

Abstract
RNA sequencing (RNA-seq) is a high throughput technology that provides unique insights into the transcriptome. It has a wide variety of applications in quantifying genes/isoforms, detecting non-coding RNA, alternative splicing, and splice junctions. It is extremely important to comprehend the entire transcriptome for a thorough understanding of the cellular system. Several RNA-seq analysis pipelines are proposed to date. However, no single analysis pipeline can capture dynamics of the entire transcriptome. Here, we compile and present a robust and most commonly used analytical pipelines covering entire spectrum of transcriptome analysis, including quality checks, aligning reads, differential gene/transcript expression analysis, cryptic splicing events discovery, and visualization. Challenges, critical parameters, and possible downstream functional analysis pipelines associated with each step are highlighted and discussed. This provides a comprehensive understanding of state-of-the-art RNA-seq analysis pipeline and a greater understanding of the transcriptome.
Keywords: RNA-seq, differential gene expression, differential isoform usage, alternative splicing, cryptic splicing
INTRODUCTION
Due to advancement in technology and drop in cost for high-throughput profiling of molecular assay and next-generation sequencing, RNA sequencing (RNA-seq) has becoming a common tool for scientists to study the transcriptomic phenomenon observed in biological samples. RNA-seq data allows one to study the system-wide transcriptional changes from a variety of aspects, ranging from expression changes in gene or isoform levels, to complex analysis like discovery of novel, alternative or cryptic splicing sites, RNA-editing sites, fusion genes, or single nucleotide variation (Conesa, Madrigal et al. 2016). These analyses had been applied in studies to understand complex neurological disease (Sztainberg, Chen et al. 2015) to basic molecular mechanisms (Lo, Chung et al. 2017).
In this article, we describe three protocols for RNA-seq data analysis. These protocols are applicable to RNA-seq data sets from the most common Illumina sequencing platform with an available reference genome. The first protocol covers the standard pipeline on RNA-seq data that interrogates the transcriptome at the gene level, which is usually referred as differentially expressed gene (DEG) analysis. This pipeline starts from the raw sequence reads, and ends with a set of differentially expressed genes. The second protocol goes beyond gene level analysis into isoforms, focusing primarily on differential expression (DE) and differential usage (DU) of isoforms. Lastly, the third protocol pinpoints specific splice junctions and decipher cryptic splicing events. A detailed motivational rationale for each protocol is given in respective sections below. Together, the three protocols help to thoroughly comprehend a transcriptome of interest from genes to junctions.
STRATEGIC PLANNING
Before starting the protocols, users should install the software listed in the software section below from download URL listed in Table 1. Following the download guides, the program will be installed so that it could be executed from any directory. Lastly, users should download required libraries, reference files such as the reference genome sequences and index files accordingly based on the experiments. Respective web resources are listed in Table 1.
Table 1.
Required tools and respective web resources
BASIC PROTOCOL 1
DIFFERENTIAL GENE EXPRESSION ANALYSIS OF RNA-SEQ
The immediate question one could ask from an experiment with RNA-seq is what genes are dysregulated due to the designed perturbation, treatment, etc. Thus, the first protocol consists of the basic pipeline for analyzing raw sequence reads of RNA data to reveal the set of significantly dysregulated genes. Specifically, this pipeline consists of five main steps, where each step corresponds to one phase of the analysis that achieve certain milestone. In the following protocol, we will use an example data of six samples from GSE72790 and provide commands based on this example. To reduce the amount of execution time in testing the protocol, we will focus on data from chromosome 19 only.
Necessary Resources
Hardware: A computer or server with access to UNIX command environment.
Software: FastQC, Tophat2, samtools, HTSeq, Rstudio, DESeq2. Web resources of respective tools are listed in table 1.
Input files: Raw sequence reads in fastq formats. We had included 12 fastq files (two files each for six samples) in the supplementary materials for this example. Each of these files contains about five million reads of 90 base-pair (bp) which should be originated from the chromosome 19 of mouse genome.
Step 1. Quality check on the raw reads.
