

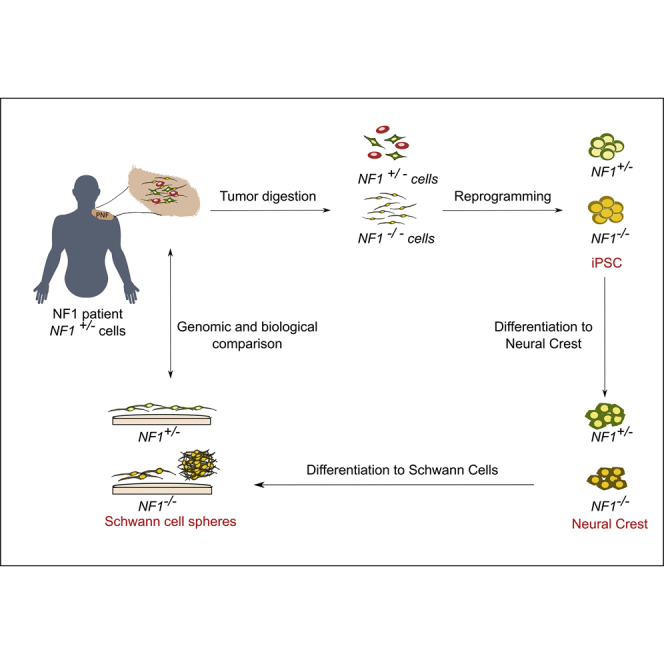
Summary
Neurofibromatosis type 1 (NF1) is a tumor predisposition genetic disease caused by mutations in the NF1 tumor suppressor gene. Plexiform neurofibromas (PNFs) are benign Schwann cell (SC) tumors of the peripheral nerve sheath that develop through NF1 inactivation and can progress toward a malignant soft tissue sarcoma. There is a lack of non-perishable model systems to investigate PNF development. We reprogrammed PNF-derived NF1(−/−) cells, descendants from the tumor originating cell. These NF1(−/−)-induced pluripotent stem cells (iPSCs) captured the genomic status of PNFs and were able to differentiate toward neural crest stem cells and further to SCs. iPSC-derived NF1(−/−) SCs exhibited a continuous high proliferation rate, poor myelination ability, and a tendency to form 3D spheres that expressed the same markers as their PNF-derived primary SC counterparts. They represent a valuable model to study and treat PNFs. PNF-derived iPSC lines were banked for making them available.
Keywords: iPSC, neurofibromatosis type 1, plexiform neurofibroma, Schwann cell, neural crest stem cell, NF1, benign tumor
Graphical Abstract
Highlights
-
•
We generated iPSCs from neurofibromatosis type 1 plexiform neurofibroma (PNF) cells
-
•
PNF-derived iPSCs were differentiated into neural crest and Schwann cells (SCs)
-
•
iPSC-differentiated NF1(−/−) SCs exhibit a high proliferation rate and form spheres
-
•
Sphere-forming SCs express the same markers as their primary PNF counterparts
In this article, Eduard Serra and colleagues describe the generation of iPSCs directly from plexiform neurofibromas (PNFs), benign Schwann cell (SC) tumors associated with neurofibromatosis type 1. iPSCs bearing the double inactivation of the NF1 gene were differentiated into SCs that exhibited a high proliferation rate, a poor myelination ability, and a tendency to form spheres, resembling PNF-derived SCs.
Introduction
Neurofibromatosis type 1 (NF1) is a tumor predisposition genetic disease (VM & Riccardi, 1992) caused by the inheritance of a mutated copy of the NF1 gene, a negative regulator of Ras (Ratner and Miller, 2015). The major disease features involve the nervous system, the skin, and the skeletal system. There is a great variability in the clinical expressivity of the disease, but the development of different tumors of the peripheral nervous system, such as cutaneous neurofibromas (CNFs), plexiform neurofibromas (PNFs) or, less frequently, malignant peripheral nerve sheath tumors (MPNSTs), constitute one of the hallmarks of the disease (Ferner, 2007).
PNFs are mainly developed in the context of NF1 and are thought to be congenital. They are identified in around 50% of NF1 individuals if MRI is used (Mautner et al., 2008). This tumor type constitutes a major source of morbidity (Prada et al., 2012) and, in some cases, undergoes malignant transformation (McCarron and Goldblum, 1998). Surgery is still the standard therapeutic option. However, complete resection can cause important functional deficiencies and sometimes can be unfeasible because of the size or location of the tumor (Packer and Rosser, 2002). Recently, the MEK inhibitor Selumetinib has been used in children with inoperable PNFs showing confirmed partial responses (Dombi et al., 2016).
Neurofibromas are composed of different cell types, mainly Schwann cells (SCs) and endoneurial fibroblasts, as well as perineurial cells and infiltrating immune cells, all embedded in an abundant collagen-rich extracellular matrix (Krone et al., 1983, Peltonen et al., 1988). PNFs arise through a biallelic inactivation of the NF1 gene (Däschner et al., 1997, Hirbe et al., 2015, Kluwe et al., 1999, Rasmussen et al., 2000). Only neurofibroma-derived SCs bear this NF1 inactivation (Kluwe et al., 1999, Li et al., 2016, Maertens et al., 2006, Muir et al., 2001, Serra et al., 2000). Like CNFs, different PNFs arising in the same individual bear different somatic NF1 mutations (Pemov et al., 2017). Also, like CNFs (Garcia-Linares et al., 2011), no recurrent gross genomic alterations or recurrent point mutations have been identified in PNFs besides the involvement of chromosome 17 in the inactivation of the NF1 locus (Beert et al., 2011, Carrió et al., 2018, Miller et al., 2009, Pemov et al., 2017). PNF progression to malignancy often occurs through the formation of a pre-malignant lesion termed atypical neurofibroma, which involves the additional loss of the CDKN2A/B locus (Beert et al., 2011, Higham et al., 2018). It has been shown in one case (Hirbe et al., 2015) that somatic NF1 inactivating mutation is shared by PNF and their subsequent MPNST and metastasis, linking the PNF and MPNST cell of origin.
Different models for PNFs have been developed, both in vitro (primary cells, immortalized cells, 3D culture models) and in vivo (genetically modified mouse models). Primary SC cultures from PNFs have been established (Wallace et al., 2000). However, these cultures are perishable after several passages, limiting their use for molecular and cellular analyses that require large amounts of cells. To overcome this problem, immortalized cell lines have been generated (Li et al., 2016), but inextricably alter the biological status of the cells. These cells have also been used to generate 3D models (Kraniak et al., 2018) to better recapitulate the natural PNF environment of SCs. In addition, different genetically modified animal models using the Cre/lox system to ablate NF1 in specific cell stages of the neural crest stem cells (NCs, for simplicity)-SC axis during development have been generated that develop PNFs (reviewed in Buchstaller et al., 2012). Furthermore, Chen et al. (2014) established a non-germline model of PNF, consisting of the transplantation of Nf1-deficient embryonic dorsal root ganglia/nerve root neurosphere cells to sciatic nerves of nude mice.
