

Abstract
Objective
To determine whether infection with human enterovirus or adenovirus, both common intestinal viruses, predicts development of coeliac disease.
Design
Case-control study nested within Norwegian birth cohort recruited between 2001 and 2007 and followed to September 2016.
Setting
Norwegian population.
Participants
Children carrying the HLA genotype DR4-DQ8/DR3-DQ2 conferring increased risk of coeliac disease.
Exposures
Enterovirus and adenovirus detected using real time polymerase chain reaction in monthly stool samples from age 3 to 36 months.
Main outcome measure
Coeliac disease diagnosed according to standard criteria. Coeliac disease antibodies were tested in blood samples taken at age 3, 6, 9, and 12 months and then annually. Adjusted odds ratios from mixed effects logistic regression model were used to assess the relation between viral infections before development of coeliac disease antibodies and coeliac disease.
Results
Among 220 children, and after a mean of 9.9 (SD 1.6) years, 25 children were diagnosed as having coeliac disease after screening and were matched to two controls each. Enterovirus was found in 370 (17%) of 2135 samples and was significantly more frequent in samples collected before development of coeliac disease antibodies in cases than in controls (adjusted odds ratio 1.49, 95% confidence interval 1.07 to 2.06; P=0.02). The association was restricted to infections after introduction of gluten. High quantity samples (>100 000 copies/μL) (adjusted odds ratio 2.11, 1.24 to 3.60; P=0.01) and long lasting infections (>2 months) (2.16, 1.16 to 4.04; P=0.02) gave higher risk estimates. Both the commonly detected enterovirus species Enterovirus A and Enterovirus B were significantly associated with coeliac disease. The association was not found for infections during or after development of coeliac disease antibodies. Adenovirus was not associated with coeliac disease.
Conclusions
In this longitudinal study, a higher frequency of enterovirus, but not adenovirus, during early childhood was associated with later coeliac disease. The finding adds new information on the role of viral infections in the aetiology of coeliac disease.
Introduction
Coeliac disease is an immune mediated disease believed to result from gluten intake and unknown environmental trigger factors in genetically susceptible individuals.1 Coeliac disease develops almost exclusively in people with the HLA-DQ2 (DQA1*05:01-DQB1*02:01) or HLA-DQ8 (DQA1*03-DQB1*03:02) haplotype. The HLA-DQ2 and HLA-DQ8 haplotypes also occur in about 40% of the general population. Although non-HLA genetic variants have also been identified, the predictive value of these variants, even in combination, is limited.2 3 As most children are exposed to gluten, this suggests that additional genetic and environmental triggers are involved in development of coeliac disease.4 The autoimmune process may start months or years before clinical manifestations,5 and the identification and confirmation of environmental triggers remain a major challenge.6
Both experimental studies and epidemiological studies based on parental reporting of illness suggest a role for infections in the development of coeliac disease, particularly gastrointestinal infections.6 7 8 9 Gastrointestinal infections are common in childhood and may impair the mucosal barrier for transfer of dietary proteins as gluten regardless of the presence of clinical symptoms.10 11 The only prospective study on coeliac disease and viral infection suggested that frequent rotavirus infections might increase the risk of development of coeliac disease antibodies in a cohort at high risk.12 Other studies with retrospective designs have studied adenovirus, enterovirus, and orthoreovirus as potential triggers of coeliac disease with conflicting or inconclusive results.9 13 14 15 Study designs including patients diagnosed as having coeliac disease and controls may be subject to bias due to reverse causality. Therefore, long term follow-up of longitudinal birth cohorts is needed.
In this study, we approached the question of potential gastrointestinal triggers by using a longitudinal birth cohort analysis of the most frequently occurring viruses: enterovirus and adenovirus. We chose these two viruses on the basis of a pilot screening of the stool virome in the cohort members. We aimed to test whether the presence of human enterovirus and adenovirus in monthly faecal samples was more common before development of coeliac disease antibodies in cases subsequently diagnosed as having coeliac disease compared with children not developing the disease.
Methods
Participants and study design
We designed a nested case-control study of coeliac disease within a birth cohort of children with the HLA-DQ2/DQ8 genotype, with monthly stool samples tested for nucleic acid of enterovirus and adenovirus. Figure 1 outlines the enrolment of the study sample, and figure 2 gives an overview of the study design. The MIDIA study, which was originally designed to study type 1 diabetes, is described in detail by Stene et al.16
Fig 1.
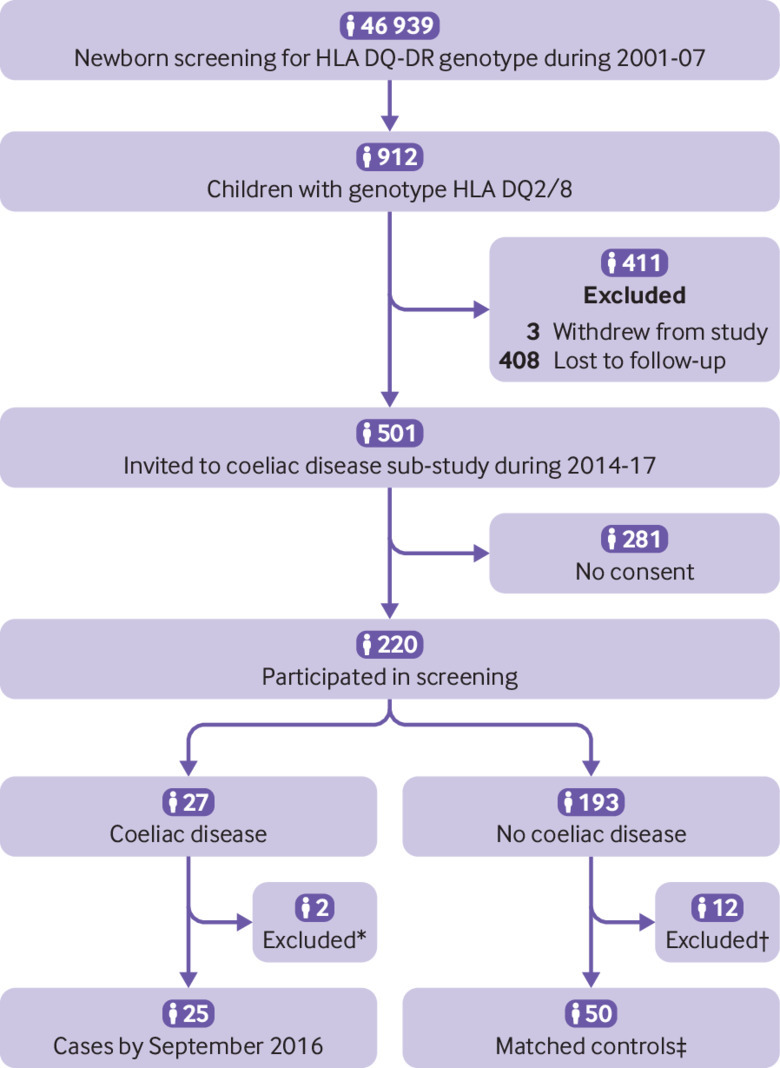
Enrolment of study sample. “Invited” children are those who were still actively participating (delivering samples, questionnaires) at start of coeliac disease sub-study and were invited to participate; “participated” are those who consented to participate and were screened for coeliac disease antibodies. *Two cases were excluded from analyses, one owing to missing stool samples and one owing to incorrect use of diagnostic criteria. †Children with positive coeliac disease antibodies at first screening test but for whom follow-up did not confirm coeliac disease diagnosis (n=10) were excluded from further analyses; control children with high antibody titres (>10 times cut-off) (n=2) in old samples but with normal values at screening were excluded and replaced by new controls selected using same matching criteria. ‡One matched control was excluded from analysis owing to missing stool samples
Fig 2.
