

Abstract
Many biological investigations require 3-D imaging of cells or tissues with nanoscale spatial resolution. We recently discovered that preserved biological specimens could be physically expanded in an isotropic fashion through a chemical process. Expansion microscopy (ExM) enables nanoscale imaging of biological specimens on conventional microscopes, decrowds biomolecules in support of signal amplification and multiplexed readout chemistries, and makes specimens transparent. We review the principles of how ExM works, as well as its applications throughout biology and medicine.
Introduction
Optical microscopy techniques make up one of the most important toolsets in the history of biology and medicine. Until recently the diffraction limit posed by physics limited the resolution of such microscopes to values of a few hundred nanometers, far greater than the size of biological molecules and the precision with which they are organized. The development of near-field imaging1,2 and a suite of far-field super-resolution microscopy techniques3,4 has enabled researchers to optically image single molecules and nanoscale structures in biology, but such technologies require expensive equipment and/or are slow in imaging speed, and thus struggle to perform 3-d imaging of extended cells and tissues such as those commonly studied in fields such as cancer biology, development, and neuroscience, or such as those used in diagnosis of disease in a clinical setting. Recently we discovered that it was possible to isotropically physically magnify a preserved biological specimen5 by synthesizing throughout such a specimen a network of swellable polyelectrolyte hydrogel (Fig. 1A), in a dense and even fashion, which in turn can smoothly expand biomolecules or labels away from each other, even in intact tissues like brain circuitry. After such physical magnification, molecules within a diffraction-limited region are isotropically separated in space to greater distances, and therefore resolvable even by conventional, ubiquitous diffraction-limited microscopes.
Figure 1. Expansion microscopy (ExM) concept and example outcomes.
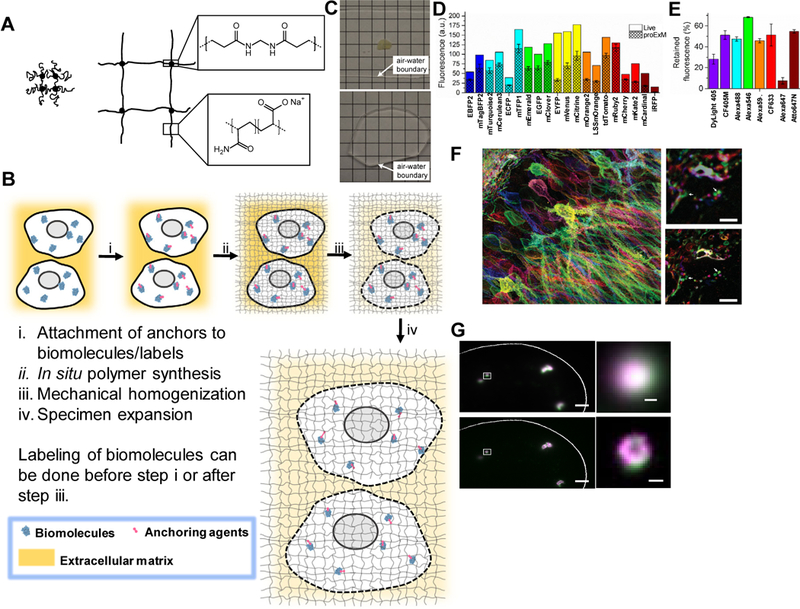
(A) Schematic of the ExM polyelectrolyte hydrogel, crosslinked sodium polyacrylate, showing the crosslinker (dot) and polymer chain (line), in the collapsed state before expansion (left) and in the expanded state after water dialysis (right). Insets show chemical structures of cross-linker and monomer components, in the synthesized polymer context. Adapted from ref. 5. (B) Diagram showing generalized workflow for ExM which involves (i) modification of biomolecules or labels (e.g., fluorescent antibodies) within a sample with a gel anchorable moiety (pink), (ii) in situ formation of the ExM polymer throughout the specimen, (iii) mechanical homogenization of the sample via heat treatment, detergent application, and/or enzymatic digestion, followed by (iv) expansion of the sample in water. Not to scale (the polymer spacing, or mesh size, is approximately 1–2 nm). (C) A 200 μm thick fixed mouse brain slice is opaque due to scattering before expansion (top) but is rendered transparent post-ExM, due to 100x dilution of the specimen contents in water (bottom). Adapted from ref. 5. (D-E) Retention of fluorescent proteins and antibody fluorescence in proExM. (D) Literature values of brightness for fluorescent proteins normalized to EGFP (open bars), compared to literature values of brightness multiplied by the observed retention percentage of each fluorescent protein with proExM (crosshatched bars, mean ± s.d.; n = 4 samples). (E) Retention of fluorescence of dyes conjugated to antibodies, after proExM (mean ± s.d., n = 3 samples). Adapted from ref.9. (F) Nanoscale imaging of intact brain circuitry with proExM. Shown is a specimen of mouse hippocampus expressing virally delivered Brainbow3.0 epitopes, stained with fluorescent antibodies, and then expanded via proExM (left); image shows maximum intensity projection of a high-resolution confocal microscopy stack. (Top, right), pre-expansion image of the boxed region of the left image. Arrows indicate features highlighted in the bottom, right image. (Bottom, right), post-expansion image of the top, right image. Adapted from ref. 9. (G) smFISH image before expansion (top panel) and post-expansion image processed with the ExFISH protocol (bottom panel), of NEAT1 lncRNA in the nucleus of a HeLa cell. Magenta and green indicate probesets binding to different parts of NEAT1. Right, a NEAT1 cluster (corresponding to the boxed regions of images on the left) imaged with smFISH (upper right) and ExFISH (bottom right). Adapted from ref. 10. Scale bars, (F, left) 50 μm (physical size post-expansion, 198 μm), (F, top right) 5 μm; (F, bottom right) 5 μm (19.8 μm); (G, top left) 2 μm; (G, bottom left) 2 μm (6.6 μm); (G, top right) 200 nm; (G, bottom right) 200 nm (660nm).
This approach, which we call expansion microscopy (ExM), brings together two different fields: the physics of swellable polyelectrolyte hydrogels, which vastly increase in size when immersed in a solvent such as water, and which were explored in depth by groups like that of Toyoichi Tanaka in the late 1970s6, and the embedding of preserved biological specimens in polymer hydrogels for imaging purposes, pioneered by teams like that of Peter Hausen and Christine Dreyer in the early 1980s7. Such gels have polymer spacings (or mesh sizes) that are quite small, ~1–2 nm8, and thus the errors introduced by such polymerization and expansion processes could in principle be quite small, on the order of the size of a biomolecule. Over the past few years, we have developed a diversity of ExM variants that are capable of enabling the effective imaging of proteins9 and RNAs10, that can physically magnify objects multiple times in succession in order to achieve very high resolution11, and that can be applied to human clinical specimens12, with other groups independently developing chemistries related to some of these techniques, including for the visualization of proteins13,14 and RNAs15 in expanded samples. We here review the progress in this rapidly developing and increasingly widely adopted technique space, discussing the principles of how ExM works, and in parallel how ExM is being applied in various biological and medical spaces, as well as future directions for the technology and its application.