Another way of obtaining imperishable cell-based model systems is the generation of induced pluripotent stem cells (iPSCs) (Takahashi and Yamanaka, 2006). iPSCs have been generated to model hereditary cancer syndromes (Papapetrou, 2016), like Fanconi anemia (Raya et al., 2009). iPSCs for NF1n have also been developed (Anastasaki et al., 2015, Larribere et al., 2015, Wegscheid et al., 2018). However, as for most other cancer syndromes, NF1 iPSCs have been generated from patient fibroblasts and not directly from cells of the associated tumors.
iPSC technology has been used to reprogram cancer cells, encountering different obstacles, such as their chromosomal and genomic composition or the necessity of remodeling their epigenetic state. Another limiting factor is the cell type to be reprogrammed. These aspects make the efficiency of generating iPSCs from cancer cells low (Kim and Zaret, 2015). Despite the low efficiency, there are several examples of iPSCs generated from cancer cells (Pan et al., 2017), mainly from established cancer cell lines (Bernhardt et al., 2017) and much less common from primary tumors (Kim et al., 2013, Kotini et al., 2017). However, the generation of iPSCs from benign tumors or pre-malignant lesions has been less explored (Papapetrou, 2016). To generate a non-perishable cell-based model system that recapitulates the genetic content and tumorigenic properties of NF1 benign PNFs, we generated iPSCs directly from PNF-derived primary cells. These iPSCs were differentiated to NCs and further to SCs. NF1(−/−) SCs obtained from PNF-derived iPSCs were extensively characterized and compared with primary NF1(−/−) SCs derived from primary tumors.
Results
Generation of PNF-Derived iPSC Lines
We obtained five different PNFs (code-named 3PNF, 5PNF, 6PNF, 7PNF, and 13PNF) from five independent patients diagnosed of NF1 according to standard diagnostic criteria (DeBella et al., 2000). For most of them, histological information is available (Carrió et al., 2018). PNFs are composed of different cell types, mainly SCs and endoneurial fibroblasts. SCs within PNFs are the only cells bearing the two NF1 alleles inactivated, one by a constitutional mutation shared by all cells of the individual, and the other by a somatic mutation specific for each PNF. Our intention was to create an imperishable cell-based model resource by reprogramming NF1(−/−) cells present in PNF descendants from the cell originating them. In addition, we planned to obtain NF1(+/−) isogenic iPSCs from the same tumors. We first determined the NF1 germline mutation of each patient by next-generation sequencing panel analysis (Castellanos et al., 2017) and also the NF1 somatic mutation of each excised PNF (Table 1; Figure S1). NF1(−/−) iPSCs were generated either from pure cultures of PNF-derived NF1(−/−) SCs (Serra et al., 2000) or directly from a short culture of PNF-dissociated cells. NF1(+/−) iPSCs were obtained by reprogramming either cultures of PNF-derived NF1(+/−) endoneurial fibroblasts, directly from PNF-dissociated cells or from skin-derived fibroblast cultures of the same patients (see Table S1 for details). Reprogramming to pluripotency was induced by retrovirus- and/or Sendai virus-mediated transduction (Ban et al., 2011, Takahashi and Yamanaka, 2006) of the patient-derived cells. Table 1 summarizes information on patient (sex, age, and germline mutation), tumor (diagnostic and NF1 somatic mutation), and iPSC (name and banking information). Further reprogramming information is summarized in Table S1.
Table 1.
Patient, Tumor, and iPSC Line Information
| Patient Information |
Tumor Information |
iPSC Lines Generated |
||||||
|---|---|---|---|---|---|---|---|---|
| Patient ID | Sex | Age (at PNF Resection) | NF1 Germline Mutation | Tumor ID | Diagnostic | NF1 Somatic Mutation | iPSC Line (Named in the Paper) | iPSC Line (Banking Name) |
| 3 | XX | 8 | c.3943C > T; p.Gln1315∗ | 3PNF | PNF with diffuse extraneural invasion | LOH (HR) whole ch.17q |
3PNFiPS(NF1+/−) | 3PNF_FiPSsv_PM |
| 3PNFiPS(NF1−/−) | 3PNF_SiPSsv_MM | |||||||
| 5 | XY | 10 | intragenic deletion (E16-35) | 5PNF | PNF with diffuse extraneural invasion | LOH (3.8Mb del) | 5PNFiPS(NF1+/−) | 5PNF_TDiPSsv_PM |
| 5PNFiPS(NF1−/−) | 5PNF_TDiPSsv_MM | |||||||
| 6 | XX | 33 | c.2946delT; p.Leu983∗ | 6PNF | PNF with diffuse extraneural invasion | c.2033dupC; p.Ile679Aspfs∗21 | 6PNFiPS(NF1+/−) | 6PNF_SiPSrv_PM |
| 7 | XX | 66 | c.2033dupC; p.Ile679Aspfs∗21 | 7PNF | PNF with diffuse extraneural invasion | LOH (1.4Mb del) | 7PNFiPS(NF1+/−) | 7PNF_TDiPSrv_PM |
| 13 | XY | 14 | c.1318C > T; p.Arg440∗ | 13PNF | PNF with diffuse extraneural invasion | LOH (HR) whole ch.17q |
13PNFiPS(NF1+/−) | not banked |
LOH, loss of heterozygosity; HR, homologous recombination. The link below will take you to the Spanish National Stem Cell Bank-Institute of Health Carlos III, where the iPSC lines have been deposited to be able to be distributed. http://www.eng.isciii.es/ISCIII/es/contenidos/fd-el-instituto/fd-organizacion/fd-estructura-directiva/fd-subdireccion-general-investigacion-terapia-celular-medicina-regenerativa/fd-centros-unidades/fd-banco-nacional-lineas-celulares/fd-lineas-celulares-disponibles/lineas-de-celulas-iPS.shtml.
Overall, we generated seven genetically different iPSC lines from five independent NF1 patients. We were able to isolate two independent NF1(−/−) iPSCs, bearing the constitutional and somatic NF1 mutations, from five distinct PNFs. From all five patients we obtained NF1(+/−) iPSCs bearing only the constitutional mutation. Thus, from two different tumors, 3PNF and 5PNF, we were able to generate isogenic iPSC lines bearing two distinct NF1 genotypes: NF1(+/−) and NF1(−/−) (Table 1).
Characterization of PNF-Derived iPSC Lines
After confirming the NF1 genetic status, selected iPSC clones representing each patient and NF1 genotype were further expanded and characterized. Figure 1 illustrates the characterization of the isogenic iPSC lines derived from 3PNF and 5PNF; the characterization of the remaining banked iPSC lines is shown in Figure S2. We selected clones that displayed a compact embryonic stem cell-like morphology, were positive for alkaline phosphatase staining, and expressed high levels of pluripotency-associated transcription factors and surface markers (Figures 1A and 1B). Moreover, selected clones showed pluripotent differentiation ability in vitro and in vivo (teratoma formation), demethylation of POU5F1 and NANOG promoters, and karyotype stability after more than 15 passages (Figures 1C–1F and S2). It is worth noting that 5PNFiPS(−/−) carried a chromosomal translocation (karyotype: 46,XYt(17; 22) (q11.2; q13.3)) also present in the parental reprogrammed SCs, as the cause of NF1 somatic inactivation (Figure S2G). Finally, we confirmed by PCR-based DNA fingerprinting analysis that the iPSC lines generated genetically matched their parental tumors (Table S2). As expected, the levels of neurofibromin were reduced in NF1(+/−) iPSCs compared with control NF1(+/+) pluripotent cells, and were absent in NF1(−/−) iPSCs (Figure 2G). Altogether, these data demonstrated that we successfully generated iPSCs from PNF-derived NF1(+/−) and NF1(−/−) cells, and indicated that reduced levels or even absence of neurofibromin did not appear to compromise somatic cell reprogramming to pluripotency, maintenance, or differentiation capacity of iPSCs.
Figure 1.

Characterization of PNF-Derived iPSC Lines
(A) Morphology and alkaline phosphatase staining of 3PNF and 5PNF iPSC colonies. Scale bars, 100 μm.
(B) Characterization of pluripotency markers. Representative images of 3PNF and 5PNF iPSC colonies stained positive for the pluripotency-associated markers NANOG, OCT4, and SOX2 (in green), and TRA-1-81, SSEA3, and SSEA4 (in red). Scale bars, 100 μm.
(C) In vitro differentiation potential of 3PNF and 5PNF iPSC lines. Generation of cell derivatives of the three primary germ layers including ectoderm (TUJ1 in green and GFAP in red), endoderm (AFP in green and FOXA2 in red) and mesoderm (SMA in green and GATA4 in red). Scale bars, 100 μm.
