

SUMMARY
Mutations affecting RNA splicing factors are the most common genetic alterations in myelodysplastic syndrome (MDS) patients and occur in a mutually exclusive manner. The basis for the mutual exclusivity of these mutations and how they contribute to MDS is not well understood. Here we report that although different spliceosome gene mutations impart distinct effects on splicing, they are negatively selected for when co-expressed due to aberrant splicing and downregulation of regulators of hematopoietic stem cell survival and quiescence. In addition to this synthetic lethal interaction, mutations in the splicing factors SF3B1 and SRSF2 share convergent effects on aberrant splicing of mRNAs that promote nuclear factor κB signaling. These data identify shared consequences of splicing-factor mutations and the basis for their mutual exclusivity.
Graphical Abstract
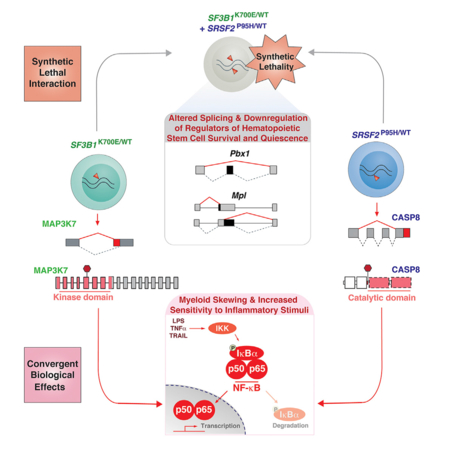
In Brief
Lee et al. report that SF3B1 and SRSF2 mutations elicit distinct effects on splicing and are synthetically lethal due to the cumulative impact on hematopoietic stem cell survival and quiescence. These mutations share convergent effects on promoting NF-κB signaling to drive myelodysplastic syndrome.
INTRODUCTION
Recurrent mutations in genes encoding spliceosome components have been identified across multiple cancer types. Spliceosomal gene mutations most commonly occur in myelodysplastic syndromes (MDS), chronic myelomonocytic leukemia (CMML), acute myeloid leukemia (AML) (Papaemmanuil et al., 2011; Yoshida et al., 2011), chronic lymphocytic leukemia (CLL) (Wang et al., 2011), and uveal melanoma (Harbour et al., 2013; Martin et al., 2013). SF3B1, U2AF1, and SRSF2 are the most commonly mutated genes. Mutations in each of these occur as heterozygous point mutations at specific residues, suggesting gain-of-function alterations. In addition, mutations in spliceosome genes are mutually exclusive with one another, presumably due to redundant effects and/or a limit on cellular tolerance of disrupted spliceosome function.
Despite insights into the effects of each mutation on pre-mRNA splicing, the basis for their mutual exclusivity, and functionally convergent effects they may have are unknown. For example, recent work identified that mutations affecting the core splicing factor SF3B1 are associated with cryptic 3′ splice site selection and altered branchpoint recognition (Darman et al., 2015; DeBoever et al., 2015). In contrast, mutations affecting SRSF2, an auxiliary splicing factor that binds exonic splicing enhancers to promote splicing, alter its RNA binding preference in a sequence-specific manner and thereby alter the efficiency of exon inclusion (Kim et al., 2015; Zhang et al., 2015). Finally, mutations affecting U2AF1 either promote or repress 3′ splice site based on sequences flanking the AG dinucleotide (Ilagan et al., 2014). Given that the effects of SF3B1, U2AF1, and SRSF2 mutations on splicing mechanisms are distinct, it is unclear why these mutations are mutually exclusive. Moreover, evaluation of the effects of these mutations on splicing and gene expression in an isogenic manner has not been performed. Here we directly assessed the functional basis for the mutual exclusivity of spliceosome gene mutations using models expressing single- or double-mutant splicing factors simultaneously. These data provide a functional explanation for the genetic configuration of spliceosome gene mutations and identify a convergent effect of these mutations on a pathway of established relevance to MDS pathogenesis.
RESULTS
Simultaneous Expression of Srsf2 and Sf3b1 Mutations Is Incompatible with Hematopoiesis
Evaluation of sequencing data from >4,000 patients with myeloid neoplasms revealed that while ~48% (1,935/4,032) have a mutation in an RNA splicing factor, only ~2% of patients (86/4,032) have >1 splicing-factor mutation (Figure 1A and Table S1). To understand the basis of this mutual exclusivity, we generated mice for inducible heterozygous expression of two of the most common splicing-factor mutations in MDS (SF3B1K700E and SRSF2P95H) simultaneously (Figure 1B). We performed noncompetitive bone marrow transplantation (BMT), whereby each mutation was induced following stable engraftment in recipient mice (Figure 1B). Bone marrow (BM) cells co-expressing Srsf2P95H and Sf3b1K700E mutations were severely defective in multi-lineage reconstitution compared with other groups (Figures 1C, 1D, and S1A–S1D). Chimerism analysis and evaluation for recombination prior to polyinosinic-polycytidylic acid (pIpC) administration confirmed minimal spontaneous excision prior to BMT (Figures S1E and S1F). In competitive BMT (Figure 1E), BM cells co-expressing Sf3b1K700E and Srsf2P95H mutations were more readily outcompeted by competitor BM cells relative to single-mutant or wild-type (WT) controls. Analyses of hematopoietic organs 6 months post BMT revealed a near-complete absence of Srsf2P95H/+ Sf3b1K700E/+ cells, which was distinct from single-mutant groups (Figures 1E, 1F, and S1C). These data provide functional evidence that co-expression of mutant RNA splicing factors is not tolerated.
Figure 1. Simultaneous Expression of Mutations in Srsf2 and Sf3b1 or Expression in the Homozygous State Is Incompatible with Hematopoiesis.
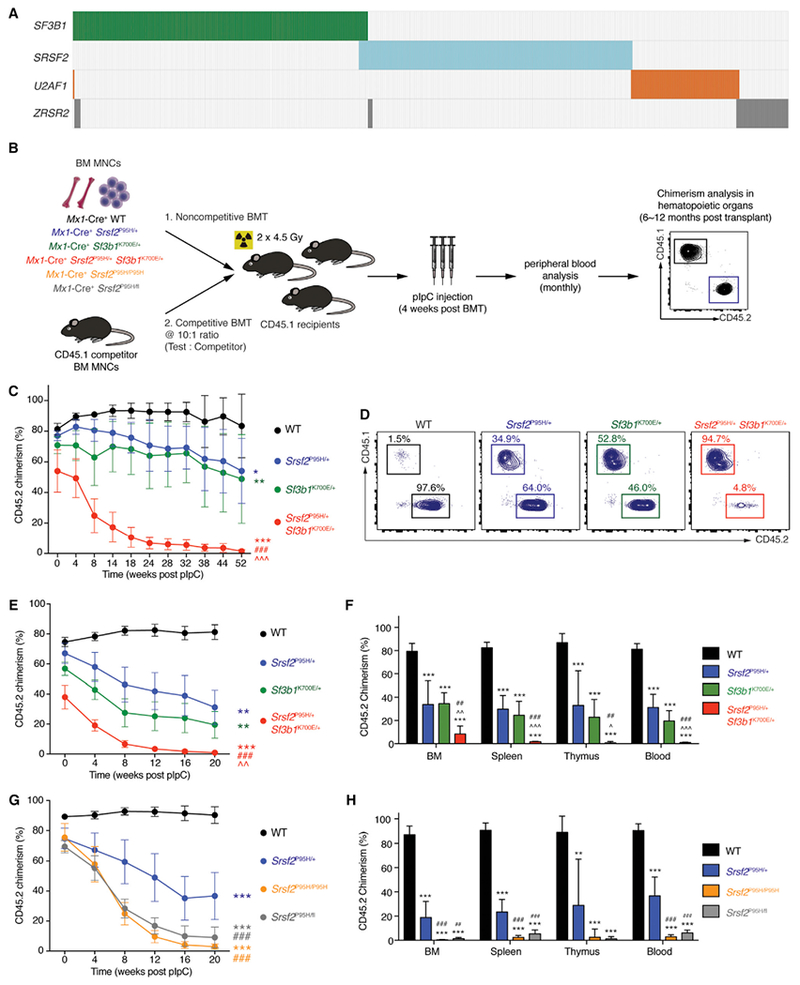
(A) Heat map of the four most commonly mutated gene sencoding RNA splicing factors across 11 studies in myeloid malignancies (Bejar et al., 2012; Damm et al., 2012; Haferlach et al., 2014; Lasho et al., 2012; Makishima et al., 2012; Meggendorfer et al., 2012; Papaemmanuil et al., 2013; Patnaik et al., 2013; Thol et al., 2012; Yoshida et al., 2011; Zhang et al., 2012). Each column represents a patient, and each colored bar represents the presence of the specified mutation.
(B) Schema of competitive and noncompetitive bone marrow transplantation (BMT) using bone marrow mononuclear cells (BM MNCs) from 8-week-old Mx1-Cre+ wild-type (WT), Mx1-Cre+ Srsf2P95H/+, Mx1-Cre+ Sf3b1K700E/+, Mx1-Cre+ Srsf2P95H/+ Sf3b1K700E/+, Mx1-Cre+ Srsf2P95H/P95H, and Mx1-Cre+ Srsf2P95H/fl mice. Polyinosinic-polycytidylic acid (pIpC) was administered to recipients 4 weeks post BMT to induce expression of mutant alleles.
(C) Percentage of CD45.2 chimerism in peripheral blood of recipients (n = 8–10 mice per genotype) in noncompetitive BMT.
(D) Representative fluorescence-activated cell sorting (FACS) plots of CD45.2+ cells in peripheral blood of recipients in noncompetitive BMT 52 weeks post pIpC.
(E) Percentage of CD45.2 chimerism in peripheral blood of recipients (n = 10 mice per genotype) in competitive BMT.
(F) Analysis of CD45.2 chimerism in the BM, spleen, thymus, and blood of recipients (n = 5–10 mice per genotype) in competitive BMT 20 weeks post pIpC.
(G) Percentage of CD45.2 chimerism in peripheral blood of recipients (n = 5–10 mice per genotype) in competitive BMT from Mx1-Cre+ WT, Mx1-Cre+ Srsf2P95H/+, Mx1-Cre+ Srsf2P95H/P95H, and Mx1-Cre+ Srsf2P95H/fl mice.
(H) Analysis of CD45.2 chimerism in BM, spleen, thymus, and blood of recipients (n = 5–10 mice per genotype) in competitive BMT 20 weeks post pIpC.
Error bars represent mean ± SD. ANOVA and Tukey’s post hoc test were used to compare groups. *p < 0.05, **p < 0.01, ***p < 0.001 versus Mx1-Cre+ WT mice; ^p < 0.05, ^^p < 0.01, ^^^p < 0.001 versus Mx1-Cre+ Sf3b1K700E/+ mice; ##p < 0.01, ###p < 0.001 versus Mx1-Cre+ Srsf2P95H/+ mice. See also Figure S1 and Table S1.
Expression of Splicing-Factor Mutations in a Homozygous or Hemizygous State Is Incompatible with Hematopoiesis
Prior studies identified that cells bearing mutant splicing factors require the WT allele for survival (Lee et al., 2016; Zhou et al., 2015). While these observations potentially explain the heterozygous nature of splicing-factor mutations in patients, the effect of expressing splicing-factor mutation in a homozygous manner was not assessed. To test this, we generated mice with conditional homozygous expression of the SRSF2P95H mutation (Mx1-Cre+ Srsf2P95H/P95H). In competitive BMT assays, hematopoietic stem and progenitor cells (HSPCs) from these mice showed severe defects in multi-lineage reconstitution relative to Mx1-Cre+ Srsf2P95H/+ or Mx1-Cre+ WT HSPCs, similar to defects seen with hemizygous Srsf2P95H expression (Mx1-Cre+ Srsf2P95H/fl) (Figures 1G, 1H, and S1G–S1I). These data firmly establish that neither hemizygous nor homozygous expression of splicing-factor mutations is tolerated, highlighting the unique dependency of spliceosome-mutant cells on residual WT spliceosome function.
Simultaneous Expression of Srsf2 and Sf3b1 Mutations Resulted in Increased Apoptosis and Reduced Quiescence of HSPCs
To identify why co-expression of Srsf2P95H and Sf3b1K700E resulted in severe HSPC dysfunction, we performed cell-cycle and apoptosis analysis. Two-weeks following recombination, there was a significant increase in lineage− Sca-1+ c-Kit+ (LSK) cells undergoing apoptosis and in the S phase of cell cycle in Mx1-Cre+ Srsf2P95H/+ Sf3b1K700E/+ mice relative to controls (Figures 2A and 2B).
Figure 2. Combined Expression of Mutations in Srsf2 and Sf3b1 Results in Hematopoietic Stem and Progenitor Cell Apoptosis and Loss of Quiescence.
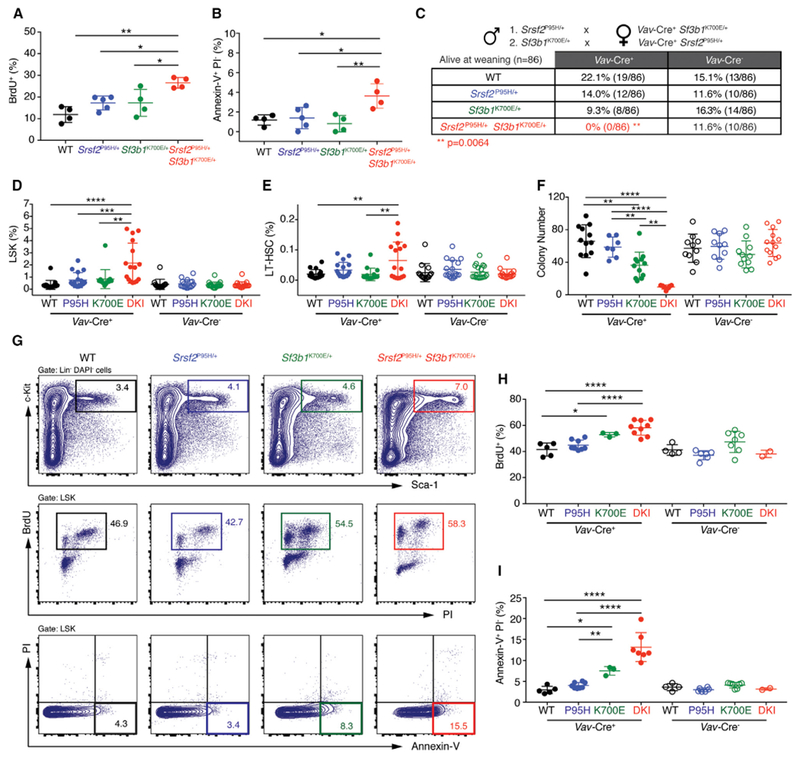
(A and B) Percentage of bromodeoxyuridine+ (BrdU+) (A) or annexin-V+ propidium iodide− (PI−) (B) LSK cells from Mx1-Cre+ WT (n = 4), Mx1-Cre+ Srsf2P95H/+ (n = 5), Mx1-Cre+ Sf3b1K700E/+ (n = 4), and Mx1-Cre+ Srsf2P95H/+ Sf3b1K700E/+ (n = 4) mice 2 weeks post pIpC administration.