All ExM protocol variants share a similar logical flow, essential for enabling an isotropic specimen expansion process that preserves biological information down to nanoscopic length scales (Fig. 1B). The design of the chemistry is aimed at insuring the greatest degree of isotropy possible during the expansion process. First, biomolecules and/or labels are covalently equipped with molecular handles (Fig. 1B, step i) that enable them to be bound to a swellable hydrogel synthesized throughout the specimen. Second, the specimen is immersed in a monomer solution (containing sodium acrylate) that reacts via free radical polymerization so as to form a densely crosslinked (via the cross-linker N-N’-methylenebisacrylamide) and highly penetrating polyelectrolyte hydrogel (sodium polyacrylate), which winds its way around and between the biomolecules and/or labels, binding to the molecular handles so that the attached biomolecules and/or labels are mechanically coupled to the polymer mesh (Fig. 1B, step ii). Third, the specimen is homogenized in mechanical properties by chemical denaturation via heat and detergent treatment, or by enzymatic digestion, as appropriate for the nature of the specimen under investigation and the molecules to be visualized (Fig. 1B, step iii). Finally, the specimen is immersed in water, which diffuses into the polyelectrolyte hydrogel through osmotic force, causing swelling that is facilitated by the highly charged nature of the polyelectrolyte mesh, which encourages further repulsion between the polymer chains and further expansion (Fig. 1B, step iv). After expansion, further application of labels, and/or amplification chemistries to improve visualization, may be applied if desired. Although the design of the protocol is aimed at the goal of isotropic expansion down to nanoscale resolution, expansion microscopy protocols have also been extensively empirically validated by comparison of images to those taken via earlier super-resolution technologies (e.g., SIM, STORM, and STED), and analyzing the distortion that results from the expansion process by a non-rigid registration algorithm, or by examination of post-expansion images of highly stereotyped structures (e.g., microtubules)5,9,11–13,16. The protocols described below all exhibit low distortions of a few percent, over length scales of dozens to hundreds of microns. The expansion process evenly pulls apart anchored biomolecules and/or labels, while preserving the spatial organization of biomolecules relative to one another, down to a precision that has been estimated to be as small as 5–10 nanometers11 (note that this is an estimate of the error in the location of gel-anchored biomolecules introduced by polymerization and expansion, and has not actually been achieved as a practical resolution to date, due to the larger size of the labels used, i.e. antibodies), and perhaps even smaller, with future versions of expansion microscopy.
Because ExM is a sample preparation method that enables ordinary, ubiquitous microscopes to image with nanoscale precision, it is easy to deploy into a diversity of contexts in biology and medicine. ExM also presents several other capabilities, in addition to enabling simple nanoscale resolution imaging on conventional high-speed microscopes. Because the final product is 99% water, with endogenous material diluted 100x or more, it is transparent (Fig. 1C5) with nearly 100% light transmission through a 200 μm-thick slice of expanded brain tissue5, which enables very fast imaging modalities such as light-sheet microscopy to become large volume nanoscopy technologies10. Because the expansion process is compatible with the visualization of a variety of biomolecules (e.g., proteins, Fig. 1D,E; RNA, Fig. 1G) with a variety of labels or stains, it can be used with many existing techniques, such as the popular “Brainbow” method of barcoding neurons with different fluorescent proteins or epitopes (Fig. 1F9), “upgrading” those technologies to help them become super-resolution imaging technologies. Most ExM protocols published to date expand a biological specimen by about 100x in volume, or ~4.5x in linear dimension, a factor set by the cross linker concentration (lowering the cross linker concentration results in greater gel expansion factors, but greater gel fragility)5. With a 4.5x linear expansion, a microscope with a diffraction limit of ~300 nm would attain an effective resolution of ~300 nm / 4.5 ~ 70 nm (e.g., Fig. 1F, Fig. 1G). It is also possible to expand specimens twice in series, enabling a ~4.5 × 4.5 ~ 20x increase in linear dimension, so that a conventional diffraction limited microscope can achieve a resolution of 300 / 20 ~ 15 nm (in practice, slightly larger due to the size of the fluorescent labels applied to the specimen)11, comparable to the best previous super-resolution methods for imaging of cells and tissues (Fig. 2). In addition, the isotropic expansion process decrowds biomolecules and/or labels away from each other, making room around them and thus facilitating the application of amplification and multiplexed-readout chemistries (Fig. 3).
Figure 2. 25nm-resolution imaging of neural circuitry and synapses with iExM.
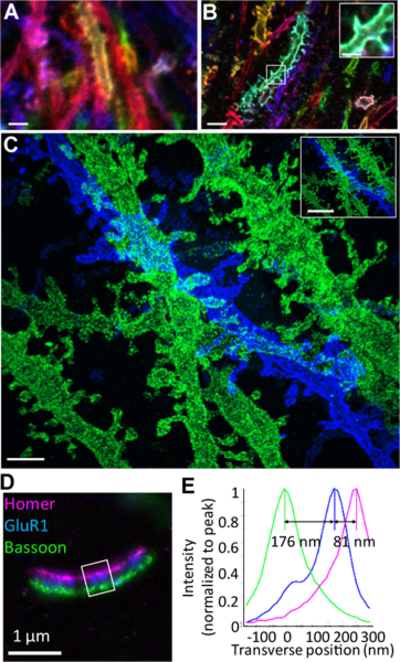
(A-C) iExM enables nanoscale imaging of brain circuitry in mouse hippocampus expressing Brainbow3.0 and immunostained with antibodies prepared for iExM expansion. (A) Confocal image of immunostained neurons in mouse hippocampus expressing Brainbow3.0, imaged without expansion. Blue, EYFP; red, TagBFP; green, mTFP. (B) As in (A) but imaged after 4.5x expansion via proExM; inset shows a magnified image of the boxed region in the main panel of B. (C) Confocal image (maximum intensity projection) of an iExM (~20x) expanded specimen of mouse hippocampus immunolabeled for EYFP (blue) and mCherry (green). Inset shows a demagnified view of (C) with the scale bar set to indicate the same distance as the scale bars in (A) and (B). (D) Nanoscale-resolution imaging of synapses using iExM. Wide-field image of a synapse from a field of cultured hippocampal neurons immunostained for Homer1 (magenta), glutamate receptor 1 (GluR1, blue), and Bassoon (green). (E) Transverse profile of the three proteins imaged in (D) (in the boxed region) after normalizing to the peak (Homer1 in magenta, GluR1 in blue, Bassoon in green). Adapted from ref. 11. Scale bars, (A) 3μm; (B) 3μm (physical size post-expansion 14μm); (B, inset) 1μm (4.5μm); (C) 1μm (20μm); (C, inset) 3μm (60μm); (D) 1μm (13μm).
Figure 3. ExM decrowds biomolecules, facilitating post-expansion staining, signal amplification, and multiplexed readout.