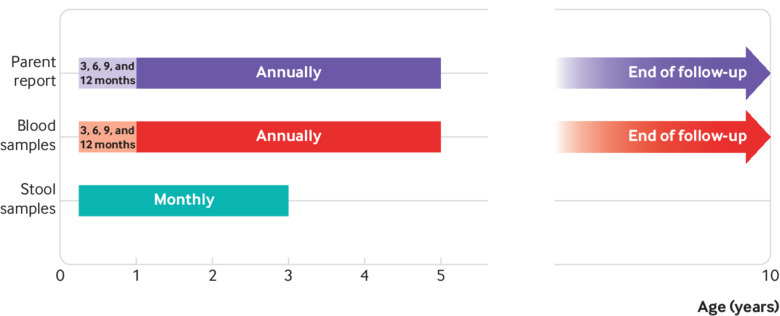
Timeline of study
Briefly, during 2001-07, 46 939 newborns throughout Norway were screened for the HLA-DQ2/DQ8 genotype conferring an increased risk of both coeliac disease and type 1 diabetes (HLA-DRB1*04:01-DQA1*03-DQB1*03:02/DRB1*03-DQA1*05:01-DQB1*02:01). This risk genotype was identified in 912 (1.9%) children, who were followed with repeated blood and faecal samples from the age of 3 months. We collected plasma samples at age 3, 6, 9, and 12 months and annually thereafter. Monthly stool samples collected at age 3-36 months were diluted in preservation buffer, centrifuged, and supernatant separated. All samples were stored at –80°C until testing.17
Children who still actively contributed with blood samples during 2014-16 (n=501) were invited to coeliac disease screening, of whom 220 consented to participate. These participants tended to have a higher prevalence of type 1 diabetes, fewer siblings, and slightly higher prevalence of family history of coeliac disease at initial recruitment around 3 months of age, than the whole cohort (supplementary table A).
Case definition and selection of matched controls
From the 220 consenting participants, we first identified those who had coeliac disease by measuring serological markers of coeliac disease in their most recent sample and by review of clinical documentation, followed by an invitation for a confirmatory blood sample and for standard paediatric diagnostic investigation for coeliac disease. We identified previously diagnosed cases by using a parental questionnaire followed by a review of medical files. For an overview of the coeliac screening process, see supplementary figure A. We diagnosed coeliac disease according to the European Society for Paediatric Gastroenterology Hepatology and Nutrition (ESPGHAN) 2012 criteria.18 By this procedure, we identified 27 cases of coeliac disease by February 2016 and confirmed that the remaining 193 participants were free from coeliac disease. Mean age at end of follow-up was 9.9 (SD 1.6) years. After evaluation for eligibility, we matched 25 cases (16 girls and 9 boys; 11% of participants) with coeliac disease to two controls each. Matching was done for duration of follow-up, date of birth, and county of residence. We excluded one matched control from analysis owing to missing stool samples. See table 1 for characteristics of the nested cases and controls. For more details about case definition, see supplementary methods.
Table 1.
Characteristics of nested cases and controls. Values are numbers (percentages) unless stated otherwise
| Characteristics | Cases (n=25) | Controls (n=49) |
|---|---|---|
| Female sex | 16 (64) | 26 (53) |
| Other children in household: | ||
| None | 7 (28) | 15 (31) |
| 1 | 10 (40) | 28 (57) |
| ≥2 | 8 (32) | 6 (12) |
| Type 1 diabetes | 4 (16) | 1 (2) |
| Family history of coeliac disease* | 7 (28) | 4 (8) |
| Mean (SD) age (months) at antibody screening test† | 99 (30) | 106 (21) |
| Mean (SD) age (months) at end of follow-up‡ | 119 (19) | 119 (19) |
| Mean (SD) age (months) at last stool sample | 32 (7) | 34 (3) |
| Stool samples§ | 703 | 1458 |
| Enterovirus: | ||
| Stool samples providing enterovirus data | 690 (98) | 1445 (99) |
| Enterovirus positive stool samples | 135 (20) | 235 (16) |
| Median (range) count of positive samples per child | 6 (0-11) | 4 (0-9) |
| Adenovirus: | ||
| Stool samples providing adenovirus data | 649 (92) | 1357 (93) |
| Adenovirus positive samples | 85 (13) | 173 (13) |
| Median (range) count of positive samples per child | 4 (0-9) | 3 (0-11) |
| Blood samples§: | ||
| Total | 326 | 593 |
| Samples providing coeliac disease antibody data¶ | 259 (79) | 374 (63) |
| Coeliac disease antibody positive samples¶ | 131 (51) | 7 (2) |
Known coeliac disease in first degree relative or half-sibling ascertained at coeliac disease screening 2014-16.