Create a directory named FastQC to store the results later. Then, call FastQC to obtain quality check metrics to inspect the quality of raw sequence reads (stored in the Fastq directory) and output the metrics to FastQC directory:
$ mkdir FastQC $ fastqc Fastq/*.fastq –o FastQC
FastQC provides a quick view on the quality of the raw sequence reads from multiple analyses, ranging from the sequence quality, GC content, to library complexity. The command above will produce a report in HTML format which could be viewed from web browser. In the report, each metric evaluated will be annotated with a green check, red cross, or yellow exclamation mark to indicate pass, fail, or caution respectively. Usually, the quality score (Fig. 1a) and A,C,G,T content (Fig. 1b) across bases could be used to decide how the reads should be groomed prior to mapping.
Figure 1. Quality check metrics on raw sequence reads from FastQC.
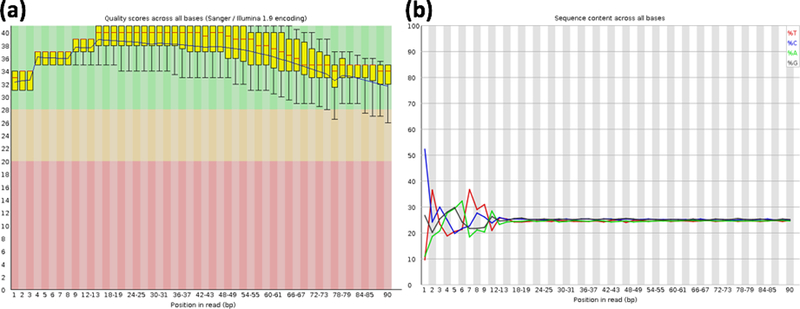
Bar plot of quality score (Phred score) for each base in the reads (a). Line plot showing the distribution of each nucleotide (A, C, G, T) in the sequence reads on each bases (b).
Step 2. Groom raw reads.
Remove sequences with low quality to get better alignment in the later steps. Based on the FastQC reports from above commands for this example, the qualities of the reads are fine except random distribution of sequence content at the 5’ end of reads. Thus, we trim 10bp from the beginning of each read:
$ awk -v s=10 -v e=0 ‘ {if (NR%2 == 0) print substr($0, s+1, length($0)-s-e); else print $0; } ‘ Fastq/MT1_1.fastq > Fastq/Trim_MT1_1.fastq
$ awk -v s=10 -v e=0 ‘ {if (NR%2 == 0) print substr($0, s+1, length($0)-s-e); else print $0; } ‘ Fastq/MT1_2.fastq > Fastq/Trim_MT1_2.fastq
$ awk -v s=10 -v e=0 ‘ {if (NR%2 == 0) print substr($0, s+1, length($0)-s-e); else print $0; } ‘ Fastq/MT2_1.fastq > Fastq/Trim_MT2_1.fastq
$ awk -v s=10 -v e=0 ‘ {if (NR%2 == 0) print substr($0, s+1, length($0)-s-e); else print $0; } ‘ Fastq/MT2_2.fastq > Fastq/Trim_MT2_2.fastq
$ awk -v s=10 -v e=0 ‘ {if (NR%2 == 0) print substr($0, s+1, length($0)-s-e); else print $0; } ‘ Fastq/MT3_1.fastq > Fastq/Trim_MT3_1.fastq
$ awk -v s=10 -v e=0 ‘ {if (NR%2 == 0) print substr($0, s+1, length($0)-s-e); else print $0; } ‘ Fastq/MT3_2.fastq > Fastq/Trim_MT3_2.fastq
We used the awk command native to Unix system here. The s=10 indicates that the first 10bp (the 5’end) will be trimmed, where e=0 indicates none will be trimmed from the end (3’ end). Users should change these according to the FastQC reports. File name for input fastq reads is specified before the > sign, where the file name for the trimmed reads should be followed after the > sign.
Note that although these are pair-end (PE) reads, the forward and reversed reads are trimmed separately. Also, users should repeat these steps for the other six fastq files for WT samples.
Usually, quality of the sequence reads at both ends of the reads are low, which can be seen from the drop in Phred score at 3’ end and random A,C,G,T concentration at the 5’ end. Although some would suggest removing the adapter sequences as well, due to the nature of next-generation sequencing, more than 99% of the adapter sequences are cleaved from the RNA fragments before sequencing and should not be present in the sequence reads. Thus, we did not include that step. However, if it happened that some adapters were not successfully cleaved and being sequenced, they should be removed prior to mapping to avoid poor mapping quality.
