

Abstract
In cardiac myocytes activation of an exchange factor activated by cAMP (Epac) leads to activation of phospholipase Cε (PLCε)-dependent hydrolysis of phosphatidylinositol 4-phosphate (PI4P) in the Golgi apparatus a process critical for development of cardiac hypertrophy. Here we show that β-adrenergic receptor (βAR) stimulation does not stimulate this pathway in the presence of the broad spectrum phosphodiesterase (PDE) inhibitor IBMX, but selective PDE3 inhibition revealed βAR-dependent PI4P depletion. On the other hand, selective inhibition of PDE2 or PDE9A blocked endothelin-1 (ET-1) and cAMP-dependent PI4P hydrolysis by PLCε. Direct activation of protein kinase A (PKA), protein kinase G (PKG), or the atrial natriuretic factor (ANF) receptor abolished PI4P hydrolysis in response to multiple upstream stimuli. These results reveal distinct pools of cyclic nucleotides that either inhibit PLCε at the Golgi through PKA/PKG, or activate PLCε at the Golgi through Epac. These data together reveal a new mechanism by which ANF and selective PDE inhibitors can protect against cardiac hypertrophy.
Keywords: Phospholipase C, Cardiac Hypertrophy, Phosphatidylinositol 4-phosphate, Phosphodiesterase, Epac, cyclic AMP, A kinase anchoring protein, Golgi apparatus
Introduction
Heart failure is a leading cause of morbidity and mortality in the western world. In response to stressors, such as hypertension or myocardial infarction, the heart undergoes pathological changes, including alterations in gene expression, increased protein synthesis, and cell growth. These changes lead to cardiac hypertrophy and ventricular remodeling, ultimately resulting in decompensation and finally heart failure. These effects are, at least in part, mediated by alterations in the circulating concentrations of neurohumoral regulators, such as epinephrine or endothelin-1 (ET-1), which signal through G-protein coupled receptor (GPCR) pathways in cardiac myocytes (1). Both Gs and Gq signaling pathways in cardiac myocytes downstream of GPCRs have been implicated in cardiac hypertrophy (1–3).
Previous studies, by our group and others, have shown that phospholipase C isoforms (PLCs), PLCβ and PLCε, are involved in cardiac hypertrophy driven by Gq coupled receptors, including α-adrenergic receptors (PLCβ and PLCε) (4, 5), and ET-1A receptors (PLCε) (6, 7). PLCβ is the canonical GPCR-regulated phospholipase activated by Gαq liberated from the G-protein heterotrimer by receptor activation, whereas PLCε is a novel member of the family activated by small GTPases (Rho, Ras and Rap family members) and Gβγ dimers from GPCR activation (8–11).
PLCε is scaffolded at the nuclear envelope by direct binding to muscle-specific A kinase anchoring protein β (mAKAPβ) in cardiac myocytes (6). ET-1 activates PLCε at this location via Gβγ dimers (12), and perhaps other upstream regulators, to regulate cardiac hypertrophy via PLCε-mediated hydrolysis of the alternate PLC substrate phosphatidylinositol-4-phosphate (PI4P) at the closely associated Golgi apparatus (7). Generation of diacylglycerol by this pathway leads to activation of protein kinase D (7), enabling phosphorylation of histone deacetylase 5 (HDAC5) and export out of the nucleus, and consequent expression of hypertrophic genes (13).
mAKAPβ scaffolds multiple proteins at the nuclear envelope including the cAMP responsive Rap exchange factor, exchange factor activated by cAMP (Epac) (14, 15). Stimulation of cardiac myocytes with the Epac-selective cAMP analogue, 8-(4-chlorophenylthio)-2′-O-methyladenosine-3′,5′-monophosphate (cpTOME), causes PLCε-dependent PI4P hydrolysis due to direct activation of PLCε by Rap1 (7). Epac has also been implicated in cardiac hypertrophy, with increased expression being seen in animal models of heart failure and Epac1-deficient animals being protected from cardiac hypertrophy (16, 17). This suggests that βAR could regulate hypertrophy in part by its ability to regulate PLCε-dependent PI4P hydrolysis in a cAMP-Epac dependent manner.
cAMP has been shown to be both prohypertrophic and protective depending on where and how the cAMP is generated in myocytes (18). Key to the maintenance of distinct compartments of cAMP in cardiac myocytes are the phosphodiesterase enzymes (PDEs) (18). This eleven-member family hydrolyzes both cAMP and cGMP, regulating their concentration and leading to the localization of cyclic nucleotide signals. PDEs have different affinities for cAMP and cGMP dependent on their isoform. PDEs 1,2,3, 10 and 11 have dual specificity for cAMP and cGMP, PDEs 4, 7 and 8 are selective for cAMP, whereas PDEs 5A, 6 and 9A are cGMP selective (19). PDE3 and PDE4 are the major isoforms in the heart responsible for regulating cyclic nucleotides (20) but PDE2, 5, 8, 9A and 10 have all been shown to be expressed in the heart (18, 19). PDE3 and PDE4 play a major role in the negative regulation of signals with knockout animal and pharmacological inhibition of PDE3 and PDE4 showing increases in cardiac hypertrophy (21–24). Conversely, the inhibition of PDE2, PDE5 and PDE9A have been shown to protect against cardiac hypertrophy (20, 25–31), with this effect being dependent on protein kinase A signaling in the case of PDE2, and PKG signaling in the case of PDE5 and PDE9A. This demonstrates that the balance of compartmentalized cyclic nucleotide signals is crucial for the maintenance of healthy myocardium.
Previously, we had demonstrated that PI4P hydrolysis in cardiac myocytes is induced by pharmacological Epac activation, and with ET-1 stimulation. ET-1 stimulation of PI4P hydrolysis required G protein βγ-mediated activation of PLCε (12). The physiological mechanism by which Epac is stimulated in this pathway, and whether Gs stimulation of cAMP production can induce PI4P hydrolysis has not been explored. In this study, we demonstrate that endogenous cAMP signals downstream of β-adrenergic stimulation activate Epac-PLCε-dependent Golgi PI4P hydrolysis through a pathway regulated by PDE3 and PDE4. In addition, we show that an opposing cAMP/cGMP pathway, regulated by PDE2 and PDE9A inhibits PI4P hydrolysis by PLCε to oppose development of hypertrophy. These data reveal a new mechanism by which compartmentalized cyclic nucleotides can promote and inhibit localized PLC activity to regulate hypertrophic signaling. Additionally, it presents further evidence for PLCε as a central player in cardiac hypertrophy and indicates that targeting PLCε signaling could lead to a new paradigm for treating heart failure.