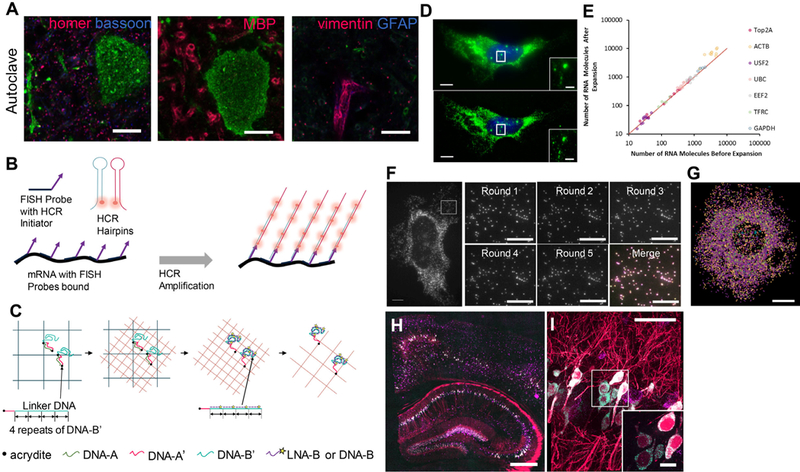
(A) Post-expansion antibody staining of protein targets in Thy1-YFP mouse brain tissue via a variant of proExM that uses high temperature detergent based (i.e., epitope-sparing) mechanical homogenization. Images show post-expansion staining against YFP (green), bassoon (blue) and homer (magenta) (left panel); YFP (green) and myelin basic protein (magenta) (middle panel); YFP (green), vimentin (magenta) and glial fibrillar acidic protein (blue) (right panel). Adapted from ref. 9. (B-C) ExM is compatible with different signal amplification techniques applied post-expansion, such as the Hybridization Chain Reaction (HCR) (B) and locked nucleic acid (LNA) based signal amplification (C). (B) Schematic for HCR-mediated signal amplification. A target RNA is labeled with FISH probes bearing HCR initiators. During amplification, metastable DNA hairpins bearing fluorophores assemble into polymer chains on the initiators, amplifying the signal from the bound FISH probe. Adapted from ref. 10. (C) Schematic showing LNA and DNA-based signal amplification for iExM. Adapted from ref. 11. (D) The decrowding of RNA molecules provided by ExFISH enables more accurate single-molecule quantification. Upper panel, pre-expansion smFISH image of ACTB RNA in a cultured HeLa cell. Inset shows zoomed-in region highlighting ACTB transcription sites in the nucleus. Lower panel, as in upper panel, but using ExFISH. (E) smFISH counts of RNA abundance for seven different transcripts before versus after expansion (n = 59 cells; each symbol represents one cell). Adapted from ref. 10. (F) ExM supports multiplexed read out of the identity of biomolecules, such as RNA, through multiple rounds of probe application and imaging. Left panel, widefield fluorescence image of ExFISH targeting GAPDH RNA in a cultured HeLa cell, with a boxed region showing five repeated cycles of staining and probe removal as shown on the right panel; lower right image shows an overlay of the five images (with each a different color: red, green, blue, magenta, yellow), showing colocalization. (G) Composite wide-field image showing ExFISH with serially delivered probes against six RNA targets in a cultured HeLa cell (NEAT1, blue; EEF2, orange; GAPDH, yellow; ACTB, purple; UBC, green; USF2, light blue). Adapted from ref. 10. (H-I) Nanoscale imaging of single RNA molecules in the mouse brain via ExFISH. (H) ExFISH post-expansion wide-field fluorescence image of a Thy1-YFP brain slice showing YFP protein (magenta), YFP mRNA (cyan) and Gad1 mRNA (purple). (I) Confocal image of mouse hippocampal tissue from (i), showing single RNA puncta. Inset, one plane of the boxed region (magenta , YFP protein; cyan, YFP mRNA; purple, Gad1 mRNA). Adapted from ref. 10. Scale bars, (A) 5μm (physical size post expansion ~21 μm); (D) 10 μm (33 μm), insets 2 μm (6.6 μm); (F) left panel, 6.6 μm (20 μm); right panel round 1–5, 3 μm (10 μm); (G) 6.6 μm (20 μm); (H) 500 μm (1450 μm); (I) 50 μm (145 μm), inset 10 μm (29 μm).
Ever since our original demonstration of isotropic cell and tissue expansion in 2015, variants have been developed by our group and by other groups to enable the imaging of proteins labeled with conventional antibodies or fused to genetically encoded fluorophores (protein retention ExM, or proExM, protocols9,13,14), to support the imaging of RNAs labeled with fluorescent in situ hybridization (FISH) probes (expansion FISH, or ExFISH10,15), to enable extremely fine resolution imaging by repeatedly expanding a sample over and over (iterative ExM, or iExM11), and to study human specimens such as those generated for pathology and diagnosis purposes (expansion pathology, or ExPath12).
Interestingly, tissue expansion was sometimes commented on in early studies of brain clearing – for example, in 2011, the Scale brain clearing protocol (involving chemicals such as urea and Triton X-100) was noted to cause 1.25× expansion of tissues17, although the nanoscale isotropy of the swelling process was not examined. Later brain clearing protocols such as CUBIC18 (involving a cocktail of small molecule chemicals including urea and Triton X-100) and CLARITY19 (involving uncharged polyacrylamide hydrogels) also commented on swelling of tissues thus treated. In these studies, however, expansion was treated as an uncontrolled and/or undesired process, and no attempts were made to design the strategy for large-scale isotropic expansion, or to analyze whether the strategy preserved nanoscale information. However, recently, an extension of CUBIC called CUBIC-X, which applies the small molecules imidazole and antipyrine to achieve swelling of CUBIC-processed tissue, was shown to be able to swell brain tissue ~2x linearly, offering subcellular resolution imaging of entire mouse brains with light-sheet microscopy (although no claims of nanoscale features, or comparison to super-resolution microscopy, was made20).
In the following sections we discuss the features of the many variants of ExM, as well as their applications, and where the toolsets are headed in the years to come.
ExM protocols and workflows
Protein Retention Expansion Microscopy (proExM)
The first version of ExM5 (which we now call ExM 1.0) required end users to synthesize their own labeling reagents for tagging proteins of interest, which limited deployability into everyday biology contexts (although it was used in one study for the visualization of biomolecules in E. coli bacteria (Fig. 4A)21). In protein retention expansion microscopy (proExM9), in contrast, only off-the shelf chemicals are needed. In proExM, applying the succinimidyl ester of 6-((acryloyl)amino)hexanoic acid (acryloyl-X, SE; abbreviated AcX for short) to a preserved specimen equips amines on proteins with a polymerizable carbon-carbon double bond (Fig. 1B)9, which in turn enables the thus-equipped proteins to be anchored to the polymer during the polymerization step (Fig. 1B). The procedures below can take, depending on the protocol used and the size of the tissue to be processed, anywhere from a day or two, to a week, with much of the time required being incubation times that permit chemical access of the tissue to occur. Fluorescent labels for visualization of proteins – e.g., fluorescent antibodies applied to specific proteins to enable them to be visualized – can be applied either before, or after, expansion.
Figure 4. Applications of ExM in biology and medicine.
