

Abstract

DNA-encoded chemical libraries (DELs) provide a high-throughput and cost-effective route for screening billions of unique molecules for binding affinity for diverse protein targets. Identifying candidate compounds from these libraries involves affinity selection, DNA sequencing, and measuring enrichment in a sample pool of DNA barcodes. Successful detection of potent binders is affected by many factors, including selection parameters, chemical yields, library amplification, sequencing depth, sequencing errors, library sizes, and the chosen enrichment metric. To date, there has not been a clear consensus about how enrichment from DEL selections should be measured or reported. We propose a normalized z-score enrichment metric using a binomial distribution model that satisfies important criteria that are relevant for analysis of DEL selection data. The introduced metric is robust with respect to library diversity and sampling and allows for quantitative comparisons of enrichment of n-synthons from parallel DEL selections. These features enable a comparative enrichment analysis strategy that can provide valuable information about hit compounds in early stage drug discovery.
Keywords: DNA-encoded libraries, data analysis, drug discovery, affinity selection
Introduction
The DNA-encoded chemical library (DEL) platform combines the strengths of combinatorial chemical synthesis and next-generation DNA sequencing to provide a high-throughput and cost-effective drug discovery strategy.1−4 Generally, a DEL is a collection of small molecules in which each member is covalently linked to a segment of synthetic DNA.5 Each DNA sequence is used as a molecular “barcode” that encodes information about the structure of its associated small molecule. Via labeling with DNA barcodes, billions of druglike molecules can be screened for binding affinity for a protein target as a complex mixture, and candidate compounds can be identified by interpreting the output of DNA sequencing. The most commonly utilized method of DEL synthesis is the so-called “DNA-recorded” strategy wherein short DNA oligomers are sequentially ligated to a growing DNA polymer that is connected via a molecular tether to a site of small molecule synthesis.1 Each of the oligomer sequences represents a specific chemical building block added or chemical transformation performed on the small molecule. The completed DNA barcode provides a recorded recipe for constructing a specific library member because the DNA sequence is constructed in parallel with the small molecule. By leveraging “split and pool” combinatorial synthesis,6 one can develop a final pool of DEL compounds that can easily reach into the millions or even billions of unique molecules. In addition, library designs incorporating different scaffolds, building blocks, and reactions can produce diverse compound libraries with a wide variety of molecular shapes, biophysical properties, and target binding profiles.7 Moreover, with the inclusion of library-identifying sequences in the DNA barcode, multiple libraries can be combined into a multilibrary pool, which greatly increases the chemical space sampled in a single DEL screen.
Hit discovery with DELs is facilitated by affinity selection experiments that involve incubating a DEL pool with an epitope-tagged protein target, separating unbound molecules, and then eluting and collecting the protein-bound molecules. Relative populations of library members are thus perturbed in a postselection DEL sample due to the distribution of binding affinities for the target protein. This “selection” process is often repeated over several iterations, with the goal of producing a postselection DEL pool that is enriched with high-affinity binders. Such changes in the composition of a DEL induced by selection experiments are monitored by DNA sequencing, decoding into molecular representations, and subsequent statistical and cheminformatic analysis. Library populations can additionally be perturbed by changing the selection conditions.8,9 For example, Wu et al. used cell-based DEL selections to identify antagonists of the NK3 tachykinin receptor by comparing the output of selections with and without a known NK3 binder.10 Similarly, Soutter et al. successfully used parallel DEL selections to identify compounds with affinity for specific binding sites to the enoyl-acyl-carrier protein reductase InhA from Mycobacterium tuberculosis.11 Cuozzo et al. used parallel selections of a DEL for Bruton’s tyrosine kinase (BTK) with varying target concentrations and the presence or absence of ATP and dasatinib to elucidate the relative binding affinities and binding mechanisms of novel BTK inhibitors.12 Other selection parameters, including the washing protocol and incubation time, can be varied to probe binding characteristics like on and off rates.13
Reports of novel analysis strategies for DEL selection data have been scarce since early publications of successful DEL screens.14,15 Traditionally, DEL selection analysis involves visualization of a two- or three-dimensional scatter plot (“cubic view”), in which each unique decoded ligand is positioned according to its component building blocks on each of the plot axes. Each point representing a unique library member is colored or sized by the number of observed molecules, or “counts”. Patterns such as lines and planes can be viewed in this representation that imply enriched chemical substructures within the library. Because these observed features are groups of conserved building blocks from a combinatorial synthetic library, these substructure groups are called n-synthons, where n is the number of cycles in the conserved group of synthetic cycles (Supporting Information section S1 further describes nomenclature for DEL analysis). Observing the enrichment of n-synthons can indicate the presence of structure–activity relationships (SARs), hint at possible binding modes, or signify truncated products from failed or incomplete chemical reactions. The interpretation of these features often depends on the specific DEL design and selection conditions. This substructure-seeking strategy has become an important part of DEL analysis and has been adopted by many other practitioners.10,12,15−19