Materials and Methods
Isolation of neonatal cardiac myocytes and adenoviral transduction
Briefly, hearts were excised from 2–4 day old Sprague-Dawley rats, ventricles separated and minced thoroughly before digestion with Collagen type II (Worthington) in Hanks buffered saline solution (HBSS) without Ca2+ or Mg2+. Following digestion, cells were collected by centrifugation into Dulbecco Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM glutamine and 2 μg/mL vitamin B12. Contaminating cells were removed by preplating cells onto tissue culture plastic for a minimum of 1hr at 37°C. NRVMs were then plated onto either glass-bottom tissue culture plates or 12 well plates coated with 0.2% gelatin and cultured in DMEM (composition as above, with additional 10 μM cytosine arabinoside). 48 hours later, cells were transferred into media supplemented with 1% FBS.
Measurement of PI4P hydrolysis
After preparation and culture of myocytes, cells were and transduced with adenovirus (50 MOI) expressing GFP-FAPP-PH overnight. The following day, expression was confirmed by epifluorescence microscopy. Imaging of GFP-FAPP-PH fluorescence was performed at room temperature on either a Nikon Eclipse TE2000U with a 40× oil-immersion lens or a LEICA DMi8 with a 100× 1.4 NA oil-immersion lens. EGFP was excited at 488 nm with an X-Cite Xled1 light source and emission monitored imaged on a backlit CMOS Photometrics Prime 95B camera. Analysis was performed using ImageJ unless otherwise stated. Data is presented as percentage of fluorescence remaining after agonist stimulation.
Measurement of myocyte cell size
NRVMs were plated into gelatin-coated 12 well plates and allowed to grow overnight. The following day, cells were infected overnight with adenovirus containing YFP, along with adenovirus expressing either SacI-K2A, the RA1 domain of PLCε, control LacZ or PLCε shRNA or control random shRNA and maintained in serum free DMEM. Subsequently, NRVMs were stimulated with hypertrophic agonists for 48 hours, as indicated. Following stimulation, cells were fixed in 4% (w/v) paraformaldehyde. Fluorescent images were taken at 20× magnification and cell area measured using NIH ImageJ software from over 50 cells from 3 separate experiments.
Measurement of Protein Synthesis
NRVMs were plated into gelatin-coated 96-well plates and allowed to grow overnight. The following day, cells were infected overnight with adenovirus containing PLCε shRNA or control random shRNA and maintained in serum free DMEM for 48hrs. NRVMs were stimulated with the indicated agonists for 48hrs. Prior to fixing the cells were treated with O-propargyl-puromycin for 2hrs. Protein synthesis assay was performed according to kit instructions (Click-iT Plus OPP Alexa Fluor 594, Invitrogen). Fluorescent images were obtained at 10× magnification and cell fluorescence was measured using NIH ImageJ software from 3 separate experiments.
Measurement of PLCε activation in COS-7 cells
COS-7 cells were plated into 12 well plates at a density of 1×105 cells/well. Cells were transfected the following day using 250 ng of each cDNA construct, unless indicated, along with a 3:1 ratio of lipofectamine 2000 reagent overnight. The following day, cells were incubated for 24 hours with 1.5 μCi of [3H]myo-inositol in inositol-free, serum-free media. The PLC assay was initiated with 10 mM LiCl incubated for 1 hour. The reaction was stopped by aspiration of media and addition of ice cold 50 mM formic acid. Total inositol phosphates (IPs) were separated by anion exchange chromatography using AG 1-X8 200–400 μm mesh, formate form, eluted with 1.2 M Ammonium Formate/100 mM Formic acid and quantitated by liquid scintillation counting. Protein expression levels were determined in parallel wells which were not radioactively labeled.
cAMP ELISA
NRVMs were plated onto 96 well plates and grown for a minimum of two days before stimulation. Cells were washed twice with HBSS (+Ca2+/Mg2+) and treatments, as indicated, were performed in HBSS buffer for 10 min before termination of assay with 0.1 M HCl. cAMP assay was performed according to kit instructions (Direct cAMP ELISA, Enzo Life Sciences)
Immunocytochemistry for α-actinin expression
NRVMs were fixed for 10 min in a solution of 4% paraformaldehyde (Sigma-Aldrich) in PBS. After blocking and permeabilization in 0.3% Triton X-100 solution containing 5% normal goat serum for 1 hour at room temperature. Primary antibody (rabbit anti-α-actinin [Sigma-Aldrich]) was incubated at a dilution of 1:500 in PBS overnight at 4°C. After three washes in PBS, cells were incubated with the secondary antibody (goat anti-rabbit Alexa Fluor 488 [Life Technologies, Grand Island, NY]) at a 1:1000 dilution in PBS for 1 h at room temperature. Cells mounted using ProLong Gold mounting media containing DAPI. Fluorescence images were captured by confocal microscopy.
Results
Endogenous cAMP can induce PI4P hydrolysis in NRVMs
Our previous observation that the Epac selective activator cpTOME induces PI4P hydrolysis in NRVMs suggests a simple pathway where cell surface β-adrenergic receptors (βARs) generate diffusible cAMP that can activate Epac/PLCε at the NE-Golgi interface. To assess the role of endogenous cAMP in regulation of PI4P signaling, NRVMs were infected with an adenovirus expressing FAPP-PH-GFP which selectively binds to PI4P in the Golgi as we and others have shown previously (7, 32) (Fig. 1A). Cells were stimulated with various compounds and the rate of depletion of PI4P associated fluorescence was measured (Fig 1A–D). As expected, cpTOME induces PI4P hydrolysis in NRVMs, as does the adenylate cyclase activator, Forskolin (Figs 1A, B and C). Surprisingly, however, activation of βARs with Isoproterenol (Iso) does not affect PI4P associated fluorescence (Fig. 1D). This is despite Isoproterenol being able to both generate cAMP (Fig. 2A) and induce PKA-dependent phosphorylation of VASP at Ser 157 (Supplemental Figure 1B) Similarly, other agonists that raise cAMP in NRVMs, including Adenosine and PGE2, had no effect on Golgi associated PI4P fluorescence (Supplemental Figure 2).
Figure 1. cpTOME and Forskolin stimulate PI4P hydrolysis in NRVMS, but Isoproterenol does not.

NRVMs were transduced with GFP-FAPP-PH adenovirus overnight before imaging in 1% FBS containing media. A) Representative images of Fsk (10μM)- and cpTOME (10μM) - induced PI4P hydrolysis. Bar-10μm B) Average data cpTOME (10μM), C) Average data Iso (1 μM), D) Average data Fsk (10μM), induced PI4P hydrolysis. Images taken from n=6 cells each from 6 separate preparations of NRVMs. All time course graphs are presented as mean ± standard error. Agonist treatments were compared to vehicle control performed on the same day and were added where indicated by the arrow. All data was analyzed by two way unpaired ANOVA with Sidak’s post-hoc test. ** p<0.001 *** p<0.0001 **** p<0.00001
Figure 2. Rolipram and Cilostamide stimulate PI4P hydrolysis in NRVMs, with Isoproterenol stimulation increasing the rate of Cilostamide-induced PI4P hydrolysis.