Step 3. Align raw reads to reference genome
Map the trimmed sequence reads to reference genome using tophat2. Specifically, we first create a directory with the sample name to store the output then calling tophat2 and output results to the directory’s sub-directory named Tophat_Out. Since this is a mouse sample, UCSC mm10 is used as the reference genome and the reference transcriptome file and bowtie index files are stored under Indexes directory in the home directory. Bowtie2 index files contain the genome sequences to be aligned to in bowtie2 format. Tophat2 uses bowtie2 as the base sequence aligner.
$ mkdir MT1 $ tophat2 -r 200 -p 8 -o MT1/Tophat_Out -G ~/Indexes/Mus_musculus/UCSC/mm10/Genes/genes.gtf ~/Indexes/Mus_musculus/UCSC/mm10/Sequence/Bowtie2Index/genome Fastq/Trim_MT1_1.fastq Fastq/Trim_MT1_2.fastq
Users should repeat these commands for other five samples, MT2, MT3, WT1, WT2, WT3, and change the directory and file names accordingly.
Note that with PE reads, the order of the input file names for R1 (forward reads) and R2 (reverse reads) should not be mixed up. Default parameters for tophat2 are used here. The insert size of 200 is used because the average fragments for the data in this example is ~400bp and common to data sequenced from Illumina HiSeq 2000. This step would take approximately 30 minutes on each sample for this example. The execution time for this step would depend on the number of reads and number of processing CPU (specified with –p parameter of tophat2) used.
Step 4. Assemble gene expression from aligned reads
Use HTSeq to quantify the number of reads mapped to each gene:
$ samtools view MT1/Tophat_Out/accepted_hits.sorted.bam | python -m HTSeq.scripts.count -q -s no - ~/Indexes/Mus_musculus/UCSC/mm10/Genes/genes.gtf > MT1/MT1.count.txt
Repeat this command to the other five samples and change the input and output file names accordingly as in last step. Upon successful execution of this step, six text files suffixed with “count.txt” will be generated with each prefixed with the sample ID and stored under the subdirectory of the sample, for example MT1/MT1.count.txt, MT2/MT2.count.txt, and so son.
Note that we need to sort the alignment file (BAM file) prior to calling HTSeq, as HTSeq requires the reads ordered by reads identifier. The output of this step will be a tab-delimited text file with two columns and about 24 thousands rows, where each row represents a gene, first column is the gene identifier, second column is the number of reads mapped to the gene.
Step 5. Differential gene expression analysis from assembled gene expression
Take the steps as suggested by DESeq2 manual to carry out the analysis. These standard steps are listed in the following five sub-steps:
5.1. Launch RStudio and load necessary library
> library(“DESeq2”)
5.2. Create necessary data object.
> sample.names <- sort(paste(c(“MT”, “WT”), rep(1:3, each=2), sep=““)) > file.names <- paste(“../”, sample.names, “/”, sample.names, “.count.txt”, sep=““) > conditions <- factor(c(rep(“MT”, 3), rep(“WT”, 3))) > sampleTable <- data.frame(sampleName=sample.names, fileName=file.names, condition=conditions) > # read in the HTSeq count data > ddsHTSeq<-DESeqDataSetFromHTSeqCount(sampleTable=sampleTable, directory=“.”, design=~ condition )
In this sub-step, we specify the sample identifiers, name of files with gene counts for each sample, and experiment condition(s) for each sample; then pass this information to DESeqDataSetFromHTSeqCount function to make a DESeqDataSet object for following analysis.