There has not yet been a consensus about how enrichment for DEL members should be measured and evaluated. Early work utilized visualization of count data in three-dimensional scatter plots. Satz proposed plotting counts of library members against varying target concentrations from multiple selections to address the issue of variable synthetic yield in a DEL.20,21 Buller et al. have described using the negative binomial distribution to model count data and determine p values for enriched compounds.3,22,23 Others have proposed a count-to-mean ratio to evaluate enrichment.24,25 Kuai et al. utilized a count-to-mean count ratio metric and demonstrated that it had advantages over a count-to-baseline measure26 because it normalizes for sequencing depth.25 Kleiner et al. evaluated enrichment as a ratio of the preselection population fraction to the postselection population fraction.27 Most recently, Amigo et al. report ranking different n-synthons by the number of standard deviations from the average count, analogous to a standard z-score metric.28
While developing our own DEL informatics and analysis pipeline, we observed that naïve measures of n-synthon enrichment were dependent on sample size and library diversity, which impeded direct quantitative comparisons of enrichment of n-synthons in DELs. We therefore initiated a search for more useful enrichment metrics, beginning by enumerating desirable properties of an enrichment function. We then developed a set of test scenarios to evaluate an enrichment metric for meeting the enumerated criteria, which included analysis of naïve library screens, selections with strong enrichment and wide structure–activity relationships (or more accurately, “structure–enrichment relationships”), selections with no significant enrichment, and comparing selections with uneven sampling of decoded library members (Supporting Information section S2). This exercise led us to consider the normalized z-score to be the most successful metric in meeting the desired criteria for an enrichment function. In the following sections, we introduce this enrichment metric, describe some of its properties, and demonstrate its use with a test system of a triazine-based DEL and selections against human soluble epoxide hydrolase (sEH).
Results and Discussion
Considerations for an Enrichment Metric
We began our investigation into DEL selection enrichment metrics by enumerating desirable characteristics of an enrichment function. First, a successful enrichment metric should be insensitive to the amount of sampling of the library pool. For most selection experiments, enrichment in target-containing samples leads to more molecules being retained after the selection compared to a non-target control (NTC) selection, presumably due to the increased number of potential binding sites and hydrophobic surfaces available. Subsequently, a target selection pool is often sequenced more deeply than an NTC pool, and therefore, comparing simple measures like molecule counts can often be misleading. Without normalizing for sampling, the pool that has been sampled more can often have features that appear to be further enriched compared to those of the smaller pool simply due to higher sampling.
An additional requirement is that the enrichment metric should be insensitive to library diversity or the magnitudes of the expected populations of n-synthons. This requirement was introduced for three reasons: (1) to enable measurement of enrichment of compounds in very large libraries where the diversity is typically severely undersampled, (2) to enable comparisons between libraries of greatly different sizes, and (3) to yield compatible enrichment measurements for different types of n-synthons within libraries. The diversities of a monosynthon (one conserved building block) and a trisynthon (three conserved building blocks) are of different orders of magnitude, which implies that their pre- and postselection populations will also be of different orders of magnitude. An ideal enrichment metric would address both extremes of populations in a similar manner so that they can be plotted together in a single visualization.
It is also desirable for an enrichment function to have quantifiable uncertainty from sampling. When there are fewer total molecules decoded after a selection, the uncertainties in the populations of various features should be higher and this should be reflected in the uncertainties in their enrichment. Quantifiable uncertainty also allows for the determination of significant differences in enrichment between two samples. This would allow analysts to be mindful of uncertainties when choosing which features to assess with resynthesis and validation assays.
Lastly, an enrichment metric should be easily interpretable. The metric should be directly proportional to enrichment, as defined as the ratio of the observed population to the expected population in an unselected sample. The metric should enable the analyst to distinguish between signal and noise and additionally to detect significant differences in enrichment when comparing multiple selections under different experimental conditions.
Normalized z-Score Metric
On the basis
of the observed behaviors of candidate enrichment metrics in several
example scenarios (Supporting Information section S2), we found the normalized z-score to be
the most successful enrichment metric. This enrichment function models
selection data with the binomial distribution, which provides the
probability of observing an event k times out of n independent samples given the probability of occurrence p. This strategy therefore approximates the DNA sequencing
process as random sampling with replacement of a DEL pool. The binomial
distribution is beneficial for analyzing combinatorial DEL data because
for low p it closely resembles the Poisson distribution
for count data and for high p it resembles the
normal distribution that is often a better fit for high count data.
Thus, the binomial distribution can model both high-diversity features
(e.g., trisynthons) and low-diversity features (e.g., monosynthons).
It is important to note that using a z-score type
metric with the binomial distribution depends not only on the expected
and observed counts and populations (Ci and pi for expected and Co and po for observed, respectively)
but also on the number of samples, n (eq 1). We therefore modified the expression
by normalizing by an additional factor of the square root of the number
of decoded samples (eq 2). The final normalized z-score showed a low sensitivity
to sampling by normalizing by 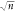 , and it additionally showed a low sensitivity
to expected probabilities by normalizing by a factor dependent on pi.