NRVMs were transduced with GFP-FAPP-PH adenovirus overnight before imaging in 1% FBS containing media. A) Time course of IBMX (300μM) or Iso (1 μM) plus IBMX induced PI4P hydrolysis. Arrow indicates time of stimulant addition. B) cAMP accumulation by PDE inhibitors. NRVMs were grown for 48hrs before being serum starved overnight and stimulated with either Iso (10 μM), IBMX (300 μM) or Iso (1 μM) plus IBMX for 15 min. Cells were then lysed and ELISA for cAMP performed according to manufacturer’s instructions. Data are representative of experiments performed three times. C) Time course PDE4 inhibitor Rolipram (10μM) or PDE3 inhibitor Cilostamide (10μM) induced PI4P hydrolysis. Phosphodiesterase inhibitors were added as indicated by arrow. D) NRVMs were grown for 48hrs before being serum starved overnight and stimulated with either Cilostamide (10μM), Rolipram (10μM) or IBMX (300μM) for 15 min and assayed for cAMP accumulation as in B. E) Time course Rolipram (10μM) or Cilostamide (10μM) induced PI4P hydrolysis in the presence of Isoproterenol (1μM). Isoproterenol and phosphodiesterase inhibitors were added simultaneously as indicated by arrow. F) NRVMs were grown for 48hrs before being serum starved overnight and stimulated with either Cilostamide (10μM), Rolipram (10μM) or IBMX (300μM) for 15 min in combination with Isoproterenol (1μM) and assayed for cAMP accumulation as in B. G) Epac inhibition blocks PDE effects. The Epac inhibitor HJC0726 (1μM) was added for 15 min before imaging. Forskolin (10 μM), Rolipram (10μM) or Cilostamide (10μM) were added at the arrows and PI4P hydrolysis was measured. For all experiments Data taken from at least 3 cells from 3 separate preparations of NRVMs. All graphs are presented as mean ± standard error. All data was analyzed by two way unpaired ANOVA with Sidak’s post-hoc test. *p<0.05 ** p<0.001 *** p<0.0001 **** p<0.00001
PI4P hydrolysis is induced by PDE3 and PDE4 inhibition in NRVMs
To understand why Forskolin generated cAMP activates PI4P hydrolysis but βAR activation does not, we explored the possibility that PDEs may prevent cAMP generated by βARs from accessing the mAKAPβ-Epac-PLCε complex. The broad specificity PDE inhibitor isobutyl-methyl xanthine (IBMX) did not affect PI4P hydrolysis either alone or in the presence of isoproterenol, despite strongly increasing the level of cAMP (Figures 2A and B). Surprisingly, however, treatment with selective PDE inhibitors, Rolipram for PDE4 or Cilostamide for PDE3, induced PI4P hydrolysis on their own (Figure 2C), despite raising cAMP to a lesser extent than IBMX (Figure 2D). Treatment with Cilostamide, but not Rolipram, was synergistic with Iso at early time points (Figure 2E). Again, this is unrelated to the amount of cAMP generated, as the amount of cAMP generated by Iso + Cilostamide was much lower than that for Iso+IBMX (Figure 2F). The effects of Fsk, Cilostamide and Rolipram on PI4P hydrolysis, are all blocked by the selective Epac1 and 2 inhibitor HJC0726 (33) (Figure 2G). These data indicate that PDE3 and PDE4 regulate a cAMP pool that controls Epac-dependent induction of PI4P hydrolysis, but that PDE3 specifically controls a βAR regulated pool of cAMP necessary for Iso-dependent signaling to PI4P hydrolysis.
Isoproterenol, PDE3, and PDE4 inhibitors induce cardiac hypertrophy in a PI4P and Golgi PLCε-dependent manner
Previous reports have indicated that Iso, Rolipram and Cilostamide induce hypertrophy of NRVMs (20). To determine if hypertrophy induction by Rolipram and Cilostamide require the PLCε-Golgi PI4P hydrolysis pathway we assessed the effects of depleting or inhibiting components of this pathway on the ability of Rolipram and Cilostamide to drive increases in NRVM cell area and protein synthesis. As we have previously shown, treatment with PLCε siRNA blocked Iso-dependent increases in cardiomyocyte cell area and protein synthesis (Figure 3A and B) (6, 7). Similarly, Cilostamide and Rolipram increased NRVM cell area and protein synthesis, which was abrogated by PLCε knockdown (Figure 3A and B). Interestingly, IBMX, which does not cause Golgi PI4P hydrolysis, does not cause NRVM hypertrophy. In addition to this, depleting PI4P with a Golgi targeted PI4P phosphatase, Sac I or inhibition of PLCε-mAKAP binding with overexpression of the RA1 domain of PLCε also inhibits increases in cell size by either Isoproterenol, Rolipram or Cilostamide (Supplemental Figure 3A and B). Taken together, this data suggests that a pool of cAMP maintained by PDE4 and PDE3 can induce NRVM hypertrophy through PI4P hydrolysis by PLCε at the Golgi apparatus when these PDE’s are inhibited.
Figure 3. Removal of PI4P or inhibiting PLCε inhibits NRVM hypertrophy in response to Iso, PDE3 or PDE4 inhibition.

A) NRVMs were infected with adenoviruses expressing either random control shRNA (Ran shRNA) or PLCε shRNA prior to stimulation with the indicated drugs for 48h and cell area was measured. B) Experiments were performed as in A, except before fixation cells were stained with O-propargyl-puromycin for 2hrs. NRVMs from at least 3 separate preparations were fixed following stimulation with indicated compounds, as described in methods. Data was analyzed using ImageJ software and size of myocytes presented in pixels2. All graphs are presented as mean ± standard error. All data was analyzed by two way unpaired ANOVA with Sidak’s post-hoc test. *p<0.05 ** p<0.001 *** p<0.0001 **** p<0.00001. For the random siRNA transduced cells, Isoproterenol, cilostamide and Rolipram all significantly increased hypertrophy relative to the vehicle treated control for both cell area (P<0.01–0.005) and protein synthesis measurements (all P<0.001).