Antibody screening test performed on both cases and controls at time of inclusion in coeliac disease sub-study of MIDIA.
At time when all 25 cases were ascertained by end of February 2016.
Discrepancy between number of stool samples and samples providing virus data and number of blood samples and samples providing data on coeliac disease antibodies was due to missing samples or failed laboratory test.
Antibodies to tissue transglutaminase.
We then determined the time interval when cases seroconverted for coeliac disease markers by retrospectively analysing biobanked plasma samples that had been collected longitudinally since age 3 months, searching for the last sample that was negative for the serological markers and the first sample indicative of coeliac disease. Of the 25 case-control groups, 15 had this seroconversion period covered by monthly stool sampling, whereas the remaining 10 seroconverted after the collection of stools was terminated (after 36 months of age). For characteristics of the children with coeliac disease, see supplementary table B.
Serological testing for coeliac disease
IgA anti-tissue transglutaminase and IgG anti-deamidated gliadin peptide (EliA Celikey IgA/EliA GliadinDP IgG, Thermo Fischer Scientific, Phadia AB; Uppsala, Sweden) were measured in the most recent blood sample from each participant at the laboratory of Akershus University Hospital, Lørenskog, Norway. Mean age at screening was 8.6 (SD 2.0) years. Screening with IgA anti-tissue transglutaminase in combination with IgG anti-deamidated gliadin peptide should better capture people with IgA deficiency and samples in which tissue transglutaminase concentrations may have been influenced by haemolysis.19 20 21 In accordance with a previous screening study and to increase sensitivity,22 we defined IgA anti-tissue transglutaminase values of 3 U/mL or higher as positive. For IgG anti-deamidated gliadin peptide, we used 7 U/mL as the cut-off value in accordance with the manufacturer’s instruction. We contacted children with positive serological results (n=20) for a second blood sample (Varelisa tissue transglutaminase IgA, Phadia, cut-off <3 U/mL (<5 U/mL until 2015); IgG anti-deamidated gliadin peptide, QUANTA Lite Gliadin IgG IIInova, cut-off <20 U/mL) which was analysed at Oslo University Hospital Ullevål, Oslo, Norway.
Detection of enterovirus and adenovirus in stool samples
All available faecal samples from cases and controls were subjected to RNA and DNA extraction using Qiagen chemistry (Qiagen, Hilden, Germany). We tested enterovirus by quantitative real time polymerase chain reaction (PCR).23 The primers detected with an equal sensitivity the Enterovirus A-D species (that is, members of species Enterovirus A, Enterovirus B, Enterovirus C, and Enterovirus D of the genus Enterovirus, family Picornaviridae, order Picornavirales; the former nomenclature of these species was Human enterovirus A-D) but did not react with human rhinoviruses (members of species Rhinovirus A-C). Adenovirus was tested using a previously published real time PCR assay.24 The threshold of enterovirus and adenovirus positivity was set to 10 copies/μL nucleic acid. We tested 2161 stool samples, of which 2135 and 2006 provided data on quantity of enterovirus and adenovirus, respectively. All samples were tested blinded as to the case-control status.
All samples with more than 100 copies/μL of enterovirus were subjected to genotyping with Sanger or next generation amplicon sequencing of PCR amplicons of the VP1 gene segment informative for the virus type. Samples with more than 10 copies/μL of adenovirus were genotyped by a similar protocol amplifying the seventh hypervariable region of its hexon gene. Primers, probes, and a detailed protocol have been described in Cinek et al.25 See also supplementary methods for more details.
Statistical analysis
We analysed the association of virus with coeliac disease primarily by using a mixed effects logistic regression model with random intercepts for each matched set and for each individual child, thus accounting for the matched design. We used faecal sample virus positivity as the dependent variable (separate models for enterovirus and adenovirus) and case-control status as an independent variable. The odds ratio for coeliac disease status is then interpreted as the odds of a faecal sample being positive for virus given that it came from a child who later developed coeliac disease, compared with the same odds for virus positivity for samples from matched controls. We included the following potential predictors of viral infection and coeliac disease as covariates in the main regression model: sex, age, age squared, season of sample collection, number of siblings (categorised as none, 1, or ≥2), and family history of coeliac disease. In the primary analysis, we included only stool samples from the matched sets collected before the case child developed coeliac disease antibodies to avoid reverse causality (fig 3).
Fig 3.
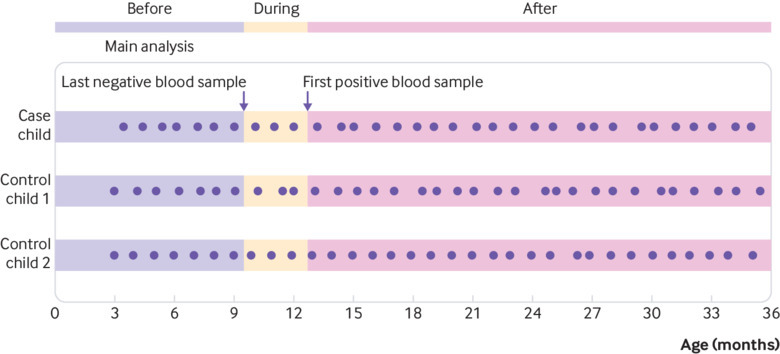
Development of coeliac disease antibodies. Figure illustrating definition of before, during, and after development of coeliac disease antibodies. Dots represent faecal samples. Primary analysis included only stool samples collected up to age at last antibody negative blood sample for cases and corresponding age for matched controls. Pre-planned sub-analyses explored same association for stool samples collected during development of coeliac disease antibodies (between last negative and first antibody positive blood sample) and samples collected after first antibody positive blood sample
In pre-planned sub-analyses, we explored the association between enterovirus and adenovirus and subsequent coeliac disease for stool samples collected during and after the development of coeliac disease antibodies (fig 3). We analysed the samples before development of coeliac disease antibodies, defining the exposure as number of infectious episodes, counting a sequence of consecutively virus positive faecal samples as a single episode (a negative stool sample is required before a new episode is defined). We speculated that a higher quantity of enterovirus, longer duration of viral shedding, or symptomatic infections would have a greater effect on development of coeliac disease. Therefore, we analysed infections with reported symptoms and defining exposures as long infectious episodes (at least two positive consecutive monthly samples) and per infection with high quantity (≥100 000 copies per μL nucleic acid). We investigated specific virus types, using an a priori determined threshold of at least 20 positive samples to be analysed. We grouped enteroviruses into Enterovirus A-D species as detailed above. We grouped adenovirus types only into specific types (for example, human adenovirus C2), as the species Adenovirus C dominated in our samples.