(D) Teratoma formation from 5PNF iPSC, showing their differentiation toward ectoderm (TUJ1 in green and GFAP in red), endoderm (AFP in green and FOXA2 in red) and mesoderm (SMA in green and GATA4 in red). Scale bars, 100 μm.
(E) Bisulphite sequencing showing demethylation of NANOG and POU5F1 promoters in the 3PNF and 5PNF iPSC lines.
(F) Karyotype of 3PNF and 5PNF iPSC lines at passage 20.
(G) Western blot analysis showing the absence of neurofibromin in 3PNFiPS(−/−) and 5PNFiPS(−/−). The human embryonic stem cell (hESC) line ES4 and a control iPSC line generated from foreskin fibroblasts (FiPS), both NF1(+/+), were used as control cell lines.
(H) Proliferation capacity of 3PNF and 5PNF iPSC lines assessed by Click-iT EdU Flow Cytometry Assay. Double-positive cells (in S phase) are represented in the graph. Bars represent means from three independent experiments.∗p < 0.05 (unpaired t tests).
Figure 2.

Genomic Characterization of PNFs, Primary Cells, and Generated iPSCs
(A) B allele frequency (BAF) data from SNP array analysis characterizing the genomic structure of five samples associated with 3PNF tumor; fibroblasts; PNF-derived Schwann cells; 3PNFiPS(+/−) and 3PNFiPS(−/−). The genome of all samples was mostly 2n, denoted by a BAF signal around 0.5. A blue shaded region indicates somatic copy neutral (CN)-loss of heterozygosity (LOH).
(B) A detailed view of BAF for chromosome 17. Somatic NF1 inactivation was produced by mitotic recombination generating CN-LOH in 17q and the reduction to homozygosity for the constitutional NF1 mutation. LOH is observed in 3PNF and in 100% of cells in 3PNF-derived Schwann cells and in 3PNFiPS(−/−). Fibroblast primary culture (3PNF fibroblasts) is an early passage and still exhibit a residual LOH due to the presence of “contaminating” tumor SCs.
(C) Summary of somatic exonic variants identified by exome sequencing. All samples associated with a PNF are represented by wide horizontal line of the same color covering all chromosomes. Color dots indicate the type of genetic variant: missense (black), frameshift (orange), in-frame deletion (purple), and non-sense (red). Position of genes containing the variants is marked with vertical lines.
PNF-Derived NF1(−/−) iPSCs Exhibit a Higher Proliferation Rate Than Control Pluripotent Cells
It has been shown that NF1-deficient cells exhibit a higher proliferation rate than their cellular counterparts carrying one or two wild-type copies of the NF1 gene (Kim et al., 1995, Kim et al., 1997, Rosenbaum et al., 1995). Consistent with this, we noticed that cultures of NF1(−/−) 3PNFiPS and 5PNFiPS needed to be split more frequently than control iPSCs or human embryonic stem cells (hESCs) maintained in parallel. To quantify the effect of the NF1 status on iPSC proliferation rate, we used a flow cytometry-based Click-iT EdU assay. We compared PNF-derived NF1(+/−) or (−/−) iPSC lines with control NF1(+/+) pluripotent stem cells (PSCs). Control cells included iPSCs from skin fibroblasts of a healthy donor (FiPS cell line) and embryonic stem cells (ES4 cell line). On average, NF1(−/−) 3PNFiPS and 5PNFiPS cell lines exhibited a 10%–15% increase in cell proliferation rate compared with control PSCs (Figure 2H). NF1(−/−) iPSCs also exhibited a higher proliferation rate than NF1(+/−) iPSCs (p < 0.05). These results indicate that cell proliferation rate in PSCs, as is the case for somatic cells, is influenced by neurofibromin activity.
PNF-Derived iPSCs Capture the Genomic Status of Their Cell of Origin
We extensively characterized the genomic content of the different iPSC lines generated from PNFs. We performed cytogenetic karyotyping, exome sequencing, and molecular karyotyping by SNP array analysis comparing tumors, NF1(−/−) SC and NF1(+/−) fibroblast cultures and iPSCs. All samples were 2n according to the cytogenetic and molecular karyotypes (Figures 1F, 2A, S2, and S3). As previously observed in CNFs (Garcia-Linares et al., 2011), the only genomic alterations present resulted from the somatic inactivation of the NF1 gene, in some cases affecting the structure of chromosome 17q (Figures 2 and S3) (Carrió et al., 2018). Gross somatic mutations affecting the NF1 gene were found in four of the tumors and consisted in either large deletions of 1.4 Mb (7PNF) and 3.8 Mb (5PNF), both involving the NF1 and SUZ12 genes, or homologous recombination (3PNF and 13PNF) generating loss of heterozygosity (LOH) in almost the entire 17q arm (Figure 2B) and bringing the constitutional NF1 mutation into homozygosity, as described previously (Serra et al., 2001, Steinmann et al., 2009). Somatic NF1 inactivation in 6PNF was due to a point mutation (Table 1; Figure S1). The same somatic NF1 inactivation was shared by PNF and its derived NF1(−/−) SC culture, but was not present in fibroblast cultures or in NF1(+/−) iPSCs (Figures 2 and S3). We also performed exome sequencing to identify the presence of small pathogenic variants. On average, we identified the presence of ten additional point mutations in the whole exome of PNF-derived iPSCs that were not present in PNFs or primary SC cultures (Figure 2C; Table S3). The low number of mutations is consistent with the reprogramming and clonal expansion of a cell already containing these mutations, which would not be detectable in the bulk cell population of PNFs or primary SC cultures. None of the identified somatic point mutations was recurrent among the five PNFs (data not shown for 13PNF). These results are in agreement with data from recent exome analysis of PNFs and CNFs (Gosline et al., 2017, Pemov et al., 2017).
Neural Crest Differentiation of PNF-Derived iPSCs
We posit that PNF-derived iPSCs constitute a non-perishable cell-based experimental system that should facilitate the identification of the PNF cell of origin as well as the development of therapeutic strategies against these types of tumors. Thus, we next set out to differentiate PNF-derived iPSCs toward the NC-SC axis. To generate NCs, we used a previously described differentiation protocol that employs chemically defined medium to activate Wnt signaling while inhibiting Activin/Nodal/transforming growth factor β signaling (Lee et al., 2007, Menendez et al., 2013) (see Supplemental Experimental Procedures for details).
Control PSCs, as well as all NF1(+/−) and NF1(−/−) PNF-derived iPSC lines tested, successfully differentiated toward NC cells when applying this protocol. Approximately 12 days after NC induction, cells adopted a stellate morphology typical of NCs (Figure 3A), which was maintained throughout the passages. To characterize the generated NCs we performed flow cytometry analysis using two specific NC markers, p75 (NGFR) and HNK1 (Lee et al., 2010), at early (7–10 days, passage 1) and late (>20 days, passage 4–5) differentiation stages (Figure 3B). Although both markers were heterogeneously expressed in early passages, NCs from both control and PNF-derived iPSCs homogeneously co-expressed high levels of p75 and HNK1 at later differentiation stages, indicating a clear NC identity. NCs cultured under these specific conditions could be maintained as a stable, self-renewing population for up to 20 passages without losing NC identity (see below), enabling the freezing and cryopreservation of NC batches for subsequent differentiation assays.
Figure 3.

PNF-Derived iPSCs Correctly Differentiate into NCs
(A) Schematic representation of the protocol used for differentiating iPSCs into NCs. Control (ES4 and FiPS) and PNF-derived iPSCs were seeded on Matrigel and cultured in NC induction medium for 20 days (see Supplemental Experimental Procedures). Representative bright-field images during the differentiation process over time (in days, D) are shown. PSC, pluripotent stem cell. Scale bar, 50 μm.