(C) Number of live mice at weaning from crossing Sf3b1K700E/+ mice to Vav-Cre+ Srsf2P95H/+ mice or by crossing Srsf2P95H/+ mice to Vav-Cre+ Sf3b1K700E/+ mice. **p = 0.0064; two-sided Chi-square test.
(D and E) Percentage of LSK (D) and long-term hematopoietic stem cells (LT-HSC; LSK CD150+ CD48−) (E) in E14.5 fetal livers from Vav-Cre+ WT (WT; n = 17), Vav-Cre+ Srsf2P95H/+ (P95H; n = 17), Vav-Cre+ Sf3b1K700E/+ (K700E; n = 15), and Vav-Cre+ Srsf2P95H/+ Sf3b1K700E/+ double-knockin (DKI; n = 16) fetuses and Vav-Cre− WT (n = 14), Vav-Cre− Srsf2P95H/+ (P95H; n = 17), Vav-Cre− Sf3b1K700E/+ (K700E; n = 19), and Vav-Cre− Srsf2P95H/+ Sf3b1K700E/+ DKI (n = 17) fetuses.
(F) Colony numbers from E14.5 fetal liver cells from Vav-Cre+ WT (n= 12), Vav-Cre+ Srsf2P95H/+ (P95H; n = 7), Vav-Cre+ Sf3b1K700E/+ (K700E; n = 12), and Vav-Cre+ Srsf2P95H/+ Sf3b1K700E/+ DKI (n = 7) fetuses and from Vav-Cre− WT (n = 10), Vav-Cre− Srsf2P95H/+ (P95H; n = 10), Vav-Cre− Sf3b1K700E/+ (K700E; n = 11), and Vav-Cre− Srsf2P95H/+ Sf3b1K700E/+ DKI (n = 13) fetuses.
(G–I) Representative FACS plots (G) and quantitation of BrdU+ (H) and annexin-V+ PI− (I) LSK cells from Vav-Cre+ WT (n = 5), Vav-Cre+ Srsf2P95H/+ (P95H; n = 8), Vav-Cre+ Sf3b1K700E/+ (K700E; n = 3), and Vav-Cre+ Srsf2P95H/+ Sf3b1K700E/+ DKI (n = 9) fetuses and from Vav-Cre− WT (n = 4), Vav-Cre− Srsf2P95H/+ (P95H; n = 6), Vav-Cre− Sf3b1K700E/+ (K700E; n = 7), and Vav-Cre− Srsf2P95H/+ Sf3b1K700E/+ DKI (n = 2) fetuses.
Error bars represent mean ± SD. ANOVA and Tukey’s post hoc test was used to compare groups. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figure S2.
Selection against co-expression of two mutant splicing factors was strongly supported by the fact that mice with hematopoietic-specific expression of Srsf2P95H and Sf3b1K700E (Vav-Cre+ Srsf2P95H/+ Sf3b1K700E/+) mutations were associated with 100% lethality at weaning, while single-mutant mice were present at the expected frequencies (Figure 2C). Analysis of 139 embryos at embryonic day 14.5 (E14.5) revealed that Vav-Cre+ Srsf2P95H/+ Sf3b1K700E/+ fetuses were detectable at the expected frequency (Figures S2A and S2B). E14.5 Vav-Cre+ Srsf2P95H/+ Sf3b1K700E/+ fetal livers had increased number of LSK cells as well as phenotypic long-term hematopoietic stem cell (LT-HSC, LSK CD150+ CD48−), multi-potent progenitor (MPP, LSK CD150− CD48−), HPC-1 (LSK CD150− CD48+), and HPC-2 (LSK CD150+ CD48+) populations compared with all other genotypes (Figures 2D, 2E, and S2C–S2G). Despite a significant increase in HSPCs, Vav-Cre+ Srsf2P95H/+ Sf3b1K700E/+ fetal liver cells had significantly impaired colony-forming ability in vitro (Figure 2F) and markedly increased percentage of cycling and apoptotic cells (Figures 2G–2I). These derangements in HSPC phenotypes were manifest by E18.5 when Vav-Cre+ Srsf2P95H/+ Sf3b1K700E/+ fetuses had near absence of hematopoietic cells in the BM as well as increased apoptotic cells in fetal liver (Figures S2H and S2I). Taken together, these data demonstrate that co-expression of splicing-factor mutations compromises hematopoiesis, driven by aberrant cell-cycle progression and increased apoptosis of HSPCs.
Srsf2 and Sf3b1 Mutations Have Largely Distinct Effects on Gene Expression
To understand the mechanistic basis for mutual exclusivity of SF3B1 and SRSF2 mutations, we performed RNA sequencing (RNA-seq) on lineage− c-Kit+ (LK) cells from Mx1-Cre+ Srsf2P95H/+ Sf3b1K700E/+ mice as well as single-mutant and WT controls (Figure 3A). The mean allelic ratio of Sf3b1K700E and Srsf2P95H expressed in double-mutant cells was 20.7% and 33.5%, markedly lower than the ~50% expression in single-mutant controls, illustrating the intolerability of combining SF3B1 and SRSF2 mutations (Figure 3B). Expression of either mutation resulted in dysregulation of hundreds of coding genes (Figure 3C and Table S2). Mx1-Cre+ Srsf2P95H/+ Sf3b1K700E/+ cells exhibited significantly more gene dysregulation than single-mutant cells, consistent with their more dramatic biological phenotype (Figure 3D). Double-mutant cells shared a greater proportion of differentially expressed genes with each single-mutant group than single mutants shared with one another (Figure 3E). Gene ontology (GO) analysis of coding genes differentially expressed within each genotype relative to WT control revealed strong signatures of impaired hematopoiesis in double-mutant cells, consistent with the multi-lineage defects in these cells (Figure 3F and Table S3).
Figure 3. Srsf2 and Sf3b1 Mutations Have Distinct and Independent Effects on Gene Dysregulation.
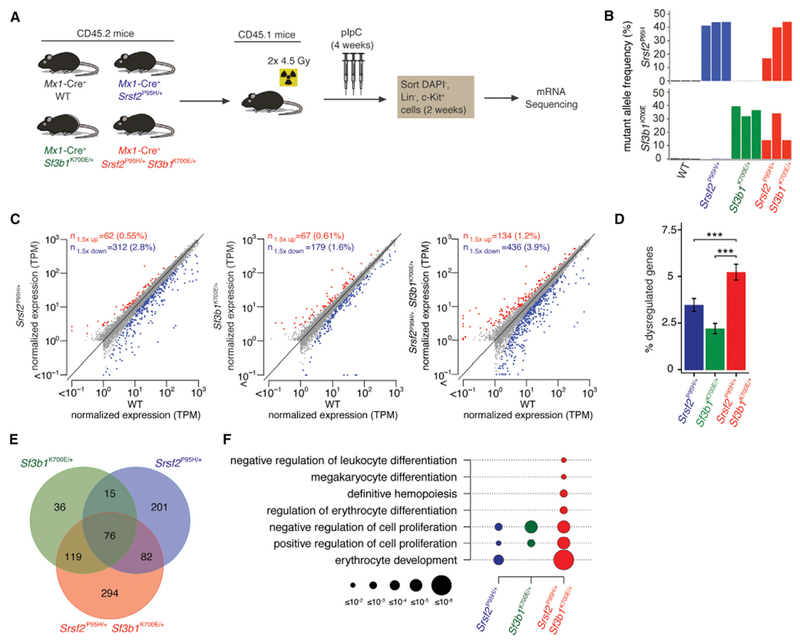
(A) Schema for RNA-seq.
(B) Expression of Srsf2P95H and Sf3b1K700E alleles as percentage of mRNAs expressed from Srsf2 and Sf3b1. Color indicates genotype; the three biological replicates are A to C from left to right.
(C) Scatterplots comparing coding gene expression in Mx1-Cre+ Srsf2P95H/+, Mx1-Cre+ Sf3b1K700E/+, and Mx1-Cre+ Srsf2P95H/+ Sf3b1K700E/+ cells relative to Mx1-Cre+WT cells for replicate B. Red and blue indicate coding genes significantly up- or down regulated, respectively, in mutant relative to Mx1-Cre+WT cells. TPM, transcripts per million (TMM-normalized).
(D) Bar plots comparing percentage of significantly dysregulated coding genes (percentage of total coding genes expressed) in Mx1-Cre+ Srsf2P95H/+, Mx1-Cre+ Sf3b1K700E/+, and Mx1-Cre+ Srsf2P95H/+ Sf3b1K700E/+ cells relative to Mx1-Cre+ WT cells for replicate B. Error bars represent mean ± SD. A two-sided binomial proportion test was used to compare groups; ***p < 0.0001.
(E) Venn diagram showing overlap between coding genes significantly dysregulated in Mx1-Cre+ Srsf2P95H/+, Mx1-Cre+ Sf3b1K700E/+, and Mx1-Cre+ Srsf2P95H/+ Sf3b1K700E/+ cells relative to Mx1-Cre+ WT cells for replicate B.
(F) GO enrichment analysis of Mx1-Cre+ Srsf2P95H/+, Mx1-Cre+ Sf3b1K700E/+, and Mx1-Cre+ Srsf2P95H/+ Sf3b1K700E/+ cells relative to Mx1-Cre+ WT cells for replicate B. Circle size indicates the magnitude of the p value for each term and comparison.
We next tested whether mutations in SF3B1 and SRSF2 have independent effects on gene expression: that is, whether double-mutant cells recapitulated the gene dysregulation observed for each single-mutant genotype relative to WT. Approximately 80% and 40% of genes dysregulated in Mx1-Cre+ Sf3b1K700E/+ and Mx1-Cre+ Srsf2P95H/+ single-mutant cells were also dysregulated in Mx1-Cre Sf3b1K700E/+ Srsf2P95H/+ double-mutant cells (Figure 3E and Table S2). That degree of recapitulation of gene dysregulation was highly similar to and statistically indistinguishable (p = 0.90 and 0.44 for Sf3b1K700E and Srsf2P95H mutations, respectively) from expected recapitulation under the assumption of independence (Figure S3A). We therefore conclude that SF3B1 and SRSF2 mutations have independent effects on gene expression even when present in the same cell. Interestingly, in addition to recapitulating the dysregulation of specific genes expected based on single-mutant cells, double-mutant cells exhibited additional gene dysregulation (Figures S3B and S3C), consistent with the severe hematopoietic phenotype of these cells. These results suggest that SF3B1 and SRSF2 mutations have independent but compound effects on gene expression when present in the same cell.
Srsf2 and Sf3b1 Mutations Have Distinct and Independent Effects on Splicing
We next assessed the consequences of single versus double mutations in Sf3b1 and Srsf2 on splicing (Table S4). Mutations in SF3B1 have been proposed to alter 3′ splice site recognition (Darman et al., 2015; DeBoever et al., 2015). In contrast, mutations in SRSF2 alter exon recognition via preferential recognition of C-rich exonic splicing enhancer (ESE) motifs relative to G-rich ESEs, while WT SRSF2 recognizes both classes of ESEs (Kim et al., 2015; Zhang et al., 2015). Our current understanding of SF3B1 and SRSF2 mutations therefore indicates that they induce distinct changes in splicing. However, the consequences of these mutations on splicing have not been compared in an isogenic context.
We used our double-mutant system to directly compare the effects of SF3B1 and SRSF2 mutations on RNA splicing. Consistent with prior studies, Sf3b1 and Srsf2 mutations affected 3′ splice site and exon recognition, respectively (Figures 4A, 4B, S4A, and S4B). We observed a modest enrichment of adenosines upstream of intron-proximal 3′ splice site promoted by the Sf3b1K700E mutation, independent of the Srsf2P95H mutation (Figure S4C). This enrichment was absent from cells expressing Srsf2P95H alone. Cassette exons promoted versus repressed in cells expressing Srsf2P95H were respectively enriched for CCNG and GGNG ESEs, independent of the presence or absence of Sf3b1K700E, but were absent from cells expressing Sf3b1K700E alone (Figures 4C and S4D). These analyses are consistent with previous reports that SF3B1 and SRSF2 mutations affect branch point recognition and ESE preference, respectively, and further demonstrate that these changes in splicing are specific to SF3B1 and SRSF2 mutations.
Figure 4. Srsf2 and Sf3b1 Mutations Have Distinct and Independent Effects on RNA Splicing.
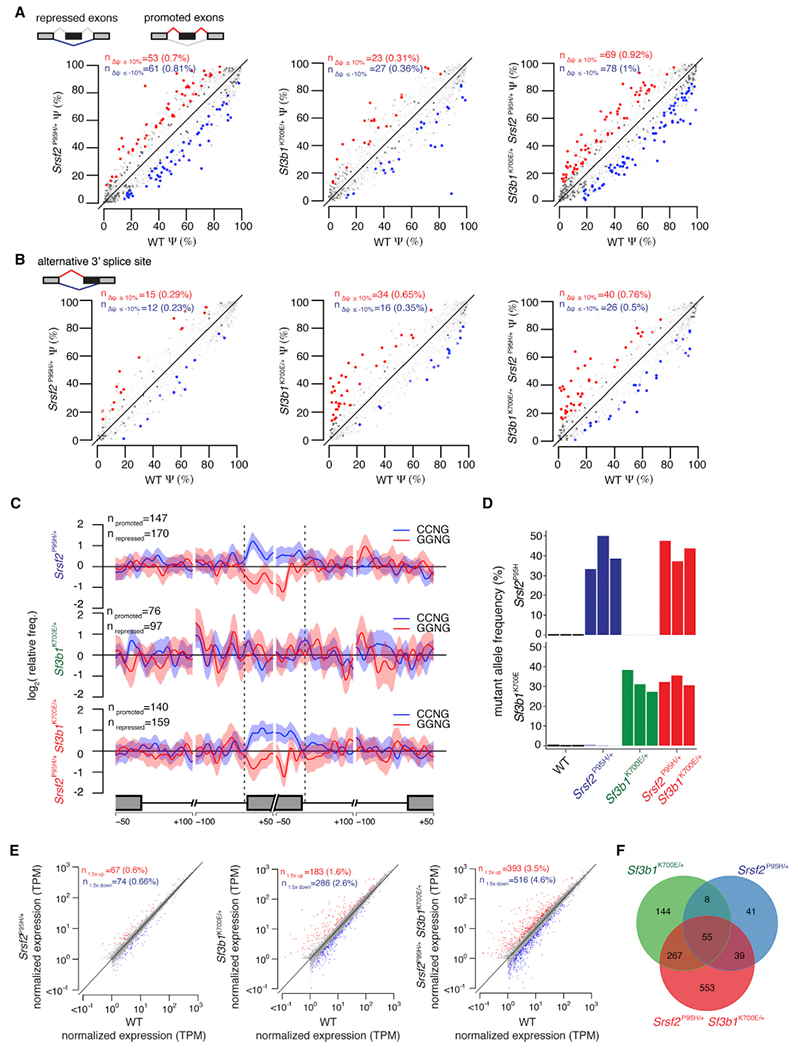
(A) Scatterplots of cassette exon inclusion in Mx1-Cre+ Srsf2P95H/+, Mx1-Cre+ Sf3b1K700E/+, and Mx1-Cre+ Srsf2P95H/+ Sf3b1K700E/+ cells relative to Mx1-Cre+ WT cells. Axes indicate the fraction of mRNAs containing each cassette exon in the indicated sample. Red and blue indicate cassette exons whose inclusion is promoted or repressed, respectively, in mutant relative to WT cells.