, and it additionally showed a low sensitivity
to expected probabilities by normalizing by a factor dependent on pi.
| 1 |
| 2 |
We investigated the interpretation of the normalized z-score by mathematical derivation and using simulated data. We found that the normalized z-score is a linear function of fold enrichment (defined as the ratio of observed to expected population fraction) but with different slopes for different values of the expected population. In eq 2, it is observed that for any specific level of enrichment (po/pi), the normalized z-score scales the enrichment by a factor of the square root of pi/qi where qi = 1 – pi. Our experience from analyzing our own DEL selection data has led us to view zn ≥ 1 for any n-synthon as being an indicator of significant enrichment (differences in DEL composition and selection protocols might require an adjusted threshold). Thus, for a three-cycle library with 1000 synthons in each cycle, a zn value of 1 is roughly equivalent to a 30-fold enrichment for a monosynthon feature, a 1000-fold enrichment for a disynthon, and a 30000-fold enrichment for a trisynthon. By scaling enrichment by a factor that is dependent on the expected population, we can plot the enrichment of different types of n-synthons in the same range for visual analysis.
As mentioned in the previous section, it is desirable for our enrichment function to have computable uncertainties due to sampling. For this purpose, we utilize the Agresti–Coull interval for the binomial distribution, which was chosen because it tends to yield conservative estimates for observed probabilities even at extreme values of p.29 Thus, observed populations of n-synthons are evaluated as
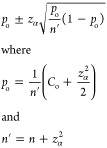 |
3 |
where zα is the 1 – α/2 quantile of the standard normal distribution (e.g., a 95% confidence interval requires α = 0.05 and zα = 1.96), n is the total number of samples (decoded ligands), and Co is the observed count for the feature. Upon combination of the Agresti–Coull estimation interval with the normalized z-score metric, the evaluated uncertainty in enrichment decreases with an increased level of sampling, due to the factor of n in eq 3. Additionally, the uncertainty decreases with an increasing expected population due to the scaling factor in eq 2. Thus, evaluated uncertainties in the enrichment of low-count, high-diversity trisynthons are generally larger than those of higher-count, lower-diversity mono- or disynthons.
Triazine DEL and Selections against Soluble Epoxide Hydrolase
As an illustrative example of our comparative enrichment analysis strategy, we have generated a DEL (hereafter termed “triazine DEL”) with a design closely following the DEL-B library previously described by Clark et al.15 The two library designs are similar in that they both link amines to a triazine core scaffold via an amino acid linker, but the specific building blocks included in each library were independently chosen. The triazine DEL contains 171 amino acids in cycle 1 that are appended to the triazine ring and 1017 amines in cycle 3 that form amide linkages with the cycle 1 amino acids. The complete library contains approximately 174 million unique molecules, and among these are close analogues of compounds previously described by Thalji et al., who measured their inhibitory activity for soluble epoxide hydrolase (sEH; encoded by the gene EPHX2).30Compounds of this series are reproduced as disynthons in the triazine DEL, as benzylic amines in cycle 3 are linked to the core triazine scaffold through isonipecotic acid in cycle 1. The inclusion of these known sEH binding ligands in our library provided positive controls for tuning selection experiments, sequencing preparation, and analysis.
The triazine DEL was first sequenced in its naïve or unselected form as a quality control procedure. Two independent library samples were taken, closing polymerase chain reaction (PCR) primers including sequencing indexes were ligated independently by different scientists in different locations, and sequencing was conducted separately for the two samples. Sample 1 resulted in approximately 9 million decoded library members, and sample 2 resulted in approximately 6 million decoded library members. Using a binomial distribution model and an approximation of equal probabilities of observation, we expected a random noise threshold of 4 counts for any specific trisynthon library member in both decoded data sets. Accordingly, in both data sets, the maximum trisynthon count was 4, but this agreement with the expected count distribution was reached only by accounting for errors in the degenerate regions of the DNA barcodes used to count unique molecules after PCR amplification (Supporting Information section S3). The enrichment of library members in the two naïve samples is compared in Figure 1, where n-synthons are colored according to their “dimension”, or value of n. Generally, most n-synthons had normalized z-score values near zero, which implies that the observed population was close to the expected population. We observed some monosynthons (blue) that were significantly under- or overpopulated, but these were similarly populated in both naïve samples. Because trisynthons (yellow) were severely undersampled in these sequencing data sets, their normalized z-score values were generally greater than zero but also were associated with much higher uncertainties. Additionally, because the observation of specific trisynthons is largely due to random selection noise, most trisynthons were observed in only one of the two data sets.
Figure 1.
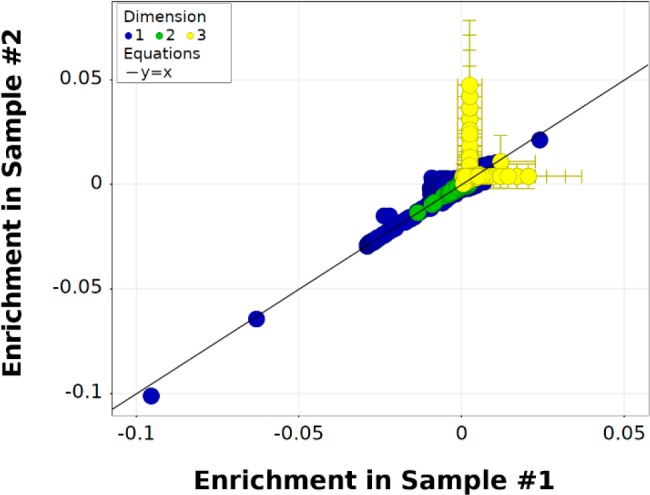
Comparison of enrichment from two independently prepared naïve samples of the triazine DEL. Observed n-synthons are colored by their value of n (their “dimension”), and enrichment is measured as normalized z-scores with their 95% confidence intervals shown as error bars (some error bars are smaller than the data point radii). The y = x line corresponding to equal enrichment between the two samples is plotted for reference.