Pharmacological activation of either PKA or PKG inhibits PI4P hydrolysis
A conundrum raised by data showing that selective inhibition of PDE3 and 4 stimulates PI4P hydrolysis, is that the broad spectrum PDE inhibitor IBMX does not affect PI4P hydrolysis. To explain these observations, we hypothesized that in addition to cAMP signaling pathways that promote PI4P hydrolysis through Epac, there may be a parallel pathway, dependent on a separate pool of cAMP that inhibits PI4P hydrolysis. A prime candidate for mediating this inhibition is PKA. Preincubation of NRVMs with either the PKA inhibitor, PKI (Figure 4A), or PKG inhibitor, rp-cGMPS (Figure 4B) reveals an IBMX dependent activation of PI4P, suggesting that IBMX treatment activates PKA, PKG and Epac, but that PKA or PKG activation is inhibitory and counters stimulation of PI4P hydrolysis by Epac. We tested whether direct activation of PKA or PKG inhibits PI4P hydrolysis using sp-cAMPS and 8-Br-cGMP, nonhydrolyzable cyclic nucleotide analogues which activate PKA and PKG, respectively. Treatment with sp-cAMPS to activate PKA completely inhibited PI4P hydrolysis in response to either cpTOME (Figure 4C) or ET-1 (Figure 4D). Similarly, treatment with 8-Br-cGMP to activate PKG inhibited both cpTOME and ET-1-dependent PI4P hydrolysis (Figure 4E and F). Since cpTOME and ET-1 activate PLCε and PI4P hydrolysis through different mechanisms it indicates that PKA and PKG must act by inhibiting a common factor in the pathway, such as PLCε itself.
Figure 4. PKA or PKG activation inhibits PI4P hydrolysis by cAMP and Gβγ-dependent pathways.

NRVMs were transduced with GFP-FAPP-PH adenovirus overnight before imaging. A) Time course of IBMX (300μM added at the arrow) induced PI4P hydrolysis in the presence of the PKA inhibitor myrPKI (1 μM) or B) PKG inhibitor, rp-cGMPS (10μM). C) Time course of cpTOME (10 μM) or D) ET-1 mediated PI4P hydrolysis in the presence of either vehicle DMSO or the PKA activator sp-cAMPs. E) Time course of cpTOME (10 μM) or F) ET-1 mediated PI4P hydrolysis in the presence of either vehicle DMSO or cGMP analogue, 8-Br-cGMP. PKI, rp-cGMPs, sp-cAMPs, or 8-Br-cGMP were added 15 mins before imaging. Data are from 4 cells from 4 separate preparations of NRVMs. All time course graphs are presented as mean ± standard error. All data was analyzed by two way unpaired ANOVA with Sidak’s post-hoc test. *p<0.05 ** p<0.001 *** p<0.0001 **** p<0.00001. Data is significantly different across complete time course or at each individual time point, as indicated.
PDE2 and PDE9A control a pool of cyclic nucleotides that inhibit PI4P hydrolysis at the Golgi
In addition to regulating signals that induce hypertrophy, particular PDE isoforms regulate cyclic nucleotide pools that restrict hypertrophic signaling (20, 25). Inhibition of the dual specificity PDE2 promotes antihypertrophic signaling in a PKA-dependent manner. Inhibitors of the cGMP specific PDE9A are antihypertrophic in several cell and animal models. Based on this data, we hypothesized that these PDEs may control cyclic nucleotide pools which inhibit PI4P hydrolysis as part of their antihypertrophic mechanism of action. In support of this hypothesis, preincubation of NRVMs with BAY60-7550, a selective PDE2 inhibitor completely inhibited stimulation of PI4P hydrolysis stimulated by either ET-1 or Forskolin (Figure 5A and B). Similarly, inhibition of PDE9A with PF04449613, prevented PI4P hydrolysis by ET-1 (Figure 5C). Interestingly, Forskolin-stimulated PI4P hydrolysis at early times (5 min) was not affected by PDE9A inhibition but sustained PI4P hydrolysis (20–30 min) was completely blocked. (Figure 5D). Inhibition of PDE5, which controls NO-regulated cGMP, does not affect PI4P hydrolysis (Supplemental Figure 2).
Figure 5. BAY60-7550, PF04449613 and atrial natriuretic peptide (ANF) inhibit the ability of ET-1 and Forskolin to stimulate PI4P hydrolysis.

A) Time course of BAY60-7550 (10 μM) or vehicle treated NRVMs after stimulation with either ET-1 (100 nM,) or B) Forskolin (10 μM) and measurement of PI4P hydrolysis. Images captured from at least n=4 cells from at least 4 separate preparations of NRVMs. C) Time course of PF-04449613 (10μM) or vehicle treated NRVMs after stimulation with either ET-1 (100nM, left) or D) Forskolin (10 μM, right) and measurement of PI4P hydrolysis. Images captured from at least n=3 cells from at least 3 separate preparations of NRVMs. E) Time course of either ET-1 (100 nM, left) or F) Forskolin (10 μM, right)–stimulated PI4P hydrolysis after incubation either ANF (10 nM) or vehicle control. Images and time courses are from at least 5 cells from 5 separate preparations of cells. NRVMs were transduced with GFP-FAPP-PH adenovirus overnight before imaging. BAY60-7550, PF04449613 and ANF were added 15 mins before samples were transferred to microscopy stage for imaging. All time course graphs are presented as mean ± standard error. All data was analyzed by two way unpaired ANOVA with Sidak’s post-hoc test. *p<0.05 ** p<0.001 *** p<0.0001 **** p<0.00001.
Atrial Natriuretic Factor (ANF) protects against hypertrophy via stimulation of the ANF receptor, which generates cGMP via its innate guanylate cyclase activity. ANF stimulation of NRVMs strongly inhibited PI4P hydrolysis stimulated by ET-1 at all time points (Figure 5E). Similar to PDE9A inhibition, ANF inhibited Forskolin-mediated PI4P hydrolysis at later times (Figure 5F). The basis for this delayed inhibition time course for Forskolin is not clear. Taken together, this data indicates that the generation of cAMP and cGMP, by both pharmacological inhibition of PDEs and receptor-mediated guanylate cyclase activation, inhibit prohypertrophic stimulation of PI4P hydrolysis at the Golgi.
PKA or PKG differentially inhibits PLCε by multiple upstream activators
The data presented thus far implicate select pools of cyclic nucleotides that activate PKA and PKG leading to phosphorylation events that inhibit PLCε-dependent PI4P hydrolysis at the Golgi apparatus. The diverse nature of the stimuli, ET-1A action at the cell surface working through Gβγ or cpTOME acting intracellularly through Epac and Rap, suggest that a component shared by these pathways, such as PLCε itself, is the target of PKA or PKG phosphorylation and inhibition. To test this, we cotransfected PLCε into Cos-7 cells along with the upstream activators in a standard assay that measures total inositol phosphates (IP) generation as a measure of PLC activation. This system is used to routinely assess the effects of regulators of PLC’s on their function and we used it here as a more direct way to determine if PLCε regulation by upstream activators can be inhibited by PKA or PKG (10, 34). Cotransfection of PLCε along with Gβγ (Figure 6A), constitutively active small GTPases, Rho G14V (Figure 6B), H-Ras G12V (Figure 6C), or Rap1a G14V (Figure 6D) all led to strong activation of PLC activity over that seen with transfection of the individual components. Transfection of these components along with active PKA catalytic domain inhibited PLCε activation by all of these components suggesting that PKA-mediated phosphorylation of PLCε inhibits its ability to be activated by upstream regulators (Figure 6A–D). Transfection with constitutively active PKG, on the other hand, had no effect on activation by Gβγ, H-Ras or Rap1, however, activation by Rho is partially inhibited in a manner similar to that seen with PKA (Figure 6E–F). Taken together, this data suggests that effects of PKA PLCε activation may be direct and that many of the effects of PKG on PLCε activation are indirect.