(B) As in (A), but for alternative 3′ splice site events. Axes indicate the fraction of mRNAs that use the intron-proximal 3′ splice site in the indicated sample. Red and blue indicate intron-proximal 3′ splice sites whose usage is promoted or repressed, respectively, in mutant relative to WT cells.
(C) Plots illustrating the spatial distribution of the CCNG and GGNG (N = any nucleotide) exonic splicing enhancers adjacent to differentially spliced cassette exons promoted versus repressed in mutant relative to WT cells. Vertical axis indicates the relative frequency of each motif, averaged over all promoted versus repressed cassette exons for the indicated genotype comparisons.
(D) Expression of Srsf2P95H and Sf3b1K700E alleles as percentage of mRNAs expressed from Srsf2 and Sf3b1 in lineage- c-Kit+ cells from fetal livers of Vav-Cre mice at E14.5. Color indicates genotype; the three biological replicates are A to C from left to right.
(E) Scatterplots comparing mean coding gene expression for all replicates in Vav-Cre+ Srsf2P95H/+, Vav-Cre+ Sf3b1K700E/+, and Vav-Cre+ Srsf2P95H/+ Sf3b1K700E/+ cells relative to Vav-Cre+ WT cells. Red and blue indicate coding genes significantly up- or down regulated, respectively, in mutant relative to Vav-Cre+ WT cells. TPM, transcripts per million (TMM-normalized).
(F) Venn diagram showing the overlap between coding genes significantly dysregulated in Vav-Cre+ Srsf2P95H/+, Vav-Cre+ Sf3b1K700E/+, and Vav-Cre+ Srsf2P95H/+ Sf3b1K700E/+ cells relative to Vav-Cre+ WT cells in all replicates.
As Sf3b1 and Srsf2 mutations cause distinct changes in splicing, even when simultaneously present, we hypothesized that the effects of these mutations on splicing were independent. Consistent with this hypothesis, double-mutant cells recapitulated the preferential effects of Sf3b1 and Srsf2 mutations on 3′ splice site recognition and cassette exon recognition, respectively, that we observed in single-mutant cells (Figure S4A). We did not observe widespread decreases in splicing efficiency, such as retention of constitutive introns, in double mutant cells (Figure S4E). Instead, Mx1-Cre+ Srsf2P95H/+ Sf3b1K700E/+ cells exhibited modest increases in mis-spliced genes relative to cells bearing single mutations in Sf3b1 or Srsf2 (Figures 4A and4B). Mx1-Cre+ Sf3b1K700E/+ Srsf2P95H/+ cells recapitulated 40% and 32% of splicing dysregulation driven by expression of the single mutations in Sf3b1K700E and Srsf2P95H, respectively (Figure S4F). We identified only seven genes mis-spliced in both Mx1-Cre+ Srsf2P95H/+ and Mx1-Cre+ Sf3b1K700E/+ single-mutant as well as double-mutant cells, and only six genes mis-spliced in both Mx1-Cre+ Srsf2P95H/+ and Mx1-Cre+ Sf3b1K700E/+ single-mutant but not double-mutant cells. We therefore conclude that SF3B1 and SRSF2 mutations typically have distinct and independent consequences on splicing.
To confirm these findings, we performed RNA-seq on purified LK cells from E14.5 Vav-Cre+ Srsf2P95H/+ Sf3b1K700E/+ fetal liver and controls. The allelic frequencies of both mutations in double-mutant cells were near 50% and comparable with those of Vav-Cre+ single-mutant controls (Figure 4D). Similar to the Mx1-Cre system, the greatest alterations in gene expression were seen in Vav-Cre+ Srsf2P95H/+ Sf3b1K700E/+ HSPCs compared with single-mutant controls, and double-mutant cells recapitulated much of the gene expression dysregulation in single-mutant cells (Figures 4E and4F). GO analysis revealed signatures of impaired hematopoiesis in double-mutant cells, consistent with those seen in Mx1-Cre mice (Figure S4G and Table S5). Moreover, effects on splicing similar to those seen in the Mx1-Cre system were seen in HSPCs from Vav-Cre mice, including preferential effects of Sf3b1 and Srsf2 mutations on 3′ splice site and cassette exon recognition, respectively (Figures S4H and S4I).
Combined Expression of Srsf2 and Sf3b1 Mutations Impairs Expression of Regulators of HSPC Survival and Increases Sensitivity to Inflammatory Stimulation
Given the largely non-overlapping effects of Srsf2P95H and Sf3b1K700E mutations on gene expression and splicing, we evaluated transcripts that exhibited concomitant dysregulation in gene expression and splicing in the double-mutant state (Figure 5A and Table S6). Multiple regulators of HSPC survival and quiescence were significantly dysregulated and mis-spliced in Srsf2P95H/+ Sf3b1K700E/+ HSPCs, including the thrombopoietin receptor c-Mpl, integrin aIIb (CD41), and the transcription factor Pbx1 (Figures 5A and S5A). After confirming reduced expression of these transcripts in LK cells in an independent cohort of Mx1-Cre+ Srsf2P95H/+ Sf3b1K700E/+ mice (Figure 5B), we evaluated the consequences of short hairpin RNA(shRNA)-mediated depletion of these mRNAs in WT HSPCs (Figures S5B and S5C). Consistent with prior published data (Alexander et al., 1996; Ficara et al., 2008; Gekas and Graf, 2013; Qian et al., 2007; Yoshihara et al., 2007), silencing any of these individual genes was associated with severe impairment of HSPC clonogenicity (Figure S5D).
Figure 5. Co-expression of Srsf2 and Sf3b1 Mutations Results in Aberrant Splicing and Expression of Key Regulators of Hematopoietic Stem Cell Survival and Quiescence.
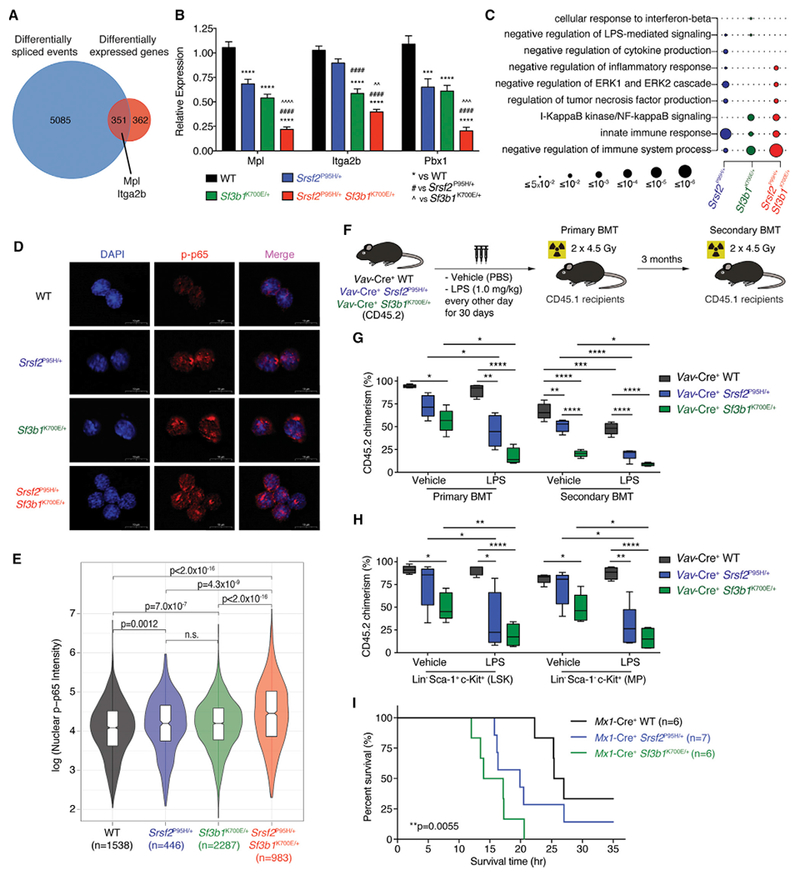
(A) Venn diagram of genes differentially expressed and spliced in Mx1-Cre+ Srsf2P95H/+ Sf3b1K700E+ cells relative to Mx1-Cre+ WT cells in any replicate.
(B) Expression of Mpl, Itga2b, and Pbx1 in lineage- c-Kit+ (LK) cells from Mx1-Cre+ Srsf2P95H/+ Sf3b1K700E/+ mice relative to control groups (n = 8–10 mice per genotype). Error bars represent mean ± SD. ***p < 0.001, ****p < 0.0001 versus Mx1-Cre+ WT; ####p < 0.0001 versus Mx1-Cre+ Srsf2P95H/+; AAp < 0.01, AAAp < 0.001, AAAAp < 0.0001 versus Mx1-Cre+ Sf3b1K700E/+.
(C) GO enrichment analysis of Mx1-Cre+ Srsf2P95H/+, Mx1-Cre+ Sf3b1K700E/+, and Mx1-Cre+ Srsf2P95H/+ Sf3b1K700E/+ cells relative to Mx1-Cre+ WT cells. Circle size indicates the magnitude of the p value for each term and comparison.
(D) Immunofluorescence of nuclear phosphorylated-p65 (p-p65) level in LK cells from Vav-Cre+ WT, Vav-Cre+ Srsf2P95H/+, Vav-Cre+ Sf3b1K700E/+, and Vav-Cre+ Srsf2P95H/+ Sf3b1K700E/+ mice following LPS stimulation ex vivo. Scale bars, 10 mm.
(E) Violin plots quantifying nuclear p-p65 intensity of LK cells from (D). ANOVA and Kruskal-Wallis ranked test was performed and adjusted for false discovery rate.
(F) Schema of competitive BMT using BM MNCs from Vav-Cre+ WT, Vav-Cre+ Srsf2P95H/+, and Vav-Cre+ Sf3b1K700E/+ mice after chronic LPS exposure.
(G) Percentage of CD45.2 chimerism in peripheral blood of recipients (n = 4–5 mice per group) from primary and secondary BMT (both at 10 weeks post BMT).
(H) Chimerism of LSK or myeloid progenitor (MP; lineage- Sca-1- c-Kit+) fractions from primary recipients 14 weeks post BMT.
(I) Kaplan-Meier analysis of Mx1-Cre+ WT, Mx1-Cre+ Srsf2P95H/+, and Mx1-Cre+ Sf3b1K700E/+ mice after a single dose of LPS (15 mg/kg) in vivo. Log-rank Mantel-Cox test was performed. p = 0.0055.
ANOVAand Tukey’s post hoc test were used to compare groups. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus Uav-Cre+WT mice(“#” denotes versus Vav-Cre+ Srsf2P95H/+ mice and “Λ” denotes versus Vav-Cre+ Sf3b1K700E/+ mice). In (G) and (H), the top and bottom lines of the box represent the upper and lower quartiles, respectively; the line inside the box represents the median; the lines above and below the box represent the maximum and minimum values, respectively. See also Figure S5 and Table S6.
In addition to gene expression changes consistent with impaired hematopoiesis in Srsf2P95H/+ Sf3b1K700E/+ cells (Figure 3F), GO analysis also revealed a strong signature associated with immune signaling that was augmented in double-relative to single-mutant cells (Figures 5C and S4G). To functionally evaluate these gene expression changes, we studied the response of Srsf2 and Sf3b1 mutant cells to lipopolysaccharide (LPS). HSPCs from Vav-Cre+ Srsf2P95H/+ and Vav-Cre+ Sf3b1K700E/+ mice exhibited increased baseline nuclear factor κB (NF-κB) activation (marked by increased nuclear accumulation of phos-phorylated p65 [p-p65]) relative to Vav-Cre+ WT HSPCs (Figures 5D and5E), and this was further enhanced following ex vivo LPS stimulation in double-mutant cells relative to single-mutant or WT cells (Figures S5E and S5F).
Given prior data identifying that chronic LPS exposure impairs repopulating potential of HSPCs (Esplin et al., 2011; Zhao et al., 2013), we next evaluated the effects of chronic inflammation on the function of Srsf2 or Sf3b1 mutant BM HSPCs (Figure 5F). Vav-Cre+ WT, Vav-Cre+ Srsf2P95H/+, and Vav-Cre+ Sf3b1K700E/+ mice were treated with LPS (1 mg/kg) every second day for 30 days followed by serial BMT into lethally irradiated recipient mice. Chronic LPS treatment had a mild effect on the repopulating potential of WT HSPCs, evidenced by similar peripheral blood and BM HSPC chimerism in primary recipients (Figures 5G and5H). In contrast, LPS-treated Vav-Cre+ Srsf2P95H/+ and Vav-Cre+ Sf3b1K700E/+ bm HSPCs showed significant reduction in repopulating ability relative to vehicle-treated counterparts, and this defect was further exacerbated following serial transplantation (Figures 5G and5H).
The above observations suggest that Srsf2 and Sf3b1 mutant HSPCs are intrinsically hypersensitive to inflammatory stimuli that contribute to defective hematopoietic function, rendering features observed in MDS patients. In addition, LPS-treated Vav-Cre+ Srsf2P95H/+ mice had enhanced myeloid skewing and reduced B lymphopoiesis relative to vehicle-treated mice or Vav-Cre+ Srsf2+/+ mice treated with LPS (Figure S5G). Moreover, acute ex vivo LPS stimulation of BM LSK cells from Vav-Cre+ Srsf2P95H/+ mice also resulted in significant increase in nuclear p-p65 relative to Vav-Cre+ Srsf2+/+ LSK cells (Figures S5H and S5I). The enhanced response to inflammatory stimuli in Srsf2 and Sf3b1 mutant mice in vivo was evident upon LPS-induced sepsis. Exposure of primary Mx1-Cre+ WT, Mx1-Cre+ Srsf2P95H/+, and Mx1-Cre+ Sf3b1K700E/+ mice with LPS (15 mg/kg) resulted in accelerated death in both Srsf2- and Sf3b1-mutant mice relative to WT controls (Figure 5I).