The triazine DEL was then evaluated in a selection against His-tagged sEH as well as an NTC sample that contained only nickel-NTA magnetic capture beads. Both postselection samples were PCR-amplified, sequenced with the Illumina HiSeq instrument, and decoded into molecular representations for analysis. Table 1 lists the reported IC50 values of selected sEH inhibitors and the enrichment of their disynthon analogues from the triazine DEL. Although correlation between enrichment and binding affinity is known to be weakened by variance in synthetic yields,21 in this case, enrichment was robust enough to clearly distinguish between the most potent and the weaker sEH binders of this series.
Table 1. Selected sEH Inhibitors Reported by Thalji et al.30 and Their Analogues in the Triazine DELa.

IC50 values are from the earlier report, and the evaluated enrichments for the DEL analogues are provided as normalized z-scores with their 95% confidence intervals.
In Figure 2, the enrichment of all observed n-synthons in the target selection is plotted against the corresponding enrichment in the NTC selection. We observed many mono- and disynthons that were significantly more enriched in the target data set than the NTC, which is consistent with target-specific binding. There were additionally some monosynthons that were significantly more enriched in the NTC than in the target data set, which implies binding affinity for the nickel-containing beads. Disynthons that correspond to two of the most potent compounds described by Thalji et al. are highlighted in green. The highlighted disynthons were observed in the target data set but not the NTC data set, and the two most potent inhibitors were indeed more enriched in the target data set than the weaker inhibitors. The remaining points in green represent disynthons from the triazine DEL with different amine and amino acid building block components. Interestingly, our selection did not highlight any specific trisynthon library member but did reveal several disynthons in the form of combinations of cycle 1 amino acids and cycle 3 amines that had significant target-specific enrichment.
Figure 2.
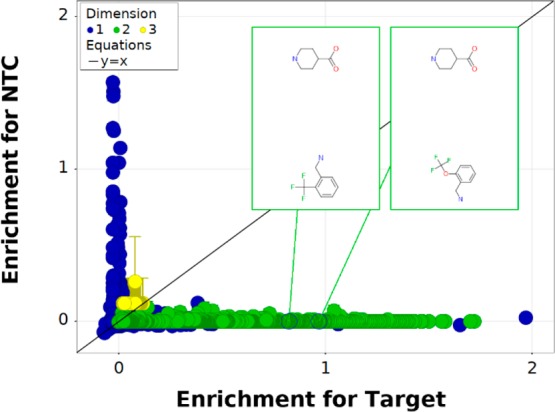
Comparative enrichment plot for a selection against sEH. Enrichment in the target data set is plotted along the horizontal axis against the measured enrichment for a control sample, an NTC, on the vertical axis. This DEL included compounds previously assayed for sEH inhibition by Thalji et al.30 as disynthons from cycles 1 and 3. Each point in the plot is a different n-synthon from the DEL, and the points highlighted in green correspond to analogues of the two most potent of the previously reported compounds. Both of these known inhibitor structures were observed in the target data but not the NTC, and the most potent inhibitors from the earlier publication were significantly more enriched in the target data set than the weaker inhibitors. The remaining points correspond to different combinations of amino acids in cycle 1 and amines in cycles 2 and 3 of the triazine DEL.
As mentioned in Considerations for an Enrichment Metric, it is important for our enrichment function to have a low sensitivity to differences in sampling between two data sets. Without this property, enrichment from different data sets could not be compared quantitatively. To investigate this property for the normalized z-score metric, we generated a subsampled data set from the sEH selection data by randomly removing approximately 90% of the decoded ligands. The initial data set contained 39177803 decoded library members, and the 10% subsampled data set contained 3916867 library members. We further generated a 1% subsampled data set by randomly removing 99% of decoded library members leaving only 392791 samples. Given that the estimated final molecule count after the selection from qPCR analysis was 4.5 × 1010, these levels of sampling correspond to sampling coverage of 0.087% for the full decoded data set and 0.0008% for the smallest subsampled data set. As shown in Figure 3, the evaluated enrichment for library members in the smaller subsampled data sets largely agree with the evaluations in the full data set. Deviations are more noticeable in the 1% subsampled data set, where the trisynthons appear to be more enriched in the smaller data set due to severe undersampling. However, the estimated uncertainties are also much larger in the smaller data set, implying that there is no significant difference between the two samples when estimated uncertainties are considered. These examples suggest that the normalized z-score metric is robust when the sampling coverage is low and when comparing two data sets with very different amounts of sampling. In contrast, other metrics we examined showed significant systematic errors when comparing two data sets with uneven sampling (Supporting Information section S2.2.4).