(B) Flow cytometry analysis for p75 and Hnk1 before and after NC differentiation. The percentage of double p75 and Hnk1-positive cells is shown inside the graph. P1, passage 1; P4-5, passages 4–5.
(C) Immunocytochemistry analysis showing that both control (ES4 and FiPS) and PNF-derived iPSCs differentiated to NCs (passage 5) express p75 (green), AP2 (green), and SOX10 (red). DAPI was used to stain cell nuclei. Scale bar, 50 μm.
(D) qRT-PCR expression analysis of pluripotent (POU5F1), NC (NGFR, SOX10, AP2), and SC (S100B) markers, in pluripotent cells (PSCs), PSCs differentiated to NCs and PNF-derived SCs. qRT-PCR values are expressed as the mean normalized relative expression (NRE) ± SEM from three independent differentiation experiments.
NC identity was further confirmed by immunofluorescence (Figure 3C) and qRT-PCR (Figure 3D) analyses of the NC markers SOX10, p75, and AP2. qRT-PCR analyses also showed that PSC-derived NCs did not express the pluripotency-associated marker OCT4 (POU5F1), or the SC lineage-specific marker S100b, present in PNF-derived SCs (Figure 3D). Moreover, we also functionally tested NC biological capacities such as migration and differentiation potential. A scratch assay showed the ability of all NCs (control and PNF derived) to start migrating already at 6 h and to be able to close the scratch in less than 24 h (Figure S4A). Furthermore, PSC-derived NCs were able to undergo further differentiation into NC-derived cell types, such as peripheral neurons and melanocytes (Figure S4B), confirming their NC multi-lineage differentiation ability.
SC Differentiation of PNF-Derived NCs
We then set up an SC differentiation protocol starting from the established NCs. We differentiated NCs from control FiPS and PNF-derived iPSC lines into SCs (Figure 4A) (see Supplemental Experimental Procedures). The differentiation process was monitored by immunocytochemistry and qRT-PCR analysis of various markers of the NC-SC lineage at different time points (7, 14, and 30 days).
Figure 4.

Schwann Cells Differentiation of iPSC-Derived NCs
(A) Top: schematic representation of the protocol used for differentiating NCs to Schwann cells (SCs). NCs were seeded on poly-L-lysine and laminin-coated plates and cultured in SC differentiation medium (see Supplemental Experimental Procedures). After 7, 14, and 30 days, SC differentiation was monitored by qRT-PCR and immunocytochemistry analysis. Representative bright-field images during the differentiation process from a control cell line are shown. Scale bars, 50 μm. Bottom: diagram showing the expression of markers associated with the NC-SC lineage. The colored horizontal bars represent the temporal window during differentiation when the corresponding marker is expressed in vivo, according to the literature (Jessen and Mirsky, 2005). SCP, Schwann cell precursor; iSC, immature Schwann cells (iSCs).
(B and C) Immunocytochemical analysis for S100b and p75 at different stages of SC differentiation (7, 14, and 30 days) in control NF1(+/+) FiPS (B) and 3PNFiPS(−/−) cells (C). DAPI was used to stain cell nuclei. Scale bars, 50 μm.
(D and E) qRT-PCR in control NF1(+/+) FiPS (D) and NF1(−/−) iPSCs (E) at five different time points during differentiation: pluripotent stage (PSC), neural crest stage (NC) and at 7, 14, or 30 days of SC differentiation. For NF1(−/−) iPSC graphs (E): light bar represents SC differentiation for 3PNF and dark bar for 5PNF. As control cells for marker expression, primary SC cultures (gray bars) from 3PNF (light gray) and 5PNF (dark gray) were used. Values are expressed as the mean NRE ± SEM from three independent differentiation experiments.
After 7 days under SC differentiation conditions, NCs from NF1(+/+) control FiPS already changed morphology, becoming more elongated. This phenotype progressed over time until reaching the typical bipolar spindle-like morphology of SCs between 14 and 30 days of differentiation (Figure 4B). SC markers such as p75 and S100b were expressed homogenously in the culture throughout the whole differentiation process (Figure 4B). qRT-PCR analysis confirmed expression of NC-SC lineage-specific markers throughout the differentiation process (Figure 4D). NGFR and SOX10, two key regulators of NC formation and SC fate determination, persisted during the entire differentiation process. Expression of SC precursor markers such as CDH19, ITG4A, and MPZ had a remarkable increase after 7 days of differentiation. GAP43 was also highly expressed. SC markers such as PLP, PMP22, and S100b were already detected after 1 week of differentiation and reached maximum expression by day 30. EGR2 (KROX20), a master regulator for myelinating SC was detected already in NCs and had a peak at 30 days of differentiation, as reported previously (Jessen and Mirsky, 2005, Reiprich et al., 2010) (Figure 4D).
At 7 days of differentiation NF1(−/−) NCs resembled control NF1(+/+) cells, both morphologically and according to SC marker expression (p75 and S100b) (Figure 4C). After 14 days of differentiation, NF1(−/−) cells already acquired the slender, elongated morphology of SCs. However, whereas control NF1(+/+) cultures progressively stopped proliferation, maintaining a homogeneous expression of SC markers, NF1(−/−) cells continued to exhibit a high proliferation capacity and heterogeneously expressed some of the markers, such as S100b (Figure 4C). This altered differentiation process of NF1(−/−) SCs was also observed by qRT-PCR analysis (Figure 4E). While markers of the NC-SC lineage were expressed in differentiation NF1(−/−) SC cultures, those markers related to SC maturation were not maintained through the differentiation process compared with control NF1(+/+).
NF1(−/−) Differentiating SCs Exhibited a Continuous High Proliferation Rate and a Lack of Myelination Capacity
NF1(−/−) differentiating SCs proliferated so much during differentiation experiments that cultures were generated with a high cell density and a natural tendency to form sphere-like structures visible to the naked eye. Spheres grew either attached to the plate surface or as free-floating cultures resembling 3D spheroids (Figures 5A and 5B). We quantified the proliferation capacity of differentiating SCs by Ki-67 immunostaining (Figure 5C), confirming a statistically significant higher proliferation rate in NF1(−/−) cells, both at 7 and at 30 days of SC differentiation, compared with control NF1(+/+) and NF1(+/−) cell lines (Figure 5D).
Figure 5.

NF1(−/−) Differentiating SCs Exhibited a Continuous High Proliferation Rate and a Lack of Myelinating Capacity
(A) Representative bright-field images after 20 days of differentiation from NC to SC for different NF1 genotypes. 3PNFiPS(−/−) and 5PNFiPS(−/−) cells exhibited a high cell density and the formation of 3D spheres. Scale bars, 50 μm.
(B) Macroscopic detail of sphere formation in 3PNiPS(−/−) and 5PNFiPS(−/−) cells during SC differentiation.
(C) Proliferation capacity of differentiating SCs. Representative immunofluorescence images of Ki-67 (green) at 7 and 30 days of differentiation. DAPI was used to stain cell nuclei. Scale bars, 50 μm.
(D) Quantification of Ki-67-positive cells (percentage over total DAPI-positive nuclei) expressed as the mean ± SE (n = 3 independent differentiation experiments). At least 300 nuclei were counted per time point and sample. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (unpaired t test).
(E and F) Myelination capacity of control NF1(+/+) FiPS (E) and NF1(−/−) iPSCs (F). Myelination was assessed by co-culturing differentiated SCs (at 7 days) with rat DRG neurons for 30 days. SC myelination capacity was measured by immunostaining for TUJ1, S100b, and MPZ. Scale bars, 50 μm.
(G) PNF-derived SC immunostained with TUJ1, S100b, and MPZ. Scale bars, 50 μm.