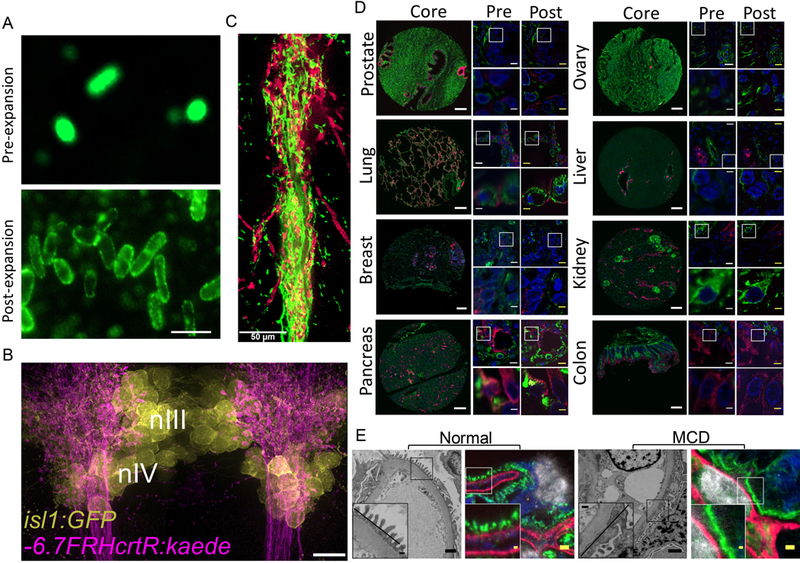
(A) Wide-field pre-expansion image of E. coli immunolabeled against membrane lipopolysaccharides (top). Wide-field post-expansion image of E. coli prepared as in (top) via ExM 1.0 (bottom). Adapted from ref. 21. (B) Image of part of the brain of a 6-day old larval zebrafish (nIII and nIV refer to the nuclei of the zebrafish brain indicated; isl1:GFP and −6.7FRHcrtR:kaede refers to the fish line utilized) using proExM. Image shows a maximum intensity projection of a ∼33-μm-thick volume of the brain immunolabeled for cells expressing GFP (yellow) and Kaede (magenta). Adapted from ref. 16. (C) proExM image of mouse brain striatal circuits showing the intertwined “bouquet” between striosomal fibers (green, mCitrine) and dopaminergic dendrites (magenta, tdTomato) in the mouse brain. Adapted from ref. 25. (D) ExPath imaging of a wide range of human tissue types. There are eight blocks of images, each corresponding to a different tissue type as labeled (e.g., prostate, lung, breast, etc.). Within each block of images for a given tissue type, there are 5 images shown. The left column within the 5 images shows the image of the tissue core, from a tissue microarray. The middle column within the 5 images shows two images, the top of which is a small field of view, and the bottom of which zooms into the area outlined in the top image by a white box, all pre-expansion. The right column within the 5 images shows the same fields of view as in the middle column, but now post-expansion after having been processed by the ExPath protocol. Blue, DAPI; green, vimentin; magenta, KRT19. Adapted from ref. 12. (E) Comparative imaging of kidney podocyte foot process in both normal and nephrotic disease states with both electron microscopy and ExPath. There are two pairs of images labeled “normal” and “MCD” (minimal change disease). Each pair of images shows one electron micrograph and one ExPath image from the same patient. Each image shows an inset, zoomed into the region outlined by the box from the main figure panel. Grey, DAPI; Blue, Collagen IV; green, vimentin; magenta, ACTN4. Adapted from ref. 12. Scale bars, (A) 2μm; (B) 10 μm (physical size post-expansion 38 μm); (C) 50 μm (D) tissue core, 200 μm; yellow scale bars have the same biological scale as the paired white scale bars: top images, 10 – 12.5 μm; bottom images, 2.5 – 3.1 μm; physical sizes post-expansion, top images, 50 μm; bottom images, 12.5 μm. (E) For electron micrographs, 1 μm; inset, 200 nm; for ExPath images, 1 μm (physical size post-expansion, 4.3 μm); inset, 200 nm.
In the former case, standard fluorescent antibodies, as are commonly used in immunohistochemistry, are applied to fixed cells or tissues before expansion occurs. Genetically encoded fluorophores, perhaps fused to proteins of interest, can also be expressed in living cells via viral delivery, transfection, or transgenesis. AcX, when applied, binds to all the proteins, including the fluorescent antibodies and/or genetically encoded fluorophores. After polymerization, the mechanical homogenization step (Fig. 1B) proceeds via application of proteinase K, a powerful protease applied at a dosage that results in the destruction of the majority of the proteins that provide structural integrity to cells and tissues but sparing the functions of the antibodies and fluorescent proteins22,23. The fact that such a dosage exists reflects the fact that many antibodies and fluorescent proteins are relatively protease resistant, compared to other proteins. Then, after gelation, digestion, and expansion, the fluorescence of the antibody labels or fluorescent proteins is often mostly retained (Fig. 1D, although the volumetric dilution due to expansion would reduce the intensity per unit volume further), enabling imaging of biomolecular identity and location.
Most fluorescent dyes commonly used with antibodies are compatible with this process, with the exception of cyanine dyes, which are destroyed during polymerization (Fig. 1D); excellent alternatives to cyanine dyes are now available in spectral bands where cyanine dyes have been popular in the past9. Most GFP-like (i.e., beta barrel-structured) fluorescent proteins are also preserved in this process (Fig. 1D); infrared fluorescent proteins based on bacteriophytochromes, in contrast, are largely destroyed9. In addition to antibodies, other protease-resistant labels like streptavidin can be applied pre-expansion as well9; this has been used to enable visualization of post-translational modifications such as S-nitrosylation that can be marked by biotin-bearing small molecule tags9.
In parallel to our development of proExM, another group independently developed a related protocol13 that uses methacrylic acid N-hydroxysuccinimidyl ester (MA-NHS, which is structurally similar to AcX) or glutaraldehyde to serve as anchors that link fluorescent proteins and antibodies to the polymer. In this study, MA-NHS was demonstrated to work well in both cultured cells and tissue slices; glutaraldehyde was used only on cultured cells in this paper. In this study, it was also shown that biotin-labeled antibodies could be used, with visualization enabled post-expansion by application of fluorescent streptavidin, which allows for utilization of fluorophores that would have been destroyed by the expansion process.
proExM protocols using AcX and MA-NHS have been validated on a wide range of fluorescent proteins and antibody-borne fluorophores, and applied to a diversity of proteins and structures (such as clathrin-coated pits, keratin fibers, actin filaments, vimentin, myelin, glial markers, chromosomes, kinetochores, mitochondria, peroxisomes, pre- and post-synaptic proteins, nuclear lamina, histones, and microtubules9,13,16) in a variety of cell and tissue types (including many kinds of cultured cells as well as mouse brain, pancreas, spleen, and lung, rhesus macaque brain, and larval zebrafish (Fig. 4B)9,13,16). This family of protocols has become rapidly utilized for the nanoscale imaging of protein identity and location in the following contexts in just the last year: examining larval zebrafish brain synaptic connections and nuclear-cytoskeletal organization16, imaging glial and gap junction organization in human brain tissue from epilepsy patients24, imaging synaptic proteins to characterize novel connections in mouse striatal brain circuitry (Fig. 4C)25, mapping the organization of kinesins relative to cytoskeletal proteins at defined points in the cell cycle in cultured cells26, visualization of melatonin receptors in purified mitochondria from mouse brain27, imaging of the Drosophila synaptonemal complex that helps chromosomes segregate during meiosis28, segmenting neurons in the mouse brain labeled with brainbow reagents29, visualizing key proteins at active zones of Drosophila synapses30, analysis of proteins indicative of a novel glial cell type in a species of planaria31, visualization of cytoskeletal filaments in the parasite Giardia32, the use of nanoparticles to perform surface-enhance Raman spectroscopy imaging of proteins in mouse brain33, and the analysis of sorting tubules in vasculature of the mouse brain34, amongst ongoing publication activity.