Figure 3.

Enrichment of n-synthons evaluated with the normalized z-score metric from the fully sampled data set compared to randomly subsampled data sets. Panel A plots the full data set against the same data with 90% of samples randomly removed, while panel B plots the full data set against the same data set with 99% of samples randomly removed. This in silico experiment simulates the effects of large differences in sampling between two decoded DEL selection samples.
Conclusions
There is much information that can be gleaned from comparing the output of parallel affinity selections of DNA-encoded chemical libraries. By observing different perturbations in the populations of library members due to differences in selection parameters, one can theoretically gain information such as binding sites, target selectivity, relative affinity, and kinetics. To enable quantitative comparison of enrichment between multiple experiments, enrichment must be measured in a way that is accurate and insensitive to sampling. We have developed an informatics and analysis pipeline that attempts to directly compare parallel DEL selection experiments by plotting measured enrichment of each n-synthon in a library in each selection against each other in a two-dimensional scatter plot. In this visualization, n-synthons that are enriched in one, neither, or both selections are obvious to the analyst. We required a novel enrichment metric that has a low sensitivity to sampling and diversity, and we chose to utilize a normalized z-score metric with the Agresti–Coull estimation interval to fulfill this need. This metric has a low sensitivity to sampling and diversity and provides a conservative error bar for uncertainties in enrichment.
Experimental Procedures
Synthesis of the Triazine DEL
The encoded split-and-pool DEL concept and the triazine DEL structure and design were adapted from a previous report.15 The triazine DEL was constructed through three cycles of chemical transformations, DNA oligomer (“codon”) ligations, and subsequent pooling. Cycle 1 consisted of codon ligation, attachment of cyanuric chloride, and nucleophilic substitution of amines and amino acids. Cycle 2 consisted of nucleophilic substitution of amines, cycle 2 codon ligation, and pooling. Cycle 3 consisted of cycle 3 codon ligation, acylation of amines, and pooling. After completion of the main build, the library was further ligated with a DNA tag to encode the library structure. Aliquots of the triazine DEL were modified with DNA oligomers containing a selection experiment identification region, a degenerate region to act as a unique molecule identifier (UMI) amplification control region, a diversity region, and a primer region for use in selection experiments. Detailed experimental procedures for the triazine DEL synthesis are provided in Supporting Information section S4.
Selection with Soluble Epoxide Hydrolase
Selection of sEH binding molecules was performed following an affinity capture-based method described previously.19 His6-tagged sEH (item no. 10011669, Cayman Chemical) at a final concentration of 1 μM was incubated with DEL at a concentration where each compound had one million copies in a model cytosolic buffer containing HEPES (20 mM, pH 7.5), potassium acetate (134 mM), sodium acetate (8 mM), sodium chloride (4 mM), magnesium acetate (0.8 mM), imidazole (10 mM), TCEP (1 mM), CHAPS (1 mM), and sheared salmon sperm DNA (1 mg/mL, Invitrogen) in a final volume of 200 μL. A non-target control selection was set up in parallel in which no protein was added to the library. The incubation lasted 45 min at room temperature with continuous shaking. The target and associated library molecules were then captured by the addition of 200 μL of prewashed HisPur Ni-NTA magnetic beads (Thermo Scientific) followed by a 10 s vortex. Beads were washed three times with cytosolic buffer without sheared salmon sperm DNA. Then the associated library molecules were eluted by heating the beads at 80 °C for 10 min. The resulting eluent in which sEH binding molecules were enriched was further added with fresh sEH protein to initiate another round of selection following the same protocol. After four rounds of selection, the encoded oligonucleotides from the last eluent were amplified using Platinum Taq DNA Polymerase High Fidelity (Invitrogen) with denaturation at 95 °C, annealing at 58 °C, and extension at 72 °C using primers that incorporate complementary sequences to the library headpiece or tailpiece along with the Illumina READ 1 or READ 2 sequences required for clustering and subsequent sequencing on an Illumina HiSeq instrument.
Analysis of DEL Selection Data
Selection output was processed using in-house data pipelines and code. Briefly, raw DNA sequences from Illumina sequencing were processed by parsing the read sequences according to the known library encoding structure and querying for perfect matches of encoding sequences. Decoded library members were then aggregated and counted using a degenerate encoding region as a unique molecular identifier and a graph-based counting algorithm similar to that of Smith et al.31 (Supporting Information section S3). Counts were aggregated across all possible n-synthon types, and enrichment was evaluated for every observed n-synthon in the library. For each enrichment evaluation, expected populations were approximated with the assumption of equal populations across n-synthons of the same class (i.e., uniform synthetic yield).
Acknowledgments
The authors thank our colleagues in the Center for Drug Discovery and foundations that have supported our expansion.
Glossary
Abbreviations
- DEL
DNA-encoded library
- NTC
non-target control
- sEH
soluble epoxide hydrolase
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscombsci.8b00116.