In addition to the proliferation rate of differentiating SCs, we also tested their ability to myelinate axons. NF1(+/+) FiPS-derived SCs, co-cultured with rat dorsal root ganglion (DRG) neurons in the presence of myelinating medium, were capable of associating and myelinating peripheral neuron axons, as demonstrated by the co-localization of S100b/myelin protein zero (MPZ)-positive cells with neuron-specific tubulin (TUJ1)-positive axons (Figure 5E). We identified fragments of myelinated axons longer than 400 μm in three independent experiments (Figure S5). These functional assays confirmed the myelinating capacity of FiPS-derived SCs and validated the protocol used to differentiate NCs into SCs. However, when we co-cultured NF1(−/−) iPSC-derived SCs with DRG neurons, they kept proliferating during the assay and were not able to properly associate and form myelinating axons, neither cells growing in monolayer nor sphere-forming cells, as happens in PNFs (Figure 5F). NF1(−/−) differentiating SCs generated either spheres or wide lanes of organized cells. In addition, NF1(−/−) cells expressed the neuronal marker TUJ1, complicating the analysis. Since TUJ1 was not expressed by NF1(+/+) differentiating SCs in the co-culture assay, we analyzed PNF-derived primary SC cultures and found that they also expressed TUJ1 (Figure 5G).
Sphere-Forming SCs from NF1(−/−) iPSCs Recapitulate the Expression Pattern of Their PNF-Derived Primary SC Counterparts
To have a better idea to which extent sphere-forming NF1(−/−) differentiating SCs from PNF-derived iPSCs recapitulated the expression of their primary PNF counterparts, we compared the expression of SC markers in NF1(−/−) spheres at 30 days of differentiation with the expression of their parental PNF-derived primary SCs (Figure 6A). In contrast to the heterogeneous expression of SC markers (s100b) exhibited by differentiating SCs growing in monolayer (Figure 4C), sphere-forming SCs homogeneously expressed all markers tested. When we analyzed the expression of p75, s100b, SOX10, GAP43, and PLP by immunofluorescence, the expression pattern of PNF-derived SCs and sphere-forming SCs were strikingly similar (Figure 6A).
Figure 6.

Sphere-Forming SCs from NF1(−/−) iPSCs Recapitulate the Expression Pattern of their PNF-Derived Primary SC Counterparts
(A) Representative immunofluorescence images showing expression of S100b, p75, SOX10, GAP43, and PLP, in 5PNF primary SCs (PNF SC) compared with sphere-forming 5PNFiPS(−/−) differentiating SCs, at 30 days of differentiation. Scale bars, 100 μm.
(B) Schematic representation of the generated PNF model.
Sphere-forming SCs bore the same genetic and genomic content as their primary SC counterparts and recapitulated both a high proliferation rate and the same expression pattern in a homogeneous manner. Taking everything together, NF1(−/−) iPSC-derived spheres represent a valuable experimental model to study PNF formation, and to test potential therapeutic options in vitro (Figure 6B).
Discussion
There exists a lack of imperishable cell-based systems to model benign tumor progression and assay therapeutic strategies. PNFs are benign SC tumors of the peripheral nervous system associated to NF1 that can progress toward a malignant soft tissue sarcoma. We have generated NF1(−/−) iPSC lines directly from PNFs, sharing the same constitutional and somatic NF1 mutations as the cell originating them. We also generated five independent NF1(+/−) iPSCs from five PNFs, two being isogenic to the NF1(−/−) iPSC lines established. These cells have the genetic and genomic content of their parental primary cells, and can be differentiated toward NCs and further to SCs. SCs derived from NF1(−/−) iPSCs exhibit a high proliferation rate, show poor ability to myelinate, and show a tendency to form spheres in culture that resemble PNFs and preserve the same expression marker profile of the NC-SC axis as their parental NF1(−/−) primary SCs.
iPSC technology has been used to reprogram cancer cells, encountering different obstacles, like the chromosomal and genomic composition of cancer cells or the necessity of remodeling their epigenetic state. The NF1(−/−) iPSCs described here may have overcome these problems since they have been generated from benign tumors. Reprogramming technology has been previously used to model hereditary cancer syndromes (Papapetrou, 2016), NF1 among them (Anastasaki et al., 2015, Larribere et al., 2015, Wegscheid et al., 2018), but never from cells of the associated tumors. PNFs have the potential to progress to malignancy. In this regard, we believe that these iPSCs could constitute an excellent model for investigating tumor progression when combined with existing DNA-editing tools (CRISPR-Cas9) to better identify the genetic and epigenetic changes required for malignant transformation.
Even though the relatively low number of samples complicates drawing strong conclusions, we noticed that the efficiency of generating NF1(−/−) iPSC lines from PNFs (also NF1(+/−)) varied depending on the tumor and on the starting cell type. Different factors could be involved, such as the culture conditions used, the different reprogramming efficiency of distinct cell types (reviewed in Ebrahimi, 2015) or the age of the PNF donor, although all these aspects would need to be further explored.
Whereas NF1(+/+) differentiating SCs progressively stopped proliferation, maintained a homogeneous expression of SC markers, and had the capacity to myelinate axons, NF1(−/−) cells continued exhibiting a high proliferation capacity and heterogeneously expressed S100b during differentiation, and exhibit a poor ability to myelinate axons. These results are consistent with the biological status of SCs within PNFs. The exact mechanism and role of the NF1 gene in relation to the altered SC differentiation is an exciting topic for further research.
The PNF-resembling spheres generated by the high proliferation capacity of differentiating SCs from PNF-derived NF1(−/−) iPSCs constitute a very promising non-perishable model for PNFs, even more so taking into account that currently there is no tumoroid model generated directly from primary PNF cells. An in vitro 3D PNF model will facilitate the testing of therapeutic agents in a PNF-resembling environment before jumping to an in vivo model, although further development will be necessary.
In the field of NF1 research, there is still an open debate regarding the cell of origin of neurofibromas (Buchstaller et al., 2012). PNFs are thought to be congenital but the identity and biological capacity of the cell type that receives the inactivation of NF1 is still not completely understood. Essential information has been obtained from the different genetically modified mouse models that develop PNFs in which NF1 ablation is driven by Cre recombinase expressed under promoters active along the NC-SC differentiation axis. The ability to differentiate PNF-derived iPSCs toward NCs and SCs could complement the information coming from genetically modified mice.
In summary, we have generated NF1(−/−) iPSCs directly from PNFs. They represent an iPSC-based non-perishable cell model system for a benign tumor. NF1(−/−) iPSCs contain the same naturally occurring mutations as their primary counterparts and preserve their proliferative properties when differentiated from NCs toward SCs. SCs differentiated from PNF-derived iPSCs have a high tendency to form spheres. This cell-based model system constitutes a great tool to investigate the PNF cell of origin, the genetic and epigenetic changes required for progression toward MPNSTs and finally, a model to test new therapeutic strategies before pre-clinical in vivo models.
Experimental Procedures
Patients, Plexiform Neurofibromas, and Tumor Processing
Tumor samples were kindly provided by NF1 patients after giving written informed consent for iPSC generation and genomic analysis studies. The study was approved by our Institutional Review Board and local ethical commitees. The patients were diagnosed according to standard diagnostic criteria (DeBella et al., 2000). Tumor specimens were obtained after surgery of five PNFs from five independent patients (two males, three females; ages 8–66 years). Immediately after excision, tumor samples were placed in DMEM medium (Gibco) containing 10% FBS (Gibco) + 1× Glx (Gibco) + 1× normocin antibiotic cocktail (InvivoGene), and shipped at room temperature to our laboratory. Tumors were processed as follows: surrounding fat tissue and skin were removed and tumors were cut into 1-mm pieces and cryopreserved in 10% DMSO (Sigma) + 90% FBS until used.