5.3. Run differential gene analysis.
> ddsHTSeq <- ddsHTSeq[rowSums(counts(ddsHTSeq)) > 10, ] > dds <-DESeq(ddsHTSeq)
Prior to differential gene analysis, filter out genes with low counts, which are analogous to genes that are not expressed. Then, use DESeq function for the differential gene analysis based on negative binomial distribution. Specifically, DESeq function is a wrapper function with multiple default analyses, including estimating the size factors and dispersions, fitting the negative binomial generalized linear model, and performing Wald tests for differential gene analysis (Love, Huber et al. 2014)
5.4. Quality checks on the samples.
> rld <- rlogTransformation(dds, blind=FALSE) > # Plot PCA plot > plotPCA(rld, intgroup=“condition”, ntop=nrow(counts(ddsHTSeq))) > # Plot correlation heatmap > cU <-cor( as.matrix(assay(rld))) > cols <- c( “dodgerblue3”, “firebrick3” )[condition] > heatmap.2(cU, symm=TRUE, col= colorRampPalette(c(“darkblue”,”white”))(100), labCol=colnames(cU), labRow=colnames(cU), distfun=function(c) as.dist(1 - c), trace=“none”, Colv=TRUE, cexRow=0.9, cexCol=0.9, key=F, font=2, RowSideColors=cols, ColSideColors=cols)
Draw PCA plot (Fig. 2a) and correlation heatmap (Fig. 2b) to visualize if the samples cluster per their conditions. In this example, samples are clustered into two groups, which are their genotypes. In cases where samples do cluster in groups but the grouping is not the experimental conditions or genotypes, it indicates that the samples are clustered by other factors. These factors could be latent biological subtypes or technical factors such as the batch effect. For example, the samples in supplemental Figure S1 are prepared in two different batches and PCA analysis shows two clusters corresponding to the batches, instead of four designed phenotype groups.
Figure 2. Visualization inspection of sample clustering.

PCA plot (a) and heatmap on correlation coefficient between samples (b) based on gene expression profiles of the six samples.
5.5. Output differential gene analysis results
#> res <- results(dds, contrast=c(“condition”, “MT”, “WT”)) > grp.mean <- sapply(levels(dds$condition), function(lvl) rowMeans(counts(dds,normalized=TRUE)[,dds$condition == lvl] ) ) > norm.counts <- counts(dds, normalized=TRUE) > all <- data.frame(res, grp.mean, norm.counts) > write.table(all, file=“DESeq2_all_rm.txt”, sep=“\t”)
Call the results function from DESeq2 package to extract the results from the differential gene analysis. These results include base means across samples, log2 fold changes, standard errors, test statistics, p-values and adjusted p-values. Then, combine the results obtained with the average expressions for each genotype (grp.mean) and normalized read counts (norm.counts) for each sample into a data table. Finally, save the data table into a tab-delimited file. Group means and normalized read counts are useful when users want to inspect how a gene is expressed in the experiment. Furthermore, users could get these values and apply to other software for more analysis.
5.6. Generate figures on significantly differentiated genes
> plotMA(res, ylim=c(−5,5), alpha = 0.01)
> topGene <- rownames(res)[res$padj <= sort(res$padj)[5] &!is.na(res$padj)]
> with(res[topGene, ], {
points(baseMean, log2FoldChange, col=“dodgerblue”, cex=1.5, lwd=2)
text(baseMean, log2FoldChange, topGene, pos=2, col=“dodgerblue”)
})
> library(pheatmap)
> sig.dat <- assay(rld)[res$padj < 0.01 & !is.na(res$padj), ]
> annC <- data.frame(condition=conditions)
> rownames(annC) <- colnames(sig.dat)
> pheatmap(sig.dat, scale=“row”, fontsize_row=9,
annotation_col = annC)
Two common plots to visualize findings from a differential gene analysis are MA plot and gene expression heatmap. The MA plot (Fig. 3a) shows how gene expressed between two genotypes (log fold-change in Y-axis) with respect to overall expression on all samples (in X-axis), highlights the significantly differentially expressed genes (DEGs, adjusted P-value < 0.01) in red, and annotates the five most statistical significant DEGs.
Figure 3. Results of differential gene expression analysis.

MA plot (a) and expression heatmap on the DEGs (adjusted P < 0.01) (b).
In Fig. 3b, heatmap is used to show the expression pattern of DEGs for all the samples and annotate the samples with their genotypes.
COMMENTARY
Background Information
Since RNA-seq first presented in studies published in year 2008 (Lister, O’Malley et al. 2008, Mortazavi, Williams et al. 2008, Nagalakshmi, Wang et al. 2008), it has becoming the default tool to use for whole transcriptome experiments in less than a decade. Various algorithms, tools, and pipelines had been developed to analyze RNA-seq data throughout these years; starting from bwa, bowtie, tophat, Cufflinks, to the new alignment free methods such as kallisto (Conesa, Madrigal et al. 2016). Among different analyses proposed, differential gene analysis, isoforms analysis, and splicing events are the three most common analyses carried out in studies with RNA-seq data, with differential gene analysis as the first and default analysis to go with.