Additionally, we adjusted the primary analysis for the timing of introduction of gluten and breast feeding. As exploratory analyses, we investigated time periods less than six months, six to 12 months, more than 12 months, after the end of breast feeding, and after the introduction of gluten. We also investigated whether infectious symptoms, as reported by parents in longitudinal questionnaires in early life, were associated with coeliac disease antibodies and whether specific symptoms were linked to infections. In sensitivity analyses, we also adjusted for type 1 diabetes.
Patient and public involvement
Patients were not involved in setting the research question or the outcome measures, nor were they involved in developing plans for recruitment, design, or implementation of the study. No patients were asked to advise on interpretation or writing up of results. We will disseminate the results of the research to study participants and the general public.
Results
Enterovirus for whole observational period
We detected enterovirus in 370 (17%) of 2135 stool samples (table 1), with 73 children having at least one positive sample. The distribution of enterovirus in cases and controls of the 25 matching groups is shown in supplementary figure B. Enterovirus showed variation with age and season, with a peak in autumn (supplementary figures C and D). Several different types were detected, occasionally as co-infections, with coxsackievirus A2 and A4 being most common (supplementary table C).
Enterovirus and coeliac disease
The frequency of enterovirus positive stool samples before development of coeliac disease antibodies was 84/429 (20%) in cases and 129/855 (15%) in matched controls (adjusted odds ratio 1.49, 95% confidence interval 1.07 to 2.06; P=0.02) (table 2). The adjusted odds ratio was 2.11 (1.24 to 3.60; P=0.01) for high quantity samples (>100 000 copies/μL), 2.16 (1.16 to 4.04; P=0.02) for long lasting infections (more than two months), and 1.27 (0.87 to 1.86; P=0.21) for infectious episodes (consecutive positive samples counted as a single episode) (supplementary table D). The frequency of enterovirus in stool samples during or after development of coeliac disease antibodies was not associated with coeliac disease (table 2).
Table 2.
Enterovirus positive stool samples and subsequent coeliac disease*
| |
Positive samples/total samples (%) | Unadjusted odds ratio (95% CI) | Adjusted† | ||
|---|---|---|---|---|---|
| Cases (n=25) | Controls (n=49) | Odds ratio (95% CI)† | P value | ||
| Before development of coeliac disease antibodies‡ | |||||
| All enteroviruses | 84/429 (20) | 129/855 (15) | 1.37 (1.01 to 1.87) | 1.49 (1.07 to 2.06) | 0.02 |
| Enterovirus A‡ | 42/387 (11) | 60/787 (8) | 1.47 (0.97 to 2.23) | 1.62 (1.04 to 2.53) | 0.03 |
| CV-A2 | 10/355 (3) | 10/737 (1) | 2.11 (0.87 to 5.11) | 2.29 (0.89 to 5.87) | 0.09 |
| CV-A4 | 5/350 (1) | 11/738 (1) | 0.96 (0.33 to 2.78) | 0.93 (0.30 to 2.86) | 0.90 |
| CV-A6 | 7/352 (2) | 9/736 (1) | 1.64 (0.60 to 4.44) | 1.59 (0.56 to 4.57) | 0.39 |
| CV-A10 | 4/349 (1) | 10/737 (1) | 0.84 (0.25 to 2.76) | 1.03 (0.30 to 3.53) | 0.96 |
| CV-A16 | 4/349 (1) | 7/734 (1) | 1.20 (0.35 to 4.14) | 1.39 (0.36 to 5.30) | 0.63 |
| Enterovirus B‡ | 34/379 (9) | 43/770 (6) | 1.74 (1.08 to 2.81) | 2.27 (1.33 to 3.88) | 0.003 |
| During/after development of coeliac disease antibodies§ | |||||
| All enteroviruses during | 30/140 (21) | 58/332 (17) | 1.24 (0.74 to 2.10) | 1.26 (0.66 to 2.39) | 0.48 |
| All enteroviruses after | 21/121 (17) | 48/258 (19) | 0.92 (0.52 to 1.62) | 0.82 (0.44 to 1.53) | 0.54 |
Number of positive/total stool samples and results from mixed effects logistic regression are given for cases and controls at different time periods. In analysis for specific genotypes, samples positive for other enteroviruses have been set to missing. Enterovirus C and Enterovirus D were too few for a meaningful analysis and are not presented.
Adjusted for sex, age, age squared, season of sample collection, number of siblings, and family history of coeliac disease.
Only enterovirus types found in more than 10 samples before development of coeliac disease antibodies are presented.
Before: before last coeliac disease antibody negative blood sample. During: between last coeliac disease antibody negative and first coeliac disease antibody positive blood sample. After: after first coeliac disease antibody positive blood sample.
Exploratory analyses of enterovirus and coeliac disease
Both the commonly identified enterovirus species Enterovirus A (adjusted odds ratio 1.62, 1.04 to 2.53; P=0.03) and Enterovirus B (2.27, 1.33 to 3.88; P=0.003) were significantly associated with later coeliac disease. Enterovirus C and Enterovirus D were detected in few or no samples (table 2).
Enterovirus infections after the first year of life showed increased estimates, whereas infections from age 3 to 6 months or from 6 to 12 months did not. Enterovirus infections after introduction of gluten were associated with coeliac disease, whereas infections before or at the time of gluten introduction were not (supplementary table E). Similarly, infections after the end of breast feeding were associated with coeliac disease, but enterovirus infections during breast feeding were not. Enterovirus infections in shorter time windows before the last sample negative for coeliac disease antibody showed borderline increased estimates (supplementary table E). We found no association between reported infectious symptoms and coeliac disease or between infectious symptoms and enterovirus positivity (supplementary tables F and G). Enterovirus positivity at the same time of reported fever or diarrhoea was associated with development of coeliac disease, but other symptoms were not (supplementary table F). Adjustment for type 1 diabetes only marginally changed the estimates (adjusted odds ratio 1.43, 1.02 to 2.02).
