

Summary
Reprogramming female mouse somatic cells into induced pluripotent stem cells (iPSCs) leads to X-chromosome reactivation. The extent to which increased X-chromosome dosage (X-dosage) in female iPSCs compared with male iPSCs leads to differences in the properties of iPSCs is still unclear. We show that chromatin accessibility in mouse iPSCs is modulated by X-dosage. Specific sets of transcriptional regulator motifs are enriched in chromatin with increased accessibility in XX or XY iPSCs. The transcriptome, growth and pluripotency exit are also modulated by X-dosage in iPSCs. To understand how increased X-dosage modulates the properties of mouse pluripotent stem cells, we used heterozygous deletions of the X-linked gene Dusp9. We show that X-dosage regulates the transcriptome, open chromatin landscape, growth, and pluripotency exit largely independently of global DNA methylation. Our results provide insights into how gene dosage modulates the epigenetic and genetic mechanisms that regulate cell identity.
Keywords: X-chromosome reactivation, mouse pluripotent stem cells, pluripotency, DNA methylation, chromatin accessibility, X-chromosome inactivation, X dosage, iPS cells, dosage compensation
Graphical Abstract
Highlights
-
•
X-chromosome dosage modulates the pluripotent chromatin accessibility landscape
-
•
Increased X-chromosome dosage slows down growth
-
•
Dusp9 heterozygous female ESCs display pluripotency exit delay
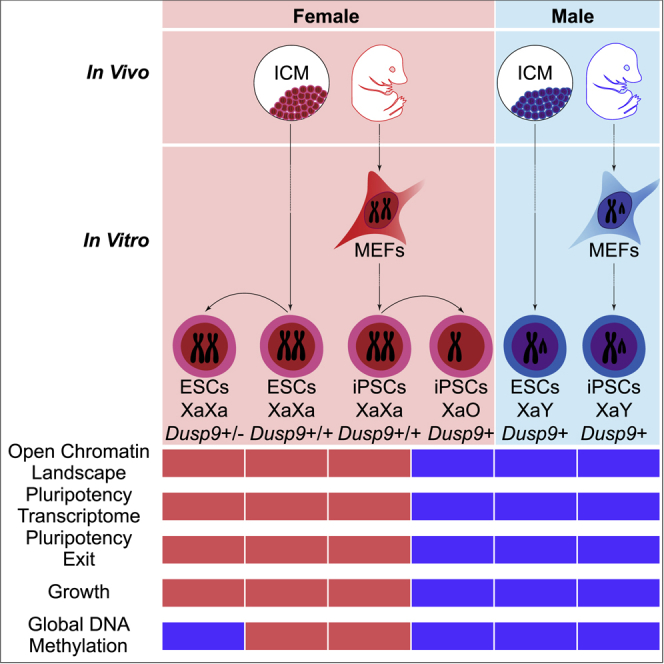
Female mouse pluripotent stem cells have two active X chromosomes while male cells have only one. Using genome-wide transcription and open chromatin analyses, Pasque and colleagues show that female and male iPSCs adopt differences in transcription, open chromatin landscape, and cellular growth. These differences can be uncoupled from global DNA hypomethylation in female ESCs through Dusp9 heterozygous deletion.
Introduction
Pluripotent stem cells (PSCs) are important for modeling development and diseases and for the design of future regenerative medicine approaches (Avior et al., 2016). A key question in the field is which mechanisms underlie the establishment and maintenance of pluripotency. Somatic cells can be reprogrammed into induced PSCs (iPSCs) by transcription factor (TF) overexpression (Takahashi and Yamanaka, 2006), and mouse embryonic stem cells (ESCs) can be derived directly from early embryos. Both cell types have the capacity to self-renew and maintain embryonic lineage differentiation potential in culture (Ying et al., 2008). It is of outstanding interest to understand which epigenetic and genetic mechanisms influence the molecular and functional properties of PSCs.
Several mammalian species including mice and human have adopted X-chromosome inactivation (XCI) as a means to compensate between the genetic imbalance of XX female and XY male cells. XCI is established during early embryogenesis following the expression of the long non-coding RNA Xist, and maintained in most somatic cells. Female cells undergo X-chromosome reactivation (XCR) in the mouse inner cell mass (ICM) resulting in two active X chromosomes (XaXa), a state maintained in female ESCs (Mak, 2004, Okamoto et al., 2004). XCR is also induced following somatic cell reprogramming to iPSCs, reviewed in Pasque and Plath (2015). XaXa is a hallmark of mouse naive pluripotency, the latter being characterized by unbiased embryonic lineage differentiation potential. Consequently, XX mouse ESCs have a higher dose of X-linked gene transcripts and hence an increased X-to-autosome gene expression ratio compared with XY cells. Increasing evidence suggests that the presence of XaXa can modulate the molecular and functional properties of mammalian PSCs (Bruck et al., 2013, Choi et al., 2017a, Choi et al., 2017b, Habibi et al., 2013, Hackett et al., 2013, Ooi et al., 2010, Ronen and Benvenisty, 2014, Schulz et al., 2014, Shirane et al., 2016, Yagi et al., 2017, Zvetkova et al., 2005). Work over the past decade showed that XX female ESCs (XX ESCs) exhibit global DNA hypomethylation affecting most genomic features including imprint control regions. More recent work showed that XX female iPSCs (XX iPSCs) also display global hypomethylation (Milagre et al., 2017, Pasque et al., 2018). Differences in global DNA methylation have been attributed to X-chromosome dosage (X-dosage), since female XO cells display male-like DNA methylation levels. Thus, mouse ESCs and iPSCs both show global DNA methylation differences due to X-dosage.
It was also discovered that XX ESCs show increased expression of several pluripotency-associated genes and display delayed pluripotency exit, suggesting that naive pluripotency features are promoted in XX ESCs (Schulz et al., 2014). Differences in transcription have also been attributed to X-dosage, since XO female ESCs (XO ESCs), or Xist-induced XCI, are associated with male-like pluripotency-associated gene expression and pluripotency exit. Despite the potential influence of X-dosage on iPSCs, X-dosage has been largely ignored in iPSC reprogramming studies so far, and it remains unclear whether X-dosage influences the molecular features of iPSCs beyond DNA methylation. Therefore, it is important to determine the potential influence of X-dosage on the molecular and cellular properties of iPSCs, which could influence mechanistic studies of reprogramming. A systematic comparison of transcriptional states, open chromatin landscapes, growth, and pluripotency exit in XX female and XY male mouse iPSCs has not yet been performed.
While several advances have been made, the molecular pathways by which XaXa modulate pluripotency remain incompletely understood (Schulz, 2017). At the mechanistic level, XaXa inhibit MAPK (mitogen-activated protein kinase) and GSK3 (glycogen synthase kinase 3) signaling (Bruck et al., 2013, Schulz et al., 2014), and global DNA hypomethylation has been attributed to reduced expression of DNMT3A and DNMT3B (Zvetkova et al., 2005), or DNMT3L (Ooi et al., 2010), or UHRF1 (Choi et al., 2017a, Milagre et al., 2017, Yagi et al., 2017) in XX ESCs/iPSCs. More recently, it was discovered that increased dosage of the X-linked MAPK inhibitor Dusp9 (dual-specificity phosphatase 9) is in part responsible for inhibiting DNMT3A/B/L and global DNA methylation in XX ESCs (Choi et al., 2017a). The expression level of Dusp9 is higher in XX ESCs than in XY ESCs, and overexpression of Dusp9 in XY ESCs induced female-like global DNA hypomethylation and a female-like proteome. Conversely, heterozygous deletion of Dusp9 in XX ESCs restored male-like global DNA methylation, suggesting that Dusp9 is responsible for MAPK-mediated DNMT3A/B repression. However, whether Dusp9 heterozygous deletion in XX ESCs has effects on the transcriptional regulatory network, open chromatin landscape, and pluripotency exit has not yet been explored. In addition, how and which X-linked genes modulate the pluripotency gene network of naive PSCs remains unclear. Furthermore, novel insights may be gained by identification of cis-regulatory elements that drive X-dosage-specific PSC states.
Here, to investigate the influence of X-dosage on iPSCs, we systematically compared multiple molecular and cellular properties of mouse XX and XY iPSCs at different passages. We found that X-dosage is associated with differences in chromatin accessibility, cell growth, the transcriptome, and pluripotency exit in early-passage iPSCs, which are subsequently resolved as a result of X-chromosome loss in female iPSCs upon prolonged culture. We further investigated the regulatory landscape of XX and XY iPSCs and ESCs. We found that thousands of chromatin regions differ in accessibility between XX and XY iPSCs. Motif discovery analysis identified that chromatin more accessible in XX iPSCs is enriched for binding sites of key pluripotency regulators including KLF/ZIC3/NANOG, suggesting stabilization of the naive pluripotency regulatory network via these regulators. By contrast, chromatin sites more accessible in XY iPSCs are enriched for activator protein 1 (AP-1) motifs, downstream effectors of signaling pathways including MAPK. We also show that XY iPSCs grow faster than XX iPSCs, irrespective of culture conditions. We further demonstrate that Dusp9 heterozygous XX ESCs maintain female-like chromatin accessibility, growth, and delayed exit from pluripotency in the presence of male-like global DNA methylation. Altogether, our study uncovers X-dosage as a previously unrecognized modulator of chromatin accessibility and of growth in PSCs. Our results clarify the effects of X-dosage on the pluripotency transcriptome, revealing the uncoupling of DNA methylation from chromatin accessibility. This provides principles for using gene dosage in designing experiments to understand the epigenetic and genetic mechanisms regulating cell identity.