Critical Parameters and Troubleshooting
The first potential problem for this pipeline can be related to the input data, the raw sequence reads. We could detect this risk from FastQC reports in the first step of the protocol. Although we could not transform the problematic sequence reads into high quality sequence reads, we could apply a more stringent threshold in grooming the reads to retain only good quality reads, in order to prevent the noisy data being propagated into downstream analysis.
The insertion size for tophat2 (Step 3 of protocol 1) is another important factor in this pipeline. As discussed above, a wrong insertion size will lead to poor mapping. Subsequently, this will affect the accuracy of quantified expressions and splice junction detection.
Lastly, reference genome index and gene annotation used are critical. Inaccurate path to these annotations will cause failure during mapping. Wrong annotation with correct path set would allow the protocol executes without error but the mapping would be poor. This could be detected from low-mappability.
Advanced Parameters
Standard Illumina RNA-seq data available is stranded paired-end reads. Note that stranded RNA-seq is not strand-specific, where reads are aligned to both the forward or reverse strand regardless if the read is an upstream read (read 1) or downstream read (read 2). In non-strand specific RNA-seq, antisense genes/ transcripts will not be able to be differentiated from the overlapping sense genes/transcripts. A significant number of antisense transcripts were observed from sense strand resulting in complimentary non-coding RNAs and such non-coding RNA are often linked to important regulatory functions (Levin, Yassour et al. 2010). Therefore, other sequencing technologies with library preparation methods such as dUTP and NSR were developed to effectively retain the strand information in sequencing reads.
If the library is prepared such that sequence reads are strand-specific RNA-seq reads, two steps should be modified as follows:
In Step 3 of protocol 1, specify --library-type to be ‘fr-firstrand’ or ‘fr-secondstrand’ according to the library preparation protocol. In the protocol above, we did not specify this parameter as tophat2 assumes --library-type to be ‘fr-unstranded’ by default.
In Step 4 of protocol 1, set -s to be ‘yes’. We set ‘-s no’ above as we have to specify that it is not strand-specific as HTSeq-count default is strand-specific assay.
Suggestions for Further Analysis
Users will obtain a list of differentially expression genes (DEGs) upon successful completion of this first protocol. The list of DEGs obtained could be used as the input for downstream analysis and functional analysis such as the enrichment of gene ontology (GO) terms (Gene Ontology 2015), gene set enrichment analysis to known pathways, network analysis to study the interactions between the DEGs (Yalamanchili, Yan et al. 2014), or even RNA-editing discovery.
Furthermore, users could integrate the transcriptomic findings from RNA-seq with genomics, ChIP-seq, or DNA-methylations to get the systems view of regulatory mechanisms.
BASIC PROTOCOL 2
BEYOND DEGs: DIFFERENTIAL EXPRESSION AND USAGE OF ISOFORMS
A single gene can code multiple proteins with different functions by re-arranging constituent exons (Yalamanchili, Xiao et al. 2012). This process of exon rearrangement (inclusion/skipping) is called splicing and different forms of the same gene are called isoforms. Standard DEG analysis presented in Protocol 1 is based on overall gene expression and could underutilize significant biological details such as the isoform specific expression. Different isoforms vary in expression in different conditions making them primary targets to explain biological anomalies that could not be explained by gene level changes (Garcia-Blanco, Baraniak et al. 2004). Isoform specific expression is found to be associated with different diseases, for example the human epidermal growth factor receptor (HER-2) in breast cancer (Menon and Omenn 2010). It is very crucial to explore expression difference of isoforms to decipher disease mechanisms and therapeutic targets (Yalamanchili, Li et al. 2014). This protocol guides through two analyses to dissect the biological phenomenon at isoform levels: 1) absolute difference in expression for each isoform, 2) change of relative abundance of isoforms of a gene across conditions.
Necessary Resources
Hardware: A computer or server with access to UNIX command environment.