Adenovirus and coeliac disease
We detected adenovirus in 258 (13%) of 2006 faecal samples (table 1), with 61 children having at least one positive sample. Adenovirus showed variation with age but did not follow a seasonal pattern (supplementary figures C and D). Several different types were detected, occasionally as co-infections, with human adenovirus C2 and C1 being most common (supplementary table H).
We found no significant difference between cases and controls in adenovirus positive stool samples before (adjusted odds ratio 0.82, 0.49 to 1.38; P=0.46), during, or after development of coeliac disease antibodies. No adenovirus types were associated with development of coeliac disease (table 3). Adenovirus was associated with fever, but adenovirus positive samples with reported symptoms were not associated with development of coeliac disease (supplementary table F).
Table 3.
Adenovirus positive stool samples and subsequent coeliac disease*
| |
Positive samples/total samples (%) | Unadjusted odds ratio (95% CI) | Adjusted† | ||
|---|---|---|---|---|---|
| Cases (n=25) | Controls (n=49) | Odds ratio (95% CI) | P value | ||
| Before development of coeliac disease antibodies‡ | |||||
| All adenoviruses | 47/390 (12) | 111/775 (14) | 0.82 (0.50 to 1.35) | 0.82 (0.49 to 1.38) | 0.46 |
| HAdV-C1 | 17/360 (5) | 60/724 (8) | 0.65 (0.19 to 2.19) | 0.66 (0.19 to 2.29) | 0.51 |
| HAdV-C2 | 20/363 (6) | 43/707 (6) | 0.88 (0.43 to 1.80) | 0.84 (0.38 to 1.87) | 0.67 |
| HAdV-C5 | 5/348 (1) | 7/671 (1) | 1.42 (0.29 to 6.96) | 2.22 (0.52 to 9.57) | 0.28 |
| During/after development of coeliac disease antibodies‡ | |||||
| All adenoviruses during | 26/139 (19) | 36/325 (11) | 1.84 (0.89 to 3.79) | 1.76 (0.87 to 3.56) | 0.12 |
| All adenoviruses after | 12/120 (10) | 26/257 (10) | 0.99 (0.41 to 2.39) | 1.29 (0.47 to 3.52) | 0.62 |
HAdV=human adenovirus.
Number of positive/total stool samples and results from mixed effects logistic regression are given for cases and controls at different time periods. In analysis for specific genotypes, samples positive for other adenoviruses have been set to missing. Adenovirus types were grouped into only specific genotypes (eg, HAdV-C2), as species Adenovirus C dominated in samples.
Adjusted for sex, age, age squared, season of sample collection, number of siblings, and family history of coeliac disease.
Before: before last coeliac disease antibody negative sample. During: between last coeliac disease antibody negative and first coeliac disease antibody positive blood sample. After: after first coeliac disease antibody positive blood sample.
Discussion
We found a significant association between exposure to enterovirus and subsequent risk of coeliac disease. Adenovirus was not associated with coeliac disease.
Strengths and limitations of study
This is the first population based study on viruses in stool samples collected longitudinally before development of coeliac disease antibody markers in children later diagnosed as having coeliac disease. We used accepted diagnostic criteria for coeliac disease. The development of coeliac disease antibodies, which is believed to be the first sign of disease, may start months or even years before clinical manifestations and diagnosis. Importantly, we analysed exposure before development of coeliac disease antibodies and thus avoided the potential reverse causation that may bias studies of infections at or after clinical diagnosis of coeliac disease. The long time period from development of coeliac disease antibodies to diagnosis of coeliac disease makes it highly unlikely that infections before development of coeliac disease antibodies prompted symptoms and diagnostic investigation for coeliac disease. Enterovirus positivity and type distribution in our study was in line with previous studies.26 We used a PCR technique, which is a highly sensitive method for detection of virus locally in the gut but does not measure the immune response as serological testing does.27 We believe that PCR was most appropriate in our study, as most infections were asymptomatic and current enterovirus serology not is designed for this kind of setting. We used a quantitative assay that enabled us to follow the dynamics of the viral load. Regular monthly stool sampling and high completeness were important strengths, because duration of viral shedding is expected to be around three to four weeks.28 29 Administration of oral poliovirus vaccine may increase the detection of enterovirus in stool samples, but this cohort received inactivated poliovirus vaccine injections.30 31
Our study also had some limitations. The participants were followed up for about 10 years, and some children (particularly among those with the shortest follow-up) would be likely to be diagnosed as having coeliac disease after that age. Even though we analysed a large number of samples, the number of children with coeliac disease was limited. Furthermore, the limited sample size for some of the sub-analyses could have led to spurious associations. Loss to follow-up and modest response rate is inevitable in most cohort studies and also raises the possibility of selection bias. The nested cases and controls tended to have a higher prevalence of type 1 diabetes, fewer siblings, and a slightly higher frequency of family history of coeliac disease than the whole cohort. This could have led to non-representativeness, but it does not necessarily bias exposure-outcome associations.32 The study was originally designed to study type 1 diabetes, with inclusion of only children with the HLA-DQ2/DQ8 genotype. Although we cannot generalise our findings beyond this genotype, we note that this genotype accommodates the two well established susceptibility haplotypes (DQ2 and DQ8) present in nearly all patients. We therefore believe that our findings are likely to apply to a sizeable proportion of patients with coeliac disease. Furthermore, the frequency of faecal enterovirus shedding did not differ significantly between people with the HLA-DQ2/DQ8 genotype and other genotypes in a previous study.33 We did our best to adjust for potential confounding factors, and we are not aware of any obvious confounding factors that could plausibly explain our findings, but unmeasured confounding factors or residual confounding can never be entirely ruled out in non-randomised studies. Further studies in other settings would therefore be necessary to corroborate our findings.