Another independent attempt at creating a fluorescent protein retention form of expansion microscopy resulted in ePACT, which while similar in some regards to the above proExM protocols, did not use an explicit protein anchoring chemical, and also used the milder enzyme collagenase for mechanical homogenization rather than proteinase K, in order to preserve YFP fluorescence in Thy1-YFP mouse brain tissue35. Although ePACT resulted in ~4x fold expansion of such brain tissue, with YFP fluorescence visible, it was accompanied by tissue destruction, including the compromising of fine processes, perhaps due to lack of protein anchoring treatment and incomplete mechanical homogenization caused by the gentler protease.
In a second style of proExM, endogenous proteins are directly anchored to the polymer hydrogel via AcX and then stained with antibodies post-expansion9. For epitopes to be retained and thus to be receptive to antibody binding post-expansion, the mechanical homogenization step does not involve protease digestion, but rather proceeds via a gentler homogenization protocol, such as treatment with a high temperature (e.g., via autoclave) detergent solution. Using a gentler protease, such as LysC (which cuts proteins at lysines), which chops proteins into parts that are smaller but of sufficient size to retain epitopes for antibody binding, can also work. After mechanical homogenization and expansion, the expanded specimen can be stained with standard antibodies allowing imaging of proteins with nanoscale resolution (Fig. 3A). An exciting possibility, which is beginning to be explored, is that post-expansion staining of specimens allows for proteins to be decrowded by the expansion process, improving accessibility and density of staining. This protocol worked for the majority of antibodies tried, but not for all of them, perhaps because epitopes were lost during autoclaving, or during LysC cleavage; this protocol also reported discontinuities in processes and white matter tracts.
A protocol similar in some regards, the Magnified Analysis of the Proteome (MAP) protocol14, developed independently and in parallel to the proExM protocol, also allows for the retention and staining of epitopes after expansion, using somewhat different fixation, polymerization and homogenization steps. In MAP, the expansion of gel-retained proteins away from each other is facilitated by reducing the amount of crosslinking between proteins during formaldehyde fixation. This reduction in the level of protein-protein crosslinking is achieved by adding a high concentration of acrylamide during fixation, which reacts to formaldehyde bound to proteins, and prevents protein-protein crosslinking. The bound acrylamide also serves to equip proteins with a gel anchorable moiety, since acrylamide can participate in free radical polymerization just as acrylate does (and indeed, was the original monomer used in the Hausen-Dreyer protocol7). After immersion of the specimen in a mixture of sodium acrylate and acrylamide, and formation of the resulting interpenetrating gel throughout the specimen, incubation in a high-temperature detergent solution caused denaturation of proteins and enabled separation and expansion upon immersion in water. Finally, epitopes were labeled after expansion with immunostaining. MAP was initially demonstrated on multiple organs, including heart, lung, liver, kidney, and brain, as well as on cerebral organoids. Post-expansion staining with MAP was demonstrated to work with over 80% of the over 100 antibodies tested, including those against microtubules, neurofilaments, and a diversity of membrane, cytoplasmic, nuclear, and synaptic proteins14. MAP also allows for multiplexed antibody staining by the iterative staining and removal of antibodies; destaining involves immersion in the high-temperature denaturation solution. While other proExM variants have focused on application to tissue sections to facilitate diffusion of reagents into specimens, MAP achieves whole-organ expansion by delivering the necessary reagents via transcardial perfusion and applying longer incubation times, an approach that potentially could be extended to other ExM methods. MAP has been applied to the visualization of the glomerular filtration barrier of the kidney in rodents36.
Going forward, it will be essential to validate and improve the performance of antibodies in order to achieve the highest fidelity staining profiles possible, especially for post-expansion administration, which is still challenging, as noted above. The use of other tags, such as Snap, Clip, and Halo tags, or the use of nanobodies, may also improve the resolution enabled by proExM protocols yet further. Recent advancement in multiplexed stimulated Raman scattering (SRS) has yielded sets of dyes that can be imaged a dozen or more at a time, and thus could also be adapted for multiplexed imaging of a large number of biomolecules simultaneously within a single ExM sample37,38. Another area of future innovation is to rethink the microscopy hardware utilized so that the largest volumes possible can be imaged, taking advantage of the fact that expansion protocols can improve resolution in a way that is complementary and orthogonal to traditional microscope design. proExM-like protocols result in specimens that can be imaged on a diversity of conventional microscopes, including confocal microscopes and epifluorescent microscopes ubiquitous in biology and medicine. For general morphological visualization of cells and tissues, it is possible to expand samples with a proExM-like protocol and then to image the resulting expanded samples in brightfield or phase contrast microscopy, to reveal the location and organization of membranes and organelles9. Such a strategy has been used, for example, to visualize tunneling nanotubes between cultured epithelial cells, which can be exploited by the influenza virus to spread from cell to cell, potentially evading the immune system39. Samples expanded with proExM have also been imaged using super-resolution microscopes, for added resolution beyond what is achievable on conventional microscopes. The use of photoswitchable fluorescent proteins as the proExM-anchored fluorescent proteins enables PALM super-resolution microscopy to be done on expanded cells, potentially enabling very high resolution imaging; the basic concept has been demonstrated using cultured cells expressing histone H3.3-Dendra2, but the fundamental resolution supported by this methodology has not been fully validated9. Other studies have applied super-resolution SIM microscopy28,32 or STED microscopy36,40 to samples (including microbes, cultured cells, mammalian tissues, and Drosophila tissues) expanded with proExM-like protocols. Finally, one paper has used the original ExM protocol to enable microscopy to be performed by inexpensive modified webcams, with the resolution improvement enabled by ExM utilized to compensate for the poorer resolution of low-cost microscopy hardware, and thus potentially enabling less expensive biological and medical imaging21.
Expansion Pathology (ExPath)
Expansion pathology (ExPath) is an extension of the pre-expansion antibody administration proExM protocol that enables a variety of tissue formats common in human clinical and pathological settings to be expanded12. Formalin-fixed paraffin-embedded (FFPE), hematoxylin and eosin (H&E)-stained, and fresh frozen thin-sliced specimens can be pre-processed so that they can enter the proExM pipeline. ExPath precedes proExM by a series of steps – xylene removal of any paraffin, antigen retrieval in hot sodium citrate – that help make clinically prepared tissues compatible with proExM. The form of proExM that follows has increased levels of EDTA which improves tissue digestion, important since clinical specimens are sometimes very heavily fixed due to long exposure to formalin. H&E stains are removed during this processing. For fresh frozen sections (e.g., acetone-fixed), one lowers the amount of AcX to avoid over-anchoring of specimens, since there are more free amines available in such specimens to bind anchors due to the lack of aldehyde fixatives occupying amines. The ExPath protocol has been applied to human skin, lung, liver, breast, prostate, pancreas, ovary, kidney, and other tissues (Fig. 4D), preserved in a variety of strategies amongst those listed above.