Results
Differences in Transcriptional Landscapes and Pluripotency Exit Correlate with the Presence of XaXa in iPSCs
To explore the importance of X-dosage on the transcriptome and pluripotency exit of mouse iPSCs, we derived XX and XY iPSC lines. We used isogenic mouse embryonic fibroblasts (MEFs) carrying a tetO inducible transgene encoding the reprogramming factors Oct4, Sox2, Klf4, and c-Myc in the Col1A locus and the reverse tetracycline transactivator (M2rtTA) in the Rosa26 locus (Figure 1A and Table S1) (Carey et al., 2010, Pasque et al., 2018). After 16 days of doxycycline (dox) treatment to induce reprogramming, 10 female and 11 male iPSC lines were expanded on feeders in the presence of serum and leukemia inhibitory factor (LIF) (S/L) in the absence of dox (Figure 1A), or adapted to dual ERK/GSK3 inhibition and LIF conditions (2i/L). This scheme allowed us to compare female and male iPSCs without the influence of differences in genetic background, reprogramming system, or derivation method. Both female and male iPSCs could be propagated over multiple passages while maintaining their morphology, indicative of self-renewal, and expressed pluripotency-associated factors NANOG and DPPA4 (Figures 1B, 1C, S1A, and S1B). As expected, the transcriptome of our iPSCs was similar to that of naive ESCs (Figure S1C). Thus, derivation of isogenic female and male iPSCs allowed us to systematically compare the transcriptome and epigenome of these cells.
Figure 1.

Two X chromosomes Modulate the Transcriptome, Cellular Growth, and Pluripotency Exit in Mouse iPSCs
(A) Scheme of female and male iPSCs derivation, characterization, and differentiation.
(B) Representative images of female and male iPSCs/ESCs grown on feeders in S/L. Scale bar, 50 μm.
(C) Immunofluorescence analysis for NANOG/DPPA4 in iPSCs grown in S/L. Representative images of all lines examined for NANOG (red), DPPA4 (green), and DAPI (blue, nuclei counterstaining) are shown. Scale bar, 50 μm.
(D) (i) Mean expression ratio to autosomes for sex chromosomes and chromosomes 8 and 9. The dosage of X- and Y-linked genes was used to infer XX, XY, XO, and partial XO (pXO) genotypes. (ii) Representative karyotype images of XX and XO iPSC lines grown in S/L.
(E) Unsupervised hierarchical clustering of top 200 most variable autosomal genes in XY, XX, pXO, and XO iPSCs. Early-passage iPSCs cluster by X-dosage, late-passage iPSCs do not.
(F) DEG analysis, identifying clear differences between XX and XY iPSCs, but not XO and XY iPSCs (log2fold ≥ log21.5, FDR ≤ 0.05).
(G) Western blot analysis for NANOG, DNMT3B, and DUSP9 protein in iPSCs grown in S/L. Lower panel: quantification using actin as a loading control. Statistical significance was analyzed using unpaired two-tailed t test. P6, 6 XY versus 6 XX iPSC lines. P13, 3 XY versus 3 XO iPSC lines (n = 1).
(H) qRT-PCR analysis for pluripotency-associated gene expression during LIF withdrawal differentiation of both early- and late-passage iPSCs. Two-way repeated-measures ANOVA with Bonferroni post tests. P8, 3 XX versus 3 XY iPSC lines. P14, 3 XO versus 3 XY iPSC lines (n = 1).
(I) Western blot analysis for NANOG during pluripotency exit. The time after LIF withdrawal is indicated (n = 1).
(J) Two XX and 2 XY iPSC lines were subject to 0, 24, 48, and 72 hr of LIF withdrawal before replating 5,000 cells/well on feeders in 12-well plates. After 5 days in 2i/L, (i) representative images of alkaline phosphatase-positive (AP+) colonies for replated XX and XY iPSCs are shown (scale bar, 5,000 μm) and (ii) the number of AP+ colonies is indicated. Results are presented as averages (±SEM) of triplicates for each cell line (n = 1). One-way ANOVA with Tukey's multiple comparisons test.
(K) Growth curves and doubling times of XX and XY iPSCs (i) and ESCs (ii) in S/L condition. Cells were counted at the indicated time points and presented as fold changes relative to day 0. P6, 3 XY versus 3 XX iPSC lines (n = 1, left panel); 1 XY versus 1 XX ESC line (n = 3, right panel). Growth curve: two-way repeated-measures ANOVA with Bonferroni post tests. Doubling time (Td): unpaired two-tailed t test.
(L) As in (K) but for XY, XX, and XO iPSCs (three lines each, n = 1).
Growth curve: two-way repeated-measures ANOVA with Bonferroni post tests. Td: unpaired two-tailed t test.
P6 XX versus P6 XY iPSCs: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; P6 XX versus P13 XO iPSCs: #p < 0.05, ##p < 0.01, ###p < 0.001.
See also Figure S1.
First, we confirmed that XX iPSCs reactivated the inactive X chromosome, a hallmark of naive pluripotency (De Los Angeles et al., 2015), using RNA sequencing (RNA-seq) analysis (Figure 1D). These results were also in agreement with an independent single-cell level assay using RNA in situ hybridization for X-linked gene Tsix (Figure S1D; Pasque et al., 2014). XX ESCs are prone to lose one of the two Xas upon extended in vitro culture (Choi et al., 2017b, Yagi et al., 2017, Zvetkova et al., 2005), and we recently showed that early-passage XX iPSCs are XaXa and become XO iPSCs upon passage (Pasque et al., 2018). To infer X-chromosome loss in our iPSC lines, we measured the average X-chromosome-to-autosome gene expression ratio using RNA-seq (Figure 1D). We found that early-passage XX iPSCs had increased X-dosage, in agreement with the XaXa state of female iPSCs (Maherali et al., 2007). However, female iPSCs at late passage showed reduced X-dosage, consistent with X-chromosome loss, and we termed these XO iPSCs. In addition, we found that X-chromosome loss in female iPSC lines displayed clonal variability. One early-passage and one late-passage female iPSC line showed partial X-dosage, consistent with partial X-chromosome loss, which we termed partial XO (pXO) iPSCs. In further support of our finding that XX iPSCs undergo X-chromosome loss rather than XCI, we designed a simple qPCR assay to determine the X/autosome genomic DNA ratio by measuring four X-linked genes (Tfe3, Bcor, Pdha1, and Mid1, located on either distal region on the X chromosome) and the autosomal gene Gapdh. We confirmed that late-passage iPSCs were XO (Figure S1E). Karyotype analyses corroborated these results (Figures 1Dii and S1F). These observations are consistent with XCR during reprogramming followed by X-chromosome loss in female iPSCs.
Using RNA-seq of XX, XY, and XO iPSCs grown in S/L, we asked whether the transcriptome of iPSCs is influenced by X-dosage. Unsupervised clustering of the top 200 most variable autosomal genes, or genes associated with stem cell maintenance, distinguished early-passage XX and XY iPSCs (Figures 1E and S1G). However, XO and XY late-passage iPSCs could not be distinguished, indicating that X-dosage rather than sex modulates the transcriptome of iPSCs. Furthermore, gene expression analysis identified 288 differentially expressed genes (DEGs) between XX and XY iPSCs, but only 4 DEGs between XO and XY iPSCs (Figure 1F, 1.5-fold, false discovery rate [FDR] = 0.05, Table S2). Using qRT-PCR, we found that in S/L, XX iPSC lines consistently expressed higher levels of pluripotency-associated genes Prdm14, Nanog, and Tcl1 compared with XY iPSCs (Figure S1H). Western blot analysis showed that XX iPSCs had increased NANOG protein levels compared with XY and XO iPSCs (Figure 1G). These marked differences between XX and XY iPSCs are consistent with patterns observed in mouse ESCs (Choi et al., 2017a, Schulz et al., 2014) (Figures S1I and S1J; Table S2), and in agreement with the notion that iPSCs are molecularly equivalent to ESCs. Despite this, X-dosage has been largely ignored in mechanistic iPSC reprogramming studies so far. Importantly, differences between XX and XY iPSCs cannot be attributed to differences in genetic background, since these differences were found when comparing cells of the same genetic background. Thus, reprogramming to iPSCs results in differences in the transcriptome of iPSCs, some of which can be attributed to differences in X-dosage.