A summary of nomenclature for DEL analysis, a comparison of our candidate enrichment metrics, a description of our unique molecule counting strategy, and details concerning the synthesis and analysis of the triazine DEL (PDF)
Author Present Address
⊥ J.B.J.M.: Dotmatics Australia Pty. Ltd., 25 Burton St., Glebe, NSW 2037, Australia.
Author Contributions
J.C.F. composed the manuscript draft and performed the DEL enrichment analysis. K.R. performed decoding of DNA sequencing data. D.R.L., J.B.J.M., and C.S.K. contributed ideas to this work, including utilizing a graph-based unique molecule counter. N.S. synthesized the triazine-focused DEL. Z.Y. performed the affinity selection of the triazine-focused DEL against sEH. M.M.M. guided portions of this work and leads the Center for Drug Discovery at the Baylor College of Medicine. All authors reviewed drafts of the manuscript.
These studies were supported by the Welch Foundation (Grant Q-0042), a Core Facility Support Award (RP160805) from the Cancer Prevention Research Institute of Texas (CPRIT), Grant OPP1160866 from The Bill and Melinda Gates Foundation, and National Institutes of Health Grant P01HD087157 from The Eunice Kennedy Shriver National Institute of Child Health and Human Development.
The authors declare no competing financial interest.
Supplementary Material
References
- Goodnow R. A. Jr; Dumelin C. E.; Keefe A. D. DNA-Encoded Chemistry: Enabling the Deeper Sampling of Chemical Space. Nat. Rev. Drug Discovery 2017, 16 (2), 131. 10.1038/nrd.2016.213. [DOI] [PubMed] [Google Scholar]
- Kleiner R. E.; Dumelin C. E.; Liu D. R. Small-Molecule Discovery from DNA-Encoded Chemical Libraries. Chem. Soc. Rev. 2011, 40 (12), 5707. 10.1039/c1cs15076f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buller F.; Mannocci L.; Scheuermann J.; Neri D. Drug Discovery with DNA-Encoded Chemical Libraries. Bioconjugate Chem. 2010, 21 (9), 1571–1580. 10.1021/bc1001483. [DOI] [PubMed] [Google Scholar]
- Shi B.; Zhou Y.; Huang Y.; Zhang J.; Li X. Recent Advances on the Encoding and Selection Methods of DNA-Encoded Chemical Library. Bioorg. Med. Chem. Lett. 2017, 27 (3), 361–369. 10.1016/j.bmcl.2016.12.025. [DOI] [PubMed] [Google Scholar]
- Brenner S.; Lerner R. A. Encoded Combinatorial Chemistry. Proc. Natl. Acad. Sci. U. S. A. 1992, 89 (12), 5381–5383. 10.1073/pnas.89.12.5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furka A.; Sebestyén F.; Asgedom M.; Dibó G. General Method for Rapid Synthesis of Multicomponent Peptide Mixtures. Int. J. Pept. Protein Res. 1991, 37 (6), 487–493. 10.1111/j.1399-3011.1991.tb00765.x. [DOI] [PubMed] [Google Scholar]
- Franzini R. M.; Randolph C. Chemical Space of DNA-Encoded Libraries. J. Med. Chem. 2016, 59 (14), 6629–6644. 10.1021/acs.jmedchem.5b01874. [DOI] [PubMed] [Google Scholar]
- Clark M. A. Selecting Chemicals: The Emerging Utility of DNA-Encoded Libraries. Curr. Opin. Chem. Biol. 2010, 14 (3), 396–403. 10.1016/j.cbpa.2010.02.017. [DOI] [PubMed] [Google Scholar]
- Wartchow C.Theoretical Considerations of the Application of DNA-Encoded Libraries to Drug Discovery. In A Handbook for DNA-Encoded Chemistry; John Wiley & Sons, Inc., 2014; pp 213–230. [Google Scholar]
- Wu Z.; Graybill T. L.; Zeng X.; Platchek M.; Zhang J.; Bodmer V. Q.; Wisnoski D. D.; Deng J.; Coppo F. T.; Yao G.; Tamburino A.; Scavello G.; Franklin G. J.; Mataruse S.; Bedard K. L.; Ding Y.; Chai J.; Summerfield J.; Centrella P. A.; Messer J. A.; Pope A. J.; Israel D. I. Cell-Based Selection Expands the Utility of DNA-Encoded Small-Molecule Library Technology to Cell Surface Drug Targets: Identification of Novel Antagonists of the NK3 Tachykinin Receptor. ACS Comb. Sci. 2015, 17 (12), 722–731. 10.1021/acscombsci.5b00124. [DOI] [PubMed] [Google Scholar]