Comparison with other studies
Coeliac disease and type 1 diabetes have several common features, and viral infections are candidate triggers in both diseases. In type 1 diabetes, enterovirus infections before islet autoimmunity, as well as persistent enterovirus infections in the gut mucosa or pancreatic tissue, have been described.34 35 36 The single study on enterovirus and coeliac disease was cross sectional but could not find persistent viral infection in intestinal biopsies from untreated coeliac disease patients.15 The study did not investigate signs of previous exposure to enterovirus. Finally, adjusting for type 1 diabetes in our analyses yielded similar results. This is consistent with the lack of association between enterovirus and diabetes autoimmunity in the same cohort.37
Adenoviruses have been proposed to precipitate coeliac disease via molecular mimicry.13 38 39 However, later studies did not confirm the association.14 40 Our negative findings are in line with the latter studies and suggest that adenovirus in general does not influence risk of coeliac disease. An important strength of our study was the prospective design, whereas the studies mentioned above were cross sectional. Although we cannot rule out moderate effects from our data, the 95% confidence interval around the odds ratios estimated from our data suggest that strong associations with adenovirus infections are unlikely.
Interpretation
Although the effect sizes are relatively small, this study suggests that infections with enterovirus in early life could be one among several key risk factors for development of a disease with lifelong consequences. Our observations suggest that, rather than a specific enterovirus species or type driving the association, several enterovirus types, high titre, and long duration infections in the period after introduction of gluten were involved. Collectively, our results are compatible with a mechanism whereby viral infections may disrupt the mucosal barrier with increased translocation of gluten peptides into the mucosa as the initial event in the loss of tolerance.41 We speculate that enteroviruses may provide a danger signal that activates dendritic cells acting as antigen presenting cells for CD4 positive gluten reactive T cells in the presence of transglutaminase modified gluten peptides.9 42 43
Patients with coeliac disease may have enteric barrier disruption before the development of autoantibodies and thus a susceptibility to enterovirus. However, we believe a more plausible explanation is that enterovirus causes impaired barrier function, which in turn increases the risk of coeliac disease. A challenge in studying the temporal association is the potential separation in time between the triggering event and disease onset as marked by appearance in the blood of coeliac disease antibodies. The results from our study suggest a certain time window between these events. As stool sampling was restricted to the period between birth and 3 years of age, we are lacking data just before the development of coeliac disease antibodies in a few cases diagnosed after 4 years of age.
Our study design is less sensitive to detection of infections occurring with lower frequency and shorter duration of viral shedding (rotavirus, orthoreovirus), so a non-specific response to several virus infections should be further investigated. If enterovirus is confirmed as a trigger factor, vaccination could reduce the risk of development of coeliac disease. Currently, except for poliovirus, non-polio enterovirus vaccines are not commercially available, but efforts are ongoing to develop such vaccines.27 44 Further research in larger sample sets are needed to confirm our results.
Conclusion
In this longitudinal study, we found that a higher frequency of enterovirus infections was associated with increased risk of coeliac disease. Given the limited number of cases, we call for corroboration in similar studies and preferably interventional studies to reach conclusions about causality.
What is already known on this topic
Recent evidence suggests that viral infections are involved in an animal model of coeliac disease
Prospective studies have shown a higher prevalence of infections in children before diagnosis of coeliac disease
Previous studies of adenoviruses and enteroviruses have been limited to cross sectional designs, and reverse causality is a possible explanation for these observations
What this study adds
Children with increased genetic risk of coeliac disease had a higher frequency of enteroviruses in stool samples before development of coeliac disease than did healthy controls
Adenovirus was not associated with coeliac disease
Identification of specific viruses as triggers of coeliac disease may have implications for preventive strategies and justify future studies to clarify mechanisms
Acknowledgments
We express our sincere gratitude to the participants. We also especially thank Nicolai Andre Lund-Blix of Oslo University Hospital, Norway, for his help with the breastfeeding data; Lisa Merete Markussen of Akershus University Hospital for help with analysing coeliac disease antibodies in historical blood samples; Kaja Klykken Aas and Nina Stensrud from the Norwegian Institute of Public Health for handling plasma and stool samples in the MIDIA project; and the public health nurses in the MIDIA project (Asbjørg Skorge Hornseth, Liv Kjeldstadli Onsrud, and Turid Wetlesen). Adriana Lourenco Caixinha, Universidade de Lisboa, is acknowledged for expert technical help in nucleic acid extraction during her IFMSA internship.
Web extra.
Extra material supplied by authors
Supplementary materials
Contributors: CRK and KS coordinated the coeliac disease sub-study in MIDIA, wrote the analysis plans, and had the primary responsibility for writing the paper. KS and LCS supervised the study, interpreted the data, and reviewed and commented on all drafts. GT did the statistical analysis, contributed with reagents/materials/analysis tools, and reviewed and commented on drafts. OC supervised the laboratory testing of virus PCR, contributed with analysis tools, and reviewed and commented on the data analysis. KC and LK did the laboratory testing of virus PCR and reviewed and commented on drafts. KSR designed the MIDIA study and reviewed and commented on the data analyses. TR, KL, and KM reviewed and contributed to the data processing. The corresponding author attests that all listed authors meet authorship criteria and that no others meeting the criteria have been omitted. KS is the guarantor.
Funding: The Research Council of Norway (grant numbers 205086/F20 to GT, 166515/V50 to KSR) and the Project for the Conceptual Development of Research Organization 00064203 (University Hospital Motol, Prague, Czech Republic). CRK received grant support from Østfold Hospital Trust, Kalnes, Norway, and KS received grant support from Oak Foundation, Geneva, Switzerland. The EliA Celikey and EliA Gliadin kits were supported by Thermo Fisher Scientific, Norway. The Norwegian Coeliac Society supported the study financially. The funders had no influence on the contents of this paper.
Competing interests: All authors have completed the ICMJE uniform disclosure form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: no support from any organisation for the submitted work other than that described above; no financial relationships with any organisations that might have an interest in the submitted work in the previous three years; no other relationships or activities that could appear to have influenced the submitted work.
Ethical approval: The study was approved by the Regional Committee for Medical Research Ethics. Written informed consent was obtained from the parents.
Data sharing: The individual raw data are provided in supplementary figure B. The Stata codes for all data handling and analyses are available on request from the corresponding author. Norwegian legislation prevents publication of the full dataset, but data supporting the presented results are available from the authors on reasonable request.