Next, we investigated the extent to which X-dosage affects exit from pluripotency in iPSCs. We subjected XX, XY, and XO iPSCs to LIF withdrawal-mediated differentiation and measured the downregulation of pluripotency-associated genes by qRT-PCR (Figures 1A and 1H). Exit from pluripotency was delayed in XX iPSCs for Prdm14, Nanog, and Tcl1, but not XY and XO iPSCs (Figure 1H). We confirmed these results using an alternative differentiation protocol that mimics epiblast differentiation (Figures S1K and S1L) (Guo et al., 2009, Schulz et al., 2014), and also at the protein level (Figure 1I). These differences had functional consequences on pluripotency exit: replating an equal number of XX or XY cells before and after LIF withdrawal followed by 2i/L culture confirmed delayed pluripotency exit in XX cells (Figure 1J). Thus, XX iPSCs functionally exit pluripotency with delayed kinetics compared with XY and XO iPSCs. Altogether, these findings show that early-passage iPSCs display previously unrecognized X-dosage specific behavior in transcriptome, including pluripotency gene expression, and in pluripotency exit kinetics, consistent with X-dosage differences in ESCs (Schulz et al., 2014).
X-Dosage Modulates Cellular Growth in Mouse iPSCs and ESCs
To determine the effect of X-dosage on cell growth, we counted the number of XX and XY iPSCs over 2 days starting from the same amount of cells. We found that XX iPSC lines grew slower than XY iPSCs, with a doubling time (Td) extended by ∼3.4 hr compared with XY iPSCs grown in S/L (Td XX iPSCs = 18.4.±1.5 hr versus Td XY iPSCs = 15.0 ± 0.9 hr) (Figure 1K). XX ESCs also grew slower than XY ESCs (Figure 1K). The delayed growth of XX iPSCs was attributed to the presence of XaXa, since XO iPSCs behaved like XY iPSCs (Figure 1L). The differences in growth of XX and XY iPSCs and ESCs did not depend on culture conditions because XX ESCs and iPSCs still grew slower than XY cells in 2i/LIF (Figure S1M). XX female mouse and human embryos show a delay in post-implantation development that has been attributed to the presence of two X chromosomes in female cells (Burgoyne et al., 1995). Our observations support the idea that the growth delay of XX female mammalian embryos is recapitulated in vitro in iPSC and ESC cultures, providing a platform to study this process.
To assess the effect of X-dosage on the cell cycle, we used EdU (5-ethynyl-2′-deoxyuridine) incorporation and propidium iodide staining in combination with flow cytometry to determine the distribution of cells over the different phases of the cell cycle. We found that the majority of both XX and XY iPSCs or ESCs reside in S phase, in line with the literature. The proportion of XX iPSCs and ESCs in S phase was larger than that of XY iPSCs and ESCs, whereas the number of XX iPSCs and ESCs in the G1 phase was smaller than that of XY cells (Figure S1Ni). To further validate these results, we introduced a fluorescence ubiquitination cell-cycle indicator (FUCCI) into XX and XY ESCs. This system provides for direct fluorescent readout of ESCs in G1 phase, G1/S transition, or S/G2/M phase (Sakaue-Sawano et al., 2008). This analysis confirmed, for XX ESCs, an increase in the proportion of cells in S phase, and a reduced proportion of XX cells in G1 phase, compared with XY ESCs (Figure S1Nii).
What might be the functional relevance of differences in cell growth between cells with one or two Xas? It has been suggested that the presence of two X chromosomes slows down development to ensure that cells progress through XCI (Schulz et al., 2014). We sought to test, in vitro, the hypothesis that reduced X-dosage provides a competitive growth advantage to cells that have undergone XCI. We mixed XX ESCs and GFP-labeled XY ESCs in different ratios and followed the proportion of labeled cells over time. We found that the increased cell growth of XY ESCs can provide a small advantage over an 8-day period (Figure S1O). Collectively, these observations support the idea that cell growth is decreased as a result of increased X-dosage in pluripotent cells in vitro and in vivo.
Influence of X-Dosage on Chromatin Accessibility Landscape in iPSCs
To assess how X-dosage differentially primes mouse PSCs for rapid exit from pluripotency and to identify additional candidate regulators, we set out to globally define the open chromatin landscape of XX, XY, and XO iPSCs. We employed an assay for transposase-accessible chromatin (omniATAC-seq) to profile genome-wide chromatin accessibility with high resolution (Corces et al., 2017). We generated ATAC-seq datasets from isogenic XX, XY, and XO iPSC lines (and XX/XY ESCs), allowing us to define the open chromatin regions and the enrichment for TF binding motifs associated with open chromatin landscapes (Figure 2 and Table S3). As expected, we observed open chromatin peaks at transcription start site proximal and distal genomic regions, suggesting enrichment in cis-regulatory sequences (Figure S2A). We also used the mean read count ratio to autosome to infer the XX, XO, and XY state of the cells (Figure S2B). We then compared autosomal chromatin accessibility globally, and found that X-dosage affects the chromatin accessibility landscape of iPSCs. Broadly, we observed a correlation between the number of Xas and open chromatin landscapes (Figure 2A). We then assessed differential accessibility between XX and XY iPSCs, and between XO and XY iPSCs. We found that most open chromatin regions were shared between XX and XY iPSCs, suggesting that XX and XY iPSCs globally display similar open chromatin landscapes. However, thousands of chromatin regions showed increased accessibility in XX or in XY iPSCs, but not between XO and XY iPSCs (>2-fold, FDR < 0.05, Figure 2B). These results further support the idea that X-dosage influences chromatin accessibility in iPSCs. We identified 2,819 and 2,363 autosomal chromatin regions that are more open in XX iPSCs or in XY iPSCs, respectively (Figure 2B, defined as “XX gain” and “XY gain” regions, Table S4), which represent differences in chromatin accessibility driven by X-dosage. We also found a strong correlation between X-dosage and open chromatin landscapes in isogenic ESCs isolated from another genetic background (Figures S2C and S2D; Table S3). In summary, these results indicate that the chromatin landscape of XX and XY iPSCs is globally similar, but also contains differentially accessible chromatin at thousands of specific genomic regions, due to differences in X-dosage.
Figure 2.

X-Dosage Modulates the Chromatin Regulatory Landscape of Mouse iPSCs
(A) ATAC-seq sample-to-sample distance heatmap showing the Euclidean distances (calculated from the rlog transformed counts, DESeq2) between iPSC samples. (i) XX versus XY iPSCs, chromatin landscapes cluster by X-dosage; (ii) XO versus XY iPSCs, the analysis cannot distinguish XO versus XY landscapes.
(B) Differential chromatin accessibility analysis between XX (or XO) and XY iPSCs. Log2 fold change (XX/XY or XO/XY) in reads per accessible region are plotted against the mean reads per ATAC-seq peak. Thousands of open chromatin regions are more open in XX iPSCs or in XY iPSCs (i), but not between XO and XY iPSCs (ii) (log2fold ≥ 1, FDR ≤ 0.05). These regions were defined as “XX gain” and “XY gain,” respectively.
(C) Venn diagrams showing the overlap between genes nearest to the “XX gain” regions and the DEGs between XX and XY iPSCs (DEGs defined in Figure 1F).
(D) Integrated genome viewer track images of ATAC-seq signal for “XX gain” example regions in all iPSCs samples. Differentially open regions are shaded.
(E) Expression of Pramel6, Pramel7, and Nlrp4c in XX, XY, and XO iPSCs as assessed by RNA-seq. CPM (counts per million) values were plotted for each gene. One-way ANOVA with Sidak's multiple comparisons test: P7 2 XX versus 3 XY iPSC lines, P13 2 XO versus 3 XY iPSC lines; ∗∗p < 0.01, ∗∗∗p < 0.001.
(F) as in (C) for “XY gain” regions.
(G) As in (D) for “XY gain” regions.
(H) as in (E) for Pou2f3 and Bmi1.
See also Figure S2.
We next assessed the correlation between differentially open chromatin and gene expression. Broadly, we observed a weak correlation between changes in chromatin accessibility and changes in gene expression (Figures 2C–2H). Most differentially open chromatin regions did not associate with DEGs (2,131/2,175 genes for XX gain regions, 1,825/1,850 genes for XY gain regions, Figures 2C and 2F). Likewise, most DEGs did not associate with differentially accessible chromatin regions (120/164 and 139/164 DEGs were not associated with changes in chromatin accessibility in XX or in XY iPSCs, respectively, Figures 2C and 2F). Nevertheless, a small fraction of differentially open chromatin regions associated with DEGs (Figures 2C and 2F; Table S3). We identified 44 genes out of 164 autosomal DEGs that associated with chromatin regions more open in XX iPSCs. Most of these genes (86%, 38/44) were transcriptionally upregulated in XX iPSCs cells (Figure 2C). For example, there were chromatin regions more accessible in XX iPSCs that associated with pluripotency-associated genes Pramel6 and Pramel7, both of which were upregulated in XX iPSCs but not in XO iPSCs (Figures 2C–2E and Table S3). Overexpression of Pramel6 and Pramel7 was found to oppose exit from pluripotency (Casanova et al., 2011) and Pramel7 was shown to mediate ground-state pluripotency (Graf et al., 2017). We also observed increased accessibility in the vicinity of Nlrp4c, Nupr1, and Tcl1 in XX iPSCs, but not XO iPSCs. These results indicate that the open chromatin landscape of iPSCs reflects specific cellular states, whereby XX-specific open chromatin could mediate stabilization of pluripotency in XX iPSCs and ESCs.