Software: Kallisto, Sleuth, Isoform Usage Two-step Analysis: IUTA, samtools, Rstudio. Web resources of respective tools are listed in Table 1.
Input files: Raw fastq reads for differentially expressed isoforms (DEIs) and aligned bam files for differential usage of isoforms. Alignment bam files must be sorted and indexed (can use the files from step 3 of protocol 1). Model reference genome in fasta format, gene annotation file in GTF format, and model transcriptome (cDNA sequences). Web resources of respective tools and files are listed in Table 1.
Step 1. Quantification of isoforms from raw reads
Typically, in most of the analysis pipelines for RNA-seq, raw reads are first mapped to reference genome and then quantified, as presented in protocol 1 above. This is highly time-consuming especially when the number of samples is large. Recent computational approaches have made quantification two magnitudes faster by pseudo-aligning reads to a reference, i.e. producing target transcripts list to each read while avoiding alignment of individual bases. This step demonstrated the use of kallisto, an alignment free near-optimal isoform quantification tool.
Kallisto pseudo-alignment is a two-step process: creating a “transcriptome index” and quantifying the transcripts. To begin, first create a directory “Index” and copy the reference genome fasta files into it. Next, build the index using kallisto index command. After building the index, kallisto quant command is used to quantify reads with respect to the reference transcriptome. See the following commands:
# Create a directory Kallisto_Analysis and copy all fastq files to it # $ cd Kallisto_Analysis # Creating Index directory # $ mkdir Index $ cp ~/genome.fa Index/Reference_Genome.fa # Building Index # $ kallisto index -i Index/transcripts.idx Index/Reference_Genome.fa # Running Kallisto # $ kallisto quant -i Index/transcripts.idx -o Kallisto_output/MT1/Quants -b 100 MT1_1.fastq MT2_2.fastq
Parameters: –i is the index built, -o is the output directory, and –b is the number of bootstraps required. Isoform quantifications are outputted to a tab-separated file (tsv) “abundance.tsv” and respective bootstrapping results to “abundance.h5”. These two files will be stored in the specified output directory (“Kallisto_output/MT1/Quants” in this example).
Repeat the last step (kallisto quant) for other five samples, MT2, MT3, WT1, WT2, and WT3 by changing the directory and file names accordingly.
Step 2. Differential expression of isoforms (DEI) analysis
Sleuth is an R package for analyzing transcript abundances quantified by kallisto. It implements statistical algorithms for differential analysis and provides exploratory data analysis interface through shiny framework in RStudio. Below has the most commonly used kallisto-sleuth pipeline:
2.1 Launch RStudio and load necessary library.
> library(“sleuth”)
2.2 Specify the kallisto results directory where each subdirectory corresponds to a sample. Here Kallisto_output have only 6 subdirectories (WT1, WT2, WT3, MT1, MT2, and MT3) one per sample.
> base_dir <- “Kallisto_Analysis”
2.3 Getting sample ID information from kallisto results directory.
> sample_id <- dir(file.path(base_dir,”Kallisto_output”))
2.4 The result can be displayed by typing:
> sample_id ## [1] “MT1” “MT2” “MT3” “WT1” “WT2” “WT3” lines beginning with ## show the output of the command
2.5 Assign file paths to sample IDs:
> kal_dirs <- sapply(sample_id, function(id) file.path(base_dir, “Kallisto_output”, id, “Quants”)) > kal_dirs ## MT1 ## “Kallisto_Analysis/Kallisto_output/MT1/Quants” ## MT2 ## “Kallisto_Analysis/Kallisto_output/MT1/Quants” ……… so on for rest of the samples
2.6 Reading experimental design from a file (sample names in column 1 and condition in column2) and adding file paths.
### samples_info.txt # sample_name condition WT1 WT WT2 WT MT1 MT … so on > s2c <- read.table(file.path(base_dir, “samples_info.txt”), header = TRUE, stringsAsFactors=FALSE) > s2c <- dplyr::select(s2c, sample = sample_name, condition) > s2c <- dplyr::mutate(s2c, path = kal_dirs)
2.7 Creating sleuth object: This triggers reading in kallisto results, normalizing est_counts, normalizing tpm values, merging in metadata, normalizing bootstrap samples and summarizing bootstraps.