Figure 6. PKA can inhibit PLCε in COS-7 cells downstream of all activators, however, PKG only inhibits Rho-dependent activation.

COS-7 cells were Cotransfected with PLCε and Gβγ (A and E), Rho (B and F), H-Ras G12V (C and G), Rap1 (D and H), and PKA catalytic subunit (A,B,C and D) or constitutively active PKG (E,F,G and H). Total IP production was measured 48 hours after transfection as indicated in Materials and Methods. Data was generated from at least 3 experiments analyzed by two way unpaired ANOVA with Sidak’s post-hoc test. *** p<0.0001 **** p<0.00001.
Discussion
Here we sought to understand how cAMP generating agonists regulate the prohypertrophic Epac-PLCε-Golgi PI4P hydrolysis pathway. Prior to these studies we had shown that Iso-induced cardiac hypertrophy in NRVMs is blocked either by deletion of PLCε by siRNA or disruption of scaffolding of PLCε to mAKAP (6). The most straightforward hypothesis was that cAMP generated by the βAR at the sarcolemma would diffuse to NE localized Epac to initiate the PLCε-PI4P hydrolysis pathway. Here we show that this is much more complex and that cAMP and cGMP signaling from distinct pools regulate this pathway in opposing ways. It is well established that functionally and spatially distinct pools of cyclic nucleotides exist in cardiac myocytes that are controlled by distinct localized pools of PDEs (31). Here we demonstrate that two functionally distinct pools impinge on PLCε-mediated PI4P hydrolysis at the Golgi, a pathway we have recently shown is important for regulation of cardiac hypertrophy. Our model for how these pathways operate in the cardiac myocytes is shown in Figure 7.
Figure 7. Model for control of PI4P hydrolysis by distinct inputs to PLCε signaling.
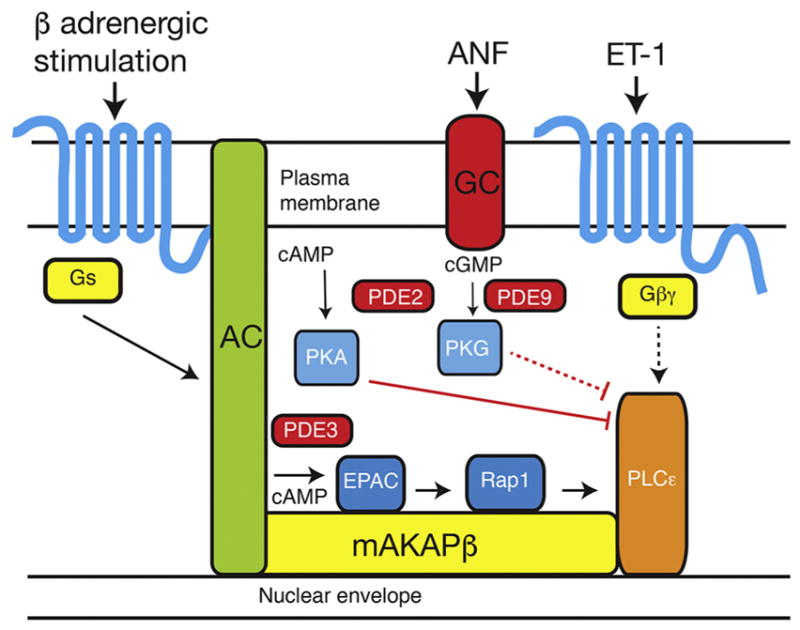
In this model, β-adrenergic receptor stimulation of adenylyl cyclase (AC) generates pools of cAMP that are differentially regulated by PDE2 and PDE3. PDE3 limits the ability of cAMP to activate Epac-PLCε, while PDE2 limits cAMP dependent inhibition of PLCε through PKA. Similarly, cGMP generated by activation of guanylyl cyclase (GC) inhibits PLCε through PKG activation, but this pathway may be indirect, as indicated by the dashed lines. PDE9 and possible PDE2 limit the ability of cGMP to act on this inhibitory PKG-dependent pathway.
Inhibition of PLCε signaling as mechanism for PDE protection against hypertrophy
One of the most exciting parts of this study is the revelation that inhibition of PLCε activity at the NE-Golgi interface may be a potential antihypertrophic target for PDE2 and PDE9A inhibition, as well as for cGMP generation by the ANF receptor. These agents have all been shown to have antihypertrophic actions in vivo and are potential antihypertrophic agents in humans (20, 25). A proposed mechanism for action for cGMP-dependent cardiac protection in response to PDE5 inhibition is through PKG dependent phosphorylation and activation of RGS2 to suppress hypertrophic Gq signaling in the heart (35). We see no effect of PDE5 inhibition on PI4P hydrolysis, yet ANF strongly inhibits this indicating that cGMP generated from ANF has a mechanism of action that may be independent of RGS2. The mechanisms for how ANF treatment, or PDE2 or PDE9A inhibition, protects against hypertrophy in a cardiac myocytes are not well defined (19). We propose that ANF treatment and PDE2 inhibition, oppose hypertrophy, at least in part, by blocking the prohypertrophic PLCε pathway. Further proof of this hypothesis will require defining the phosphorylation sites on PLCε responsible for its inhibition and showing that these sites must be phosphorylated in vivo for ANF or PDE inhibitors to impart cardioprotection.
How does PKG activation inhibit PLCε?
We show that cotransfection of PKA with PLCε leads to inhibition of its ability to be activated by multiple upstream regulators. This implies that PLCε is phosphorylated in a PKA-dependent manner to inhibit its activation in the face of prohypertrophic signaling pathways that would activate PLCε. The phosphorylation site on PLCε mediating these effects remains to be determined but the data suggests a direct mechanism for how PLCε is inhibited by specific subcellular pools of cAMP. PKG on the other hand is more limited in its ability to inhibit PLCε regulation by upstream factors with a specific effect on Rho signaling thus the inhibition by PKG is likely to be indirect and that PKG and PKA have different mechanisms for inhibition of PI4P hydrolysis.
How does Iso regulate hypertrophy if Epac is protected by PDE?