PNF-Derived SCs and Fibroblasts Cultures
PNF-derived SCs and fibroblasts were isolated as described previously (Serra et al., 2000). In brief, PNF pieces that were preserved in liquid nitrogen were thawed and digested with 160 U/mL collagenase type 1 and 0.8 U/mL dispase (Worthington, Lakewood, NJ) for 16 h at 37°C. Dissociated cells were washed and seeded onto 0.1 mg/mL poly-L-lysine (Sigma) and 4 μg/mL laminin (Gibco)-coated dishes in Schwann cell medium (SCM) and maintained at 37°C under a 10% CO2 atmosphere. SCM is DMEM (Gibco) with 10% FBS, 500 U/mL penicillin/500 μg/mL streptomycin (Gibco), 0.5 mM 3-iso-butyl-1-methilxantine (Sigma), 2.5 mg/mL insulin (Sigma), 10 nM heregulin-β1 (PeproTech), and 0.5 μM forskolin (Sigma). One day after plating, culture medium was replaced by SCM without forskolin for an additional 2–3 days. This process was repeated in cycles and cells were passaged as needed with trypsin 0.05% (Gibco). SC purity was assessed by performing S100β staining as described previously (Serra et al., 2000). To isolate fibroblasts, dissociated cells were plated in DMEM 10% FBS media and passaged when necessary.
Reprogramming of SCs, Fibroblasts, and Digested Tumors
Between 1 × 104 and 2 × 104 cells were reprogrammed through the retroviral delivery of human cDNA coding for OCT4, SOX2, KLF4, and cMYC transcription factors as described previously (Raya et al., 2009). For non-integrative reprogramming, a Cytotune-iPS 2.0 Sendai Reprogramming Kit (Thermo Fisher Scientific) was used according to the manufacturer's protocol. Approximately 3 or 4 weeks after transduction, colonies displaying embryonic stem cell-like morphology and behavior were selected for further characterization and genotyping. iPSC established lines were grown on dishes coated with growth factor-reduced Matrigel (BD Biosciences) in mTESR1 medium (STEMCELL Technologies). See Supplemental Information for a detailed description of iPSC characterization.
Differentiation toward NCs and SCs
Neural crest differentiation was performed as described by Menendez et al. (2013) with some modifications. In brief, 9 × 104 cells/cm2 were plated onto Matrigel-coated plates in mTESR medium. The following day, the medium was replaced with hESC maintenance medium: DMEM:F12 (Gibco) 1:1; 5 mg/mL BSA (Sigma); 500 U/mL penicillin/500 μg/mL streptomycin (Gibco); 2 mM GlutaMAX (Gibco); 1× MEM non-essential amino acids (Gibco); 1× trace elements A; 1× trace elements B; 1× trace elements C (Corning); 2-mercaptoethanol (Gibco); 10 μg/mL transferrin (Sigma); 50 μg/mL sodium L-ascorbate (Sigma); 10 ng/mL heregulin-β1 (PeproTech); 10 ng/mL activin A (PeproTech); 200 μg/mL LONG R3 IGFR (PeproTech); 8 ng/mL basic fibroblast growth factor 2 (PeproTech). Next day, the medium was replaced with neural crest induction/differentiation medium: hESC medium without activin and supplemented with 2 μM CHIR9902 (STEMCELL Technologies) and 20 μM SB432542 (STEMCELL Technologies), and was replaced every day. NCs were maintained in this medium and split with Accutase (Thermo Fischer Scientific) when necessary.
For SC differentiation NCs were plated onto 0.1 mg/mL poly-L-lysine (Sigma) and 4 μg/mL laminin (Gibco)-coated plates and cultured in SC differentiation medium: DMEM:F12 (3:1); 500 U/ml penicillin/500 μg/mL streptomycin antibiotics (Gibco); 5 μM forskolin (Sigma); 50 ng/mL heregulin-β1; 2% N2 supplement (Gibco); 1% FBS (Gibco). The medium was replaced twice a week.
Additional Experimental Procedures
Additional experimental procedures can be found in Supplemental Information.
Repositories
The iPSC lines generated have been banked banked and are currently distributed by the Spanish National Stem Cell Bank-Institute of Health Carlos III in compliance with the informed consent signed by the patient (see Table 1).
Author Contributions
E.S. conceived the study. M.C., Y.R.-P., A.R., and E.S. designed the study and wrote the manuscript that was revised, corrected, and improved by all authors. M.C., H.M., and Y.R.-P. performed most of the experimental work. B.G. performed bioinformatic analysis. I.R. and E.C. performed NF1 genetic analysis. E.T., S.J.-D., J.B., and L.V. performed experimental work. I.B. and C.L. provided scientific input. M.C. and B.G. generated the figures for the paper. All authors approved the final version of the manuscript.
Acknowledgments
This work has mainly been supported by an agreement from the Johns Hopkins University School of Medicine and the Neurofibromatosis Therapeutic Acceleration Program (NTAP). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the Johns Hopkins University School of Medicine. The work has also been partially supported by the Spanish Ministry of Science and Innovation, Carlos III Health Institute (ISCIII) (PI14/00577; PI17/00524; RD16/0011/0024; and PIE14/00061) Plan Estatal de I. D. i 2013–16; co-financed by the FEDER program; and by the Government of Catalonia (2017-SGR-899) and CERCA Program/Generalitat de Catalunya. We thank the patients and their physicians who contributed the original samples. We would like to specially thank the Asociación de Afectados de Neurofibromatosis for their constant support and coordination with patients. We also give thanks for the support of ACNefi and the Spanish Association Against Cancer (AECC) for recognizing our group with one of its awards. We thank the IGTP core facilities and their staff for their contribution and technical support: Translational Genomics Core Facility; High Content Genomics and Bioinformatics Core Facility; Flow Cytometry (Gerard Requena and Marco A. Fernández); Microscopy Core facility (Gerard Requena). We also thank the National Center for Genomic Analysis (CNAG). Finally, we would like to thank Dr. Jaishri Blakeley, Dr. Sharad Verma, and people at NTAP for their constant support and appreciate insightful discussions of the different PIs funded by the NTAP Cell Culture Initiative.
Published: January 31, 2019
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, five figures, and five tables and can be found with this article online at https://doi.org/10.1016/j.stemcr.2019.01.001.
Contributor Information
Ángel Raya, Email: araya@cmrb.eu.
Eduard Serra, Email: eserra@igtp.cat.
Accession Numbers
Data are available at the Synapse repository with accession number syn17413894 (DOI: 10.7303/syn17413894.1) (https://www.synapse.org/#!Synapse:syn17413894/tables/).
Supplemental Information
This Table Completes Figure 2.