ExPath is compatible with antibody staining, as demonstrated by labeling of many different markers (vimentin, keratin, etc.) in a diversity of tissues, as well as post-expansion DNA FISH against genes (as demonstrated by labeling of Her2 in Her2- and Her2+ breast cancer samples from human patients). ExPath reduces autofluorescence by up to an order of magnitude, by diluting or eliminating contributors to autofluorescence while preserving information covalently anchored to the polymer. We demonstrated that ExPath could resolve structures in human specimens that traditionally require electron microscopy imaging in the pathology lab, for example podocyte foot processes that are effaced in human patients with nephrotic kidney diseases, which usually require electron microscopy for a diagnosis (Fig. 4E). It is important to note that electron microscopy has superior resolution over current expansion microscopy protocols, but the processing time of ExPath is significantly shorter than that for electron microscopy, and the requirement for skills and equipment to perform ExPath is much less demanding in comparison with those required for electron microscopy. We also found that, applied to problem of classifying early breast lesions, for which agreement between pathologists can be as low as 50%41, we were able to greatly improve the performance of a machine learning diagnosis classifier on expanded human breast biopsies, over the performance of the same classifier on unexpanded samples. These initial clinical studies were small, so future studies involving large numbers of patients will be essential for understanding the potential value of ExPath in the clinic.
ExFISH: nanoscale imaging of RNA with expansion microscopy
One powerful attribute of ExM is that the various steps (Fig. 1B) can easily be extended to enable the visualization of new molecule classes, or the incorporation of new amplification and analysis chemistries (Fig. 3B). For example, RNA molecules can be anchored to the swellable polymer by applying the molecule LabelX, a reagent formed from the reaction of two commercially available reagents10. LabelX possesses an alkylating group which reacts primarily to guanine bases in RNA as well as DNA, as well as a carbon-carbon bond that can be incorporated into the gel. Thus, specimens treated with LabelX can then be expanded in a way that retains RNA identity and position. The ability to stain RNA after expansion opens the door to the use of various fluorescent in situ hybridization (FISH) strategies to interrogate RNA location and identity; we call this suite of technologies expansion FISH (ExFISH) protocols. In cultured cells, we have shown that standard single-molecule FISH (smFISH) can be performed after expansion to stain RNA, allowing for the imaging of the nanoscale organization of RNA molecules such as long non-coding RNAs (Figure 1G). The ability of expansion to decrowd RNA molecules enables better smFISH quantification of RNA abundance (Fig. 3C). Furthermore, expansion supports multiplexed smFISH by allowing for the efficient delivery and removal of probes, making it possible to read out the identity and location of multiple RNA molecules over time (Fig. 3D). Another group developed a simple method for visualizing RNAs by applying gel-anchorable fluorescent FISH probes before expansion, which are then anchored to the polymer and expanded away from each other15, and also obtained improved signal-to-noise ratio and more accurate RNA accounts than in the unexpanded state. This method does not directly retain RNAs themselves, but instead retains the probes applied, and exemplifies how the core ExM philosophy can easily be adapted to alternative staining, labeling, and/or processing steps as desired.
In the future, thanks to the decrowding effect of ExM, we anticipate ExFISH being used with a diversity of signal amplification strategies to enable better imaging of single RNA molecules in large, intact tissues. Signal amplification techniques such as hybridization chain reaction (HCR)42–44 and rolling circle amplification (RCA)45,46 produce bright signals by forming large assemblies of fluorophores which can be easily detected. ExM invites the use of such signal amplification techniques by isotropically separating biomolecules to create room around them for such large assemblies of fluorophores. For instance, we used HCR with ExFISH to amplify FISH stains (Fig. 3B), which allowed for single molecule-resolution visualization of RNA with nanoscale precision in thick brain tissues (Fig. 3E). HCR-ExFISH enables the super-resolved localization of RNA molecules in small compartments, such as neural synapses, while allowing for them to be imaged over large regions of tissue. HCR has also been used to amplify the fluorescence connected to an antibody, enabling protein visualization with a 3-order-of-magnitude boost in brightness over conventional antibody staining44. Future investigations of how ExFISH protocols could be used for visualization of the 3-D configuration of the genome will be of great interest in the probing of epigenetic states, chromatin conformation, and other important determinants of cell fate, function, and health.
Iterative Expansion Microscopy (iExM)
In iterative expansion microscopy (iExM), a specimen is expanded (Fig. 1B), and then a second swellable gel is synthesized in the space opened up by the first expansion, and the sample expanded again11. To achieve, this, the gel synthesized for the first expansion step is made with a chemically cleavable cross-linker, so that after the second gel is made, the first gel can be cleaved, enabling further expansion to proceed. Also, biomolecules or labels must be transferred from the first gel to the second gel, to insure no loss of information. One way to do this is to initially stain samples with antibodies bearing oligonucleotide barcodes, which are chemically anchored to the first-round expansion polymer. After the first round of expansion, complementary oligonucleotide strands are added, which hybridize to the gel-anchored initial barcodes. These complementary strands are then chemically anchored to the second-round expansion gel, and expanded away from each other further. These strands can then be labeled with further hybridizing probes bearing fluorophores (Fig. 3C), enabling amplification that is greatly helpful given the ~10,000x volumetric dilution of biomolecules or labels relative to the starting condition.
The resolution offered by iExM is comparable to the resolution of the best super-resolution methods that have been applied to cells and tissues (Supplementary Table 1), and thus enables powerful 3D imaging with nanoscale precision of thick specimens, on conventional microscopes. The ~25 nm resolution enabled by iExM can enable, for example, the resolving of pre- and postsynaptic proteins such as neurotransmitter receptors and scaffolding proteins within synapses (Fig. 2D-E). Of particular interest is the idea that iExM may enable, in the future, detailed reconstruction of dense brain circuitry (see ref. 47 for a theoretical study of this possibility). Although the spatial resolution of iExM does not yet approach that of electron microscopy, the inherent multicolor nature of optical microscopy can allow for multiple kinds of tags, each labeled with different colors, to be used in an intact tissue nanoscopy context. Thus information represented by one color could, if insufficient for tracing a neural circuit, be error corrected by the information represented by a second color47. Preliminary studies using Brainbow-labeled mouse cortex suggests that iExM can support the visualization of spines and other compartments along neural processes over extended 3D volumes (Fig. 2C).
Going forward, an interesting question is how different ExM protocols might be combined. For example, can the iExM protocol be combined with ExFISH, so that ultraprecise volumetric visualization of RNAs in extended intact tissues is possible? Can iExM be combined with the post-expansion-antibody-administration forms of proExM/MAP, so that epitopes can be decrowded for denser and higher resolution antibody staining? And can the procedure be automated, so that extremely high throughput processing of cells and tissues is possible? In the next few years, these questions represent opportunities for biotechnological innovation, and in turn for downstream biological and medical discovery.