A question that arises from this study is how Iso stimulates PLCε-dependent hypertrophy if PLCε-Golgi signaling is not observed except under conditions where PDE 3 is inhibited. Some possible explanations are that hypertrophy of cardiac cells proceeds over the course of a 48h treatment while our measurements of PI4P hydrolysis is over the course of 30 min. It has been shown that during hypertrophy development PDEs change both with respect to the level of expression and their localization, changing the nature of the cyclic nucleotide pools (36–38). It is possible that during 48h of chronic βAR stimulation that cAMP that previously could not access Epac at the NE could gain access to this subcellular compartment due to reorganization or changes in expression of PDEs.
Epac regulation of hypertrophy
Previous work has suggested that Epac overexpression and/or stimulation is prohypertrophic. Knockout mice with cardiomyocyte specific deletion of Epac1 are protected from isoproterenol induced cardiac hypertrophy (39) (17). In these studies, it was suggested that the mechanism for this protection was through prevention of cAMP induced autophagy. As has been discussed, we have shown that PLCε pathway at the Golgi-NE interface is a critical pathway involved in promoting hypertrophy (6, 7). Since this pathway is strongly activated by Epac in cardiac myocytes we believe that this is another potential mechanism by which βAR signals can promote hypertrophy.
In summary, we have shown that opposing prohypertrophic and antihypertrophic pools of cyclic nucleotides control the recently identified prohypertrophic PLCε-PI4P hydrolysis pathway at the Golgi apparatus. These data suggest a previously unappreciated potential mechanism of action for agents that protect against hypertrophy including ANF, and inhibitors of PDE9A and PDE2. A limitation to this study is that while neonatal myocytes are relatively simple to manipulate, and recapitulate many aspects of cardiac hypertrophy, they do not represent a fully differentiated adult myocyte. We previously demonstrated that ET-1 stimulates Golgi PI4P hydrolysis in adult myocytes, but more work will be required extend the observations from this work to differentiated adult myocytes. Overall, understanding this mechanism in more detail may lead to novel approaches to treatment of heart failure.
Supplementary Material
Supplemental Figure 1. Neonatal myocytes are healthy and are able to generate PKA signals downstream of Isoproterenol. A) NRVMs were fixed, permeabilized and stained for α-actinin and with DAPI. B) NRVMs were treated with either vehicle, 10μM Isoproterenol or 10μM Forskolin in the presence of 300μM IBMX for 10 min before lysis. Samples were run on 4–20% SDS-PAGE gels and blotted for either VASP pSer157 or β-tubulin, as indicated.
Supplemental Figure 2. Neither PGE2 nor adenosine induce PI4P hydrolysis in NRVMs NRVMs were transduced with GFP-FAPP-PH adenovirus overnight before imaging in 1% FBS containing media. A) Time course of either PGE2 (1μM) or B) Adenosine (10μM) and vehicle controls. Images and time courses are from at least 4 cells from 4 separate preparations of cells.
Supplemental Figure 3. Removal of PI4P or inhibiting PLCε inhibits NRVM hypertrophy in response to Iso, PDE3 or PDE4 inhibition. A) NRVMs were infected with adenovirus expressing either lacZ as a negative control (NC) or a Golgi localized PI4P phosphatase SacI-K2A to deplete PI4P followed by treatment with the indicated drugs for 48h and cell area was measured. B) Experiments were performed as in A except cells were infected with adenoviruses expressing either lacZ as a negative control (NC) or the RA1 domain from PLCε to block PLCε scaffolding to mAKAP at the nuclear envelope. NRVMs from at least 3 separate preparations were fixed following stimulation with indicated compounds, as described in methods. Data was analyzed using ImageJ software and size of myocytes presented in pixels2. All graphs are presented as mean ± standard error. All data was analyzed by two way unpaired ANOVA with Sidak’s post-hoc test. *p<0.05 ** p<0.001 *** p<0.0001 **** p<0.00001. For the lacZ transduced control cells, Isoproterenol, cilostamide and Rolipram all significantly increased cell area relative to the vehicle treated control, p<0.05–0.002. IBMX did not significantly increase cell area.
Supplemental Figure 4. PDE5 inhibition has no effect on PI4P hydrolysis by ET-1 or Forskolin. NRVMs were transduced with GFP-FAPP-PH adenovirus overnight before imaging in 1% FBS containing media. A) Time course of either ET-1 (100nM, left) or B) Forskolin (10μM, right)–stimulated PI4P hydrolysis after incubation either Sildenifil (10μM) or vehicle control. Images and time courses are from at least 4 cells from 4 separate preparations of cells.
Highlights.
We demonstrate how compartmentalized cyclic nucleotides may impact cardiac hypertrophy through regulation of phospholipase C dependent phosphoinositide hydrolysis in the Golgi apparatus. We identified a pool of cyclic nucleotides that regulate PLC activation and a separate pool of cyclic nucleotides that oppose PLC activation. We propose that nucleotide pools opposing PLC activation are anti-hypertrophic.
Acknowledgments
This study was funded by U.S. National Institutes of Health (NIH) research grant R01GM0535336 to A.V.S and grant number T32-HL007853 from the NIH to L.M.B. The authors declare no conflicts of interest.
Footnotes
Author Contributions
C. Nash and A. Smrcka designed and conducted the research. C. Nash, L. Brown and S. Malik conducted the experiments. C. Nash wrote the manuscript which was revised and approved by all of the authors. X. Cheng provided essential inhibitors.