- Soutter H. H.; Centrella P.; Clark M. A.; Cuozzo J. W.; Dumelin C. E.; Guie M.-A.; Habeshian S.; Keefe A. D.; Kennedy K. M.; Sigel E. A.; Troast D. M.; Zhang Y.; Ferguson A. D.; Davies G.; Stead E. R.; Breed J.; Madhavapeddi P.; Read J. A. Discovery of Cofactor-Specific, bactericidal Mycobacterium tuberculosis InhA Inhibitors Using DNA-Encoded Library Technology. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (49), E7880–E7889. 10.1073/pnas.1610978113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuozzo J. W.; Centrella P. A.; Gikunju D.; Habeshian S.; Hupp C. D.; Keefe A. D.; Sigel E. A.; Soutter H. H.; Thomson H. A.; Zhang Y.; Clark M. A. Discovery of a Potent BTK Inhibitor with a Novel Binding Mode by Using Parallel Selections with a DNA-Encoded Chemical Library. ChemBioChem 2017, 18 (9), 864–871. 10.1002/cbic.201600573. [DOI] [PubMed] [Google Scholar]
- Hale S. P.Screening Large Compound Collections. In A Handbook for DNA-Encoded Chemistry; John Wiley & Sons, Inc., 2014; pp 281–317. [Google Scholar]
- Mannocci L.; Zhang Y.; Scheuermann J.; Leimbacher M.; De Bellis G.; Rizzi E.; Dumelin C.; Melkko S.; Neri D. High-Throughput Sequencing Allows the Identification of Binding Molecules Isolated from DNA-Encoded Chemical Libraries. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (46), 17670–17675. 10.1073/pnas.0805130105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark M. A.; Acharya R. A.; Arico-Muendel C. C.; Belyanskaya S. L.; Benjamin D. R.; Carlson N. R.; Centrella P. A.; Chiu C. H.; Creaser S. P.; Cuozzo J. W.; Davie C. P.; Ding Y.; Franklin G. J.; Franzen K. D.; Gefter M. L.; Hale S. P.; Hansen N. J. V.; Israel D. I.; Jiang J.; Kavarana M. J.; Kelley M. S.; Kollmann C. S.; Li F.; Lind K.; Mataruse S.; Medeiros P. F.; Messer J. A.; Myers P.; O'Keefe H.; Oliff M. C.; Rise C. E.; Satz A. L.; Skinner S. R.; Svendsen J. L.; Tang L.; van Vloten K.; Wagner R. W.; Yao G.; Zhao B.; Morgan B. A. Design, Synthesis and Selection of DNA-Encoded Small-Molecule Libraries. Nat. Chem. Biol. 2009, 5 (9), 647–654. 10.1038/nchembio.211. [DOI] [PubMed] [Google Scholar]
- Encinas L.; O’Keefe H.; Neu M.; Remuiñán M. J.; Patel A. M.; Guardia A.; Davie C. P.; Pérez-Macıas N.; Yang H.; Convery M. A.; Messer J. A.; Pérez-Herrán E.; Centrella P. A.; Álvarez-Gómez D.; Clark M. A.; Huss S.; O’Donovan G. K.; Ortega-Muro F.; McDowell W.; Castañeda P.; Arico-Muendel C. C.; Pajk S.; Rullás J.; Angulo-Barturen I.; Álvarez-Ruız E.; Mendoza-Losana A.; Ballell Pages L.; Castro-Pichel J.; Evindar G. Encoded Library Technology as a Source of Hits for the Discovery and Lead Optimization of a Potent and Selective Class of Bactericidal Direct Inhibitors of Mycobacterium Tuberculosis InhA. J. Med. Chem. 2014, 57 (4), 1276–1288. 10.1021/jm401326j. [DOI] [PubMed] [Google Scholar]
- Deng H.; Zhou J.; Sundersingh F. S.; Summerfield J.; Somers D.; Messer J. A.; Satz A. L.; Ancellin N.; Arico-Muendel C. C.; Bedard K. L.; Beljean A.; Belyanskaya S. L.; Bingham R.; Smith S. E.; Boursier E.; Carter P.; Centrella P. A.; Clark M. A.; Chung C.; Davie C. P.; Delorey J. L.; Ding Y.; Franklin G. J.; Grady L. C.; Herry K.; Hobbs C.; Kollmann C. S.; Morgan B. A.; Kaushansky L. J.; Zhou Q. Discovery, SAR, and X-Ray Binding Mode Study of BCATm Inhibitors from a Novel DNA-Encoded Library. ACS Med. Chem. Lett. 2015, 6 (8), 919–924. 10.1021/acsmedchemlett.5b00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris P. A.; King B. W.; Bandyopadhyay D.; Berger S. B.; Campobasso N.; Capriotti C. A.; Cox J. A.; Dare L.; Dong X.; Finger J. N.; Grady L. C.; Hoffman S. J.; Jeong J. U.; Kang J.; Kasparcova V.; Lakdawala A. S.; Lehr R.; McNulty D. E.; Nagilla R.; Ouellette M. T.; Pao C. S.; Rendina A. R.; Schaeffer M. C.; Summerfield J. D.; Swift B. A.; Totoritis R. D.; Ward P.; Zhang A.; Zhang D.; Marquis R. W.; Bertin J.; Gough P. J. DNA-Encoded Library Screening Identifies Benzo[b][1,4]oxazepin-4-Ones as Highly Potent and Monoselective Receptor Interacting Protein 1 Kinase Inhibitors. J. Med. Chem. 2016, 59 (5), 2163–2178. 10.1021/acs.jmedchem.5b01898. [DOI] [PubMed] [Google Scholar]
