015;11:1114–1129. doi: 10.1080/15548627.2015.1034406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mavrakis KJ, Wolfe AL, Oricchio E, Palomero T, de Keersmaecker K, McJunkin K, Zuber J, James T, Chang K, Khan AA. Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nat Cell Biol. 2010;12 doi: 10.1038/ncb2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vakana E, Arslan AD, Szilard A, Altman JK, Platanias LC. Regulatory effects of sestrin 3 (SESN3) in BCR-ABL expressing cells. PLoS One. 2013;8 doi: 10.1371/journal.pone.0078780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang X, Yao X, Qin C, Luo P, Zhang J. Investigation of the molecular mechanisms underlying metastasis in prostate cancer by gene expression profiling. Exp Ther Med. 2016;12:925–932. doi: 10.3892/etm.2016.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang N, Liang X, Yu W, Zhou S, Fang M. Differential Expression of MicroRNA-19b Promotes Proliferation of Cancer Stem Cells by Regulating the TSC1/mTOR Signaling Pathway in Multiple Myeloma. Cell Physiol Biochem. 2018;50:1804–1814. doi: 10.1159/000494821. [DOI] [PubMed] [Google Scholar]
- 86.Zhang Y, Qu S, Wang Q, Li J, Xu Z, Qin T, Huang G, Xiao Z. A novel fusion of PDGFRB to TSC1, an intrinsic suppressor of mTOR-signaling pathway, in a chronic eosinophilic leukemia patient with t(5;9)(q32;q34) Leuk Lymphoma. 2018:1–3. doi: 10.1080/10428194.2018.1427855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mehra R, Vats P, Cao X, Su F, Lee ND, Lonigro R, Premkumar K, Trpkov K, McKenney JK, Dhanasekaran SM. Somatic Bi-allelic Loss of TSC Genes in Eosinophilic Solid and Cystic Renal Cell Carcinoma. Eur Urol. 2018;74:483–486. doi: 10.1016/j.eururo.2018.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pikman Y, Alexe G, Roti G, Conway AS, Furman A, Lee ES, Place AE, Kim S, Saran C, Modiste R. Synergistic Drug Combinations with a CDK4/6 Inhibitor in T-cell Acute Lymphoblastic Leukemia. Clin Cancer Res. 2017;23:1012–1024. doi: 10.1158/1078-0432.CCR-15-2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee YR, Chen M, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat Rev Mol Cell Biol. 2018;19:547–562. doi: 10.1038/s41580-018-0015-0. [DOI] [PubMed] [Google Scholar]
- 90.Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dafou D, Dofou D. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007;67:9006–9012. doi: 10.1158/0008-5472.CAN-07-1320. [DOI] [PubMed] [Google Scholar]
- 91.Li Y, Deutzmann A, Choi PS, Fan AC, Felsher DW. BIM mediates oncogene inactivation-induced apoptosis in multiple transgenic mouse models of acute lymphoblastic leukemia. Oncotarget. 2016;7:26926–26934. doi: 10.18632/oncotarget.8731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Aleem E, Arceci RJ. Targeting cell cycle regulators in hematologic malignancies. Front Cell Dev Biol. 2015;3 doi: 10.3389/fcell.2015.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Landais S, Landry S, Legault P, Rassart E. Oncogenic potential of the miR-106-363 cluster and its implication in human T-cell leukemia. Cancer Res. 2007;67:5699–5707. doi: 10.1158/0008-5472.CAN-06-4478. [DOI] [PubMed] [Google Scholar]
- 94.Dylla L, Jedlicka P. Growth-promoting role of the miR-106a~363 cluster in Ewing sarcoma. PLoS One. 2013;8 doi: 10.1371/journal.pone.0063032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li X, Zhou Q, Tao L, Yu C. MicroRNA-106a promotes cell migration and invasion by targeting tissue inhibitor of matrix metalloproteinase 2 in cervical cancer. Oncol Rep. 2017;38:1774–1782. doi: 10.3892/or.2017.5832. [DOI] [PubMed] [Google Scholar]