Disclosures
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 2.D’Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, Dorn GW., II Transgenic Gαq Overexpression Induces Cardiac Contractile Failure in Mice. Proc Natl Acad Sci U S A. 1997;94:8121–8126. doi: 10.1073/pnas.94.15.8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams JW, Sakata Y, Davis MG, Sah VP, Wang Y, Liggett SB, Chien KR, Brown JH, Dorn GW., II Enhanced Gαq signaling: A common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc Natl Acad Sci U S A. 1998;95:10140–10145. doi: 10.1073/pnas.95.17.10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Filtz TM, Grubb DR, McLeod-Dryden TJ, Luo J, Woodcock EA. Gq-initiated cardiomyocyte hypertrophy is mediated by phospholipase Cβ1b. FASEB J. 2009;23:3564–3570. doi: 10.1096/fj.09-133983. [DOI] [PubMed] [Google Scholar]
- 5.Grubb DR, Iliades P, Cooley N, Yu YL, Luo J, Filtz TM, Woodcock EA. Phospholipase Cβ1b associates with a Shank3 complex at the cardiac sarcolemma. FASEB J. 2011;25:1040–1047. doi: 10.1096/fj.10-171470. [DOI] [PubMed] [Google Scholar]
- 6.Zhang L, Malik S, Kelley GG, Kapiloff MS, Smrcka AV. Phospholipase Cε scaffolds to muscle-specific A kinase anchoring protein (mAKAPβ) and integrates multiple hypertrophic stimuli in cardiac myocytes. J Biol Chem. 2011;286:23012–23021. doi: 10.1074/jbc.M111.231993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang L, Malik S, Pang J, Wang H, Park K, Yule D, Blaxall B, Smrcka A. Phospholipase Cε Hydrolyzes Perinuclear Phosphatidylinositol 4-Phosphate to Regulate Cardiac Hypertrophy. Cell. 2013;153:216–227. doi: 10.1016/j.cell.2013.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smrcka AV, Brown JH, Holz GG. Role of phospholipase Cε in physiological phosphoinositide signaling networks. Cell Signal. 2012;24:1333–1343. doi: 10.1016/j.cellsig.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harden TK, Sondek J. Regulation of phospholipase C isozymes by ras superfamily GTPases. Ann Rev Pharmacol Toxicol. 2006;46:355–379. doi: 10.1146/annurev.pharmtox.46.120604.141223. [DOI] [PubMed] [Google Scholar]
- 10.Kelley GG, Reks SE, Ondrako JM, Smrcka AV. Phospholipase Cε: a novel Ras effector. EMBO J. 2001;20:743–754. doi: 10.1093/emboj/20.4.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song C, Hu CD, Masago M, Kariya K, Yamawaki-Kataoka Y, Shibatohge M, Wu D, Satoh T, Kataoka T. Regulation of a Novel Human Phospholipase C, PLCε, through Membrane Targeting by Ras. J Biol Chem. 2001;276:2752–2757. doi: 10.1074/jbc.M008324200. [DOI] [PubMed] [Google Scholar]
- 12.Malik S, deRubio RG, Trembley M, Irannejad R, Wedegaertner PB, Smrcka AV. G protein βγ subunits regulate cardiomyocyte hypertrophy through a perinuclear Golgi phosphatidylinositol 4-phosphate hydrolysis pathway. Mol Biol Cell. 2015;26:1188–1198. doi: 10.1091/mbc.E14-10-1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol. 2004;24:8374–8385. doi: 10.1128/MCB.24.19.8374-8385.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kritzer MD, Li J, Passariello CL, Gayanilo M, Thakur H, Dayan J, Dodge-Kafka K, Kapiloff MS. The scaffold protein muscle A-kinase anchoring protein β orchestrates cardiac myocyte hypertrophic signaling required for the development of heart failure. Circ Heart Fail. 2014;7:663–672. doi: 10.1161/CIRCHEARTFAILURE.114.001266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature. 2005;437:574–578. doi: 10.1038/nature03966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morel E, Marcantoni A, Gastineau M, Birkedal R, Rochais F, Garnier A, Lompre AM, Vandecasteele G, Lezoualc’h F. cAMP-Binding Protein Epac Induces Cardiomyocyte Hypertrophy. Circul Res. 2005;97:1296–1304. doi: 10.1161/01.RES.0000194325.31359.86. [DOI] [PubMed] [Google Scholar]
- 17.Laurent AC, Bisserier M, Lucas A, Tortosa F, Roumieux M, De Regibus A, Swiader A, Sainte-Marie Y, Heymes C, Vindis C, Lezoualc’h F. Exchange protein directly activated by cAMP 1 promotes autophagy during cardiomyocyte hypertrophy. Cardiovasc Res. 2015;105:55–64. doi: 10.1093/cvr/cvu242. [DOI] [PubMed] [Google Scholar]
- 18.Stangherlin A, Zaccolo M. Phosphodiesterases and subcellular compartmentalized cAMP signaling in the cardiovascular system. American Journal of Physiology-Heart and Circulatory Physiology. 2012;302:H379–H390. doi: 10.1152/ajpheart.00766.2011. [DOI] [PubMed] [Google Scholar]
- 19.Kokkonen K, Kass DA. Nanodomain Regulation of Cardiac Cyclic Nucleotide Signaling by Phosphodiesterases. Annu Rev Pharmacol Toxicol. 2017;57:455–479. doi: 10.1146/annurev-pharmtox-010716-104756. [DOI] [PubMed] [Google Scholar]
- 20.Zoccarato A, Surdo NC, Aronsen JM, Fields LA, Mancuso L, Dodoni G, Stangherlin A, Livie C, Jiang H, Sin YY, Gesellchen F, Terrin A, Baillie GS, Nicklin SA, Graham D, Szabo-Fresnais N, Krall J, Vandeput F, Movsesian M, Furlan L, Corsetti V, Hamilton G, Lefkimmiatis K, Sjaastad I, Zaccolo M. Cardiac Hypertrophy Is Inhibited by a Local Pool of cAMP Regulated by Phosphodiesterase 2. Circ Res. 2015;117:707–719. doi: 10.1161/CIRCRESAHA.114.305892. [DOI] [PubMed] [Google Scholar]
- 21.Ding B, Abe Ji, Wei H, Huang Q, Walsh RA, Molina CA, Zhao A, Sadoshima J, Blaxall BC, Berk BC, Yan C. Functional Role of Phosphodiesterase 3 in Cardiomyocyte Apoptosis: Implication in Heart Failure. Circulation. 2005;111:2469–2476. doi: 10.1161/01.CIR.0000165128.39715.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu H, Maurice DH. Expression of cyclic GMP-inhibited phosphodiesterases 3A and 3B (PDE3A and PDE3B) in rat tissues: differential subcellular localization and regulated expression by cyclic AMP. Br J Pharmacol. 1998;125:1501–1510. doi: 10.1038/sj.bjp.0702227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haworth RS, Cuello F, Avkiran M. Regulation by phosphodiesterase isoforms of protein kinase A-mediated attenuation of myocardial protein kinase D activation. Basic Res Cardiol. 2011;106:51–63. doi: 10.1007/s00395-010-0116-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jorgensen C, Yasmeen S, Iversen HK, Kruuse C. Phosphodiesterase4D (PDE4D)--A risk factor for atrial fibrillation and stroke? J Neurol Sci. 2015;359:266–274. doi: 10.1016/j.jns.2015.11.010. [DOI] [PubMed] [Google Scholar]
- 25.Lee DI, Zhu G, Sasaki T, Cho GS, Hamdani N, Holewinski R, Jo SH, Danner T, Zhang M, Rainer PP, Bedja D, Kirk JA, Ranek MJ, Dostmann WR, Kwon C, Margulies KB, Van Eyk JE, Paulus WJ, Takimoto E, Kass DA. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature. 2015;519:472–476. doi: 10.1038/nature14332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, Zile MR Investigators, P.-H., and Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med. 2014;371:993–1004. doi: 10.1056/NEJMoa1409077. [DOI] [PubMed] [Google Scholar]