- Litovchick A.; Dumelin C. E.; Habeshian S.; Gikunju D.; Guié M.-A.; Centrella P.; Zhang Y.; Sigel E. A.; Cuozzo J. W.; Keefe A. D.; Clark M. A. Encoded Library Synthesis Using Chemical Ligation and the Discovery of sEH Inhibitors from a 334-Million Member Library. Sci. Rep. 2015, 5 (1), 10916. 10.1038/srep10916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satz A. L. DNA Encoded Library Selections and Insights Provided by Computational Simulations. ACS Chem. Biol. 2015, 10 (10), 2237–2245. 10.1021/acschembio.5b00378. [DOI] [PubMed] [Google Scholar]
- Satz A. L. Simulated Screens of DNA Encoded Libraries: The Potential Influence of Chemical Synthesis Fidelity on Interpretation of Structure–Activity Relationships. ACS Comb. Sci. 2016, 18 (7), 415–424. 10.1021/acscombsci.6b00001. [DOI] [PubMed] [Google Scholar]
- Buller F.; Zhang Y.; Scheuermann J.; Schäfer J.; Bühlmann P.; Neri D. Discovery of TNF Inhibitors from a DNA-Encoded Chemical Library Based on Diels-Alder Cycloaddition. Chem. Biol. 2009, 16 (10), 1075–1086. 10.1016/j.chembiol.2009.09.011. [DOI] [PubMed] [Google Scholar]
- Buller F.; Steiner M.; Scheuermann J.; Mannocci L.; Nissen I.; Kohler M.; Beisel C.; Neri D. High-Throughput Sequencing for the Identification of Binding Molecules from DNA-Encoded Chemical Libraries. Bioorg. Med. Chem. Lett. 2010, 20 (14), 4188–4192. 10.1016/j.bmcl.2010.05.053. [DOI] [PubMed] [Google Scholar]
- Decurtins W.; Wichert M.; Franzini R. M.; Buller F.; Stravs M. A.; Zhang Y.; Neri D.; Scheuermann J. Automated Screening for Small Organic Ligands Using DNA-Encoded Chemical Libraries. Nat. Protoc. 2016, 11 (4), 764–780. 10.1038/nprot.2016.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuai L.; O’Keeffe T.; Arico-Muendel C. Randomness in DNA Encoded Library Selection Data Can Be Modeled for More Reliable Enrichment Calculation. SLAS Discovery 2018, 23 (5), 405–416. 10.1177/2472555218757718. [DOI] [PubMed] [Google Scholar]
- Satz A. L.; Hochstrasser R.; Petersen A. C. Analysis of Current DNA Encoded Library Screening Data Indicates Higher False Negative Rates for Numerically Larger Libraries. ACS Comb. Sci. 2017, 19 (4), 234–238. 10.1021/acscombsci.7b00023. [DOI] [PubMed] [Google Scholar]
- Kleiner R. E.; Dumelin C. E.; Tiu G. C.; Sakurai K.; Liu D. R. In Vitro Selection of a DNA-Templated Small-Molecule Library Reveals a Class of Macrocyclic Kinase Inhibitors. J. Am. Chem. Soc. 2010, 132 (33), 11779–11791. 10.1021/ja104903x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amigo J.; Rama-Garda R.; Bello X.; Sobrino B.; de Blas J.; Martin-Ortega M.; Jessop T. C.; Carracedo Á.; Loza M. I. G.; Dominguez E. tagFinder: A Novel Tag Analysis Methodology That Enables Detection of Molecules from DNA-Encoded Chemical Libraries. SLAS Discovery 2018, 23 (5), 397–404. 10.1177/2472555217753840. [DOI] [PubMed] [Google Scholar]
- DasGupta A.; Cai T. T.; Brown L. D. Interval Estimation for a Binomial Proportion. Statist. Sci. 2001, 16 (2), 101–133. 10.1214/ss/1009213286. [DOI] [Google Scholar]
- Thalji R. K.; McAtee J. J.; Belyanskaya S.; Brandt M.; Brown G. D.; Costell M. H.; Ding Y.; Dodson J. W.; Eisennagel S. H.; Fries R. E.; Gross J. W.; Harpel M. R.; Holt D. A.; Israel D. I.; Jolivette L. J.; Krosky D.; Li H.; Lu Q.; Mandichak T.; Roethke T.; Schnackenberg C. G.; Schwartz B.; Shewchuk L. M.; Xie W.; Behm D. J.; Douglas S. A.; Shaw A. L.; Marino J. P. Discovery of 1-(1,3,5-Triazin-2-Yl)piperidine-4-Carboxamides as Inhibitors of Soluble Epoxide Hydrolase. Bioorg. Med. Chem. Lett. 2013, 23 (12), 3584–3588. 10.1016/j.bmcl.2013.04.019. [DOI] [PubMed] [Google Scholar]
- Smith T.; Heger A.; Sudbery I. UMI-Tools: Modeling Sequencing Errors in Unique Molecular Identifiers to Improve Quantification Accuracy. Genome Res. 2017, 27 (3), 491–499. 10.1101/gr.209601.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.