Chromatin regions more accessible in XY iPSCs associated with multiple genes involved in embryonic development and morphogenesis (several Hoxb genes, Pax2, Pax7, Krt8, Krt18, Sox1, Sox11, and Gata4, Table S3). Twenty-five genes associated with chromatin regions more open in XY iPSCs, 72% of which (18/25) were upregulated in XY iPSCs (Figure 2F). Examples of upregulated genes include Pou2f3 and Pbx1 (Figures 2G and 2H). Within the 25 DEGs associated with XY gain chromatin regions in iPSCs, 7 genes were downregulated in XY iPSCs (Bmi1). In summary, these findings indicate that chromatin more open in XY or in XX iPSCs is associated with several lineage specification/differentiation-related and pluripotency-associated genes, respectively.
Motif Analysis Reveals Potential Regulators of X-Dosage-Mediated Cell States
To identify TFs involved in modulating iPSCs as a result of differences in X-dosage, we searched for known TF motifs enriched in chromatin more open in XX or in XY iPSCs. Motif enrichment analysis of chromatin regions more open in XX iPSCs revealed a strong enrichment for the binding motif of TFs such as regulatory factor X (RFX) (10%), KLF5 (48.14%), ZIC3 (26.32%), and NANOG (26.92%) (Figure 3A). RFX proteins encode TFs expressed in many tissues including brain and testes (Choksi et al., 2014). KLF5 and NANOG have been functionally implicated in ESC self-renewal (reviewed in Martello and Smith, 2014). ZIC3 is a pluripotency-associated factor required to maintain pluripotency (Lim et al., 2010). Interestingly, Zic3 is located on the X chromosome, raising the possibility that ZIC3 dosage could drive X-linked driven stabilization of pluripotency in XX iPSCs (see below). In summary, several top TF motifs enriched in chromatin with increased accessibility in XX iPSCs belong to pluripotency-associated factors, suggesting that the identified pluripotency-associated TFs participate in stabilizing the pluripotency transcriptional regulatory network of XX iPSCs.
Figure 3.

Identification of Candidate Regulators Mediating the Effects of X-Dosage on Open Chromatin
(A–C) TF motifs enriched in chromatin regions more open in XX or XY iPSCs.
(D) Venn diagram showing the overlap between genes associated the ATAC-seq regions more open in XX iPSCs that contain a motif for KLF5, RFX, or ZIC. The number of genes associated with all three motifs is indicated.
See also Figure S2.
By contrast, pluripotency-associated TF motifs were less represented from the top motifs enriched in chromatin with increased accessibility in XY iPSCs, with a few exceptions (Figures 3B and 3C). Instead, within chromatin more open in XY iPSCs, motif enrichment analysis revealed binding motifs of multiple TFs of the AP-1 family such as JUN/AP-1 (39.27%), FOSL2 (43.76%), and ATF3 (51.08%) (Figures 3B and 3C). JUN/AP-1 is a transcriptional activator complex involved in regulating many processes (Shaulian, 2010) including cell growth and differentiation in response to a variety of stimuli including the MAPK pathway (Karin, 1995, Yang et al., 2012). FOSL2 is a member of the AP-1 complex (Shaulian, 2010). Collectively, these findings reveal that X-dosage modulates chromatin accessibility in iPSCs. As expected, we made similar observations in ESCs (Figures S2C–S2I). In addition, open chromatin regions that are common between XX and XY iPSCs still showed enrichment of pluripotency-related TFs (Figures S2I and S2J). We propose that the differential enrichment of TF binding sites in open chromatin regions modulated by X-dosage provides a molecular link between transcriptional regulators, stabilization of pluripotency in XX PSCs, and rapid exit from pluripotency in XY PSCs.
To identify the putative target genes, we searched for genes associated with open chromatin regions enriched for specific motifs, then determined the target genes shared for open chromatin containing more than one motif. In chromatin more open in XX iPSCs, we found that 67 genes were associated with binding motifs for all three TF motifs RFX, KLF, and ZIC (Figure 3D). Taken together, these analyses allowed the identification of TFs that regulate a large number of cis-regulatory regions, thereby improving our understanding on how X-dosage can drive two distinct PSC states.
Zic3 and Tfe3 Dosage Do Not Explain X-Dosage Differences in Transcription and Pluripotency Exit
We sought to test whether X-linked pluripotency-associated genes with enriched motifs identified in chromatin more open in XX iPSCs stabilize pluripotency in XX PSCs. Our motif discovery analysis identified the X-linked gene Zic3 within the top motifs enriched in chromatin more open in XX iPSCs and ESCs (Figures 3A and S2F). Western blot analysis showed that XX iPSCs and ESCs express higher ZIC3 protein than XY iPSCs and ESCs (Figure 4A). In addition, the increased Zic3 transcript levels of XX iPSCs were restored to XY levels in XO iPSCs (Figure S3A). Moreover, Zic3 was reported to prevent endodermal lineage specification and to act as a transcriptional activator of Nanog expression (Lim et al., 2010), further suggesting that it could have a role in stabilizing pluripotency in XX ESCs. To test the hypothesis that increased Zic3 dosage stabilizes pluripotency in XX ESCs, we overexpressed Zic3 in XY iPSCs and asked whether it induced XX-like features (Figure S3B). We achieved 3-fold overexpression of ZIC3 protein tagged in N- or C-terminal with hemagglutinin (HA) (Figure S3C). We then subjected the cells to LIF withdrawal. We found that overexpression of Zic3 with an N-terminal HA tag, but not with the C-terminal HA tag, delayed pluripotency exit during LIF withdrawal differentiation (Figures S3B–S3E). These results suggested that increased Zic3 dosage might be responsible for the pluripotency exit delay of XX PSCs. Using an independent approach, we generated Zic3 heterozygous deletions in XX ESCs to reduce Zic3 dosage, which is a more stringent method to test whether Zic3 dosage stabilizes pluripotency in XX ESCs (Figures 4B–4D, S3F, and S3G). Two independent Zic3+/− XX ESC clones maintained XX-like expression of Prdm14, Nanog, and Tcl1 and also maintained female-like delayed exit from pluripotency (Figure 4E). We performed similar experiments for another additional X-linked gene involved in pluripotency, Tfe3, and obtained similar results as for Zic3 (Figures 4F–4H and S3H–S3J). These results support the idea that the dosage of Zic3 and Tfe3 does not explain the differences in pluripotency gene expression and exit from pluripotency between XX and XY ESCs.
Figure 4.

Effects of Zic3/Tfe3 Heterozygous Deletion on Pluripotency Exit
(A) Western blot analysis for ZIC3 in iPSCs and ESCs grown in S/L. Right: quantification using actin as loading control. Three XY versus 3 XX iPSC lines, and 1 XY versus 1 XX ESC line. ∗∗p < 0.01, unpaired two-tailed t test. Data from a representative experiment from at least three independent experiments.
(B) Scheme of heterozygous Zic3 deletion strategy in XX ESCs followed by LIF withdrawal.
(C) Western blot analysis for ZIC3 in two independent Zic3+/− ESC lines, Zic3+/+ ESCs and XY ESCs all grown in S/L (n = 1). KO, knockout; WT, wild-type.
(D) qPCR analysis for X-chromosome DNA copy number. X copy numbers are presented as the average ratio of genomic DNA (gDNA) quantities for four X-linked genes (Tfe3, Bcor, Pdha1, and Mid1) to gDNA quantities for autosomal gene Gapdh (n = 2).
(E) qRT-PCR analysis for pluripotency-associated gene expression during LIF withdrawal in the two independent Zic3+/− XX ESC lines, the Zic3+/+ XX parental ESC line, and an XY ESC line (n = 2).
(F) Scheme of heterozygous Tfe3 deletion strategy in XX ESCs followed by LIF withdrawal.
(G) qRT-PCR analysis for Tfe3 expression in the Tfe3+/− XX ESC line, the Tfe3+/+ XX parental ESC line, and an XY ESC line (n = 1).
(H) As in (E) for Tfe3+/− ESCs.
Results are presented as averages (±SEM) of biological duplicates (n = 1). See also Figures S3 and S4.
Dkc1, Otud6a, Fhl1, Zfp185, and Scml2 Dosage Do Not Explain X-Dosage-Specific Differences in Pluripotency Exit
We sought to find the X-linked regulators that drive stabilization of pluripotency in XX PSCs. We analyzed RNA-seq and published proteomics data of XX and XY ESCs (Choi et al., 2017a). We selected X-linked candidate factors with (1) increased expression in XX ESCs over XY or XO ESCs and ranked by expression ratio, (2) evidence that the genes are subject to XCI (Table S4), and (3) literature consistent with a possible role in stabilizing pluripotency. The selected candidate genes were Dkc1, Otud6a, Fhl1, Zfp185, and Scml2. We overexpressed their cDNAs in XY iPSCs (Figure S4). To test the effect of overexpression on pluripotency exit, we induced differentiation by LIF withdrawal and measured pluripotency gene expression at 24 and 48 hr. We found that overexpression of Dkc1, Otud6a, Fhl1, Zfp185, or Scml2 was not sufficient to induce a delay in pluripotency exit (Figure S4). Collectively, these findings do not support a significant role for these X-linked pluripotency-associated genes in stabilizing pluripotency in XX ESCs.
Heterozygous Dusp9 Deletion in XX ESCs Induces Male-like DNA Methylation yet Maintains a Female-like Transcriptome, Open Chromatin Landscape, Growth, and Delayed Pluripotency Exit
In an effort to understand the mechanisms by which X-dosage affects PSC properties, we generated Dusp9 heterozygous deletions in XX ESCs, resulting in two independent Dusp9+/− XX ESC clones (Figures 5A, 5B, S5A, and S5B). To ensure the maintenance of two Xas in Dusp9+/− ESCs, we used polymorphic Musculus/Castaneus (Mus/Cas) ESCs, known to be less susceptible to X-chromosome loss (Choi et al., 2017a, Lee and Lu, 1999). We confirmed that Dusp9+/− ESCs maintained two Xas (Figures S5C and S5D). Dusp9+/− XX ESCs showed male-like global DNA methylation (Figure S5E), corroborating recent findings (Choi et al., 2017a). To determine whether Dusp9+/− XX ESCs with male-like DNA methylation acquire male-like transcription, we analyzed the expression of stem cell maintenance-related genes using RNA-seq in Dusp9+/− XX ESCs, Dusp9+/+ XX ESCs, and Dusp9+ XY ESCs, all sharing a Mus/Cas background to exclude potential strain-specific effects. Principal component analysis (PCA) placed Dusp9+/− XX ESCs away from both Dusp9+/+ XX ESCs and Dusp9+ XY ESCs, indicating that Dusp9+/− XX ESCs do not adopt a male-like transcriptional state (Figure 5C). We corroborated this finding using unsupervised clustering of stem cell maintenance-related gene expression, whereby Dusp9+/− XX ESCs clustered together with Dusp9+/+ XX ESCs, and away from Dusp9+ XY ESCs (Figure 5D). Unsupervised clustering analysis also showed the activation of most MAPK target genes in Dusp9+/− XX ESCs, in agreement with the function of Dusp9 as an MAPK inhibitor (Figure 5E) (Li et al., 2012). Furthermore, we found more DEGs between Dusp9+/+ XX ESCs and XY ESCs, and less DEGs between Dusp9+/− XX ESCs and Dusp9+/+ XX ESCs, with little overlap between the two sets of DEGs (Figures 5F and 5G). The only exception was the Pramel7 gene, the expression of which was reduced to XY levels in Dusp9+/− XX ESCs, indicating that transcription of Pramel7 is influenced by Dusp9 dosage (Table S4). Overall these results indicate that male-like DNA methylation can be induced in the absence of male-like transcription in Dusp9+/− XX ESCs. These experiments raise the possibility that distinct molecular features modulated by X-dosage in ESCs might be controlled by different regulators.
Figure 5.

The Open Chromatin Landscape, Growth, and Pluripotency Exit Delay of XX ESCs Are Maintained in the Presence of Male-like Global DNA Methylation
(A) Scheme of Dusp9 heterozygous deletion in XX ESCs followed by LIF withdrawal differentiation. KO, knockout; WT, wild-type.
(B) (i) Western blot analysis for DUSP9 in Dusp9+/− ESCs, Dusp9+/+ ESCs, and XY ESCs grown in S/L. (ii) Quantification of DUSP9 levels using actin as a loading control (n = 2).
(C) PCA of stem cell maintenance genes (RNA-seq data) for the Dusp9+/−, Dusp9+/+, and XY ESCs grown in S/L conditions.
(D) Unsupervised hierarchical clustering of stem cell maintenance genes for the Dusp9+/−, Dusp9+/+, and XY ESCs.
(E) As in (D) for MAPK pathway-related genes (defined in Schulz et al., 2014).
(F) DEG analysis, identifying clear differences between Dusp9+/+ XX and XY ESCs, but much less between Dusp9+/− XX and Dusp9+/+ XX ESCs.
(G) Overlap between the DEGs of Dusp9+/+ versus XY ESCs and Dusp9+/− versus Dusp9+/+ ESCs for upregulated genes (up) and downregulated genes (down). Dusp9 heterozygous deletion maintains a female-like transcriptome.
(H) ATAC-seq sample-to-sample distance heatmap in Dusp9+/+, Dusp9+/−, and XY ESCs. Dusp9+/− ESCs maintain a Dusp9+/+-like open chromatin landscape.
(I) Differential chromatin accessibility analysis between Dusp9 heterozygous mutant and wild-type XX ESCs. Log2 fold change (mutant/wild-type) in reads per accessible region are plotted against the mean reads per ATAC-seq peak. Dusp9 heterozygous mutants maintain a female-like open chromatin landscape.
(J) Growth curves and doubling times of Dusp9+/−, Dusp9+/+, and XY ESCs in S/L condition. Cells were counted at the indicated time points and presented as fold changes relative to day 0, averages (±SEM) of biological duplicates. Data from a representative experiment from at least 4 independent experiments.
(K) qRT-PCR for Prdm14, Nanog, Tcl1, and Dusp9 expression before and after LIF withdrawal (n = 3).
See also Figure S5.
Next, we sought to determine whether chromatin accessibility is affected by reduced DUSP9 dosage in XX ESCs. ATAC-seq revealed that Dusp9+/− XX ESCs maintained a female-like open chromatin landscape (Figure 5H). We only observed very few differences in chromatin accessibility between Dusp9+/− XX ESCs and Dusp9+/+ XX ESCs (Figure 5I), while the same analysis identified thousands of regions differentially accessible in XX and XY ESCs (Figure S2D). This finding indicates that the effects of X-dosage on chromatin accessibility can be largely dissociated from X-dosage effects on global DNA methylation levels.
We then measured the growth of Dusp9+/− XX ESCs, and found that the cells grew as slowly as their parental Dusp9+/+ XX ESCs, both of which grew slower than XY ESCs (Figures 5J and 1K). Therefore, reducing Dusp9 dosage and inducing global DNA methylation in XX ESCs is not sufficient to induce male-like cellular growth. We propose that X-dosage-specific growth and global DNA methylation are regulated by different pathways in mouse PSCs.
To study the effects of Dusp9 heterozygous deletion in XX ESCs on pluripotency exit, we subjected Dusp9+/− XX ESCs, Dusp9+/+ XX ESCs, and XY ESCs to LIF withdrawal differentiation for 48 hr followed by qRT-PCR analysis. The delay in pluripotency exit as judged by Prdm14, Nanog, and Tcl1 expression was maintained in Dusp9+/− XX cells relative to Dusp9+/+ XX cells (Figure 5K). In further support of the finding that Dusp9+/− XX ESCs maintain a delay in pluripotency exit, RNA-seq analysis showed that multiple pluripotency-associated genes behaved similarly in Dusp9+/− XX and Dusp9+/+ XX cells undergoing differentiation (Figure S5F). Therefore, mechanistically, reducing Dusp9 dosage is compatible with female-like pluripotency exit. We conclude that reducing the dosage of Dusp9 in XX ESCs is not sufficient to induce a male-like transcriptome or accelerate pluripotency exit to a male-like state, despite changes in the expression level of multiple genes in the MAPK signaling pathway and despite male-like DNA methylation. In addition, Dusp9 overexpression in XY ESCs did not induce a female-like delay in differentiation (Figures S5G–S5L) despite inducing female-like global DNA hypomethylation (Choi et al., 2017a).
Altogether, these results indicate that most changes in open chromatin, growth, and pluripotency exit as a result of differences in X-dosage are regulated independently of global DNA methylation in XX ESCs. Hence, mechanistically, heterozygous Dusp9 deletion molecularly uncouples global DNA methylation from the open chromatin landscape, growth, and the pluripotency exit delay of XX ESCs. Importantly, this points toward the existence of other pathways and X-linked genes involved in mediating the effects of X-dosage in PSCs.
Large-Fragment Heterozygous Deletions
Two models emerged to explain delayed pluripotency exit in XX PSCs. In the first model, a single X-linked gene is responsible for delayed pluripotency exit. In the second model, multiple X-linked genes act together to delay pluripotency exit. To test these models, we generated a series of large-fragment (LF) heterozygous deletions of the X chromosome in XX ESCs, which were confirmed by genotyping and Sanger sequencing (Figure S6), then induced differentiation. There was a partial rescue of the pluripotency exit delay of XX ESCs in multiple, but not all, LF deletions (Figure 6 and Table S1). The partial rescue was gene specific and fragment specific. These results favor model 2, in which multiple X-linked genes participate in delayed pluripotency exit in XX PSCs.
Figure 6.

Multiple X-Linked Genes Modulate the Pluripotency Exit Delay of XX PSCs
(A–C) Scheme of large-fragment (LF) heterozygous deletions in XX ESCs followed by LIF withdrawal differentiation. (A) LF1, (B) LF2, (C) LF3. WT, wild-type.
(D–F) qRT-PCR for Prdm14, Nanog, and Tcl1 expression before and after LIF withdrawal in 2–3 independent LF+/− XX ESC lines, the parental XX ESC line, and an XY ESC line (n = 2). (D) LF1, (E) LF2, (F) LF3.
See also Figure S6.
Discussion
Induction of naive pluripotency during reprogramming to iPSCs and during in vivo development in ICM leads to XCR in murine female cells (Maherali et al., 2007, Mak, 2004, Okamoto et al., 2004). The consequences of X-dosage imbalance between female (XX) and male (XY) cells on mouse iPSCs, and the regulatory mechanisms at the basis of distinct X-dosage-specific features in mouse PSCs, remain incompletely understood. In this study, we addressed these questions by analyzing the transcriptome, growth properties, chromatin accessibility landscape, and pluripotency exit of isogenic female and male iPSCs. We identified X-dosage as a factor influencing the molecular and cellular properties of iPSCs. By employing epigenomic analyses we found that X-dosage modulates open chromatin in iPSCs and ESCs. Furthermore, using genome editing we found that modulation of the transcriptome, open chromatin, cell growth, and pluripotency exit by X-dosage is largely independent of global DNA methylation. We provide evidence favoring a model in which multiple X-linked genes delay pluripotency exit.
One outcome of our study is that the number of Xas correlates with differences in the transcriptome and in pluripotency exit in iPSCs, in addition to differences in DNA methylation (Milagre et al., 2017, Pasque et al., 2018). Reprogramming somatic cells to iPSCs is an important system for studying erasure of epigenetic memory and pluripotency. Sex does not appear to influence the efficiency of iPSC generation, since we previously showed that female and male cells reprogram with similar efficiencies in this system (Pasque et al., 2014). However, we have now established that the presence of two Xas, as a result of reprogramming to pluripotency in female cells, is associated with the slower growth of XX iPSCs, an altered transcriptome including increased pluripotency-associated gene expression, and delayed pluripotency exit. These differences are caused by changes in X-dosage, since XO iPSCs revert to an XY iPSC phenotype. The influence of X-dosage on the growth of mouse PSCs, and on the transcriptome and pluripotency exit of iPSCs, is consistent with studies in mouse ESCs (Schulz, 2017, Schulz et al., 2014, Zvetkova et al., 2005) and human ESCs (Bruck et al., 2013, Ronen and Benvenisty, 2014), and in post-implantation mammalian embryos (Schulz, 2017). However, X-dosage is currently largely ignored in most reprogramming studies, in which neglecting sex-specific differences could have a negative impact on interpretation of results. The notion that X-dosage influences the molecular and cellular properties of iPSCs is further supported by the loss of sex-specific differences concomitant with loss of one X chromosome in female iPSCs, in agreement with previous observations in ESCs (Choi et al., 2017a, Schulz et al., 2014, Zvetkova et al., 2005) and iPSCs (Pasque et al., 2018). The important point is that studies of reprogramming to iPSCs should consider the number of Xas as a modulator of the transcriptome, and cells of different sex should be studied separately, but also considered together.
The influence of X-dosage on the heterogeneity of ESCs also remained unclear. Our analysis of single-cell RNA-seq data for XX and XY ESCs in S/L and 2i/L (Chen et al., 2016) revealed that both XX and XY ESCs reside in a metastable state, with Nanog-high and Nanog-low cells (Figure S7). More XX ESCs express Tcl1 than XY ESCs, in agreement with increased pluripotency-associated gene expression in XX ESCs (Schulz et al., 2014). Differences between the transcriptome of XX and XY ESCs persist in 2i/L, despite more homogeneous pluripotency-associated gene expression. Altogether, this analysis reveals that XX PSCs in S/L maintain a metastable state with a bias toward increased expression of specific pluripotency-associated genes, whereas transcriptomic differences between XX and XY PSCs persist in 2i/L.
Here we show that the presence of two Xas in iPSCs and ESCs is associated with delayed cellular growth. One possible interpretation is that the delayed growth of female post-implantation mammalian embryos (Burgoyne et al., 1995) is recapitulated in mouse PSCs. Since the growth differences of XX and XY/XO PSCs are maintained after dual GSK3B and ERK inhibition, additional pathways are likely involved. One hypothesis is that there could be a competitive growth advantage of cells that have undergone XCI in the post-implantation mammalian embryo to select against remaining cells that may fail to undergo XCI, and maintain two Xas. Our in vitro experiment suggests that XCI could indeed provide a small growth advantage. However, this hypothesis remains to be tested in vivo.
To better understand what drives the features of the pluripotent regulatory network that are modulated by X-dosage in PSCs, we explored the open chromatin landscapes of female and male PSCs. While both female and male iPSCs/ESCs possessed globally similar open chromatin landscapes, thousands of chromatin regions were differentially accessible in XX and XY PSCs. These differentially accessible regions may underlie differences in the transcriptional regulatory network and functional properties of XX and XY PSCs. Decoding differentially accessible chromatin regions, we identified pluripotency-associated genes Pramel6 and Pramel7 with increased accessibility in XX iPSCs. Pramel7 has been associated with naive pluripotency (Graf et al., 2017), and overexpression of Pramel6 and Pramel7 both compromise pluripotency exit (Casanova et al., 2011). We went further by identifying a catalog of cis-regulatory regions including promoters that are modulated by X-dosage in iPSCs and ESCs. These observations indicate that X-dosage can modulate chromatin accessibility in mouse PSCs.
Decoding differentially accessible chromatin allowed us to distinguish distinct sets of enriched TF binding motifs in XX and XY ESCs. Specifically, motifs for KLF5, ZIC3, and NANOG were enriched in chromatin more open in XX iPSCs, all of which have been implicated in pluripotency (reviewed in Martello and Smith, 2014). These results suggest that the stabilization of pluripotency in XX ESCs may be mediated by these core master regulators. In particular, Zic3 is a known pluripotency factor (Lim et al., 2010), encoded on the X chromosome, and is not dosage compensated in XX PSCs. However, Zic3 heterozygous deletion had no effect on stabilization of pluripotency. Although no TF chromatin immunoprecipitation sequencing (ChIP-seq) data are available for XX iPSCs to date, our in silico analyses identified a high-confidence set of direct putative KLF5, ZIC3, and NANOG targets in XX iPSCs, including known pluripotency-associated genes. Moreover, the specific putative regulatory region associated with Pramel6, which becomes more accessible in XX iPSCs, overlaps with ChIP-seq binding sites of OCT4, SOX2, and NANOG in XY ESCs (not shown), further suggesting that increased binding of master pluripotency regulators takes place at these more accessible regions in XX iPSCs. Our results raise the possibility that pluripotency is stabilized in XX iPSCs by binding of core pluripotency factors to a subset of regulatory elements whose accessibility is influenced by X-dosage.
In contrast to the XX state, chromatin more open in XY iPSCs identified AP-1 TFs as candidate regulators, which have not previously been implicated in X-dosage-specific regulation of pluripotency. JUN/AP-1 control many cellular processes including proliferation, apoptosis, and differentiation in response to a variety of stimuli including the MAPK pathway (reviewed in Shaulian and Karin, 2002). The role of AP-1 TFs in the context of X-dosage in iPSCs warrants future study.
A previous study showed that Dusp9 modulates DNA hypomethylation and the proteome in XX female mouse ESCs (Choi et al., 2017a). However, the effects of reducing Dusp9 dosage in XX ESCs on growth, transcription, and pluripotency exit were unknown. An important outcome of our analyses is that Dusp9 heterozygous XX ESCs maintain a female-like open chromatin landscape, growth, and delayed pluripotency exit concomitant with male-like global DNA methylation levels. These results suggest that chromatin accessibility, growth, and delayed pluripotency exit can be regulated independently of global DNA methylation levels in mouse PSCs. This result was unexpected for two reasons. First, Dusp9 overexpression in ESCs was reported to induce a female-like proteome, including activation of naive pluripotency marker PRDM14. Second, reducing the expression of DNMTs in male ESCs is associated, at least in part, with delayed pluripotency exit (Schulz et al., 2014). However, Choi et al. (2017a) reported that the ICM of female and male embryos shows comparable DNA methylation, despite delayed female development, suggesting that DNA hypomethylation and stabilization of pluripotency can be uncoupled both in vivo and in vitro. Our results therefore suggest that global DNA methylation levels are regulated, at least in part, by distinct X-linked genes, different from those regulating the open chromatin landscape and stabilization of pluripotency in PSCs (Dusp9 for DNA methylation levels, other gene(s) for chromatin accessibility and delayed pluripotency exit and growth). Choi et al. (2017a) reported that Dusp9 overexpression in male ESCs increases the expression of PRDM14, ROR2, and TFCP2L1. In our study, 3.5- to 3.7-fold overexpression of DUSP9 protein in male ESCs, achieving DUSP9 protein level comparable with that in XX ESCs, did not lead to an increase in Prdm14 transcript level. This may be explained by differences in the level or method of Dusp9 overexpression (inducible system versus piggyBac), or the assay used to judge expression of pluripotency markers (mass spectrometry versus qRT-PCR). Interestingly, Pramel7 overexpression in ESCs was shown to induce DNA hypomethylation through the degradation of DNA methylation maintenance factor UHRF1 (Graf et al., 2017). At the same time, we found that reducing Dusp9 dosage in XX ESCs reduces Pramel7 expression to male levels. The results suggest that Pramel7 may be downstream of Dusp9, and may participate in the control of DNA methylation by X-dosage.
Our results do not support a model in which a single X-linked gene stabilizes pluripotency in XX PSCs. First, single heterozygous deletions of Zic3, Tfe3, or overexpression of Zic3, Dkc1, Otud6a, Fhl1, Zfp185, and Scml2 had little effect. Second, distinct LF heterozygous deletions suggested that multiple X-linked genes participate in delaying pluripotency exit in XX PSCs. Therefore, identifying additional X-linked regulators that mediate the effects of X-dosage in PSCs requires future investigations. An interesting additional candidate is the recently identified X-linked transient octamer binding factor 1 (TOBF1) (Chakraborty et al., 2017), since it was shown to sustain pluripotency. It is also possible that other regulators of the Erk pathway are involved. A previous study in human ESCs reported that human primed PSCs with eroded XCI and increased expression of the MAPK/ERK downstream effector ELK-1 have decreased expression of TRA-1-60, a marker of the differentiated state (Bruck et al., 2013). However, human primed PSCs studies are likely not compatible with mouse naive PSC studies because cells reside in distinct pluripotent states.
To conclude, we revealed that global DNA methylation can be uncoupled from delayed pluripotency exit in XX mouse ESCs. Furthermore, our study shows that X-dosage-specific differences in cell growth, open chromatin landscape, transcription, and pluripotency exit in iPSCs correlate with the number of Xas. We also reveal a mechanism by which multiple genomic regions on the X chromosome are responsible for delaying pluripotency exit. Using information from the genome, the epigenome, and the transcriptome we gained insights into modulation of the open chromatin landscape and the transcriptional regulatory network of iPSCs by X-dosage. Furthermore, better understanding how X-dosage modulates pluripotency will have important implications for disease modeling and regenerative medicine.
Experimental Procedures
Detailed information on cell line derivation, culture conditions, differentiation, plasmids, immunofluorescence, RNA fluorescence in situ hybridization, qPCR, qRT-PCR, and western blot, as well as clonal, cell growth, and cell-cycle assays are available in Supplemental Experimental Procedures.
Statistical Analysis
Statistical tests were performed using GraphPad Prism 5 (GraphPad Software). Unpaired two-tailed t test, one-way ANOVA with multiple comparisons test, or two-way repeated-measures ANOVA were used as indicated. The number of independent experiments performed is indicated (n). All data are presented as the mean ± SEM. p values of <0.05 were considered statistically significant.
RNA-Seq and Analysis
Total RNA was extracted from 2–3 independent cell lines for each cell type (Table S1) and used for construction of a single-end indexed, stranded poly(A) mRNA-seq library. DEG analysis and clustering analysis were performed in R.
ATAC-Seq and Analysis
OmniATAC-seq was performed as described by Corces et al. (2017) from 2–3 independent cell lines for each cell type (Table S1). Analysis of HiSeq4000 single-end reads used a pipeline from the Kundaje lab (version 0.3.3) (Lee et al., 2016). Differential chromatin accessibility analysis was performed using the DiffBind package. Motif analysis was performed using the HOMER package (v4.9.1).
Author Contributions
Conception and design, J.S. and V.P.; Experiments, J.S., N.D.G., L.V., M.E.B., T.O., I.T., and V.P.; Analyses, J.S.; ATAC-seq and RNA-seq analyses, A.J. and J.S.; Writing, V.P. and J.S. with input from all authors; Supervision, V.P.
Acknowledgments
We thank Stein Aerts, Konrad Hochedlinger, Frederic Lluis Vinas, Edda Schulz, and Edith Heard for discussions; Rudolf Jaenisch for providing mice; Qiaolin Deng for providing Mus/Cas ESCs; Rita Khoueiry and Michela Bartoccetti for help with derivation of Mus/Cas ESCs; Ye-Guang Chen for the Dusp9 plasmids; Miguel Branco for the Scml2 plasmids; José Silva for the PiggyBac plasmid; Mitchell Guttman and Jesse Engreitz for the pZB-sg3 plasmid; Ali Brivanlou and Ariel Waisman for the FUCCI plasmids; Stein Aerts, Kristofer Davie, Liesbeth Minnoye, and Xinlong Luo for help with bioinformatics and ATAC-seq; and Kenjiro Shirane and Hiroyuki Sasaki for providing processed data. We thank Constantinos Chronis, Kian Koh, Kathrin Plath, and Edda Schulz for comments on an early version of the manuscript. We are grateful to the help provided by the KU Leuven FACS Core, Genomics Core, and Mouse Facility; Metabolomics Core, VIB/KU Leuven; and the Stem Cell Institute, Leuven. This work was supported by The Research Foundation Flanders (FWO) (Odysseus return grant G0F7716N to V.P.), the KU Leuven Research Fund (BOFZAP starting grant StG/15/021BF to V.P., C1 grant C14/16/077 to V.P., and project financing), and FWO PhD fellowship to A.J. (1158318N).
Published: January 10, 2019
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and five tables and can be found with this article online at https://doi.org/10.1016/j.stemcr.2018.12.004.
Contributor Information
Juan Song, Email: juan.song@kuleuven.be.
Vincent Pasque, Email: vincent.pasque@kuleuven.be.
Accession Numbers
The GEO accession number for the RNA-seq and ATAC-seq data reported in this paper is GEO: GSE110215. The single-cell RNA-seq data (GEO: GSE74155) was from Chen et al. (2016), and V6.5 ESC and MEF RNA-seq (GEO: GSE90894) from Chronis et al. (2017).
Supplemental Information
References
- Avior Y., Sagi I., Benvenisty N. Pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Mol. Cell Biol. 2016;17:170–182. doi: 10.1038/nrm.2015.27. [DOI] [PubMed] [Google Scholar]
- Bruck T., Yanuka O., Benvenisty N. Human pluripotent stem cells with distinct X inactivation status show molecular and cellular differences controlled by the X-Linked ELK-1 gene. Cell Rep. 2013;4:262–270. doi: 10.1016/j.celrep.2013.06.026. [DOI] [PubMed] [Google Scholar]
- Burgoyne P.S., Thornhill A.R., Boudrean S.K., Darling S.M., Bishop C.E., Evans E.P. The genetic basis of XX-XY differences present before gonadal sex differentiation in the mouse. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1995;350:253–260. doi: 10.1098/rstb.1995.0159. discussion 260–261. [DOI] [PubMed] [Google Scholar]
- Carey B.W., Markoulaki S., Beard C., Hanna J., Jaenisch R. Single-gene transgenic mouse strains for reprogramming adult somatic cells. Nat. Methods. 2010;7:56–59. doi: 10.1038/nmeth.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova E.A., Shakhova O., Patel S.S., Asner I.N., Pelczar P., Weber F.A., Graf U., Sommer L., Bürki K., Cinelli P. Pramel7 mediates LIF/STAT3-dependent self-renewal in embryonic stem cells. Stem Cells. 2011;29:474–485. doi: 10.1002/stem.588. [DOI] [PubMed] [Google Scholar]
- Chakraborty D., Paszkowski-Rogacz M., Berger N., Ding L., Mircetic J., Fu J., Iesmantavicius V., Choudhary C., Anastassiadis K., Stewart A.F. lncRNA Panct1 maintains mouse embryonic stem cell identity by regulating TOBF1 recruitment to Oct-Sox sequences in early G1. Cell Rep. 2017;21:3012–3021. doi: 10.1016/j.celrep.2017.11.045. [DOI] [PubMed] [Google Scholar]
- Chen G., Schell J.P., Benitez J.A., Petropoulos S., Yilmaz M., Reinius B., Alekseenko Z., Shi L., Hedlund E., Lanner F. Single-cell analyses of X Chromosome inactivation dynamics and pluripotency during differentiation. Genome Res. 2016;26:1342–1354. doi: 10.1101/gr.201954.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J., Clement K., Huebner A.J., Webster J., Rose C.M., Brumbaugh J., Walsh R.M., Lee S., Savol A., Etchegaray J.-P. DUSP9 modulates DNA hypomethylation in female mouse pluripotent stem cells. Cell Stem Cell. 2017;20:706–719.e7. doi: 10.1016/j.stem.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J., Huebner A.J., Clement K., Walsh R.M., Savol A., Lin K., Gu H., Di Stefano B., Brumbaugh J., Kim S.-Y. Prolonged Mek1/2 suppression impairs the developmental potential of embryonic stem cells. Nature. 2017;548:219–223. doi: 10.1038/nature23274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choksi S.P., Lauter G., Swoboda P., Roy S. Switching on cilia: transcriptional networks regulating ciliogenesis. Development. 2014;141:1427–1441. doi: 10.1242/dev.074666. [DOI] [PubMed] [Google Scholar]