Discussion
In the sections above we have tried to document the status of specific ExM protocols, and where they are headed. We now offer some general comments on the state and future of the entire tool suite, in terms of fundamental performance and limitations. Compared to conventional Super-Resolution Microscopy (SRM) methods, ExM offers numerous technical advantages, such as the ability to perform 3D nanoscale imaging of thick specimens, as well as speed and ease of use (Supplementary Table 1, Supplementary Note 1). The clearest disadvantage of ExM is its incompatibility with live samples. In contrast to SRM methods, ExM poses unique considerations owing to its physical mechanism. The expansion achieved is designed to be isotropic, and measurements of isotropy with different ExM methods have shown that distortions arising from the expansion process are minimal (Supplementary Table 1, Supplementary Note 1). In addition, ExM yields transparent samples as a byproduct of its physical mechanism. In comparison to current clearing techniques, ExM clears samples due to the expansion process, which results in a hydrogel mostly composed of water (Supplementary Table 2, Supplementary Note 2).
Going forward, while iExM offers 3D nanoscale imaging in tissues with resolution comparable to the best performing SRM techniques, it may be possible to achieve even higher levels of expansion and resolution by improving polymer composition and processing protocols. Already a proof-of-concept triple expansion has been demonstrated, resulting in ~53x linear expansion, although the resolution was not validated11. By selecting polymers with higher swelling ratio in pure water, such as DMAA48, higher expansion factors may be possible, and thus potentially higher resolution. Such precision might actually exceed that of existing SRM technologies, especially if antibodies or other labels are delivered post-expansion, thus reducing the resolution error introduced by exogenous tags in a fashion not possible with other methods.
Another area of future interest is the combination of multiple protocols. A unified protocol that enabled the visualization of DNA, RNA, proteins, and lipids would be able to reveal the organization of complexes of multiple kinds of biomolecules. Already the combined use of protein-anchoring AcX and RNA-labeling LabelX has been used to visualize fluorescent proteins and FISH-labeled RNAs in the same sample10, and the ExPath protocol has been used to perform DNA FISH in concert with antibody labeling of proteins in the same sample12. Combining multimodal anchoring and labeling strategies with iExM is also an exciting future direction, potentially enabling a diversity of biomolecules to be visualized within a single sample with extremely precise resolution.
The aqueous nature of expanded specimens and the decrowding of biomolecules may open the door for highly multiplexed readout of molecular information with nanoscale precision. ExFISH has already been used for serial-staining with multiple FISH probes for multiplexed imaging of different transcripts after RNA anchoring and expansion10. It may be possible to implement in intact tissues the use of barcoded, combinatorial RNA-FISH multiplexing approaches49,50, such as MERFISH51–53 and seqFISH54, in which coded FISH probes are administered serially to yield the location of exponentially increasing numbers of transcripts over many cycles of hybridization. The ability to follow individual transcripts over many rounds of hybridization would be facilitated by the decrowding of transcipts from each other. Already a publication has appeared using expansion microscopy followed by MERFISH to visualize, with 10x higher density, members of a ~130-RNA library with near-100% detection efficiency in cultured cells53. The covalent anchoring of RNA to the polymer may also help support controlled enzymatic reactions such as fluorescent in situ sequencing (FISSEQ) of RNA45,46. Such approaches would yield transcriptome data in conjunction with cell morphology, protein locations, and other biomolecular information, with nanoscale precision.
Such techniques could also be used to identify any biomolecular labels that are conjugated to oligonucleotide barcodes that are retained within the gel (e.g., by using DNA PAINT style probes55). For instance, it may be possible to deliver antibodies for various targets, each bearing a unique oligonucleotide tag that can be read-out after expansion via multiplexed FISH or in situ sequencing, thus providing location and identity information for very large numbers of proteins. This scheme could be extended to label any biomolecule with an oligonucleotide barcode that can be identified after expansion, thereby enabling the nanoscale mapping of biomolecules throughout specimens in a highly multiplexed fashion.
Supplementary Material
ACKNOWLEDGEMENTS
ESB acknowledges NIH 1R01NS102727, the Open Philanthropy Project, DARPA, John Doerr, NIH 1R01EB024261, NSF Grant 1734870, the MIT Aging Brain Initiative/Ludwig Foundation, the Cancer Research UK Grand Challenge, the HHMI-Simons Faculty Scholars Program, NIH 1R41MH112318, NIH 1R01MH110932, IARPA D16PC00008, U. S. Army Research Laboratory and the U. S. Army Research Office under contract/grant number W911NF1510548, NIH 1RM1HG008525, US-Israel Binational Science Foundation Grant 2014509, the MIT Media Lab, MIT Brain and Cognitive Sciences Department, and McGovern Institute, and NIH Director’s Pioneer Award 1DP1NS087724. A.T.W. acknowledges the Hertz Foundation Fellowship.
Footnotes
COMPETING FINANCIAL INTERESTS
ESB is a co-inventor on multiple patents relating to ExM and is also a co-founder of a company (http://extbio.com/) commercializing ExM. YZ and ATW are inventors on several inventions relating to expansion microscopy.
Contributor Information
Asmamaw T Wassie, Department of Brain and Cognitive Sciences, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA.; Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA. McGovern Institute, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA. Media Lab, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA.
Yongxin Zhao, Media Lab, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA.; Department of Biological Sciences, Carnegie Mellon University, Pittsburgh, Pennsylvania
Edward S Boyden, Department of Brain and Cognitive Sciences, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA.; Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA. McGovern Institute, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA. Media Lab, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA. Koch Institute, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA. Center for Neurobiological Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA.