SF3B1 Mutations Promote Mis-splicing of MAP3K7, Resulting in Hyperactivation of NF-κB Signaling
The above results identify that spliceosomal mutations are intolerable when co-expressed but they are independently more sensitive to inflammatory stimuli that converge on NF-κB signaling. To understand how spliceosome gene mutations activate immune signaling pathways, we first focused on events that are mis-spliced within SF3B1-mutant hematopoietic cells in both human and mouse (Table S7). Although recent studies reported few such shared mis-spliced events (Mupo et al., 2016; Obeng et al., 2016), we identified a larger set of 205 transcripts mis-spliced in both Sf3b1-mutant murine hematopoietic cells and SF3S1-mutant MDS patients (Figure 6A). One of the most robust mis-spliced events was MAP3K7, for which an intron-proximal 3′ splice site was promoted by mutant SF3B1 in both human and mouse hematopoietic cells. Reanalysis of RNA-seq data from CLL patient cohorts (DeBoever et al., 2015) and isogenic cell lines with endogenous mutations in SF3B1, as well as RT-PCR and Sanger sequencing of cDNA from SF3B1-mutant cells, validated this isoform (Figures 6B, 6C, and S6A). This aberrant 3′ splice site recognition occurred in exon 5 of MAP3K7, which encodes part of the kinase domain, and is predicted to result in an out-of-frame transcript that undergoes nonsense-mediated decay (Figures S6B and S6C). Consistent with this, we observed reduced MAP3K7 protein in isogenic cell lines (Figures 6D and6E) as well as patient BM mononuclear cells (MNCs) and peripheral blood MNCs from MDS and CLL patients, respectively (Figures 6F and 6G), with SF3B1 mutations versus those lacking splicing-factor mutations.
Figure 6. SF3B1 Mutations Promote Mis-splicing of MAP3K7, Resulting in Hyperactivation of NF-κB Signaling.
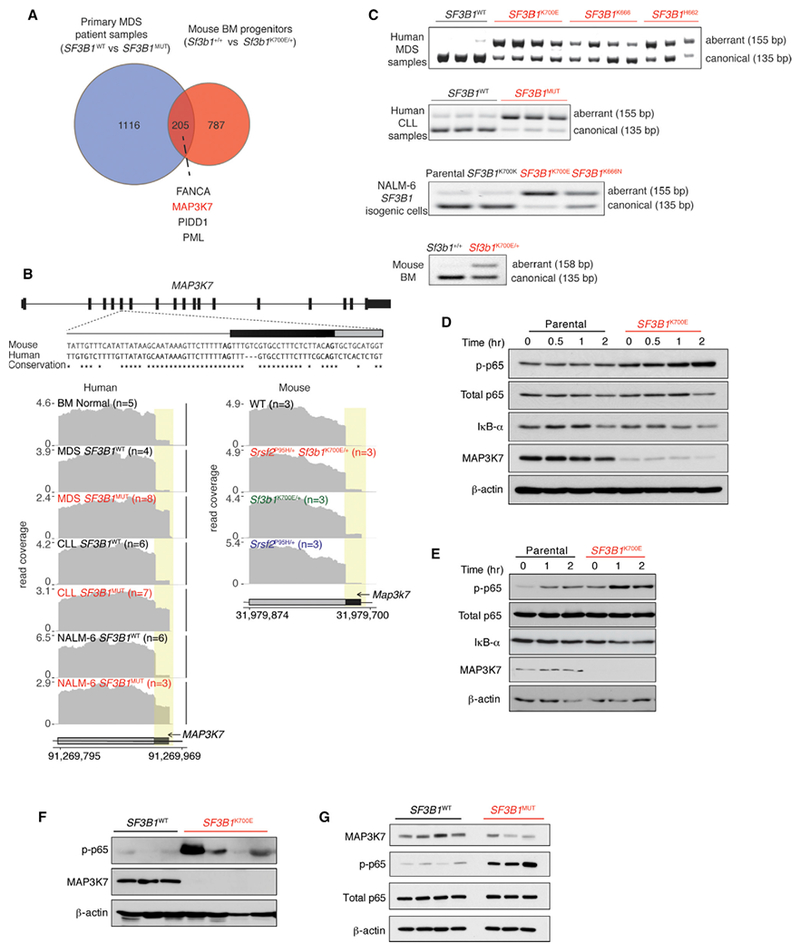
(A) Venn diagram illustrating overlap of differentially spliced genes in MDS patient samples mutant versus WT for SF3B1 and murine hematopoietic progenitors in Sf3b1K700E/+ versus Sf3b1+/+ mice.
(B) From top to bottom, conservation of mouse and human MAP3K7 sequences adjacent to the competing 3 splice site affected by SF3B1 mutations, and RNA-seq coverage plots in human and mouse samples.
(C) RT-PCR of the MAP3K7 competing 3′ splice site in MDS and CLL patient samples with or without SF3B1 mutations as well asisogenic human and mouse cells.
(D and E) Immuno blot of phosphorylated p65 (p-p65), IκB-α, MAP3K7, and loading controls in isogenic K562 (D) and NALM-6 (E) cells. “Time (hr)” refers to hours following LPS (5 mg/mL) exposure.
(F and G) Immunoblot analysis of p-p65, MAP3K7, and loading controls in BM MNCs from MDS (F) and peripheral blood MNCs from CLL patients (G) with or without SF3B1 mutations.
MAP3K7 encodes a kinase that mediates tumor necrosis factor κ (TNFκ), interleukin-1 β (IL-1β), and Toll-like receptor signaling through the NF-κB, JNK, and MAPK pathways. The effects of MAP3K7 loss have been extensively studied and can result in loss or promotion of inflammation depending on cellular context (Ajibade et al., 2012; Lamothe et al., 2012; Sato et al., 2005; Tang et al., 2008; Vink et al., 2013; Xin et al., 2017). Here, we observed that SF3B1K700E human myeloid or lymphoid leukemia cells stimulated with LPS had enhanced NF-κB activation compared with SF3B1 WT controls (Figures 6D, 6E, S6D, and S6E). Increased p-p65 level was also evident at baseline in isogenic cells as well as primary patient BM MNCs from SF3B1-mutant MDS and peripheral blood MNCs from SF3B1-mutant CLL relative to SF3B1 WT counterparts (Figures 6F and6G). Increased nuclear p-p65 levels (Figure 7A) and enhanced NF-κB transcriptional activity using a reporter assay (Figures 7B and S7A) were also evident in SF3B1K700E cells compared with SF3B1 WT cells following LPS or TNFα stimulation. This was further confirmed by marked induction in NF-κB targets IL-1β and TNFα following LPS treatment in Sf3b1K700E NALM-6 cells relative to parental and SF3B1K700K control cells (Figure 7C).
Figure 7. MAP3K7 Loss Results in Hyperactive NF-κB Signaling in SF3B1-Mutant Cells.
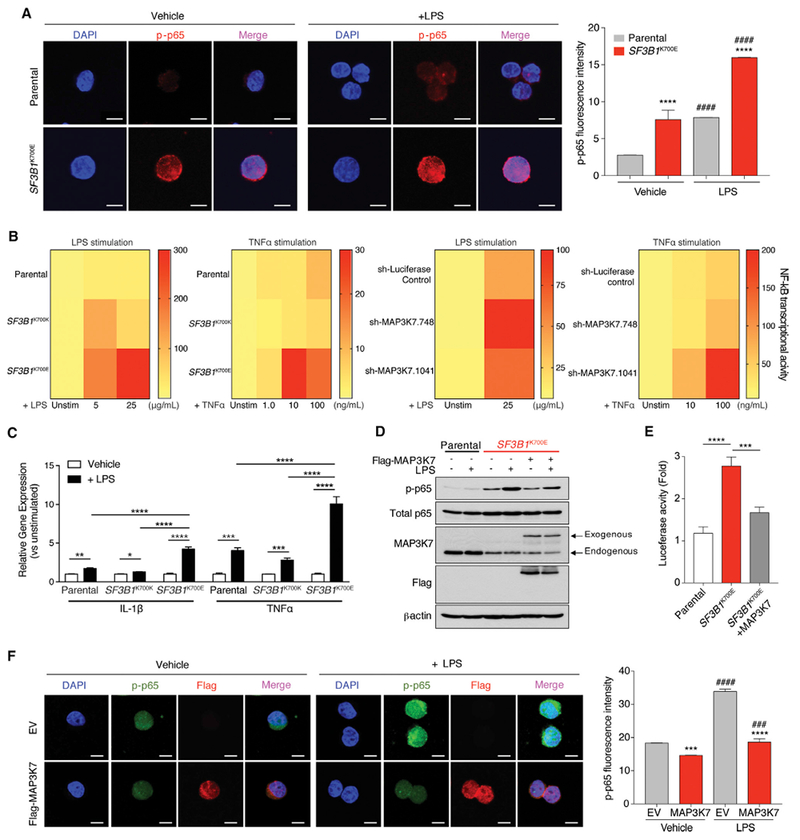
(A) Immunofluorescence of phosphorylated p65 (p-p65) in K562 cells with or without SF3B1K700E mutation 2 hr following LPS stimulation. Quantitation of p-p65 intensity is shown on the right (n = 3 independent experiments). Scale bars, 10 mm.
(B) Heat map of NF-κB reporter signal in NALM-6 SF3B1-isogenic cells (left two panels) or parental NALM-6 cells with MAP3K7 shRNAs (right two panels) following LPS or TNF a stimulation for 24 hr.
(C) qRT-PCR analysis of IL-1β and TNF a 8 hr post LPS stimulation in NALM-6 SF3B1-isogenic cells (n = 2 independent experiments).
(D) Immunoblot of p-p65 in K562 SF3B1-isogenic cells ± FLAG-MAP3K7 cDNA and/or LPS (5 mg/mL) exposure for 2 hr.
(E) Quantitation of NF-κB reporter signal in cells from (D) (n = 3 independent experiments).
(F) Immunofluorescence of nuclear p-p65 and FLAG (MAP3K7) in cells from (D) (quantitation of p-p65 intensity [n = 3 independent experiments] on the right). Scale bars, 10 μm.
Error bars represent mean ± SD. ANOVA followed by Tukey’s post hoc test were used to compare groups. *p < 0.05, **p < 0.005, ***p < 0.0002, ****p < 0.0001, ####p < 0.0001 versus Vehicle; ###p < 0.001 versus Vehicle. See also Figure S7.
Given that a number of aberrant splicing and gene expression events occur in SF3B1-mutant cells, we next sought to understand the contribution of MAP3K7 loss to hyperactivated NF-κB signaling. First, hyperactive NF-κB signaling was confirmed using shRNA-mediated downregulation of MAP3K7 in NALM-6 and K562 parental cells at the level of p-p65 signaling and NF-κB transcriptional activity at both baseline and after LPS stimulation (Figures 7B and S7B). In addition, re-expression of MAP3K7 in K562 SF3B1K700E cells resulted in a significant decrease in p-p65 in both resting state as well as following LPS exposure based on immunoblotting, NF-κB luciferase reporter, and nuclear p-p65 level (Figures 7D, 7F, and S7C). At a biological level, restoration of Map3k7 expression in Sf3b1K700E/+ HSPCs resulted in mild rescue of HSPC clonogenicity (Figure S7D). Overall, these data suggest that the effects of Sf3b1K700E mutation on induction of NF-κB signaling are indeed, in part, mediated through aberrant splicing of MAP3K7.
SRSF2 Mutations Promote Aberrant Splicing of Caspase-8, Resulting in a Truncated Protein that Hyperactivates NF-κB Signaling
Consistent with the distinct effects of SF3B1 and SRSF2 mutations on splicing, MAP3K7 aberrant splicing was restricted to SF3B1-mutant cells (Figure S8A). We therefore searched for aberrant splicing events in SRSF2-mutant cells that might affect NF-κB signaling. One such event was aberrant splicing of caspase-8, encoded by CASP8, that was recurrently mis-spliced in AML and CMML patients with SRSF2 mutations, but not those bearing SF3B1 mutations (Figures 8A and S8B). Caspase-8 is a cysteine protease that initiates death-receptor-mediated apoptosis (Shu et al., 1997; Thome et al., 1997) in addition to regulating necroptosis and serving as a key activator of NF-κB (Chaudhary et al., 2000; Hu et al., 2000; Shikama et al., 2003). SRSF2 mutations repressed a cassette exon of caspase-8, as was evident by RNA-seq, RT-PCR, qRT-PCR, and cDNA sequencing from cell lines and patient samples (Figures 8A, 8B, and S8C–S8E). CASP8 normally encodes a 54/55-kDa protein containing two death-effector domains (DED) at the N terminus and a C-terminal catalytic domain. Exclusion of this cassette exon results in an mRNA encoding a truncated caspase-8 protein lacking the C-terminal catalytic domains, which was readily detectable in SRSF2-mutant cells using an N-terminal anti-caspase-8 antibody (Figures 8C and8D). Although several caspase-8 isoforms have been described (Xu et al., 2009; Yuan et al., 2012), the specific isoform detected in SRSF2-mutant cells is distinct from those previously described.
Figure 8. SRSF2 Mutations Promote Aberrant Splicing of Caspase-8, Resulting in Expression of a Truncated Protein that Hyperactivates NF-κB Signaling.
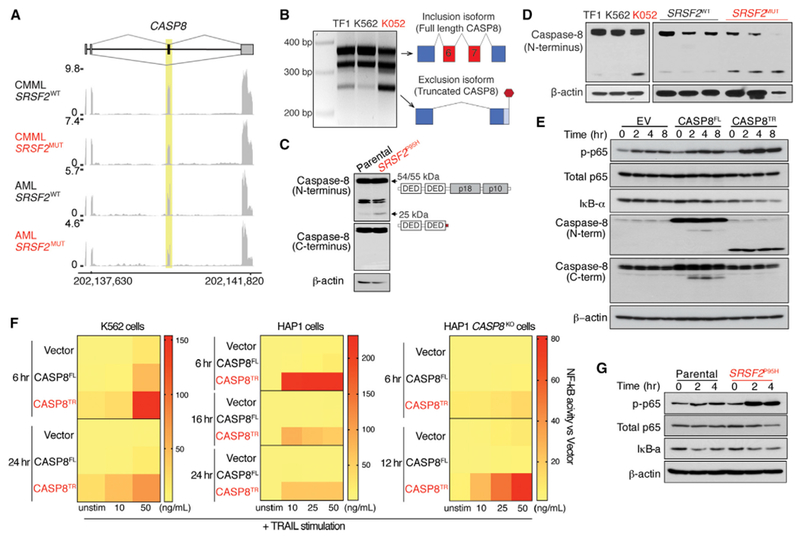
(A) RNA-seq coverage plots of caspase-8 splicing in CMML and AML patients WT or mutant (MUT) for SRSF2.
(B) RT-PCR analysis of caspase-8 splicing in SRSF2 WT (TF1 and K562) or SRSF2-mutant (K052) cells.
(C and D) Immunoblot of caspase-8 using an N-terminal anti-caspase-8 antibody in K562 SRSF2-isogenic cells (C) or human leukemia cells (left panel) and primary AML patient samples (right panel) WT or mutant for SRSF2 (D).
(E) Immunoblot of phosphorylated p65 (p-p65), IκB-α, caspase-8 in K562 cells expressing empty vector (EV), full-length caspase-8 (CASP8FL), or the truncated caspase-8 isoform (CASP8TR) after exposure to TRAIL (50 ng/mL).
(F) Heatmap of NF-κB reporter signal following TRAIL stimulation in K562, HAP1, or CASP8KO HAP1 cells expressing EV, CASP8FL, or CASP8TR.
(G) Immunoblot of p-p65 and IκB-α in K562 cells with or without Srsf2P95H following exposure to TRAIL (50 ng/mL).
See also Figure S8.
Previously described DED-only forms of caspase-8 have been suggested to serve either as competitive inhibitors or promoters of apoptosis (Xu et al., 2009; Yuan et al., 2012). Given this, we tested the effect of this SRSF2-mutant-specific truncated isoform (hereafter referred to as CASP8TR) in an SRSF2 WT cell line followed by TRAIL (TNF-related apoptosis-inducing ligand) stimulation to engage the death-receptor pathway. Overexpression of both caspase-8 full-length (CASP8FL) and CASP8TR isoforms resulted in robust expression of proteins at the expected size (Figures 8E and S8F) with no effect on cell growth (Figure S8G). Moreover, both isoforms promoted TRAIL-mediated cell death, suggesting that the truncated isoform did not affect cell death relative to CASP8FL (Figure S8H). Given this, we next evaluated the effect of overexpressing CASP8TR on NF-κB transcriptional activity, signaling, and nuclear localization. In both K562 and HAP1 cells with endogenous caspase-8 expression, overexpression of the CASP8TR, but not the CASP8FL isoform, resulted in robust induction of NF-κB activity upon increasing concentration of TRAIL (Figures 8E, 8F, S8I, and S8J). These data were also confirmed in K562 cells with or without endogenous Srsf2P95H mutation (Figures 8G, S8K, and S8L). To rule out an effect of endogenous caspase-8, we assessed the effect of CASP8TR on cell death and NF-κB activity in CASP8KO HAP1 cells. Consistent with the lack of the catalytic domain, CASP8KO HAP1 cells expressing CASP8TR isoform were unable to undergo TRAIL-mediated cell death (Figures S8M and S8N); however, the CASP8TR isoform was able to induce robust NF-κB signaling in the absence of WT caspase-8 (Figures 8F, S8O, and S8P).
DISCUSSION
Mutations affecting RNA splicing factors are the most common genetic alterations in MDS but the basis for their significant enrichment in this disease remains largely unexplained. Here we identify that two of the most commonly mutated splicing factors in MDS converge on activation of innate immune signaling through aberrant splicing of mRNAs encoding distinct enzymes.
Although the initial description of spliceosomal mutations predicted that the mutually exclusive pattern of these mutations might be due to a common impact on MDS pathogenesis (Yoshida et al., 2011), the data here identify that these mutations are mutually exclusive, in part due to synthetic lethal interaction. This provides one of the few examples of mutually exclusive oncogenic alterations due to synthetic lethal interactions in cancer. Interestingly, co-expression of SF3B1K700E and SRSF2P95H mutations resulted in impaired HSPC self-renewal, differentiation, and survival, but this is not due to widespread inhibition of splicing efficiency. Instead we identified aberrant splicing and dysregulation of key regulators of HSPC function in double mutant cells, including the thrombopoietin receptor c-Mpl, the homeodomain transcription factor Pbx1, and integrin αIIb. Hematopoietic-specific deletion of these factors individually has previously been shown to result in failure of hematopoiesis due to reduced HSC self-renewal, increased apoptosis, and loss of quiescence (Ficara et al., 2008; Gekas and Graf, 2013; Qian et al., 2007; Yoshihara et al., 2007), all features characteristic of the double-mutant state.
Multiple lines of evidence implicate innate immune signaling in MDS pathogenesis (Basiorka et al., 2016; Fang et al., 2014; Wei et al., 2013), including experiments demonstrating increased innate immune signaling contributes to MDS development in vivo (Fang et al., 2017; Varney et al., 2015). Increased activation of Toll-like receptor and IL-1 receptor signaling with downstream activation of MAPK and NF-κB pathways is widely reported in MDS, but the mechanistic basis for this activation have mostly been restricted to MDS with deletion of chromosome 5q (Fang et al., 2014, 2017; Starczynowski, 2014; Varney et al., 2015). Our work identifies a mechanism for hyperactivated NF-κB signaling in a wider spectrum of MDS.
Although recent studies of SF3B1K700E mutation identified relatively few mRNAs mis-spliced in both mouse and human cells (Mupo et al., 2016; Obeng et al., 2016), we identified a much greater overlap of aberrantly spliced transcripts shared across mouse and human SF3B1K700E mutant hematopoietic cells than previously reported. This includes aberrant splicing of MAP3K7 through the use of an alternative 3′ splice site promoted by mutant SF3B1. As down regulation of Map3k7 in myeloid cells promotes myeloid neoplasms, this finding is likely to be relevant to SF3B1-mutant MDS. For example, in vivo knockdown of Map3k7 resulted in splenomegaly, myeloproliferation, and extramedullary hematopoiesis, as well as increased immune activation that was exacerbated by LPS (Vink et al., 2013). Similarly, myeloid-specific deletion of Map3k7 heightened the response to inflammatory stimuli and causes a clonal myeloid leukemia (Ajibade et al., 2012; Eftychi et al., 2012; Lamothe et al., 2012; Xin et al., 2017). Interestingly, the pro-inflammatory and leukemogenic effects of Map3k7 loss appear to be restricted to myeloid lineages (Ajibade et al., 2012), in stark contrast to pan-hematopoietic deletion of Map3k7, which results in complete failure of hematopoiesis (Tang et al., 2008). It will therefore be important to understand whether there is a differential requirement for hematopoiesis in SF3B1 mutant cells given the partial loss of MAP3K7 induced by mutant SF3B1.
While aberrant splicing and downregulation of MAP3K7 was specific to SF3B1 mutant cells, cells expressing mutant SRSF2 shared similar elevated innate immune signaling and hypersensitivity to LPS. This led us to identify a gain-of-function effect of SRSF2 mutations through generation of a C-terminal truncated caspase-8 isoform that promotes NF-κB signaling. This is consistent with prior work showing that N-terminal prodomain-only containing isoforms of procaspase-8 activate NF-κB signaling through interactions with upstream regulators of NF-κB, a function not mediated by canonical full-length caspase-8 (Chaudhary et al., 2000; Hu et al., 2000; Shikama et al., 2003).
Together, our data demonstrate that different splicing-factor mutations alter distinct targets at the level of pre-mRNA splicing that nonetheless converge on the same downstream signaling node to hyperactivate innate immune signaling.
STAR*METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-B220 Alexa Fluor 700 (RA3-6B2) | eBioscience | Cat# 56-0452-82; RRID: AB_891458 |
| Anti-CD19 PECy7 (1D3) | eBioscience | Cat# 25-0193-82; RRID: AB_657663 |
| Anti-CD3 BV605 (17A2) | BioLegend | Cat# 100237; RRID: AB_2562039 |
| Anti-CD4 BV711 (GK1.5) | BioLegend | Cat# 100447; RRID: AB_2564586 |
| Anti-CD8a PerCP/Cy5.5 (53-6.7) | BioLegend | Cat# 100734; RRID: AB_2075238 |
| Anti-Gr-1 PECy7 (RB6-8C5) | eBioscience | Cat# 25-5931-82; RRID: AB_469663 |
| Anti-CD11b/Mac-1 PE (M1/70) | eBioscience | Cat# 12-0112-82; RRID: AB_465546 |
| Anti-NK1.1 APCCy7 (PK136) | BioLegend | Cat# 108724; RRID: AB_830871 |
| Anti-Ter119 | BioLegend | Cat# 116223; RRID: AB_2137788 |
| Anti-c-Kit APC (2B8) | eBioscience | Cat# 17-1171-82; RRID: AB_469430 |
| Anti-Sca-1 PECy7 (D7) | eBioscience | Cat# 25-5981-82; RRID: AB_469669 |
| Anti-FcγRII/III Alexa Fluor 700 (93) | eBioscience | Cat# 56-0161-82; RRID: AB_493994 |
| Anti-CD34 FITC (RAM34) | eBioscience | Cat# 11-0341-82; RRID: AB_465021 |
| Anti-CD45.1 PerCP/Cy5.5 (A20) | BioLegend | Cat# 110728; RRID: AB_893346 |
| Anti-CD45.2 BV605 (104) | BioLegend | Cat# 109841; RRID: AB_2563485 |
| Anti-CD48 PerCP/Cy5.5 (HM48-1) | BioLegend | Cat# 103422; RRID: AB_2075051 |
| Anti-CD150 PE(9D1) | eBioscience | Cat# 12-1501-82; RRID: AB_465873 |
| Anti-CD44 FITC (IM7) | eBioscience | Cat# 11-0441-82; RRID: AB_465045 |
| Anti-IgM PE (II/41) | eBioscience | Cat# 12-5790-82; RRID: AB_465940 |
| Anti-CD25 PE (PC61.5) | eBioscience | Cat # 12-0251-82; RRID: AB_465607 |
| Anti-CD43 FITC (S11) | BioLegend | Cat# 143204; RRID: AB_10960745 |
| Anti-BrdU FITC Flow Kit | BD Pharmingen | Cat# 559619; RRID: AB_2617060 |
| Annexin V FITC Apoptosis Detection Kit | BD Pharmingen | Cat# 556547 |
| Anti-NF-κB/p65 (D14E12) | Cell Signaling Technology | Cat# 8242; RRID: AB_10859369 |
| Anti-phospho NF-κB/p65-Ser536 (93H1) | Cell Signaling Technology | Cat# 3033; RRID: AB_331284 |
| Anti-IκB-α, rabbit polyclonal | Cell Signaling Technologies | Cat# 9242; RRID: AB_823540 |
| Anti-TAK1/MAP3K7 (D94D7), rabbit monoclonal | Cell Signaling Technologies | Cat# 5206; RRID: AB_10694079 |
| Anti-Caspase-8, N-terminal (E6), rabbit monoclonal | Abcam | Cat# ab32125; RRID: AB_2068469 |
| Anti-Caspase-8, C-terminal (12F5), mouse monoclonal | Enzo Life Science | Cat# ALX-804-242-C100; RRID: AB_2050949 |
| Anti-β-actin (AC-15), mouse monoclonal | Sigma-Aldrich | Cat# A5441; RRID: AB_476744 |
| Anti-Flag (M2), mouse monoclonal | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| Anti-mouse CD45, rat monoclonal | BD Pharmingen | Cat# 550539; RRID: AB_2174426 |
| Anti-Myeloperoxidase, rabbit polyclonal | Dako | Cat# A0398; RRID: AB_2335676 |
| Anti-Cleaved Caspase-3, rabbit polyclonal | Cell Signaling Technologies | Cat# 9661; RRID: AB_2341188 |
| Anti-mouse Alexa Fluor 488, goat polyclonal | ThermoFisher Scientific | Cat# A-11001; RRID: AB_2534069 |
| Anti-rabbit Alexa Fluor 594, goat polyclonal | ThermoFisher Scientific | Cat# A-11012; RRID: AB_2534079 |
| Prolong Gold Antifade mounting agent | ThermoFisher Scientific | Cat# P36930 |
| Biological Samples | ||
| MDS and CLL primary patient samples | MSKCC | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Escherichia coli 0111:B5 LPS | Sigma Aldrich | Cat# L2880 |
| Polybrene | Millipore | Cat# TR-1003-G |
| Recombinant TRAIL (soluble, human) | Enzo Life Science | Cat# ALX-201-073-3020 |
| Recombinant human TNFα | PeproTech | Cat# 300-01A |
| Recombinant mouse IL-3 | R&D | Cat# 403-ML |
| Recombinant mouse IL-6 | R&D | Cat# 406-ML |
| Recombinant mouse SCF | R&D | Cat# 455-MC |
| Recombinant mouse TPO | R&D | Cat# 488-TO |
| Normal goat serum | ThermoFisher Scientific | Cat# PCN500 |
| Actinomycin D | Life Technologies | Cat# 11805-017 |
| Critical Commercial Assays | ||
| MethoCult™ GF M3434 | StemCell Technologies | Cat# M3434 |
| NF-κB Cignal™ Reporter Assay | Qiagen | Cat# CCS-013L |
| Deposited Data | ||
| RNAseq data | This paper | GSE97452 |
| Experimental Models: Cell Lines | ||
| Human: NALM-6 cells, Parental, SF3B1K700K, SF3B1K700E, SF3B1K666N | Horizon Discovery | Cat# N/A |
| Human: K562 cells, Parental, SF3B1K700E, SRSF2P95H | Horizon Discovery | Cat# N/A |
| Human: HAP1 Parental cells | Horizon Discovery | Cat# C631 |
| Human: HAP1 CASP8KO cells | Horizon Discovery | Cat# HZGHC001511c007 |
| Human: 293T cells | ClonTech | Cat# 632180 |
| Human: 293 GPII cells | Clontech | Cat# 631530 |
| Experimental Models: Organisms/Strains | ||
| Mice: Mx1-Cre transgenic mice (C.Cg-Tg(Mx1-cre)1Cgn/J) | The Jackson Laboratory | Cat# JAX: 005673 |
| Mice: Vav-Cre transgenic mice (B6.Cg-Tg(Vav1-icre)A2Kio/J) | The Jackson Laboratory | Cat# JAX: 008610 |
| Mice: Srsf2P95H/+ (B6J.B6NTac(SJL)-Srsf2tm1.1Oaw/J) | Kim et al. 2015 | |
| Mice: Srsf2fl/fl (B6;129S4-Srsf2tm1Xdfu/J) | The Jackson Laboratory | Cat# JAX: 018019 |
| Mice: Sf3b1K700E//+ | Obeng et al. 2016 | |
| Oligonucleotides | ||
| Please Refer to Table S8 | Table S8 | |
| Recombinant DNA | ||
| pVSV.G | Addgene | Cat# 12259 |
| psPAX2 | Addgene | Cat# 12260 |
| MSCV-IRES-GFP empty vector | Addgene | Cat# 52107 |
| MSCV-Flag-CASP8FL-IRES-GFP | This paper | |
| MSCV-Flag-CASP8TR-IRES-GFP | This paper | |
| MSCV-Flag-mMap3k7-IRES-GFP | This paper | |
| MSCV-Flag-hMAK3K7-IRES-GFP | This paper | |
| T3G-dsRED-mirE-PGK-Neo-IRES-rtTA3 (LT3RENIR) | Fellmann et al., 2013 | |
| pMSCV-LTR-mirE-PGK-SV40-IRES-GFP (MLS-E) | Fellmann et al., 2013 | |
| pLKO.Empty Vector-Puro | Kim et al. 2015 | |
| pLKO.sh-UPF1-Puro | Kim et al. 2015 | |
| pNL3.2.NF-κB-RE Reporter Plasmid | Promega | Cat# N1111 |
| Software and Algorithms | ||
| Bowtie v1.0.0 | Langmead et al. 2009 | |
| RSEM v1.2.4 | Li and Dewey, 2011 | |
| TopHat v2.0.8b | Trapnell et al. 2009 | |
| TMM method | Robinson and Oshlack 2010 | |
| Wagenmakers’s Bayesian framework | Wagenmakers et al. 2010 | |
| MISO v2.0 | Katz et al. 2010 | |
| GOseq | Young et al. 2010 | |
| Bioconductor | Huber et al. 2015 | |
| Other | ||
| FASTQ files from published RNA-seq studies of patients with MDS | Dolatshad et al. 2015 | GSE63569 |
| FASTQ files from published RNA-seq studies of patients with CLL | Darman et al. 2015 | GSE72790 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Omar Abdel-Wahab (abdelwao@mskcc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All animals were housed at Memorial Sloan Kettering Cancer Center (MSKCC). All animal procedures were completed in accordance with the Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committees at MSKCC. Generation and genotyping of the Srsf2P95H/+, Sf3b1K700E/+ and Srsf2fl/+ as well as the Mx1-Cre and Vav-Cre transgenic mice have been previously described (Kim et al., 2015; Obeng et al., 2016). 8-week-old female CD45.1 C57BL/6J mice (The Jackson Laboratory) were used as recipients for bone marrow transplantation assays.
Primary Human MDS and CLL Samples
Studies were approved by the Institutional Review Boards of Memorial Sloan Kettering Cancer Center and conducted in accordance to the Declaration of Helsinki protocol. Patients provided samples after their informed consent and primary human de-identified MDS, AML, and CLL samples derived from whole peripheral blood or BM mononuclear cells were utilized.
Cell Lines
The NALM-6 isogenic cell lines (NALM-6 cells engineered to express the single mutations SF3B1K700E or SF3B1K700K from the endogenous locus) were cultured in RPMI/10% FCS and K562 isogenic cell lines (engineered to express SF3B1K700E or Srsf2P95H mutations from each respective endogenous locus) were cultured in IMDM/10% FCS. HAP1 and CASP8KO HAP1 cells (obtained from Horizon Discovery) were cultured in IMDM/10% FCS.
METHOD DETAILS
Peripheral Blood Analysis
Blood was collected by submandibular bleeding using heparinized microhematocrit capillary tubes (Thermo Fisher Scientific). Automated peripheral blood counts were obtained using a Pro Cyte Dx Hematology Analyzer (IDEXX).
Bone Marrow Transplantation Assays
Primary mouse bone marrow (BM) cells were isolated from Mx1-Cre+ wild-type (WT), Mx1-Cre+ Srsf2P95H/+, Mx1-Cre+ Sf3b1K700E/+, Mx1-Cre+ Srsf2P95H/+ Sf3b1K700E/+, Mx1-Cre+ Srsf2P95H/P95H or Mx1-Cre+ Srsf2P95H/fl mice (aged 8 weeks) into cold phosphate-buffered saline (PBS), without Ca2+ and Mg2+, and supplemented with 2% bovine serum albumin (BSA) to generate single cell suspensions. Red blood cells (RBCs) were removed using ammonium chloride-potassium bicarbonate (ACK) lysis buffer, resuspended in PBS/2% BSA, and filtered through a 40mm cell strainer. Total nucleated cells were quantified by the Vi-Cell XR cell counter (Beckman Coulter). For competitive transplantation experiments, a total of 1.8 × 106 BM cells from donor mice (Mx1-Cre+ WT, Mx1-Cre+ Srsf2P95H/+, Mx1-Cre+ Sf3b1K700E/+, Mx1-Cre+ Srsf2P95H/+ Sf3b1K700E/+, Mx1-Cre+ Srsf2P95H/P95H, Mx1-Cre+ Srsf2P95H/fl, Vav-Cre+ WT, Vav-Cre+ Srsf2P95H/+, and Vav-Cre+ Sf3b1K700E/+ CD45.2+ mice were mixed with 0.2 × 106 wild-type CD45.1+ BM and transplanted via tail vein injection into 8-week old lethally irradiated (2 × 450 cGy) CD45.1+ recipient mice. For noncompetitive transplantation experiments, 2 × 106 total BM cells from Mx1-Cre+ WT, Mx1-Cre+ Srsf2P95H/+, Mx1-Cre+ Sf3b1K700E/+, or Mx1-Cre+ Srsf2P95H/+ Sf3b1K700E/+ mice were injected into lethally irradiated (2 ×450 cGy) CD45.1+ recipient mice. To induce the conditional alleles on Mx1-Cre background, mice were treated with 3 doses of polyinosinic:polycytidylic acid (pIpC; 12mg/kg/day; GE Healthcare) every other day via intra-peritoneal injection. Peripheral blood chimerism of mature blood cell lineages was assessed routinely by flow cytometry.
In Vivo LPS Stimulation Experiment
For in vivo LPS stimulation, Escherichia coli 055:B5 LPS (Sigma Aldrich) was used. For chronic LPS exposure, Vav-Cre+WT, Vav-Cre+ Srsf2P95H/+ and Vav-Cre+ Sf3b1K700E/+ received intra-peritoneal injection of LPS (1 mg/kg) every other day for 30 days. For acute LPS exposure, Mx1-Cre+ WT, Mx1-Cre+ Srsf2P95H/+ and Mx1-Cre+ Sf3b1K700E/+ mice that have received pIpC for 8 weeks prior to activate the mutant alleles were given a single dose of LPS (15 mg/kg) via intra-peritoneal injection.
In Vitro Colony-Forming Assays
Single-cell suspension was prepared from E14.5 fetal livers, and 25,000 cells from each embryo were plated in duplicates in cytokine-supplemented methylcellulose medium (Metho Cult™ GF M3434; Stem Cell Technologies), and colonies were enumerated 10-14 days later. To assess the effect of shRNA-mediated knockdown of target genes, 8-12 week-old C57BL/6 male mice were treated with 5-fluorouracil (150 mg/kg) via intra-peritoneal injection. Five days after injection, BM cells were harvested from the legs (femora and tibiae) and hip bones, and lineage-depletion was performed with biotin-conjugated antibodies against B220 (RA3-6B2), CD19 (1D3), CD3 (17A2), CD4 (GK1.5), CD8a (53-6.7), CD11b (M1/70), Gr-1 (RB6-8C5), NK1.1 (PK136) and Ter119, labeled with anti-biotin MicroBeads (130-090-485; Miltenyi Biotec), and lineage-negative (Lin−) cells were magnetically separated using MACS columns according to the manufacturer’s instructions. Lin− BM cells were cultured overnight in IMDM/10% FCS supplemented with mIL-3 (10 ng/mL), mIL-6 (10 ng/mL) and mSCF (50 ng/mL). The next day, cells were subjected to spinfection (2,700 rpm for 1 hr) with retroviral supernatants containing shRNAs or cDNAs of interests in the presence of polybrene (5 mg/mL; Millipore). 24 hr after spinfection, cells that were successfully infected with retrovirus were marked with GFP, and were purified by flow cytometry. FACS-sorted cells were cultured in cytokine-supplemented methylcellulose medium (MethoCult™ GF M3434; StemCell Technologies), and colonies were enumerated 10-14 days later.
Flow Cytometry Analyses
Cells were incubated with antibodies in PBS/2% BSA (without Ca2+ and Mg2+) for 45 min on ice. For hematopoietic stem and progenitor cell analysis from adult mouse bone marrow, cells were stained with a lineage cocktail of monoclonal antibodies including B220 (RA3-6B2), CD19 (1D3), CD3 (17A2), CD4 (GK1.5), CD8a (53-6.7), CD11b (M1/70), Gr-1 (RB6-8C5), NK1.1 (PK136) and Ter119, allowing for mature lineage exclusion from the analysis. For fetal liver analysis, CD11b (M1/70) was excluded from the lineage depletion cocktail. Cells were also stained with antibodies against c-Kit (2B8), Sca-1 (D7), FcγRII/III (93), CD34 (RAM34), CD45.1 (A20), CD45.2 (104), CD48 (HM48-1) and CD150 (9D1). DAPI was used to exclude dead cells. The composition of mature hematopoietic cell lineages in the bone marrow, spleen, thymus and peripheral blood was assessed using a combination of antibodies against B220, CD19, CD3, CD4, CD8a, CD11b, CD25 (PC61.5), CD44 (IM7), Gr-1, IgM (Il/41), CD43 (S11). All FACS sorting was performed on FACS Aria, and analysis was performed on an LSR Fortessa (BD Biosciences). Data analysis was performed using the FlowJo software.
Cell Cycle and Apoptosis Analyses
For apoptosis assays, freshly harvested bone marrow or fetal liver cells were first stained with antibodies against cell surface markers of interests, and then stained with FITC-conjugated Annexin-V in Annexin-V binding buffer (BD Pharmingen) according to manufacturer instructions. For assessment of cell cycle status in adult bone marrow HSPCs, BrdU (1 mg/kg) was administered via intra-peritoneal injection to adult mice 48 hr prior to sacrifice. For cell cycle analysis of E14.5 fetal HSPCs, BrdU (1 mg/kg) was administered to pregnant mice via intra-peritoneal injection 3 hr prior to harvesting fetal livers. Assessment of BrdU incorporation was performed following manufacturer instructions (BD Pharmingen) and data was acquired on a LSR Fortessa (BD Biosciences).
Histological Analysis
Tissues, embryos and pups were fixed in 4% paraformaldehyde, processed routinely in alcohol and xylene, embedded in paraffin, sectioned at 5-micron thickness, and stained with hematoxylin-eosin (H&E). Multiple sections were obtained through the head in the coronal plane, trunk in the transverse plane, and fore and hind limbs in the longitudinal plane. Immunohistochemistry (IHC) was performed on a Leica Bond RX automated stainer (Leica Biosystems, Buffalo Grove, IL). Following HIER at pH 6.0, the primary antibody against mouse CD45 (BD Pharmingen; 550539), myeloperoxidase (Dako; A0398), and cleaved caspase-3 (Cell Signaling; 9661) were applied at a concentration of 1:250,1:1000, and 1:250 respectively, followed by application of a polymer detection system (DS9800, Novocastra Bond Polymer Refine Detection, Leica Biosystems) in which the chromogen was 3,3 diaminobenzidine tetrachloride (DAB) and counterstain was hematoxylin. For quantification of cleaved caspase-3 by image analysis, whole slide digital images were generated on a slide scanner (Pannoramic 250 Flash III, 3DHistech, 20×/0.8NA objective, Budapest, Hungary) at a resolution of 0.2431 mm per pixel. Staining quantification was performed with Qu Path 0.1.2 software (Centre for Cancer Research & Cell Biology, Queen’s University Belfast, UK). The region of interest (ROI) was defined as the liver parenchyma. The number of DAB positive cells per mm2 was measured with the positive cell detection algorithm. ROI selection and algorithm optimization and validation, and qualitative examination of all H&E and IHC slides were performed by a board-certified veterinary pathologist (S.M.).
Immunoblot
For immunoblotting, the following antibodies were used: NF-κB/p65 (CST; 8242), phosphorylated NF-κB/p65-Ser536 (CST; 3033), IκB-α (CST; 9242), TAK1/MAP3K7 (CST; 5206), Caspase-8, N-terminal (Abcam; clone E6), Caspase-8, C-terminal (Enzo Life Science; clone 12F5), Flag (Sigma-Aldrich; F-1804), β-actin (Sigma-Aldrich; A-5441).
Immunofluorescence
Following stimulation with LPS (Sigma-Aldrich), TNFα (Pepro Tech), or TRAIL (Enzo Life Science), cells were fixed with 4% paraformaldehyde/PBS for 10 min at room temperature (RT), permeabilized with PBS-T (PBS/1% BSA/0.2% Triton-X) for 15 min, blocked with PBS-T/5% goat serum (Thermo Fisher PCN500) for 1 hr at RT, and incubated with primary antibodies (1:50 dilution for p-p65 and 1:100 for Flag) in PBS-T/5% goat serum overnight at 4°C. Cells were washed three times with PBS-T for 10 minutes at RT with gentle agitation, and were incubated with goat anti-rabbit Alexa Fluor 594 (Thermo Fisher Scientific; A-11012) or goat anti-mouse Alexa Fluor 488 (Thermo Fisher Scientific; A-11001) secondary antibodies (1:500 dilution in PBS-T/5/% normal goat serum) for 2 hr at RT in the dark. Cells were then washed twice with PBS-T for 10 min at RT, and counter-stained with DAPI (0.5 mg/mL; Sigma-Aldrich; D-9542) for 20 min at RT. Cells were coverslipped with Pro Long Gold anti-fade (Thermo Fisher Scientific; P36930). Images were captured using either a confocal microscope (Leica TCS SP5, Upright; Leica Micro Systems), or were digitally scanned with a Pannoramic Confocal Scanner (3DHistech, Budapest Hungary) using a 20×/0.8NA objective. The projected images were exported into tif format using Case Viewer software (3DHistech) and analyzed with Image J/FIJI. A macro was written that segments each nucleus using the DAPI channel and measures the p-p65 intensity within the nucleus after appropriate threshold was set.
Caspase-8 and MAP3K7 Constructs
MSCV-Flag-CASP8FL-IRES-GFP, MSCV-Flag-CASP8TR-IRES-GFP, MSCV-Flag-MAK3K7-IRES-GFP (human), MSCV-Flag-Map3k7-IRES-GFP (mouse) and MSCV-IRES-GFP empty vector constructs were used for overexpression studies. Retroviral supernatants were produced by transfecting 293 GPII cells with cDNA constructs and the packaging plasmid VSV.G using XtremeGene9 (Roche), and were used to transduce HAP1, CASP8KO HAP1 and K562 parental and SF3B1K700E isogenic cells in the presence of polybrene (5 mg/mL; Millipore). Successfully transduced cells expressing GFP were purified by flow cytometry. Cells were stimulated with LPS (Sigma-Aldrich), TNFα (Pepro Tech) or TRAIL (Enzo Life Science).
mRNA Stability Assay
For mRNA half-life measurement using qRT-PCR, UPF1 shRNA and control lentivirus infected K562 SF3B1K700E cells were treated with 2.5 mg/ml Actinomycin D (Life Technologies) and harvested at 0, 2, 4, 6, and 8 hr (following protocols established previously (t Hoen et al., 2011)). MAP3K7 inclusion, MAP3K7 exclusion and 18s rRNA mRNA levels were measured by qRT-PCR.
Luciferase Reporter Assay
We generated K562, NALM-6 SF3B1 isogenic cells, HAP1 and CASP8KO HAP1 cells expressing the luciferase reporter for NF-κB response elements by following the manufacturer instructions (Cignal™ Reporter Assay, Qiagen). Cells were stimulated with LPS, TNFα or TRAIL as described above, and NF-κB activity was assessed by luciferase intensity using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer instructions. To verify that the luciferase reporter assay was not aberrantly activated by basal leakiness, we used another NF-κB reporter plasmids (Promega; N1111) with known NF-κB response elements (RE), and performed mutagenesis in the NF-κB-RE using the QuiK Change II Site-Directed Mutagenesis Kit (Agilent Technologies; #200522). There are five putative NF-κB-RE binding sites in this reporter plasmid (5′-GGGRNTTTCC-3′, where R is a purine, Y is a pyrimidine and N is any nucleotide). To mutagenize the binding sequence, the “TTTC” sequence was mutated to “AAAA”. The primers used to create the two mutant plasmids are:
Mutant-Fwd
GGTACCTGAGCTCGCTAGCGGGAAAAAACGGGGACAAAACGGGAAAAAACGGGGACAAAACGGGAAAAAACAGATCTGGCCTCGGCGGCCAAGCTTA.
Mutant-Rev
TAAGCTTGGCCGCCGAGGCCAGATCTGTTTTTTCCCGTTTTGTCCCCGTTTTTTCCCGTTTTGTCCCCGTTTTTTCCCGCTAGCGAGCTCAGGTACC.
RT-PCR and Quantitative RT-PCR (qRT-PCR)
Total RNA was isolated using RNeasy Mini kit (Qiagen). For cDNA synthesis, total RNA was reverse transcribed to cDNA with SuperScript VILO cDNA synthesis kit (Life Technologies). The resulting cDNA was diluted 10-20 fold prior to use. Quantitative RT-PCR (qRT-PCR) was performed in 10 mL reactions with either SYBR Green PCR Master Mix orTaqman Gene Expression Master Mix with AmpErase (ThermoFisher Scientific). All qRT-PCR analysis was performed on an Applied Biosystems QuantStudio 6 Flex Cycler (ThermoFisher Scientific). Relative gene expression levels were calculated using the comparative CT method.
Primers used in RT-PCR reactions were:
MAP3K7 (human) – Fwd: GATGGAATATGCTGAAGGGG, Rev: CACTCCTTGGGAACACTGTA
Map3k7 (mouse) – Fwd: GATGGAATATGCAGAGGGGG, Rev: CACTCCTTGGGAACACTGTA
CASP8 (human) – Fwd: GAACTTCAGACACCAGGC, Rev: CTTTGTCCAAAGTCTTTGCTG
Primers used in qRT-PCR reactions were:
CASP8 exclusion isoform (aberrant):
Fwd: GATGAATTTTCAAATGACTTTGGAC
Rev: TGATCAGACAGTATCCCCGAG CASP8 inclusion isoform (canonical):
Fwd: TGATGAATTTTCAAATGGGGAGGA
Rev: ATCCTGTTCTCTTGGAGAGTCC
MAP3K7 mRNA half-life experiment (human):
Fwd (common): GCGTTTATTGTAGAGCTTCGG
Rev (canonical): GCACCATGCAGCACATTATATAAAG
Rev (aberrant): CATGCAGCACTGCGAAAGAAAG
Taqman probes were used for gene expression analysis of TNF (Hs00174128_m1), IL-1β (Hs01555410_m1), GAPDH (Hs02786624_g1), Mpl (Mm00440310_m1), Pbx1 (Mm04207617_m1), Itga2b (Mm00439741_m1), and Hprt (Mm03024075_m1).
shRNA Experiments
NALM-6 parental cells carrying the NF-κB luciferase reporter were transduced with a doxycycline-inducible lentiviral vector, T3G-dsRED-mirE-PGK-Neo-IRES-rtTA3 (Fellmann et al., 2013), expressing shRNAs for MAP3K7 ora non-targeting renilla or firefly luciferase control. Transduced cells were selected with G418 (0.5 mg/mL; Thermo Fisher Scientific), and the short hairpins were induced with the addition of doxycycline (2.0 mg/mL; Sigma Aldrich). All mouse shRNAs used in clonogenic assays were cloned into the retroviral pMSCV-LTR-mirE-PGK-SV40-IRES-GFP (MLS-E) backbone (Fellmann et al., 2013). All shRNAs were designed using the Splash RNA algorithm (Pelossof et al., 2017). The short hairpin sequences are:
sh-MAP3K7.748: TTAGGTAAATTTTTTATCAGTG
sh-MAP3K7.1041: TTTTCAACAATTTTGATTCTAA
sh-Luciferase control: TTAATCAGAGACTTCAGGCGGT
sh-Ren.713 control: CAGGAATTATAATGCTTATCTA
sh-Mpl.2121: TTATATAATAAACAGTGTCTAA
sh-Mpl.2368: TCAAATAAATAGATGACAGCAA
sh-Pbx1.824: TTCATCCAAACTCTGGTCTGTG
sh-Pbx1.1393: TCATTCAGAATTTCTGTGGCTT
sh-Itga2b.2279: TTCTCTTTCTTCTGAGTGCAGA
sh-Itga2b.3380: TTAGGAAAAGGGATGCACCCGG
sh-UPF1 (TRCN0000022254): CCGGGCATCTTATTCTGGGTAATAACTCGAGTTATTACCCAGAATAAGATGCTTTTT
mRNA Isolation, Sequencing, and Analysis
RNA was extracted from sorted mouse cell populations using Qiagen R Neasy columns. Poly(A)-selected, unstranded Illumina libraries were prepared with a modified Tru Seq protocol. 0.5× AM Pure XP beads were added to the sample library to select for fragments <400 bp, followed by 1× beads to select for fragments >100 bp. These fragments were then amplified with PCR (15 cycles) and separated by gel electrophoresis (2% agarose). 300 bp DNA fragments were isolated and sequenced on the Illumina HiSeq 2000 (~100M 101 bp reads per sample).
Genome Annotations
Genome annotations for the human (NCBI GRCh37/UCSC hg19) and mouse (NCBI GRCm38/UCSC mm10) genomes were created as previously described (Dvinge et al., 2014). Genome annotations from the Ensembl (Flicek et al., 2013) and UCSC (Meyer et al., 2013) databases were merged with splicing event annotations from MISO v2.0 (Katz et al., 2010). An additional annotation of all possible combinations of annotated 5′ and 3′ splice sites found in the merged annotation was created for read mapping. Constitutive introns were defined as those whose associated 5′ and 3′ splice sites were alternatively spliced in the UCSC annotation.
RNA-seq Read Mapping
RNA-seq reads were sequentially mapped to the transcriptome and genome as previously described (Dvinge et al., 2014). Reads were first mapped to the transcriptome using Bowtie v1.0.0 (Langmead et al., 2009) and RSEM v1.2.4 (Li and Dewey, 2011). The resulting read alignments were then filtered to require that reads spanning splice junctions overlapped the flanking exons by at least six nt. The remaining unaligned reads were then mapped to the genome and splice junctions using TopHat v2.0.8b(Trapnell et al., 2009), where reads were only allowed to align to the splice junctions present in the file of all possible combinations of annotated 5′ and 3′ splice sites described above. The resulting read alignments were then merged with the output of RSEM’s alignment to create a final file of aligned reads.
Differential Gene Expression Analysis
Gene expression analysis was performed using the gene expression estimates computed by RSEM in units of transcripts per million (TPM). Those estimates were then further normalized using the TMM method (Robinson and Oshlack, 2010), with a reference set of all protein-coding genes. Differentially expressed genes were defined as those with an associated Bayes factor ≥ 100 (computed using Wagenmakers’s Bayesian framework (Wagenmakers et al., 2010)) and an associated fold-change ≥ 1.5.
Differential Splicing Analysis
Isoform ratios for annotated splicing events (cassette exons, competing 5′ and 3′ splice sites, and annotated retained introns) were calculated using MISO v2.0 (Katz et al., 2010). Splicing of constitutively spliced introns and junctions was quantified using only junction-spanning reads, as previously described (Hubert et al., 2013). Differential splicing in the murine data was identified by comparing samples from different genotypes for a single replicate in a pairwise fashion. The analysis was restricted to splicing events with at least 20 informative reads, where an informative read is defined as a read that distinguishes between isoforms. Differentially spliced events were defined as those with an associated Bayes factor was ≥ 5 (computed using Wagenmakers’s Bayesian framework (Wagenmakers et al., 2010)) and absolute change in isoform ratio of ≥ 10%. Differential splicing in the human patient cohorts was identified using a group statistical test to identify differences between patient samples with or without defined splicing factor mutations. Differentially spliced events were defined as those with an associated p value ≥ 0.05 (computed using the Wilcoxon rank-sum test) and an absolute change in median per-group isoform ratio of ≥ 10%.
Gene Ontology (GO) Enrichment Analysis
GO enrichment analysis was performed using the GOseq method (Young et al., 2010) to correct for sequencing depth biases. The background set of genes was defined as all protein-coding genes. False discovery rates were calculated using the Wallenius method and corrected using the Benjamini-Hochberg method. We restricted reporting of enriched terms to those with at least two ancestors and fewer than 500 associated genes.
Motif Enrichment Analysis and Sequence Logos
The relative enrichment of different ESEs was computed by comparing motif occurrence within and adjacent to cassette exons that were promoted versus repressed in cells or samples with versus without defined splicing factor mutations. These analyses were performed using all cassette exons that were differentially spliced in at least one mouse replicate for a given genotype comparison. 95% confidence intervals were calculated by bootstrapping with 500 resampling steps. Sequence logos centered on intron-proximal or intron-distal 3′ splice sites were created using all competing 3′ splice sites that were differentially spliced in at least one mouse replicate for a given genotype comparison. The analysis was restricted to events with canonical GT and AG dinucleotides at the 5′ and 3′ splice sites. These analyses relied upon the Genomic Ranges package in Bioconductor (Huber et al., 2015).
Analysis of Expected and Observed Gene Expression Convergence
Expected gene expression programs for Mx1-Cre+ Srsf2P95H/+ Sf3b1K700E/+ samples were calculated by computing the mean of individual gene expression levels between Mx1-Cre+ Srsf2P95H/+ and Mx1-Cre+ Sf3b1K700E/+ samples for each replicate. This method of computing the expected gene expression program was motivated by the assumption that mutations in Srsf2 and Sf3b1 have independent consequences for individual gene expression. Gene dysregulation for the expected gene expression program was then calculated as described previously for the actual, observed gene expression values.
The numbers of dysregulated genes that were convergent between the expected gene expression program and the two Mx1-Cre+ Srsf2P95H/+ and Mx1-Cre+ Sf3b1K700E/+ single mutants was then determined for each replicate, again using a method identical to that for the actual, observed gene expression values (Figure S4A). A one-sided binomial proportion test was then used to test whether there were more dysregulated genes that were observed than expected (Figure S4B).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analyses
All data are presented as mean±standard deviation, unless otherwise stated. The replicate for each experiment was stated in the figure legend or indicated in the figure. Statistical significance was determined by analysis of variance (ANOVA) after testing for normal distribution and equal variance, followed by Tukey’s post-hoc test for multiple group comparisons. A p value of <0.05 was considered statistically significant. For non-normally distributed data, a non-parametric test (Kruskal-Wallis) was used, followed by multiple group comparisons using false-discovery rate (FDR). For Kaplan Meier survival analysis, Mantel-Cox log-ranked test was used to determine statistical significance. For offspring frequency analysis, a Chi-Square test was performed to test the difference between observed and expected frequencies from different genotypes. No blinding or randomization was used. Unless otherwise noted, all immunoblot quantitation and immunofluorescence image quantitation were representative of at least three biological replicates from independent experiments. Data were plotted using Graph Pad Prism 7 software.
DATA AND SOFTWARE AVAILABILITY
Publicly Available RNA-seq Data
FASTQ files from published RNA-seq studies of patients with MDS (Dolatshad et al., 2015) and CLL(Darman et al., 2015) were downloaded from GEO series GSE63569 and GSE72790.
Accession Codes
Gene Expression Omnibus: The accession number for all newly generated RNA-seq data reported in this paper are deposited into the GEO database (accession number GSE97452).
Supplementary Material
Highlights.
Mutations in SF3B1 and SRSF2 have a synthetic lethal interaction
Mutations in RNA splicing factors are not tolerated in a homozygous state
Mutations in SF3B1 and SRSF2 have distinct effects on pre-mRNA splicing
Both SF3B1 and SRSF2 mutations result in hyperactive NF-κB signaling
Significance.
Mutual exclusivity of different mutations that affect a single pathway in cancer is commonly thought to indicate convergent effects of these mutations. RNA splicing-factor mutations constitute the most common class of alterations in patients with myelodysplastic syndromes and are also frequent in several additional cancer types, and occur as heterozygous mutations at restricted residues in a mutually exclusive manner. The mutual exclusivity of spliceosomal mutations suggests synthetic lethal and/or convergent biological effects of these mutations; however, there is currently no functional evidence supporting either of these possibilities. Here we report that individual spliceosomal mutations have non-overlapping effects on splicing and are mutually exclusive due to both synthetic lethal interactions and convergent effects on hyperactivation of innate immune signaling.
ACKNOWLEDGMENTS
This work was supported by the Leukemia and Lymphoma Society (S.C.-W.L., D. I., R.K.B., O.A.-W.), the NCI K99CA218896(S.C.-W.L.),Aplastic Anemia and MDS International Foundation (A.Y.), Lauri Strauss Leukemia Foundation (A.Y.), US Dept. of Defense Bone Marrow Failure Research Program grant W81XWH-12-1-0041 (R.K.B., O.A.-W.), the National Research Foundation of Korea (NRF) grant funded by the Korean Government (Young Researcher Program, NRF-2017R1C1B2001991) (E.K.), the Settlement Research Fund (1.160104.01) of UNIST (Ulsan National Institute of Science and Technology) (E.K.), the NRF grant (the Individual Research in Basic Science and Engineering program, NRF-2017R1D1A1B03034094) (E.J.), the American Society of Hematology (S.C.-W.L., J.T., B.H.D., O.A.-W.), the Edward P. Evans Foundation (R.K.B., O.A.-W.), the Taub Foundation (O.A.-W.), grant R01 HL128239 (R.K.B., O.A.-W.), the Ellison Medical Foundation grant AG-NS-1030-13 (R.K.B.), grant R01 DK103854 (R.K.B.), the Starr Foundation grants I8-A8-075 and I9-A9-059 (O.A.-W.), and The Pershing Square Sohn Foundation. We would like to acknowledge the Molecular Cytology, Flow Cytometry, and Laboratory of Comparative Pathology core facilities for their technical assistance, and the MSK Cancer Center Support grant P30 CA008748.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes eight figures and eight tables and can be found with this article online at https://doi.org/10.1016/j.ccell.2018.07.003.
DECLARATION OF INTERESTS
J.P., M.S., S.B., and P.G.S. are employees of H3 Biomedicine.
REFERENCES
- Ajibade AA, Wang Q, Cui J, Zou J, Xia X, Wang M, Tong Y, Hui W, Liu D, Su B, et al. (2012). TAK1 negatively regulates NF-kappaB and p38 MAP kinase activation in Gr-1 +CD11b+ neutrophils. Immunity 36, 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander WS, Roberts AW, Nicola NA, Li R, and Metcalf D (1996). Deficiencies in progenitor cells of multiple hematopoietic lineages and defective megakaryocytopoiesis in mice lacking the thrombopoietic receptor c-Mpl. Blood 87,2162–2170. [PubMed] [Google Scholar]
- Basiorka AA, McGraw KL, Eksioglu EA, Chen X, Johnson J, Zhang L, Zhang Q, Irvine BA, Cluzeau T, Sallman DA, et al. (2016). The NLRP3 in-flammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 128, 2960–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejar R, Stevenson KE, Caughey BA, Abdel-Wahab O, Steensma DP, Galili N, Raza A, Kantarjian H, Levine RL, Neuberg D, et al. (2012). Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J. Clin. Oncol. 30, 3376–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary PM, Eby MT, Jasmin A, Kumar A, Liu L, and Hood L (2000). Activation of the NF-kappaB pathway by caspase 8 and its homologs. Oncogene 19, 4451–4460. [DOI] [PubMed] [Google Scholar]
- Damm F, Kosmider O, Gelsi-Boyer V, Renneville A, Carbuccia N, Hidalgo-Curtis C, Della Valle V, Couronne L, Scourzic L, Chesnais V, et al. (2012). Mutations affecting mRNA splicing define distinct clinical phenotypes and correlate with patient outcome in myelodysplastic syndromes. Blood 119,3211–3218. [DOI] [PubMed] [Google Scholar]
- Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, et al. (2015). Cancer-associated SF3B1 hotspot mutations induce cryptic 3′ splice site selection through use of a different branch point. Cell Rep. 13, 1033–1045. [DOI] [PubMed] [Google Scholar]
- DeBoever C, Ghia EM, Shepard PJ, Rassenti L, Barrett CL, Jepsen K, Jamieson CH, Carson D, Kipps TJ, and Frazer KA (2015). Transcriptome sequencing reveals potential mechanism of cryptic 3′ splice site selection in SF3B1-mutated cancers. PLoS Comput. Biol. 11, e1004105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolatshad H, Pellagatti A, Fernandez-Mercado M, Yip BH, Malcovati L, Attwood M, Przychodzen B, Sahgal N, Kanapin AA, Lockstone H, et al. (2015). Disruption of SF3B1 results in deregulated expression and splicing of key genes and pathways in myelodysplastic syndrome hematopoietic stem and progenitor cells. Leukemia 29, 1092–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge H, Ries RE, Ilagan JO, Stirewalt DL, Meshinchi S, and Bradley RK (2014). Sample processing obscures cancer-specific alterations in leukemic transcriptomes. Proc. Natl. Acad. Sci. USA 111, 16802–16807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eftychi C, Karagianni N, Alexiou M, Apostolaki M, and Kollias G (2012). Myeloid TAKI [corrected] acts as a negative regulator of the LPS response and mediates resistance to endotoxemia. PLoS One 7, e31550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esplin BL, Shimazu T, Welner RS, Garrett KP, Nie L, Zhang Q, Humphrey MB, Yang Q, Borghesi LA, and Kincade PW (2011). Chronic exposure to a TLR ligand injures hematopoietic stem cells. J. Immunol. 186, 5367–5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Barker B, Bolanos L, Liu X, Jerez A, Makishima H, Christie S, Chen X, Rao DS, Grimes HL, et al. (2014). Myeloid malignancies with chromosome 5q deletions acquire a dependency on an intrachromosomal NF-kappaB gene network. Cell Rep. 8, 1328–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Bolanos LC, Choi K, Liu X, Christie S, Akunuru S, Kumar R, Wang D, Chen X, Greis KD, et al. (2017). Ubiquitination of hnRNPA1 by TRAF6 links chronic innate immune signaling with myelodysplasia. Nat. Immunol. 18, 236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, Lai DY, Barbosa IA, Kwon JS, Guan Y, et al. (2013). An optimized microRNA backbone for effective single-copy RNAi. Cell Rep. 5, 1704–1713. [DOI] [PubMed] [Google Scholar]
- Ficara F, Murphy MJ, Lin M, and Cleary ML (2008). Pbx1 regulates selfrenewal of long-term hematopoietic stem cells by maintaining their quiescence. Cell Stem Cell 2, 484–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, et al. (2013). Ensembl 2013. Nucleic Acids Res. 41, D48–D55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gekas C, and Graf T (2013). CD41 expression marks myeloid-biased adult hematopoietic stem cells and increases with age. Blood 121, 4463–4472. [DOI] [PubMed] [Google Scholar]
- Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T, et al. (2014). Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 28, 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, and Bowcock AM (2013). Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat. Genet. 45, 133–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu WH, Johnson H, and Shu HB (2000). Activation of NF-kappaB by FADD, casper, and caspase-8. J. Biol. Chem. 275, 10838–10844. [DOI] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. (2015). Orchestrating high-throughput genomic analysis with bioconductor. Nat. Methods 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert CG, Bradley RK, Ding Y, Toledo CM, Herman J, Skutt-Kakaria K, Girard EJ, Davison J, Berndt J, Corrin P, et al. (2013). Genome-wide RNAi screens in human brain tumor isolates reveal a novel viability requirement for PHF5A. Genes Dev. 27, 1032–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, and Bradley RK (2014). U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 25, 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, and Burge CB (2010). Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 7, 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, et al. (2015). SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell 27, 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamothe B, Lai Y, Hur L, Orozco NM, Wang J, Campos AD, Xie M, Schneider MD, Lockworth CR, Jakacky J, et al. (2012). Deletion of TAK1 in the myeloid lineage results in the spontaneous development of mye-lomonocytic leukemia in mice. PLoS One 7, e51228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasho TL, Jimma T, Finke CM, Patnaik M, Hanson CA, Ketterling RP, Pardanani A, and Tefferi A (2012). SRSF2 mutations in primary myelofibrosis: significant clustering with IDH mutations and independent association with inferior overall and leukemia-free survival. Blood 120, 4168–4171. [DOI] [PubMed] [Google Scholar]
- Lee SC, Dvinge H, Kim E, Cho H, Micol JB, Chung YR, Durham BH, Yoshimi A, Kim YJ, Thomas M, et al. (2016). Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat. Med. 22, 672–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, Przychodzen B, Bupathi M, Guinta K, Afable MG, et al. (2012). Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 119, 3203–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Masshofer L, Temming P, Rahmann S, Metz C, Bornfeld N, van de Nes J, Klein-Hitpass L, Hinnebusch AG, Horsthemke B, et al. (2013). Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat. Genet. 45, 933–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meggendorfer M, Roller A, Haferlach T, Eder C, Dicker F, Grossmann V, Kohlmann A, Alpermann T, Yoshida K, Ogawa S, et al. (2012). SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood 120, 3080–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer LR, Zweig AS, Hinrichs AS, Karolchik D, Kuhn RM, Wong M, Sloan CA, Rosenbloom KR, Roe G, Rhead B, et al. (2013). The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res. 41, D64–D69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mupo A, Seiler M, Sathiaseelan V, Pance A, Yang Y, Agrawal AA, lorio E, Bautista R, Pacharne S, Tzelepis K, et al. (2016). Hemopoietic-specific Sf3b1-K700E knock-in mice display the splicing defect seen in human MDS but develop anemia without ring sideroblasts. Leukemia 31, 720–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, Schneider RK, Lord AM, Wang L, Gambe RG, et al. (2016). Physiologic expression of Sf3b1(K700E) causes impaired erythropoiesis, aberrant splicing, and sensitivity to therapeutic spliceosome modulation. Cancer Cell 30, 404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L,Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, et al. (2011). Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med. 365, 1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, Yoon CJ, Ellis P, Wedge DC, Pellagatti A, et al. (2013). Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 122, 3616–3627, quiz 3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patnaik MM, Lasho TL, Finke CM, Hanson CA, Hodnefield JM, Knudson RA, Ketterling RP, Pardanani A, and Tefferi A (2013). Spliceosome mutations involving SRSF2, SF3B1, and U2AF35 in chronic myelomonocytic leukemia: prevalence, clinical correlates, and prognostic relevance. Am. J. Hematol. 88, 201–206. [DOI] [PubMed] [Google Scholar]
- Pelossof R, Fairchild L, Huang CH, Widmer C, Sreedharan VT, Sinha N, Lai DY, Guan Y, Premsrirut PK, Tschaharganeh DF, et al. (2017). Prediction of potent shRNAs with a sequential classification algorithm. Nat. Biotechnol. 35, 350–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian H, Buza-Vidas N, Hyland CD, Jensen CT, Antonchuk J, Mansson R, Thoren LA, Ekblom M, Alexander WS, and Jacobsen SE (2007). Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell Stem Cell 1, 671–684. [DOI] [PubMed] [Google Scholar]
- Robinson MD, and Oshlack A (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Matsumoto K, Takeuchi O, and Akira S (2005). Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 6, 1087–1095. [DOI] [PubMed] [Google Scholar]
- Shikama Y, Yamada M, and Miyashita T (2003). Caspase-8 and caspase-10 activate NF-kappaB through RIP, NIK and IKKalpha kinases. Eur. J. Immunol. 33, 1998–2006. [DOI] [PubMed] [Google Scholar]
- Shu HB, Halpin DR, and Goeddel DV (1997). Casper is a FADD- and caspase-related inducer of apoptosis. Immunity 6, 751–763. [DOI] [PubMed] [Google Scholar]
- Starczynowski DT (2014). Errant innate immune signaling in del(5q) MDS. Blood 124, 669–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- t Hoen PA, Hirsch M, de Meijer EJ, de Menezes RX, van Ommen GJ, and den Dunnen JT (2011). mRNA degradation controls differentiation state-dependent differences in transcript and splice variant abundance. Nucleic Acids Res. 39, 556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M, Wei X, Guo Y, Breslin P, Zhang S, Zhang S, Wei W, Xia Z, Diaz M, Akira S, and Zhang J (2008). TAK1 is required for the survival of hematopoietic cells and hepatocytes in mice. J. Exp. Med. 205, 1611–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thol F, Kade S, Schlarmann C, Loffeld P, Morgan M, Krauter J, Wlodarski MW, Kolking B, Wichmann M, Gorlich K, et al. (2012). Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood 119, 3578–3584. [DOI] [PubMed] [Google Scholar]
- Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schroter M, et al. (1997). Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 386, 517–521. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varney ME, Niederkorn M, Konno H, Matsumura T, Gohda J, Yoshida N, Akiyama T, Christie S, Fang J, Miller D, et al. (2015). Loss of Tifab, a del(5q) MDS gene, alters hematopoiesis through derepression of Toll-like receptor-TRAF6 signaling. J. Exp. Med. 212, 1967–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink PM, Smout WM, Driessen-Engels LJ, de Bruin AM, Delsing D, Krajnc-Franken MA, Jansen AJ, Rovers EF, van Puijenbroek AA, Kaptein A, et al. (2013). In vivo knockdown of TAK1 accelerates bone marrow proliferation/differentiation and induces systemic inflammation. PLoS One 8, e57348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenmakers EJ, Lodewyckx T, Kuriyal H, and Grasman R (2010). Bayesian hypothesis testing for psychologists: a tutorial on the Savage-Dickey method. Cogn. Psychol. 60, 158–189. [DOI] [PubMed] [Google Scholar]
- Wang L, Lawrence M, Wan Y, Stojanov P, Sougnez C, Stevenson K, Werner L, Sivachenko A, DeLuca D, Zhang L, et al. (2011). SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N. Engl. J. Med. 365,2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Dimicoli S, Bueso-Ramos C, Chen R, Yang H, Neuberg D, Pierce S, Jia Y, Zheng H, Wang H, et al. (2013). Toll-like receptor alterations in myelodysplastic syndrome. Leukemia 27, 1832–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin J, Breslin P, Wei W, Li J, Gutierrez R, Cannova J, Ni A, Ng G, Schmidt R, Chen H, et al. (2017). Necroptosis in spontaneously-mutated hematopoietic cells induces autoimmune bone marrow failure in mice. Haematologica 102, 295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Tang K, Wang M, Rao Q, Liu B, and Wang J (2009). A new caspase-8 isoform caspase-8s increased sensitivity to apoptosis in Jurkat cells. J. Biomed. Biotechnol. 2009, 930462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. (2011). Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478, 64–69. [DOI] [PubMed] [Google Scholar]
- Yoshihara H, Arai F, Hosokawa K, Hagiwara T, Takubo K, Nakamura Y, Gomei Y, Iwasaki H, Matsuoka S, Miyamoto K, et al. (2007). Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell 1, 685–697. [DOI] [PubMed] [Google Scholar]
- Young MD, Wakefield MJ, Smyth GK, and Oshlack A (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11, R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan RT, Young S, Liang J, Schmid MC, Mielgo A, and Stupack DG (2012). Caspase-8 isoform 6 promotes death effector filament formation independent of microtubules. Apoptosis 17, 229–235. [DOI] [PubMed] [Google Scholar]
- Zhang J, Lieu YK, Ali AM, Penson A, Reggio KS, Rabadan R, Raza A, Mukherjee S, and Manley JL (2015). Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc. Natl. Acad. Sci. USA 112, E4726–E4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SJ, Rampal R, Manshouri T, Patel J, Mensah N, Kayserian A, Hricik T, Heguy A, Hedvat C, Gonen M, et al. (2012). Genetic analysis of patients with leukemic transformation of myeloproliferative neoplasms shows recurrent SRSF2 mutations that are associated with adverse outcome. Blood 119,4480–4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Ling F, Wang HC, and Sun XH (2013). Chronic TLR signaling impairs the long-term repopulating potential of hematopoietic stem cells of wild type but not Id1 deficient mice. PLoS One 8, e55552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Derti A, Ruddy D, Rakiec D, Kao I, Lira M, Gibaja V, Chan H, Yang Y, Min J, et al. (2015).A chemical genetics approach for the functional assessment of novel cancer genes. Cancer Res. 75, 1949–1958. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Publicly Available RNA-seq Data
FASTQ files from published RNA-seq studies of patients with MDS (Dolatshad et al., 2015) and CLL(Darman et al., 2015) were downloaded from GEO series GSE63569 and GSE72790.
Accession Codes
Gene Expression Omnibus: The accession number for all newly generated RNA-seq data reported in this paper are deposited into the GEO database (accession number GSE97452).