- 27.Mehel H, Emons J, Vettel C, Wittkopper K, Seppelt D, Dewenter M, Lutz S, Sossalla S, Maier LS, Lechene P, Leroy J, Lefebvre F, Varin A, Eschenhagen T, Nattel S, Dobrev D, Zimmermann WH, Nikolaev VO, Vandecasteele G, Fischmeister R, El-Armouche A. Phosphodiesterase-2 is up-regulated in human failing hearts and blunts β-adrenergic responses in cardiomyocytes. J Am Coll Cardiol. 2013;62:1596–1606. doi: 10.1016/j.jacc.2013.05.057. [DOI] [PubMed] [Google Scholar]
- 28.Mongillo M, Tocchetti CG, Terrin A, Lissandron V, Cheung YF, Dostmann WR, Pozzan T, Kass DA, Paolocci N, Houslay MD, Zaccolo M. Compartmentalized phosphodiesterase-2 activity blunts β-adrenergic cardiac inotropy via an NO/cGMP-dependent pathway. Circ Res. 2006;98:226–234. doi: 10.1161/01.RES.0000200178.34179.93. [DOI] [PubMed] [Google Scholar]
- 29.Takimoto E, Belardi D, Tocchetti CG, Vahebi S, Cormaci G, Ketner EA, Moens AL, Champion HC, Kass DA. Compartmentalization of cardiac β-adrenergic inotropy modulation by phosphodiesterase type 5. Circulation. 2007;115:2159–2167. doi: 10.1161/CIRCULATIONAHA.106.643536. [DOI] [PubMed] [Google Scholar]
- 30.Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, Bedja D, Gabrielson KL, Wang Y, Kass DA. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005;11:214–222. doi: 10.1038/nm1175. [DOI] [PubMed] [Google Scholar]
- 31.Zaccolo M, Movsesian MA. cAMP and cGMP signaling cross-talk: role of phosphodiesterases and implications for cardiac pathophysiology. Circ Res. 2007;100:1569–1578. doi: 10.1161/CIRCRESAHA.106.144501. [DOI] [PubMed] [Google Scholar]
- 32.Balla T, Szentpetery Z, Kim YJ. Phosphoinositide Signaling: New Tools and Insights. Physiology. 2009;24:231–244. doi: 10.1152/physiol.00014.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu Y, Chen H, Boulton S, Mei F, Ye N, Melacini G, Zhou J, Cheng X. Biochemical and pharmacological characterizations of ESI-09 based EPAC inhibitors: defining the ESI-09 “therapeutic window”. Sci Rep. 2015;5:9344. doi: 10.1038/srep09344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kelley GG, Reks SE, Smrcka AV. Hormonal regulation of phospholipase Cε through distinct and overlapping pathways involving G12 and ras family proteins. Biochem J. 2004;378:129–139. doi: 10.1042/BJ20031370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takimoto E, Koitabashi N, Hsu S, Ketner EA, Zhang M, Nagayama T, Bedja D, Gabrielson KL, Blanton R, Siderovski DP, Mendelsohn ME, Kass DA. Regulator of G protein signaling 2 mediates cardiac compensation to pressure overload and antihypertrophic effects of PDE5 inhibition in mice. J Clin Invest. 2009;119:408–420. doi: 10.1172/JCI35620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fields LA, Koschinski A, Zaccolo M. Sustained exposure to catecholamines affects cAMP/PKA compartmentalised signalling in adult rat ventricular myocytes. Cell Signal. 2016;28:725–732. doi: 10.1016/j.cellsig.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mokni W, Keravis T, Etienne-Selloum N, Walter A, Kane MO, Schini-Kerth VB, Lugnier C. Concerted regulation of cGMP and cAMP phosphodiesterases in early cardiac hypertrophy induced by angiotensin II. PLoS One. 2010;5:e14227. doi: 10.1371/journal.pone.0014227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abi-Gerges A, Richter W, Lefebvre F, Mateo P, Varin A, Heymes C, Samuel JL, Lugnier C, Conti M, Fischmeister R, Vandecasteele G. Decreased expression and activity of cAMP phosphodiesterases in cardiac hypertrophy and its impact on β-adrenergic cAMP signals. Circ Res. 2009;105:784–792. doi: 10.1161/CIRCRESAHA.109.197947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Métrich M, Lucas A, Gastineau M, Samuel JL, Heymes C, Morel E, Lezoualc’h F. Epac Mediates β-Adrenergic Receptor Induced–Cardiomyocyte Hypertrophy. Circul Res. 2008;102:959–965. doi: 10.1161/CIRCRESAHA.107.164947. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Neonatal myocytes are healthy and are able to generate PKA signals downstream of Isoproterenol. A) NRVMs were fixed, permeabilized and stained for α-actinin and with DAPI. B) NRVMs were treated with either vehicle, 10μM Isoproterenol or 10μM Forskolin in the presence of 300μM IBMX for 10 min before lysis. Samples were run on 4–20% SDS-PAGE gels and blotted for either VASP pSer157 or β-tubulin, as indicated.
Supplemental Figure 2. Neither PGE2 nor adenosine induce PI4P hydrolysis in NRVMs NRVMs were transduced with GFP-FAPP-PH adenovirus overnight before imaging in 1% FBS containing media. A) Time course of either PGE2 (1μM) or B) Adenosine (10μM) and vehicle controls. Images and time courses are from at least 4 cells from 4 separate preparations of cells.
Supplemental Figure 3. Removal of PI4P or inhibiting PLCε inhibits NRVM hypertrophy in response to Iso, PDE3 or PDE4 inhibition. A) NRVMs were infected with adenovirus expressing either lacZ as a negative control (NC) or a Golgi localized PI4P phosphatase SacI-K2A to deplete PI4P followed by treatment with the indicated drugs for 48h and cell area was measured. B) Experiments were performed as in A except cells were infected with adenoviruses expressing either lacZ as a negative control (NC) or the RA1 domain from PLCε to block PLCε scaffolding to mAKAP at the nuclear envelope. NRVMs from at least 3 separate preparations were fixed following stimulation with indicated compounds, as described in methods. Data was analyzed using ImageJ software and size of myocytes presented in pixels2. All graphs are presented as mean ± standard error. All data was analyzed by two way unpaired ANOVA with Sidak’s post-hoc test. *p<0.05 ** p<0.001 *** p<0.0001 **** p<0.00001. For the lacZ transduced control cells, Isoproterenol, cilostamide and Rolipram all significantly increased cell area relative to the vehicle treated control, p<0.05–0.002. IBMX did not significantly increase cell area.
Supplemental Figure 4. PDE5 inhibition has no effect on PI4P hydrolysis by ET-1 or Forskolin. NRVMs were transduced with GFP-FAPP-PH adenovirus overnight before imaging in 1% FBS containing media. A) Time course of either ET-1 (100nM, left) or B) Forskolin (10μM, right)–stimulated PI4P hydrolysis after incubation either Sildenifil (10μM) or vehicle control. Images and time courses are from at least 4 cells from 4 separate preparations of cells.