REFERENCES
- 1.Dunn RC Near-field scanning optical microscopy. Chem. Rev. 99, 2891–928 (1999). [DOI] [PubMed] [Google Scholar]
- 2.Dürig U, Pohl DW & Rohner F Near-field optical-scanning microscopy. J. Appl. Phys. 59, 3318–3327 (1986). [Google Scholar]
- 3.Hell SW Far-field optical nanoscopy. in 2010 23rd Annual Meeting of the IEEE Photonics Society, PHOTINICS 2010 3–4 (2010). doi: 10.1109/PHOTONICS.2010.5698725 [DOI] [Google Scholar]
- 4.Huang B, Bates M & Zhuang X Super-Resolution Fluorescence Microscopy. Annu. Rev. Biochem. 78, 993–1016 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen F, Tillberg PW & Boyden ES Expansion microscopy. Science (80-. ). 347, 543–548 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanaka T et al. Phase Transitions in Ionic Gels. Phys. Rev. Lett. 45, 1636–1639 (1980). [Google Scholar]
- 7.Hausen P & Dreyer C The Use of Polyacrylamide as an Embedding Medium for Immunohistochemical Studies of Embryonic Tissues. Stain Technol. 56, 287–293 (1981). [DOI] [PubMed] [Google Scholar]
- 8.Cohen Y, Ramon O, Kopelman IJ & Mizrahi S Characterization of inhomogeneous polyacrylamide hydrogels. J. Polym. Sci. Part B Polym. Phys. 30, 1055–1067 (1992). [Google Scholar]
- 9.Tillberg PW et al. Protein-retention expansion microscopy of cells and tissues labeled using standard fluorescent proteins and antibodies. Nat. Biotechnol. 34, 987–992 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen F et al. Nanoscale imaging of RNA with expansion microscopy. Nat. Methods 13, 679–684 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang J-B et al. Iterative expansion microscopy. Nat. Methods 14, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao Y et al. Nanoscale imaging of clinical specimens using pathology-optimized expansion microscopy. Nat Biotech 35, 757–764 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chozinski TJ et al. Expansion microscopy with conventional antibodies and fluorescent proteins. Nat. Methods (2016). doi: 10.1038/nmeth.3833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ku T et al. Multiplexed and scalable super-resolution imaging of three-dimensional protein localization in size-adjustable tissues. Nat Biotech 34, 973–981 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsanov N et al. SmiFISH and FISH-quant - A flexible single RNA detection approach with super-resolution capability. Nucleic Acids Res. 44, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freifeld L et al. Expansion microscopy of zebrafish for neuroscience and developmental biology studies. Proc. Natl. Acad. Sci. 201706281 (2017). doi: 10.1073/pnas.1706281114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hama H et al. Scale: a chemical approach for fluorescence imaging and reconstruction of transparent mouse brain. Nat. Neurosci. 14, 1481–8 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Susaki EA et al. Whole-brain imaging with single-cell resolution using chemical cocktails and computational analysis. Cell 157, 726–739 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Chung K et al. Structural and molecular interrogation of intact biological systems. Nature 497, 332–7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murakami TC et al. A three-dimensional single-cell-resolution whole-brain atlas using CUBIC-X expansion microscopy and tissue clearing. Nat. Neurosci. 21, 625 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Zhang YS et al. Hybrid Microscopy: Enabling Inexpensive High-Performance Imaging through Combined Physical and Optical Magnifications. Sci. Rep. 6, 22691 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aoki T, Tsuchida S, Yahara T & Hamaue N Novel assays for proteases using green fluorescent protein-tagged substrate immobilized on a membrane disk. Anal. Biochem. 378, 132–137 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Nicholls SB & Hardy JA Structural basis of fluorescence quenching in caspase activatable-GFP. Protein Sci. 22, 247–257 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deshpande T et al. Subcellular reorganization and altered phosphorylation of the astrocytic gap junction protein connexin43 in human and experimental temporal lobe epilepsy. Glia 1–12 (2017). doi: 10.1002/glia.23196 [DOI] [PubMed] [Google Scholar]
- 25.Crittenden JR et al. Striosome–dendron bouquets highlight a unique striatonigral circuit targeting dopamine-containing neurons. Proc. Natl. Acad. Sci. 113, 11318–11323 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Decarreau J et al. The tetrameric kinesin Kif25 suppresses pre-mitotic centrosome separation to establish proper spindle orientation. Nat. Cell Biol. 19, (2017). [DOI] [PubMed] [Google Scholar]
- 27.Suofu Y et al. Dual role of mitochondria in producing melatonin and driving GPCR signaling to block cytochrome c release. Proc. Natl. Acad. Sci. 201705768 (2017). doi: 10.1073/pnas.1705768114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cahoon CK et al. Superresolution expansion microscopy reveals the three-dimensional organization of the Drosophila synaptonemal complex. Proc. Natl. Acad. Sci. 201705623 (2017). doi: 10.1073/pnas.1705623114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sümbül U et al. Automated scalable segmentation of neurons from multispectral images. Adv. Neural Inf. Process. Syst. (2016). [Google Scholar]
- 30.Mosca TJ, Luginbuhl DJ, Wang IE & Luo L Presynaptic LRP4 promotes synapse number and function of excitatory CNS neurons. Elife 6, 1–29 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang IE, Lapan SW, Scimone ML, Clandinin TR & Reddien PW Hedgehog signaling regulates gene expression in planarian glia. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halpern AR, Alas GCM, Chozinski TJ, Paredez AR & Vaughan JC Hybrid Structured Illumination Expansion Microscopy Reveals Microbial Cytoskeleton Organization. ACS Nano acsnano.7b07200 (2017). doi: 10.1021/acsnano.7b07200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Artur CG et al. Plasmonic nanoparticle-based expansion microscopy with surface-enhanced Raman and dark-field spectroscopic imaging. Biomed. Opt. Express 9, 603 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Villaseñor R, Schilling M, Sundaresan J, Lutz Y & Collin L Sorting Tubules Regulate Blood-Brain Barrier Transcytosis. Cell Rep. 21, 3256–3270 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Treweek JB et al. Whole-body tissue stabilization and selective extractions via tissue-hydrogel hybrids for high-resolution intact circuit mapping and phenotyping. Nat. Protoc. 10, 1860–1896 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Unnersjö-Jess D et al. Confocal super-resolution imaging of the glomerular filtration barrier enabled by tissue expansion. Kidney Int. in press, (2018). [DOI] [PubMed] [Google Scholar]
- 37.Wei L et al. Super-multiplex vibrational imaging. Nature 544, 465–470 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu F et al. Supermultiplexed optical imaging and barcoding with engineered polyynes. Nat. Methods 15, 194–200 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar A et al. Influenza virus exploits tunneling nanotubes for cell-to-cell spread. Sci. Rep. 7, 40360 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao M et al. Expansion Stimulated Emission Depletion Microscopy (ExSTED). ACS Nano acsnano.8b00776 (2018). doi: 10.1021/acsnano.8b00776 [DOI] [PubMed] [Google Scholar]
- 41.Elmore JG et al. Diagnostic Concordance Among Pathologists Interpreting Breast Biopsy Specimens. JAMA 313, 1122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi HMT et al. Programmable in situ amplification for multiplexed imaging of mRNA expression. Nat. Biotechnol. 28, 1208–12 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Choi HMT, Beck VA & Pierce NA Next-Generation in Situ Hybridization Chain Reaction: Higher Gain, Lower Cost, Greater Durability. ACS Nano 8, 4284–4294 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin R et al. A hybridization-chain-reaction-based method for amplifying immunosignals. Nat. Methods 15, 275–278 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Lee JH et al. Highly Multiplexed Subcellular RNA Sequencing in Situ. Science (80-. ). 343, 1360–1363 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ke R et al. In situ sequencing for RNA analysis in preserved tissue and cells. Nat. Methods 10, 857–60 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Yoon Y-G et al. Feasibility of 3D Reconstruction of Neural Morphology Using Expansion Microscopy and Barcode-Guided Agglomeration. Front. Comput. Neurosci. 11, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Truckenbrodt S et al. X10 Expansion Microscopy Enables 25 nm Resolution on Conventional Microscopes. doi.org 172130 (2017). doi: 10.1101/172130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lubeck E & Cai L Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat. Methods 9, 743–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M & Cai L Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 11, 360–1 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen KH, Boettiger AN, Moffitt JR, Wang S & Zhuang X Spatially resolved, highly multiplexed RNA profiling in single cells. Sci. 348, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moffitt JR et al. High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization. Proc. Natl. Acad. Sci. 113, 201612826 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang G, Moffitt JR & Zhuang X Multiplexed imaging of high-density libraries of RNAs with MERFISH and expansion microscopy. Sci. Rep. 8, 4847 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shah S et al. In Situ Transcription Profiling of Single Cells Reveals Spatial Organization of Cells in the Mouse Hippocampus. Neuron 92, 342–357 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Y et al. Rapid sequential in situ multiplexing with DNA-Exchange-Imaging in Neuronal Cells and Tissues. Nano Lett. acs.nanolett.7b02716 (2017). doi: 10.1021/acs.nanolett.7b02716 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.