Transparency: The lead author (the manuscript’s guarantor) affirms that this manuscript is an honest, accurate, and transparent account of the study being reported; that no important aspects of the study have been omitted; and that any discrepancies from the study as planned (and, if relevant, registered) have been explained.
References
- 1. Green PH, Cellier C. Celiac disease. N Engl J Med 2007;357:1731-43. 10.1056/NEJMra071600 [DOI] [PubMed] [Google Scholar]
- 2. Romanos J, Rosén A, Kumar V, et al. PreventCD Group Improving coeliac disease risk prediction by testing non-HLA variants additional to HLA variants. Gut 2014;63:415-22. 10.1136/gutjnl-2012-304110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mårild K, Vistnes M, Tapia G, et al. Midpregnancy and cord blood immunologic biomarkers, HLA genotype, and pediatric celiac disease. J Allergy Clin Immunol 2017;139:1696-8. 10.1016/j.jaci.2016.10.016 [DOI] [PubMed] [Google Scholar]
- 4. Liu E, Lee HS, Aronsson CA, et al. TEDDY Study Group Risk of pediatric celiac disease according to HLA haplotype and country. N Engl J Med 2014;371:42-9. 10.1056/NEJMoa1313977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Agardh D, Lee HS, Kurppa K, et al. TEDDY Study Group Clinical features of celiac disease: a prospective birth cohort. Pediatrics 2015;135:627-34. 10.1542/peds.2014-3675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Abadie V, Sollid LM, Barreiro LB, Jabri B. Integration of genetic and immunological insights into a model of celiac disease pathogenesis. Annu Rev Immunol 2011;29:493-525. 10.1146/annurev-immunol-040210-092915 [DOI] [PubMed] [Google Scholar]
- 7. Mårild K, Kahrs CR, Tapia G, Stene LC, Størdal K. Infections and risk of celiac disease in childhood: a prospective nationwide cohort study. Am J Gastroenterol 2015;110:1475-84. 10.1038/ajg.2015.287 [DOI] [PubMed] [Google Scholar]
- 8. Kemppainen KM, Lynch KF, Liu E, et al. TEDDY Study Group Factors That Increase Risk of Celiac Disease Autoimmunity After a Gastrointestinal Infection in Early Life. Clin Gastroenterol Hepatol 2017;15:694-702.e5. 10.1016/j.cgh.2016.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bouziat R, Hinterleitner R, Brown JJ, et al. Reovirus infection triggers inflammatory responses to dietary antigens and development of celiac disease. Science 2017;356:44-50. 10.1126/science.aah5298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Webb A, Starr M. Acute gastroenteritis in children. Aust Fam Physician 2005;34:227-31. [PubMed] [Google Scholar]
- 11. Schumann M, Siegmund B, Schulzke JD, Fromm M. Celiac Disease: Role of the Epithelial Barrier. Cell Mol Gastroenterol Hepatol 2017;3:150-62. 10.1016/j.jcmgh.2016.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stene LC, Honeyman MC, Hoffenberg EJ, et al. Rotavirus infection frequency and risk of celiac disease autoimmunity in early childhood: a longitudinal study. Am J Gastroenterol 2006;101:2333-40. 10.1111/j.1572-0241.2006.00741.x [DOI] [PubMed] [Google Scholar]
- 13. Kagnoff MF, Paterson YJ, Kumar PJ, et al. Evidence for the role of a human intestinal adenovirus in the pathogenesis of coeliac disease. Gut 1987;28:995-1001. 10.1136/gut.28.8.995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mahon J, Blair GE, Wood GM, Scott BB, Losowsky MS, Howdle PD. Is persistent adenovirus 12 infection involved in coeliac disease? A search for viral DNA using the polymerase chain reaction. Gut 1991;32:1114-6. 10.1136/gut.32.10.1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mercalli A, Lampasona V, Klingel K, et al. No evidence of enteroviruses in the intestine of patients with type 1 diabetes. Diabetologia 2012;55:2479-88. 10.1007/s00125-012-2591-4 [DOI] [PubMed] [Google Scholar]
- 16. Stene LC, Witsø E, Torjesen PA, et al. Islet autoantibody development during follow-up of high-risk children from the general Norwegian population from three months of age: design and early results from the MIDIA study. J Autoimmun 2007;29:44-51. 10.1016/j.jaut.2007.04.003 [DOI] [PubMed] [Google Scholar]
- 17. Cinek O, Witsø E, Jeansson S, et al. Longitudinal observation of enterovirus and adenovirus in stool samples from Norwegian infants with the highest genetic risk of type 1 diabetes. J Clin Virol 2006;35:33-40. 10.1016/j.jcv.2005.03.007 [DOI] [PubMed] [Google Scholar]
- 18. Husby S, Koletzko S, Korponay-Szabó IR, et al. ESPGHAN Working Group on Coeliac Disease Diagnosis. ESPGHAN Gastroenterology Committee. European Society for Pediatric Gastroenterology, Hepatology, and Nutrition European Society for Pediatric Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease. J Pediatr Gastroenterol Nutr 2012;54:136-60. 10.1097/MPG.0b013e31821a23d0 [DOI] [PubMed] [Google Scholar]
- 19. Bürgin-Wolff A, Gaze H, Hadziselimovic F, et al. Antigliadin and antiendomysium antibody determination for coeliac disease. Arch Dis Child 1991;66:941-7. 10.1136/adc.66.8.941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bürgin-Wolff A, Dahlbom I, Hadziselimovic F, Petersson CJ. Antibodies against human tissue transglutaminase and endomysium in diagnosing and monitoring coeliac disease. Scand J Gastroenterol 2002;37:685-91. 10.1080/00365520212496 [DOI] [PubMed] [Google Scholar]
- 21. Arguelles-Grande C, Norman GL, Bhagat G, Green PH. Hemolysis interferes with the detection of anti-tissue transglutaminase antibodies in celiac disease. Clin Chem 2010;56:1034-6. 10.1373/clinchem.2009.141242 [DOI] [PubMed] [Google Scholar]