- Chronis C., Fiziev P., Papp B., Butz S., Bonora G., Sabri S., Ernst J., Plath K. Cooperative binding of transcription factors orchestrates reprogramming. Cell. 2017;168:442–459.e20. doi: 10.1016/j.cell.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces M.R., Trevino A.E., Hamilton E.G., Greenside P.G., Sinnott-Armstrong N.A., Vesuna S., Satpathy A.T., Rubin A.J., Montine K.S., Wu B. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods. 2017;14:959–962. doi: 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Los Angeles A., Ferrari F., Xi R., Fujiwara Y., Benvenisty N., Deng H., Hochedlinger K., Jaenisch R., Lee S., Leitch H.G. Hallmarks of pluripotency. Nature. 2015;525:469–478. doi: 10.1038/nature15515. [DOI] [PubMed] [Google Scholar]
- Graf U., Casanova E.A., Wyck S., Dalcher D., Gatti M., Vollenweider E., Okoniewski M.J., Weber F.A., Patel S.S., Schmid M.W. Pramel7 mediates ground-state pluripotency through proteasomal-epigenetic combined pathways. Nat. Cell Biol. 2017;19:763–773. doi: 10.1038/ncb3554. [DOI] [PubMed] [Google Scholar]
- Guo G., Yang J., Nichols J., Hall J.S., Eyres I., Mansfield W., Smith A. Klf4 reverts developmentally programmed restriction of ground state pluripotency. Development. 2009;136:1063–1069. doi: 10.1242/dev.030957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habibi E., Brinkman A.B., Arand J., Kroeze L.I., Kerstens H.H.D., Matarese F., Lepikhov K., Gut M., Brun-Heath I., Hubner N.C. Whole-genome bisulfite sequencing of two distinct interconvertible DNA methylomes of mouse embryonic stem cells. Cell Stem Cell. 2013;13:360–369. doi: 10.1016/j.stem.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Hackett J.A., Dietmann S., Murakami K., Down T.A., Leitch H.G., Azim Surani M. Synergistic mechanisms of DNA demethylation during transition to ground-state pluripotency. Stem Cell Reports. 2013;1:518–531. doi: 10.1016/j.stemcr.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- Lee J.T., Lu N. Targeted mutagenesis of Tsix leads to nonrandom X inactivation. Cell. 1999;99:47–57. doi: 10.1016/s0092-8674(00)80061-6. [DOI] [PubMed] [Google Scholar]
- Lee J., Christoforo G., Christoforo G., Foo C.S., Probert C., Kundaje A., Boley N., kohpangwei, Dacre M., Kim D. kundajelab/atac_dnase_pipelines: 0.3.3. 2016. [DOI]
- Li Z., Fei T., Zhang J., Zhu G., Wang L., Lu D., Chi X., Teng Y., Hou N., Yang X. BMP4 signaling acts via dual-specificity phosphatase 9 to control ERK activity in mouse embryonic stem cells. Cell Stem Cell. 2012;10:171–182. doi: 10.1016/j.stem.2011.12.016. [DOI] [PubMed] [Google Scholar]
- Lim L.S., Hong F.H., Kunarso G., Stanton L.W. The pluripotency regulator Zic3 is a direct activator of the Nanog promoter in ESCs. Stem Cells. 2010;28:1961–1969. doi: 10.1002/stem.527. [DOI] [PubMed] [Google Scholar]
- Maherali N., Sridharan R., Xie W., Utikal J., Eminli S., Arnold K., Stadtfeld M., Yachechko R., Tchieu J., Jaenisch R. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 2007;1:55–70. doi: 10.1016/j.stem.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Mak W. Reactivation of the paternal X chromosome in early mouse embryos. Science. 2004;303:666–669. doi: 10.1126/science.1092674. [DOI] [PubMed] [Google Scholar]
- Martello G., Smith A. The nature of embryonic stem cells. Annu. Rev. Cell Dev. Biol. 2014;30:647–675. doi: 10.1146/annurev-cellbio-100913-013116. [DOI] [PubMed] [Google Scholar]
- Milagre I., Stubbs T.M., King M.R., Spindel J., Santos F., Krueger F., Bachman M., Segonds-Pichon A., Balasubramanian S., Andrews S.R. Gender differences in global but not targeted demethylation in iPSC reprogramming. Cell Rep. 2017;18:1079–1089. doi: 10.1016/j.celrep.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto I., Otte A.P., Allis C.D., Reinberg D., Heard E. Epigenetic dynamics of imprinted X inactivation during early mouse development. Science. 2004;303:644–649. doi: 10.1126/science.1092727. [DOI] [PubMed] [Google Scholar]
- Ooi S.K., Wolf D., Hartung O., Agarwal S., Daley G.Q., Goff S.P., Bestor T.H. Dynamic instability of genomic methylation patterns in pluripotent stem cells. Epigenetics Chromatin. 2010;3:17. doi: 10.1186/1756-8935-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasque V., Plath K. X chromosome reactivation in reprogramming and in development. Curr. Opin. Cell Biol. 2015;37:75–83. doi: 10.1016/j.ceb.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasque V., Tchieu J., Karnik R., Uyeda M., Sadhu Dimashkie A., Case D., Papp B., Bonora G., Patel S., Ho R. X chromosome reactivation dynamics reveal stages of reprogramming to pluripotency. Cell. 2014;159:1681–1697. doi: 10.1016/j.cell.2014.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasque V., Karnik R., Chronis C., Petrella P., Langerman J., Bonora G., Song J., Vanheer L., Sadhu Dimashkie A., Meissner A. X chromosome dosage influences DNA methylation dynamics during reprogramming to mouse iPSCs. Stem Cell Rep. 2018;10:1537–1550. doi: 10.1016/j.stemcr.2018.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronen D., Benvenisty N. Sex-dependent gene expression in human pluripotent stem cells. Cell Rep. 2014;8:923–932. doi: 10.1016/j.celrep.2014.07.013. [DOI] [PubMed] [Google Scholar]
- Sakaue-Sawano A., Kurokawa H., Morimura T., Hanyu A., Hama H., Osawa H., Kashiwagi S., Fukami K., Miyata T., Miyoshi H. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132:487–498. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- Schulz E.G. X-chromosome dosage as a modulator of pluripotency, signalling and differentiation? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017;372 doi: 10.1098/rstb.2016.0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz E.G., Meisig J., Nakamura T., Okamoto I., Sieber A., Picard C., Borensztein M., Saitou M., Blüthgen N., Heard E. The two active X chromosomes in female ESCs block exit from the pluripotent state by modulating the ESC signaling network. Cell Stem Cell. 2014;14:203–216. doi: 10.1016/j.stem.2013.11.022. [DOI] [PubMed] [Google Scholar]
- Shaulian E. AP-1—the Jun proteins: oncogenes or tumor suppressors in disguise? Cell. Signal. 2010;22:894–899. doi: 10.1016/j.cellsig.2009.12.008. [DOI] [PubMed] [Google Scholar]
- Shaulian E., Karin M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002;4:E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- Shirane K., Kurimoto K., Yabuta Y., Yamaji M., Satoh J., Ito S., Watanabe A., Hayashi K., Saitou M., Sasaki H. Global landscape and regulatory principles of DNA methylation reprogramming for germ cell specification by mouse pluripotent stem cells. Dev. Cell. 2016;39:87–103. doi: 10.1016/j.devcel.2016.08.008. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Yagi M., Kishigami S., Tanaka A., Semi K., Mizutani E., Wakayama S., Wakayama T., Yamamoto T., Yamada Y. Derivation of ground-state female ES cells maintaining gamete-derived DNA methylation. Nature. 2017;548:224–227. doi: 10.1038/nature23286. [DOI] [PubMed] [Google Scholar]
- Yang S.-H., Kalkan T., Morrisroe C., Smith A., Sharrocks A.D. A genome-wide RNAi screen reveals MAP kinase phosphatases as key ERK pathway regulators during embryonic stem cell differentiation. PLoS Genet. 2012;8:e1003112. doi: 10.1371/journal.pgen.1003112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Q.-L., Wray J., Nichols J., Batlle-Morera L., Doble B., Woodgett J., Cohen P., Smith A. The ground state of embryonic stem cell self-renewal. Nature. 2008;453:519–523. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zvetkova I., Apedaile A., Ramsahoye B., Mermoud J.E., Crompton L.A., John R., Feil R., Brockdorff N. Global hypomethylation of the genome in XX embryonic stem cells. Nat. Genet. 2005;37:1274–1279. doi: 10.1038/ng1663. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.