References
- Anastasaki C., Woo A.S., Messiaen L.M., Gutmann D.H. Elucidating the impact of neurofibromatosis-1 germline mutations on neurofibromin function and dopamine-based learning. Hum. Mol. Genet. 2015;24:3518–3528. doi: 10.1093/hmg/ddv103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban H., Nishishita N., Fusaki N., Tabata T., Saeki K., Shikamura M., Takada N., Inoue M., Hasegawa M., Kawamata S., Nishikawa S. Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc. Natl. Acad. Sci. U S A. 2011;108:14234–14239. doi: 10.1073/pnas.1103509108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beert E., Brems H., Daniels B., De Wever I., Van Calenbergh F., Schoenaers J., Debiec-Rychter M., Gevaert O., De Raedt T., Van Den Bruel A. Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosomes Cancer. 2011;50:1021–1032. doi: 10.1002/gcc.20921. [DOI] [PubMed] [Google Scholar]
- Bernhardt M., Novak D., Assenov Y., Orouji E., Knappe N., Weina K., Reith M., Larribere L., Gebhardt C., Plass C. Melanoma-derived iPCCs show differential tumorigenicity and therapy response. Stem Cell Rep. 2017;8:1379–1391. doi: 10.1016/j.stemcr.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchstaller J., Clapp D.W., Parada L.F., Zhu Y. Cell of origin and the contribution of microenvironment in NF1 tumorigenesis and therapeutic implications. In: Upadhyaya M., Cooper D.N., editors. Neurofibromatosis Type 1:Molecular and Cellular Biology. Springer Berlin Heidelberg; 2012. pp. 549–568. [Google Scholar]
- Carrió M., Gel B., Terribas E., Carolina Zucchiatti A., Moliné M., Rosas I., Teulé A., Ramón y Cajal S., López-Gutiérrez J.C., Blanco I. Analysis of intra-tumor heterogeneity in neurofibromatosis type 1 plexiform neurofibromas and neurofibromas with atypical features: correlating histological and genomic findings. Hum. Mutat. 2018;39:1112–1125. doi: 10.1002/humu.23552. [DOI] [PubMed] [Google Scholar]
- Castellanos E., Gel B., Rosas I., Tornero E., Santín S., Pluvinet R., Velasco J., Sumoy L., Del Valle J., Perucho M. A comprehensive custom panel design for routine hereditary cancer testing: preserving control, improving diagnostics and revealing a complex variation landscape. Sci. Rep. 2017;7:39348. doi: 10.1038/srep39348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Liu C., Patel A.J., Liao C.-P., Wang Y., Le L.Q. Cells of origin in the embryonic nerve roots for NF1-associated plexiform neurofibroma. Cancer Cell. 2014;26:695–706. doi: 10.1016/j.ccell.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Däschner K., Assum G., Eisenbarth I., Krone W., Hoffmeyer S., Wortmann S., Heymer B., Kehrer-Sawatzki H. Clonal origin of tumor cells in a plexiform neurofibroma with LOH in NF1 intron 38 and in dermal neurofibromas without LOH of the NF1 gene. Biochem. Biophys. Res. Commun. 1997;234:346–350. doi: 10.1006/bbrc.1997.6645. [DOI] [PubMed] [Google Scholar]
- DeBella K., Szudek J., Friedman J.M. Use of the National Institutes of Health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics. 2000;105(3 Pt 1):608–614. doi: 10.1542/peds.105.3.608. [DOI] [PubMed] [Google Scholar]
- Dombi E., Baldwin A., Marcus L.J., Fisher M.J., Weiss B., Kim A., Whitcomb P., Martin S., Aschbacher-Smith L.E., Rizvi T.A. Activity of selumetinib in neurofibromatosis type 1-related plexiform neurofibromas. N. Engl. J. Med. 2016;375:2550–2560. doi: 10.1056/NEJMoa1605943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi B. Reprogramming barriers and enhancers: strategies to enhance the efficiency and kinetics of induced pluripotency. Cell Regen. (Lond) 2015;4:10. doi: 10.1186/s13619-015-0024-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferner R.E. Neurofibromatosis 1 and neurofibromatosis 2: a twenty-first century perspective. Lancet Neurol. 2007;6:340–351. doi: 10.1016/S1474-4422(07)70075-3. [DOI] [PubMed] [Google Scholar]
- Garcia-Linares C., Fernández-Rodríguez J., Terribas E., Mercadé J., Pros E., Benito L., Benavente Y., Capellà G., Ravella A., Blanco I. Dissecting loss of heterozygosity (LOH) in neurofibromatosis type 1-associated neurofibromas: importance of copy neutral LOH. Hum. Mutat. 2011;32:78–90. doi: 10.1002/humu.21387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosline S.J.C., Weinberg H., Knight P., Yu T., Guo X., Prasad N., Jones A., Shrestha S., Boone B., Levy S.E. A high-throughput molecular data resource for cutaneous neurofibromas. Sci. Data. 2017;4:170045. doi: 10.1038/sdata.2017.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higham C.S., Dombi E., Rogiers A., Bhaumik S., Pans S., Connor S.E.J., Miettinen M., Sciot R., Tirabosco R., Brems H. The characteristics of 76 atypical neurofibromas as precursors to neurofibromatosis 1 associated malignant peripheral nerve sheath tumors. Neuro. Oncol. 2018;20:818–825. doi: 10.1093/neuonc/noy013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirbe A.C., Dahiya S., Miller C.A., Li T., Fulton R.S., Zhang X., McDonald S., DeSchryver K., Duncavage E.J., Walrath J. Whole exome sequencing reveals the order of genetic changes during malignant transformation and metastasis in a single patient with NF1-plexiform neurofibroma. Clin. Cancer Res. 2015;21:4201–4211. doi: 10.1158/1078-0432.CCR-14-3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen K.R., Mirsky R. The origin and development of glial cells in peripheral nerves. Nat. Rev. Neurosci. 2005;6:671–682. doi: 10.1038/nrn1746. [DOI] [PubMed] [Google Scholar]
- Kim H.A., Ling B., Ratner N. Nf1-deficient mouse Schwann cells are angiogenic and invasive and can be induced to hyperproliferate: reversion of some phenotypes by an inhibitor of farnesyl protein transferase. Mol. Cell. Biol. 1997;17:862–872. doi: 10.1128/mcb.17.2.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.A., Rosenbaum T., Marchionni M.A., Ratner N., DeClue J.E. Schwann cells from neurofibromin deficient mice exhibit activation of p21ras, inhibition of cell proliferation and morphological changes. Oncogene. 1995;11:325–335. [PubMed] [Google Scholar]
- Kim J., Hoffman J.P., Alpaugh R.K., Rhim A.D., Reichert M., Stanger B.Z., Furth E.E., Sepulveda A.R., Yuan C.-X., Won K.-J. An iPSC line from human pancreatic ductal adenocarcinoma undergoes early to invasive stages of pancreatic cancer progression. Cell Rep. 2013;3:2088–2099. doi: 10.1016/j.celrep.2013.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Zaret K.S. Reprogramming of human cancer cells to pluripotency for models of cancer progression. EMBO J. 2015;34:739–747. doi: 10.15252/embj.201490736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluwe L., Friedrich R.E., Mautner V.F. Allelic loss of the NF1 gene in NF1-associated plexiform neurofibromas. Cancer Genet. Cytogenet. 1999;113:65–69. doi: 10.1016/s0165-4608(99)00006-0. [DOI] [PubMed] [Google Scholar]
- Kotini A.G., Chang C.-J., Chow A., Yuan H., Ho T.-C., Wang T., Vora S., Solovyov A., Husser C., Olszewska M. Stage-specific human induced pluripotent stem cells map the progression of myeloid transformation to transplantable leukemia. Cell Stem Cell. 2017;20:315–328.e7. doi: 10.1016/j.stem.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraniak J.M., Chalasani A., Wallace M.R., Mattingly R.R. Development of 3D culture models of plexiform neurofibroma and initial application for phenotypic characterization and drug screening. Exp. Neurol. 2018;299:289–298. doi: 10.1016/j.expneurol.2017.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krone W., Jirikowski G., Mühleck O., Kling H., Gall H. Cell culture studies on neurofibromatosis (von Recklinghausen). II. Occurrence of glial cells in primary cultures of peripheral neurofibromas. Hum. Genet. 1983;63:247–251. doi: 10.1007/BF00284658. [DOI] [PubMed] [Google Scholar]