- 96.Gutierrez A, Sanda T, Grebliunaite R, Carracedo A, Salmena L, Ahn Y, Dahlberg S, Neuberg D, Moreau LA, Winter SS. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood. 2009;114:647–650. doi: 10.1182/blood-2009-02-206722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Paganin M, Grillo MF, Silvestri D, Scapinello G, Buldini B, Cazzaniga G, Biondi A, Valsecchi MG, Conter V, Te Kronnie G. The presence of mutated and deleted PTEN is associated with an increased risk of relapse in childhood T cell acute lymphoblastic leukaemia treated with AIEOP-BFM ALL protocols. Br J Haematol. 2018;182:705–711. doi: 10.1111/bjh.15449. [DOI] [PubMed] [Google Scholar]
- 98.Leslie NR, Downes CP. PTEN function: how normal cells control it and tumour cells lose it. Biochem J. 2004;382:1–11. doi: 10.1042/BJ20040825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li D, Ilnytskyy Y, Kovalchuk A, Khachigian LM, Bronson RT, Wang B, Kovalchuk O. Crucial role for early growth response-1 in the transcriptional regulation of miR-20b in breast cancer. Oncotarget. 2013;4:1373–1387. doi: 10.18632/oncotarget.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhu J, Chen L, Zou L, Yang P, Wu R, Mao Y, Zhou H, Li R, Wang K, Wang W. MiR-20b, -21, and -130b inhibit PTEN expression resulting in B7-H1 over-expression in advanced colorectal cancer. Hum Immunol. 2014;75:348–353. doi: 10.1016/j.humimm.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 101.Aylon Y, Michael D, Shmueli A, Yabuta N, Nojima H, Oren M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev. 2006;20:2687–2700. doi: 10.1101/gad.1447006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tranchant R, Quetel L, Tallet A, Meiller C, Renier A, de Koning L, de Reynies A, Le Pimpec-Barthes F, Zucman-Rossi J, Jaurand MC. Co-occurring Mutations of Tumor Suppressor Genes. Clin Cancer Res. 2017;23:3191–3202. doi: 10.1158/1078-0432.CCR-16-1971. [DOI] [PubMed] [Google Scholar]
- 103.Takahashi Y, Miyoshi Y, Takahata C, Irahara N, Taguchi T, Tamaki Y, Noguchi S. Down-regulation of LATS1 and LATS2 mRNA expression by promoter hypermethylation and its association with biologically aggressive phenotype in human breast cancers. Clin Cancer Res. 2005;11:1380–1385. doi: 10.1158/1078-0432.CCR-04-1773. [DOI] [PubMed] [Google Scholar]
- 104.Lee KH, Goan YG, Hsiao M, Lee CH, Jian SH, Lin JT, Chen YL, Lu PJ. MicroRNA-373 (miR-373) post-transcriptionally regulates large tumor suppressor, homolog 2 (LATS2) and stimulates proliferation in human esophageal cancer. Exp Cell Res. 2009;315:2529–2538. doi: 10.1016/j.yexcr.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 105.Jiménez-Velasco A, Román-Gómez J, Agirre X, Barrios M, Navarro G, Vázquez I, Prósper F, Torres A, Heiniger A. Downregulation of the large tumor suppressor 2 (LATS2/KPM) gene is associated with poor prognosis in acute lymphoblastic leukemia. Leukemia. 2005;19:2347–2350. doi: 10.1038/sj.leu.2403974. [DOI] [PubMed] [Google Scholar]
- 106.Hong S, Yu S, Li J, Yin Y, Liu Y, Zhang Q, Guan H, Li Y, Xiao H. MiR-20b Displays Tumor-Suppressor Functions in Papillary Thyroid Carcinoma by Regulating the MAPK/ERK Signaling Pathway. Thyroid. 2016;26:1733–1743. doi: 10.1089/thy.2015.0578. [DOI] [PubMed] [Google Scholar]
- 107.Kortum RL, Sommers CL, Alexander CP, Pinski JM, Li W, Grinberg A, Lee J, Love PE, Samelson LE. Targeted Sos1 deletion reveals its critical role in early T-cell development. Proc Natl Acad Sci U S A. 2011;108:12407–12412. doi: 10.1073/pnas.1104295108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ebinu JO, Stang SL, Teixeira C, Bottorff DA, Hooton J, Blumberg PM, Barry M, Bleakley RC, Ostergaard HL, Stone JC. RasGRP links T-cell receptor signaling to Ras. Blood. 2000;95:3199–3203. [PubMed] [Google Scholar]
- 109.Pierre S, Bats AS, Coumoul X. Understanding SOS (Son of Sevenless) Biochem Pharmacol. 2011;82:1049–1056. doi: 10.1016/j.bcp.2011.07.072. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures & Supplementary Data.
Supplementary Tables