- 22. Myléus A, Ivarsson A, Webb C, et al. Celiac disease revealed in 3% of Swedish 12-year-olds born during an epidemic. J Pediatr Gastroenterol Nutr 2009;49:170-6. 10.1097/MPG.0b013e31818c52cc [DOI] [PubMed] [Google Scholar]
- 23. Honkanen H, Oikarinen S, Pakkanen O, et al. Human enterovirus 71 strains in the background population and in hospital patients in Finland. J Clin Virol 2013;56:348-53. 10.1016/j.jcv.2012.11.018 [DOI] [PubMed] [Google Scholar]
- 24. Claas EC, Schilham MW, de Brouwer CS, et al. Internally controlled real-time PCR monitoring of adenovirus DNA load in serum or plasma of transplant recipients. J Clin Microbiol 2005;43:1738-44. 10.1128/JCM.43.4.1738-1744.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cinek O, Kramna L, Mazankova K, et al. Virus genotyping by massive parallel amplicon sequencing: adenovirus and enterovirus in the Norwegian MIDIA study. J Med Virol 2018;10: [Epub ahead of print]. 10.1002/jmv.25361. [DOI] [PubMed] [Google Scholar]
- 26. Tapia G, Cinek O, Rasmussen T, et al. Human enterovirus RNA in monthly fecal samples and islet autoimmunity in Norwegian children with high genetic risk for type 1 diabetes: the MIDIA study. Diabetes Care 2011;34:151-5. 10.2337/dc10-1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baggen J, Thibaut HJ, Strating JRPM, van Kuppeveld FJM. Publisher Correction: The life cycle of non-polio enteroviruses and how to target it. Nat Rev Microbiol 2018;16:391. 10.1038/s41579-018-0022-3 [DOI] [PubMed] [Google Scholar]
- 28. Pallansch MA, Roos RP. Enteroviruses: polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses. In: Knipe DM, Howley PM, eds. Fields Virology. 7th ed Lippincott Williams &Wilkins, 2007: 839-94. [Google Scholar]
- 29. Witsø E, Palacios G, Cinek O, et al. High prevalence of human enterovirus a infections in natural circulation of human enteroviruses. J Clin Microbiol 2006;44:4095-100. 10.1128/JCM.00653-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Viskari H, Oikarinen S, Hoppu S, et al. Live attenuated enterovirus vaccine (OPV) is not associated with islet autoimmunity in children with genetic susceptibility to type 1 diabetes: prospective cohort study. Diabetologia 2018;61:203-9. 10.1007/s00125-017-4410-4 [DOI] [PubMed] [Google Scholar]
- 31.Norwegian Institute of Public Health. Vaksinasjonsveilederen for helsepersonell (information about the norwegian vaccine program). 2017 [cited 6 February 2018]. https://www.fhi.no/nettpub/vaksinasjonsveilederen-for-helsepersonell/vaksiner-mot-de-enkelte-sykdommene/poliovaksinasjon-poliomyelitt---vei/.
- 32. Nilsen RM, Vollset SE, Gjessing HK, et al. Self-selection and bias in a large prospective pregnancy cohort in Norway. Paediatr Perinat Epidemiol 2009;23:597-608. 10.1111/j.1365-3016.2009.01062.x [DOI] [PubMed] [Google Scholar]
- 33. Witsø E, Cinek O, Aldrin M, et al. Predictors of sub-clinical enterovirus infections in infants: a prospective cohort study. Int J Epidemiol 2010;39:459-68. 10.1093/ije/dyp333 [DOI] [PubMed] [Google Scholar]
- 34. Oikarinen M, Tauriainen S, Oikarinen S, et al. Type 1 diabetes is associated with enterovirus infection in gut mucosa. Diabetes 2012;61:687-91. 10.2337/db11-1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Honkanen H, Oikarinen S, Nurminen N, et al. Detection of enteroviruses in stools precedes islet autoimmunity by several months: possible evidence for slowly operating mechanisms in virus-induced autoimmunity. Diabetologia 2017;60:424-31. 10.1007/s00125-016-4177-z [DOI] [PubMed] [Google Scholar]
- 36. Krogvold L, Edwin B, Buanes T, et al. Detection of a low-grade enteroviral infection in the islets of langerhans of living patients newly diagnosed with type 1 diabetes. Diabetes 2015;64:1682-7. 10.2337/db14-1370 [DOI] [PubMed] [Google Scholar]
- 37. Tapia G, Cinek O, Rasmussen T, et al. Human enterovirus RNA in monthly fecal samples and islet autoimmunity in Norwegian children with high genetic risk for type 1 diabetes: the MIDIA study. Diabetes Care 2011;34:151-5. 10.2337/dc10-1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kagnoff MF, Austin RK, Hubert JJ, Bernardin JE, Kasarda DD. Possible role for a human adenovirus in the pathogenesis of celiac disease. J Exp Med 1984;160:1544-57. 10.1084/jem.160.5.1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lähdeaho ML, Parkkonen P, Reunala T, Mäki M, Lehtinen M. Antibodies to E1b protein-derived peptides of enteric adenovirus type 40 are associated with celiac disease and dermatitis herpetiformis. Clin Immunol Immunopathol 1993;69:300-5. 10.1006/clin.1993.1184 [DOI] [PubMed] [Google Scholar]
- 40. Lawler M, Humphries P, O’Farrelly C, et al. Adenovirus 12 E1A gene detection by polymerase chain reaction in both the normal and coeliac duodenum. Gut 1994;35:1226-32. 10.1136/gut.35.9.1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schumann M, Siegmund B, Schulzke JD, Fromm M. Celiac disease: role of the epithelial barrier. Cell Mol Gastroenterol Hepatol 2017;3:150-62. 10.1016/j.jcmgh.2016.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sollid LM, Jabri B. Triggers and drivers of autoimmunity: lessons from coeliac disease. Nat Rev Immunol 2013;13:294-302. 10.1038/nri3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Matzinger P. The danger model: a renewed sense of self. Science 2002;296:301-5. 10.1126/science.1071059 [DOI] [PubMed] [Google Scholar]
- 44. Stone VM, Hankaniemi MM, Svedin E, et al. A Coxsackievirus B vaccine protects against virus-induced diabetes in an experimental mouse model of type 1 diabetes. Diabetologia 2018;61:476-81. 10.1007/s00125-017-4492-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials