

 This work is licensed under a
This work is licensed under a Summary
Intracranial germinomas are rare tumors affecting mostly patients at young age. Therefore, molecular data on its etiopathogenesis are scarce. We present a clinical case of a male patient of 25 years with an intracranial germinoma and a 16p11.2 microdeletion. His initial complaints were related to obesity, loss of facial hair and polydipsia. He also had a history of social-interaction difficulties during childhood. His blood tests were consistent with hypogonadotropic hypogonadism and secondary adrenal insufficiency, and he had been previously diagnosed with hypothyroidism. He also presented with polyuria and polydipsia and the water deprivation test confirmed the diagnosis of diabetes insipidus. His sellar magnetic resonance imaging (MRI) showed two lesions: one located in the pineal gland and other in the suprasellar region, both with characteristics suggestive of germinoma. Chromosomal microarray analysis was performed due to the association of obesity with social disability, and the result identified a 604 kb 16p11.2 microdeletion. The surgical biopsy confirmed the histological diagnosis of a germinoma. Pharmacological treatment with testosterone, hydrocortisone and desmopressin was started, and the patient underwent radiotherapy (40 Gy divided in 25 fractions). Three months after radiotherapy, a significant decrease in suprasellar and pineal lesions without improvement in pituitary hormonal deficiencies was observed. The patient is currently under follow-up. To the best of our knowledge, we describe the first germinoma in a patient with a 16p11.2 deletion syndrome, raising the question about the impact of this genetic alteration on tumorigenesis and highlighting the need of molecular analysis of germ cell tumors as only little is known about their genetic background.
Learning points:
Central nervous system germ cell tumors (CNSGTs) are rare intracranial tumors that affect mainly young male patients. They are typically located in the pineal and suprasellar regions and patients frequently present with symptoms of hypopituitarism.
The molecular pathology of CNSGTs is unknown, but it has been associated with gain of function of the KIT gene, isochromosome 12p amplification and a low DNA methylation.
Germinoma is a radiosensitive tumor whose diagnosis depends on imaging, tumor marker detection, surgical biopsy and cerebrospinal fluid cytology.
16p11.2 microdeletion syndrome is phenotypically characterized by developmental delay, intellectual disability and autism spectrum disorders.
Seminoma, cholesteatoma, desmoid tumor, leiomyoma and Wilms tumor have been described in a few patients with 16p11.2 deletion.
Bifocal germinoma was identified in this patient with a 16p11.2 microdeletion syndrome, which represents a putative new association not previously reported in the literature.
Background
Intracranial germ cell tumors (IGCTs) are rare neoplasms with an overall incidence of 0.6 and 1.0 per million per year in the United States and in Europe, respectively, and have a peak incidence near the time of puberty (1). These tumors have a male-to-female ratio of 3–4:1 and about 50% are present in the pineal gland (1). IGCTs are classified histologically in two main groups: pure germinoma and non-germinomatous germ cell tumors (1, 2). Germinoma is the most common subtype, and it is present in about two-thirds of patients (1). Their diagnosis and management are complex due to their heterogeneous clinical presentation, variable tumor location and different treatment approaches and outcomes. Furthermore, the scarcity of this neoplasms and, as so, their low availability for molecular analysis has hampered the understanding of the pathogenesis of IGCTs. Studies have demonstrated that KIT/RAS signaling pathway is frequently overactive in more than 50% of these tumors (1), the amplification of 12p is present in approximately 50% of CNSGTs (3) and, regarding epigenetic modifications, more than 60% of germinomas were clustered in a low methylation profile (3).
Case presentation
A 25-year-old man was referred to our endocrinology outpatient clinic because of grade III obesity. He complained of significant weight gain in the last 10 years and was medicated with 50 µg/day of levothyroxine for hypothyroidism without a defined etiology. He admitted a reduced libido without erectile dysfunction, polydipsia (>5 litter of water/day) and polyuria. His mother mentioned that he had social-interaction difficulties since he was a child. On physical examination, he presented a BMI of 46 kg/m2, abdominal perimeter of 132 cm, loss of facial hair and bilateral reduced testicular volume (right testicular volume of 12 mL and left testicular volume of 10 mL, as assessed by Prader orchidometer). He also presented macrocephaly, a broad forehead, a broad and prominent nasal bridge, anteverted nares and a small brachydactyly of the fourth and fifth fingers.
Investigation
The biochemical endocrine work-up showed the following results: follicle-stimulating hormone (FSH) <0.3 mIU/mL (<15), luteinising hormone (LH) <0.1 mIU/mL (<9), total testosterone 0.4 ng/mL (2.7–11), prolactin 16 ng/mL (<18), thyroid-stimulating hormone (TSH) 0.47 µIU/mL (0.4–4) and free thyroxine (FT4) 0.8 ng/dL (0.8–1.9), under 50 µg/day of levothyroxine. His growth hormone (GH) and insulin-like growth factor 1 (IGF-1) plasmatic levels were within the normal range (Table 1); analytical tests revealed an adrenocorticotropic hormone (ACTH) plasmatic level at 8 AM of 26 pg/mL (9–52) and a cortisol plasmatic level at 8 AM of 8.8 µg/dL (5–25). A Synacthen test with 250 µg ACTH injected intramuscularly revealed a subnormal adrenal reserve response with a cortisol peak at 60 min of 19 µg/dL (reference range: >20 µg/dL). He presented a urinary density of 1.003 (1.010–1.030) and osmolality of 104 (300–900 mosmol/kg), urinary sodium of 154 (40–220 mmol/24 h), plasmatic osmolality of 278 (260–302 mosmol/kg) and plasmatic sodium of 139 (136–146 mmol/L) with a 24-h diuresis of approximately 8500 mL. He performed a water deprivation test that confirmed the diagnosis of central diabetes insipidus.
Table 1.
Analytical evaluation.
| Parameter | Result | Reference range |
|---|---|---|
| Plasma | ||
| FSH | <0.3 mIU/mL | <15 |
| LH | <0.1 mIU/mL | <9 |
| Total testosterone | 0.4 ng/mL | 2.7–11 |
| Prolactin | 16.0 ng/mL | <18 |
| TSH | 0.47 µIU/mL | 0.4–4 |
| FT4 | 0.8 ng/dL | 0.8–1.9 |
| ACTH (8 AM) | 26 pg/mL | 9–52 |
| Cortisol (8 AM) | 8.8 µg/dL | 5–25 |
| GH | <0.1 µg/L | <1 |
| IGF-1 | 128 ng/mL | 117–329 |
| Osmolality | 278 mosmol/kg | 260–302 |
| Sodium | 139 mmol/L | 136–146 |
| α-Fetoprotein | 1.0 ng/mL | <8.6 |
| β-hCG | <2.0 mIU/mL | <10 |
| Urine | ||
| Density | 1.003 | 1.010–1.030 |
| Osmolality | 104 mosmol/kg | 300–900 |
| Sodium | 154 mmol/24 h | 40–220 |
| Cerebrospinal fluid | ||
| α-Fetoprotein | <0.2 ng/mL | <1.5 |
| β-hCG | 8.8 mIU/mL | <1 |
ACTH, adrenocorticotropic hormone; FSH, follicle-stimulating hormone; FT4, free thyroxine; GH, growth hormone; IGF-1, Insulin-like growth factor 1; LH, luteinising hormone; TSH, thyroid-stimulating hormone; β-hCG, β subunit of human chorionic gonadotropin.
The sellar and parasellar MRI showed two solid expansive lesions: one with 13 mm in the suprasellar region and another with 9 mm with a pineal location, isointense to the brain and with a homogeneous contrast enhancement on T1-weighted image, suggestive of a germinoma (Fig. 1).
Figure 1.
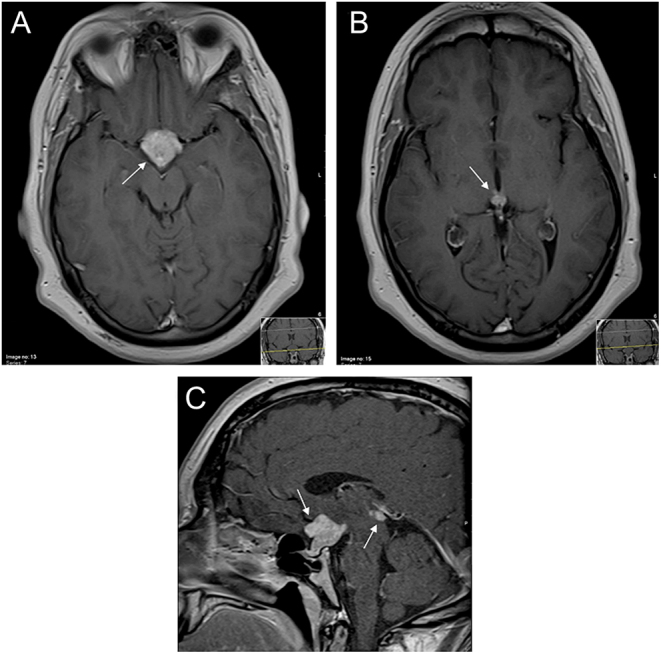
Bifocal germinoma with parasellar (A) and pineal (B) lesion, showing homogeneous contrast enhancement on axial T1-weighted image and on sagittal T1-weighted image (C).
Tumor markers α-fetoprotein and β-subunit of human chorionic gonadotropin (β-hCG) were not elevated in the serum and β-hCG in cerebrospinal fluid was slightly elevated in the initial work-up (Table 1). The patient performed a testicular ultrasound that revealed decreased testicular size (right testicular dimensions of 27 × 21 × 13 mm and left testicular dimensions of 22 × 14 × 13 mm) without further alterations. Computed tomography of chest, abdomen and pelvis was reported as normal. The mineral bone densitometry revealed a T-score of −1.4 s.d. and a Z-score of −1.4 s.d. in lumbar spine and a T-score of 1.0 s.d. and a Z-score of 1.0 s.d. in total femur. Conventional cytogenetics showed a 46,XY karyotype and the molecular cytogenetic evaluation through Agilent comparative genomic hybridization (CGH) microarray demonstrated a deletion of 604 kb on the short arm of chromosome 16 arr[hg19] 16p11.2(29 592 783–30 197 341)x1, compatible with the syndrome of 16p11.2 microdeletion (Fig. 2).
Figure 2.

Array comparative genomic hybridization analysis shows a 604 kb deletion on the short arm of chromosome 16 arr[hg19] 16p11.2(29 592 783–30 197 341) × 1.
An open biopsy of the suprasellar lesion was performed via right pterional craniotomy and histological study revealed a germ cell tumor of germinoma type, with associated lymphocytic inflammatory infiltrate mainly composed of B lymphocytes (CD20+) and T lymphocytes (CD4+ and CD8+). The immunohistochemical analysis demonstrated positivity of the tumor cells for placental alkaline phosphatase (PLAP), CD117, octamer-binding transcription factor 4 (OCT 4) and sal-like protein 4 (SALL4), and it was cytokeratin CAM 5.2 and β-hCG negative.
Treatment
Treatment with testosterone in a dosage of 125 mg each 4 weeks, hydrocortisone 10 mg at wake time and 5 mg in the afternoon and desmopressin 0.06 mg/day was started. Levothyroxine dosage was titrated up to 175 µg/day according to patients’ periodical biochemical analysis, to a target of normal FT4 level (0.8–1.9 ng/dL). His testosterone levels were within the normal range after 12 weeks of hormonal replacement (6.9 ng/mL, reference range: 2.7–11) and a significant improvement on his polyuria and polydipsia complaints was observed. After a multidisciplinary discussion, radiotherapy of the ventricular system and tumor locations was initiated in a total dose of 24 Gy in 15 fractions followed by a primary tumor boost of 16 Gy in ten fractions, without adverse reactions.
Outcome and follow-up
Radiotherapy was well tolerated. Three months after the completion of this treatment, the patient achieved a significant radiological improvement: his MRI revealed a substantial reduction of the suprasellar lesion, which currently involves only the pituitary stalk and hypothalamo-hypophyseal junctional area, and the pineal lesion was not observed (Fig. 3).
Figure 3.

Sellar MRI after radiotherapy, revealing a substantial reduction of the suprasellar and pineal lesions on axial T1-weighted image (A and B) and on sagittal T1-weighted image (C).
At this time, the patient reported weakness and fatigue. Biochemical evaluation was performed, after withdrawing hydrocortisone for 24 h under supervision and revealed morning serum cortisol of 0.6 μg/dL (5–25), ACTH 9.5 pg/mL (9–52), FSH <0.1 mIU/mL (<15), LH <0.1 mIU/mL (<9.0), total testosterone 1.7 ng/mL (2.7–11.0), TSH <0.004 µIU/mL, FT4 1.2 ng/dL (0.8–1.9), GH <0.1 µg/mL (<1), IGF-1 62 ng/mL (117–329). The patient reported improvement in polydipsia and polyuria complaints and, for this reason, he decided to suspend desmopressin therapy 2 weeks before biochemical tests. He presented a 24-h diuresis of approximately 4700 mL and a urinary density of 1.003 (1.010–1.030), urinary osmolality of 107 (300–900 mosmol/kg), plasmatic osmolality of 295 (260–302 mosmol/kg) and plasmatic sodium of 150 (136–146 mmol/L). Patients’ hydrocortisone dosage was increased to 10 mg at wake time, 5 mg at lunch and 5 mg in the afternoon, and he was advised to restart desmopressin 0.06 mg per day. He remains under surveillance and regular follow-up appointments were scheduled.
Discussion
We present an unusual case of a primary intracranial germinoma in a patient with 16p11.2 microdeletion syndrome, an association not previously reported in the literature.
Intracranial germinomas are rare tumors occurring mostly in children and young adults. Their first clinical manifestations may be related to hypopituitarism, and they are frequently diagnosed by clinical endocrinologists. The diagnosis is usually made by stereotactic or endoscopic biopsy in conjunction with blood and cerebrospinal fluid tumor markers, specially α-fetoprotein and β-hCG, which help in differential diagnosis (3). These tumors usually have an excellent response to radiotherapy, contrary to other ICGTs, which makes their histological diagnosis a fundamental step in the choice of patient’s treatment and follow-up (4). Regarding associated endocrine manifestations, studies suggest that patients do not recover completely after radiotherapy; in fact, pituitary deficiencies may even increase in severity, and hormonal replacement treatment is usually necessary for their entire life (5). Owing to their low incidence and non-surgical treatment options, ICGTs remain one of the less explored brain tumors of young patients. However, the elucidation of these tumors’ pathogenesis is essential to identify new biological markers useful for diagnosis and follow-up, as well as to unveil new therapeutic targets for refractory cases (6). Therefore, recent studies have focused on the molecular pathology of CNSGTs. The most remarkable chromosome alteration is the amplification of 12p, present in more than half of CNSGTs (3); the gain-of-function in the KIT gene, a proto-oncogene, represents the main genetic change, and the KIT protein is overexpressed in approximately 60% of CNSGTs (3).
The human 16p11.2 microdeletion has a population prevalence of approximately 1/2000 (7) and is associated with variable clinical features that include learning difficulties/intellectual disability, social impairment, autism, delayed language, obesity/overweight and minor dysmorphic facial features (8). A variety of rare clinical features have been associated with this deletion and tumors as seminoma, cholesteatoma, desmoid tumor, leiomyoma and Wilms tumor have been described in a few patients, suggesting either fortuitous associations or low penetrance through unmasking of recessive mutations (7, 9). The scarcity of ICGTs and the lack of an in-depth characterization of their genotype make it difficult to understand the true mechanisms beyond these associations. To the best of our knowledge, there are currently no studies reporting an association between 16p11.2 deletion and germ cell tumors. Therefore, we may hypothesize that this deletion may be involved in promoting cell proliferation, contributing to tumorigenesis. In fact, some of the genes within the deleted area (Table 2) are associated with cell cycle proliferation and cellular replication, namely mitogen-activated protein kinase 3 (MAPK3) (10).
Table 2.
List of the deleted genes.
| Gene symbol | Region location (GRCg37) |
|---|---|
| SMG1P2 | chr16:29 556 332–29 625 038 |
| MIR3680-2 | chr16:29 610 500–29 610 586 |
| SLC7A5P1 | chr16:29 624 424–29 625 038 |
| CA5AP1 | chr16:29 629 996–29 647 652 |
| SPN | chr16:29 674 271–29 681 828 |
| QPRT | chr16:29 690 329–29 710 020 |
| RN7SKP127 | chr16:29 742 372–29 742 725 |
| C16orf54 | chr16:29 753 784–29 757 340 |
| ZG16 | chr16:29 789 561–29 792 969 |
| KIF22 | chr16:29 802 034–29 816 706 |
| MAZ | chr16:29 817 417–29 822 504 |
| LOC100289283 | chr16:29 821 745–29 823 178 |
| PRRT2 | chr16:29 823 409–29 827 202 |
| PAGR1 | chr16:29 827 528–29 833 816 |
| MVP | chr16:29 831 715–29 859 360 |
| CDIPT | chr16:29 869 677–29 874 609 |
| CDIPT-AS1 | chr16:29 875 004–29 879 374 |
| SEZ6L2 | chr16:29 882 480–29 910 585 |
| ASPHD1 | chr16:29 912 147–29 917 377 |
| KCTD13 | chr16:29 917 657–29 937 553 |
| TMEM219 | chr16:29 973 351–29 984 373 |
| TAOK2 | chr16:29 985 188–30 003 582 |
| HIRIP3 | chr16:30 003 642–30 007 417 |
| INO80E | chr16:30 007 530–30 017 115 |
| DOC2A | chr16:30 016 835–30 024 917 |
| C16orf92 | chr16:30 034 655–30 036 023 |
| FAM57B | chr16:30 035 744–30 042 186 |
| ALDOA | chr16:30 064 411–30 081 741 |
| PPP4C | chr16:30 087 297–30 096 698 |
| TBX6 | chr16:30 097 114–30 103 205 |
| YPEL3 | chr16:30 103 635–30 107 537 |
| LOC101928595 | chr16:30 107 751–30 116 777 |
| LOC100506914 | chr16:30 107 751–30 116 841 |
| GDPD3 | chr16:30 116 131–30 124 878 |
| MAPK3 | chr16:30 125 426–30 134 630 |
| CORO1A | chr16:30 194 731–30 200 397 |
ALDOA, aldolase, fructose-bisphosphate A; ASPHD1, aspartate beta-hydroxylase domain-containing 1; C16orf54, chromosome 16 open reading frame 54; C16orf92, chromosome 16 open reading frame 92; CA5AP1, carbonic anhydrase 5A pseudogene 1; CDIPT, CDP-diacylglycerol-inositol 3-phosphatidyltransferase; CDIPT-AS1, CDIPT antisense RNA 1; CORO1A, coronin 1A; DOC2A, double C2-like domain-containing protein alpha; FAM57B, family with sequence similarity 57 member B; GDPD3, glycerophosphodiester phosphodiesterase domain-containing 3; HIRIP3, HIRA-interacting protein 3; INO80E, INO80 complex subunit E; KCTD13, potassium channel tetramerization domain-containing 13; KIF22, kinesin family member 22; MAPK3, mitogen-activated protein kinase 3; MAZ, MYC-associated zinc finger protein (purine-binding transcription factor); MIR3680-2, microRNA 3680-2; MVP, major vault protein; PAGR1, PAXIP1 associated glutamate rich protein 1; PPP4C, protein phosphatase 4 catalytic subunit; PRRT2, proline-rich transmembrane protein 2; QPRT, quinolinate phosphoribosyltransferase; RN7SKP127, 7SK small nuclear pseudogene 127; SEZ6L2, seizure related 6 homolog like 2; SLC7A5P1, solute carrier family 7 member 5 pseudogene 1; SMG1P2, nonsense mediated mRNA decay associated PI3K related kinase pseudogene 2; SPN, sialophorin; TAOK2, TAO kinase 2; TBX6, T-box transcription factor TBX6; TMEM219, transmembrane protein 219; YPEL3, Yippee-like 3; ZG16, zymogen granule protein 16.
The limited available data and the phenotypic heterogeneity of the syndrome are important pitfalls that need to be overcome to get a clear picture on this relationship. Future studies should evaluate the influence of additional genetic and environmental factors in shaping the phenotype of this syndrome. A comprehensive knowledge of the molecular mechanisms involved may have a relevant impact on patient’s diagnosis, treatment and follow-up and may help in the management of endocrine insufficiencies, with the potential to reduce the undesirable effects of current therapeutic approaches.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Patient consent
A written informed consent was obtained from the patient for publication of the submitted article and accompanying images.
Author contribution statement
M V is one of the patients’ physician, drafted the manuscript and conducted the literature review. L G is currently the patients’ main physician and critically revised the manuscript. J R-S was one of the patients’ physician and conducted the molecular cytogenetic evaluation. L B and I P conducted the initial evaluation of the patient and critically revised the manuscript. D O, M M and F C critically revised the manuscript.
References
- 1.Wang L, Yamaguchi S, Burstein MD, Terashima K, Chang K, Ng HK, Nakamura H, He Z, Doddapaneni H, Lewis L, et al Novel somatic and germline mutations in intracranial germ cell tumours. Nature 2014. 511 241–245. ( 10.1038/nature13296) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Phi JH, Wang KC, Kim SK. Intracranial germ cell tumor in the molecular era. Journal of Korean Neurosurgical Society 2018. 61 333–342. ( 10.3340/jkns.2018.0056) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kong Z, Wang Y, Dai C, Yao Y, Ma W, Wang Y. Central nervous system germ cell tumors: a review of the literature. Journal of Child Neurology 2018. 33 610–620. ( 10.1177/0883073818772470) [DOI] [PubMed] [Google Scholar]
- 4.Martens T, Rotermund R, Zu Eulenburg C, Westphal M, Flitsch J. Long-term follow-up and quality of life in patients with intracranial germinoma. Neurosurgical Review 2014. 37 445–450; discussion 51. ( 10.1007/s10143-014-0544-8) [DOI] [PubMed] [Google Scholar]
- 5.Osorio DS, Allen JC. Management of CNS germinoma. CNS Oncology 2015. 4 273–279. ( 10.2217/cns.15.13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fukushima S, Yamashita S, Kobayashi H, Takami H, Fukuoka K, Nakamura T, Yamasaki K, Matsushita Y, Nakamura H, Totoki Y, et al Genome-wide methylation profiles in primary intracranial germ cell tumors indicate a primordial germ cell origin for germinomas. Acta Neuropathologica 2017. 133 445–462. ( 10.1007/s00401-017-1673-2) [DOI] [PubMed] [Google Scholar]
- 7.Zufferey F, Sherr EH, Beckmann ND, Hanson E, Maillard AM, Hippolyte L, Macé A, Ferrari C, Kutalik Z, Andrieux J, et al A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. Journal of Medical Genetics 2012. 49 660–668. ( 10.1136/jmedgenet-2012-101203) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loviglio MN, Leleu M, Mannik K, Passeggeri M, Giannuzzi G, Van Der Werf I, Waszak SM, Zazhytska M, Roberts-Caldeira I, Gheldof N, et al Chromosomal contacts connect loci associated with autism, BMI and head circumference phenotypes. Molecular Psychiatry 2017. 22 836–849. ( 10.1038/mp.2016.84) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D’Angelo D, Lebon S, Chen Q, Martin-Brevet S, Snyder LG, Hippolyte L, Hanson E, Maillard AM, Faucett WA, Macé A, et al Defining the effect of the 16p11.2 duplication on cognition, behavior, and medical comorbidities. JAMA Psychiatry 2016. 73 20–30. ( 10.1001/jamapsychiatry.2015.2123) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.man OMii. National Center for Biotechnology Information. (available at: https://www.ncbi.nlm.nih.gov/omim/?term=601795). Accessed on 31 October 2018. [Google Scholar]