- Larribere L., Wu H., Novak D., Galach M., Bernhardt M., Orouji E., Weina K., Knappe N., Sachpekidis C., Umansky L. NF1 loss induces senescence during human melanocyte differentiation in an iPSC-based model. Pigment Cell Melanoma Res. 2015;28:407–416. doi: 10.1111/pcmr.12369. [DOI] [PubMed] [Google Scholar]
- Lee G., Kim H., Elkabetz Y., Al Shamy G., Panagiotakos G., Barberi T., Tabar V., Studer L. Isolation and directed differentiation of neural crest stem cells derived from human embryonic stem cells. Nat. Biotechnol. 2007;25:1468–1475. doi: 10.1038/nbt1365. [DOI] [PubMed] [Google Scholar]
- Lee G., Chambers S.M., Tomishima M.J., Studer L. Derivation of neural crest cells from human pluripotent stem cells. Nat. Protoc. 2010;5:688–701. doi: 10.1038/nprot.2010.35. [DOI] [PubMed] [Google Scholar]
- Li H., Chang L.-J., Neubauer D.R., Muir D.F., Wallace M.R. Immortalization of human normal and NF1 neurofibroma Schwann cells. Lab. Invest. 2016;96:1105–1115. doi: 10.1038/labinvest.2016.88. [DOI] [PubMed] [Google Scholar]
- Maertens O., Brems H., Vandesompele J., De Raedt T., Heyns I., Rosenbaum T., De Schepper S., De Paepe A., Mortier G., Janssens S. Comprehensive NF1 screening on cultured Schwann cells from neurofibromas. Hum. Mutat. 2006;27:1030–1040. doi: 10.1002/humu.20389. [DOI] [PubMed] [Google Scholar]
- Mautner V.-F., Asuagbor F.A., Dombi E., Funsterer C., Kluwe L., Wenzel R., Widemann B.C., Friedman J.M. Assessment of benign tumor burden by whole-body MRI in patients with neurofibromatosis 1. Neuro. Oncol. 2008;10:593–598. doi: 10.1215/15228517-2008-011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarron K.F., Goldblum J.R. Plexiform neurofibroma with and without associated malignant peripheral nerve sheath tumor: a clinicopathologic and immunohistochemical analysis of 54 cases. Mod. Pathol. 1998;11:612–617. [PubMed] [Google Scholar]
- Menendez L., Kulik M.J., Page A.T., Park S.S., Lauderdale J.D., Cunningham M.L., Dalton S. Directed differentiation of human pluripotent cells to neural crest stem cells. Nat. Protoc. 2013;8:203–212. doi: 10.1038/nprot.2012.156. [DOI] [PubMed] [Google Scholar]
- Miller S.J., Jessen W.J., Mehta J., Hardiman A., Sites E., Kaiser S., Jegga A.G., Li H., Upadhyaya M., Giovannini M. Integrative genomic analyses of neurofibromatosis tumours identify SOX9 as a biomarker and survival gene. EMBO Mol. Med. 2009;1:236–248. doi: 10.1002/emmm.200900027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir D., Neubauer D., Lim I.T., Yachnis A.T., Wallace M.R. Tumorigenic properties of neurofibromin-deficient neurofibroma Schwann cells. Am. J. Pathol. 2001;158:501–513. doi: 10.1016/S0002-9440(10)63992-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer R.J., Rosser T. Therapy for plexiform neurofibromas in children with neurofibromatosis 1: an overview. J. Child Neurol. 2002;17:638–641. doi: 10.1177/088307380201700816. [DOI] [PubMed] [Google Scholar]
- Pan X.-Y., Tsai M.-H., Wuputra K., Ku C.-C., Lin W.-H., Lin Y.-C., Kishikawa S., Noguchi M., Saito S., Lin C.-S., Yokoyama K.K. Application of cancer cell reprogramming technology to human cancer research. Anticancer Res. 2017;37:3367–3377. doi: 10.21873/anticanres.11703. [DOI] [PubMed] [Google Scholar]
- Papapetrou E.P. Patient-derived induced pluripotent stem cells in cancer research and precision oncology. Nat. Med. 2016;22:1392–1401. doi: 10.1038/nm.4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltonen J., Jaakkola S., Lebwohl M., Renvall S., Risteli L., Virtanen I., Uitto J. Cellular differentiation and expression of matrix genes in type 1 neurofibromatosis. Lab. Invest. 1988;59:760–771. [PubMed] [Google Scholar]
- Pemov A., Li H., Patidar R., Hansen N.F., Sindiri S., Hartley S.W., Wei J.S., Elkahloun A., Chandrasekharappa S.C., Boland J.F. The primacy of NF1 loss as the driver of tumorigenesis in neurofibromatosis type 1-associated plexiform neurofibromas. Oncogene. 2017;36:3168–3177. doi: 10.1038/onc.2016.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prada C.E., Rangwala F.A., Martin L.J., Lovell A.M., Saal H.M., Schorry E.K., Hopkin R.J. Pediatric plexiform neurofibromas: impact on morbidity and mortality in neurofibromatosis type 1. J. Pediatr. 2012;160:461–467. doi: 10.1016/j.jpeds.2011.08.051. [DOI] [PubMed] [Google Scholar]
- Rasmussen S.A., Overman J., Thomson S.A., Colman S.D., Abernathy C.R., Trimpert R.E., Moose R., Virdi G., Roux K., Bauer M. Chromosome 17 loss-of-heterozygosity studies in benign and malignant tumors in neurofibromatosis type 1. Genes. Chromosomes Cancer. 2000;28:425–431. [PubMed] [Google Scholar]
- Ratner N., Miller S.J. A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat. Rev. Cancer. 2015;15:290–301. doi: 10.1038/nrc3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raya Á., Rodríguez-Pizà I., Guenechea G., Vassena R., Navarro S., Barrero M.J., Consiglio A., Castellà M., Río P., Sleep E. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature. 2009;460:53–59. doi: 10.1038/nature08129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiprich S., Kriesch J., Schreiner S., Wegner M. Activation of Krox20 gene expression by Sox10 in myelinating Schwann cells. J. Neurochem. 2010;112:744–754. doi: 10.1111/j.1471-4159.2009.06498.x. [DOI] [PubMed] [Google Scholar]
- Rosenbaum T., Boissy Y.L., Kombrinck K., Brannan C.I., Jenkins N.A., Copeland N.G., Ratner N. Neurofibromin-deficient fibroblasts fail to form perineurium in vitro. Development. 1995;121:3583–3592. doi: 10.1242/dev.121.11.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra E., Rosenbaum T., Winner U., Aledo R., Ars E., Estivill X., Lenard H.G., Lázaro C. Schwann cells harbor the somatic NF1 mutation in neurofibromas: evidence of two different Schwann cell subpopulations. Hum. Mol. Genet. 2000;9:3055–3064. doi: 10.1093/hmg/9.20.3055. [DOI] [PubMed] [Google Scholar]
- Serra E., Rosenbaum T., Nadal M., Winner U., Ars E., Estivill X., Lázaro C. Mitotic recombination effects homozygosity for NF1 germline mutations in neurofibromas. Nat. Genet. 2001;28:294–296. doi: 10.1038/90148. [DOI] [PubMed] [Google Scholar]
- Steinmann K., Kluwe L., Friedrich R.E., Mautner V.-F., Cooper D.N., Kehrer-Sawatzki H. Mechanisms of loss of heterozygosity in neurofibromatosis type 1-associated plexiform neurofibromas. J. Invest. Dermatol. 2009;129:615–621. doi: 10.1038/jid.2008.274. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- VM, Riccardi . Second Edition. Johns Hopkins University Press; 1992. Neurofibromatosis: Phenotype, Natural History, and Pathogenesis. [Google Scholar]
- Wallace M.R., Rasmussen S.A., Lim I.T., Gray B.A., Zori R.T., Muir D. Culture of cytogenetically abnormal Schwann cells from benign and malignant NF1 tumors. Genes. Chromosomes Cancer. 2000;27:117–123. [PubMed] [Google Scholar]
- Wegscheid M.L., Anastasaki C., Gutmann D.H. Human stem cell modeling in neurofibromatosis type 1 (NF1) Exp. Neurol. 2018;299:270–280. doi: 10.1016/j.expneurol.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This Table Completes Figure 2.