# Creating object > so <- sleuth_prep(s2c, ~ condition)
2.8 Fitting full models and testing using likelihood ratio test.
# Fit full model > so <- sleuth_fit(so) # Fit reduced model. In this case, the reduced model is the intercept-only model: > so <- sleuth_fit(so, ~1, ‘reduced’) # Test # > so <- sleuth_lrt(so, ‘reduced’, ‘full’)
2.9 Writing results to a file and exploratory data analysis and visualization in sleuth Shiny app.
# Write results to a file # > results_table <- sleuth_results(so, ‘reduced:full’, test_type = ‘lrt’) > write.table(results_table,file=“Out.txt”,sep=“\t”) # Visualization in Shiny# > sleuth_live(so)
Sleuth outputs a tab delimited text file. Each row corresponds to a transcript. User can screen for statistically significant differentially expressed transcripts by applying a q-values cutoff, column 4 in the example output (provided as supplementary file). Sleuth also provides an intuitive exploratory data analysis and visualization interface. As described in protocol 1, several analytical plots like PCA, heatmap, MA, scatter plots, density and fragment distribution can be plotted using sleuth shiny app. Representative plots are shown in Figure 4. PCA and heatmaps plot are discussed in protocol 1 above. Scatter plots and density plots useful in comparing transcriptome expressions of any two samples (Drăghici 2012).
Figure 4. Representative plots from sleuth analysis shiny app.

(a) PCA, (b) Heatmap, (c) Scatterplot, and (d) Density plots.
Step 3. Differential usage of isoforms analysis
Cells often express different isoforms of a gene in different concentrations. This is one of the key mechanisms controlling cell fate and tissue differentiation. Any imbalance in expression proportions could lead to adverse effect in several diseases including cancer, for example imbalance between the long and short isoforms of B-cell lymphoma affects lung cancer prognosis (Dou, Xu et al. 2010). Standard analysis of individual isoforms cannot completely decipher the shift in isoform proportions. It is very possible to have expression level differences with no change in relative abundance. Following steps demonstrate how to identify differential isoform usage using Isoform Usage Two-step Analysis ( IUTA) (Niu, Huang et al. 2014).
3.1 First, create a directory named DUI and copy all the alignment (bam) files by renaming them to sample names. Bam files from step 3 of protocol 1 can be used here. Use samtools to sort and index all bam files.
$ mkdir DUI $ cp MT1/Tophat_Out/accepted_hits.sorted.bam.bam DUI/MT1.bam $ samtools sort MT1.bam MT1.sorted $ samtools index MT1.sorted.bam
Repeat this step for the other five samples MT2, MT3, WT1, WT2, and WT3; and change the directory and file name accordingly.
3.2 Launch RStudio and load IUTA library.
> library(“IUTA”)
3.3 Set input, output and other parameters
# Output directory # > outdir <- “IUTA_OUT” # number of core to use # > ncores <- 6 # Control files # > bam.list.1<-c(‘WT1.sorted.bam’,’WT2.sorted.bam’,’WT3.sorted.bam’) > rep.info.1 = rep(1,length(bam.list.1)) # Treatment files # > bam.list.2<-c(‘MT1.sorted.bam’,’MT2.sorted.bam’,’MT3.sorted.bam’) > rep.info.2 = rep(1,length(bam.list.2))
3.4 Provide reference transcriptome annotations in gtf format.
> transcript.info <- ‘/Projectdirectory/genes.gtf’
3.5 Run IUTA command with desired parameters.
> IUTA(bam.list.1, bam.list.2, transcript.info,rep.info.1,rep.info.2,output.dir =outdir, output.na = FALSE, genes.interested = “all”, strand.specific = rep(“1.5”,length(rep.info.1)+length(rep.info.2)), gene.filter.chr = c(“_”, “M”, “Un”), mapq.cutoff = NA, alignment.per.kb.cutoff = 10, IU.for.NA.estimate = “even”, sample.FLD = FALSE, FLD = “empirical”, mean.FL.normal = NA, sd.FL.normal = NA, number.samples.EFLD = 1e+06, isoform.weight.cutoff = 1e-4, test.type = “SKK”, log.p = FALSE, fwer = 1e-2, mc.cores.user = ncores)
A few important parameters for IUTA are: alignment.per.kb.cutoff to specify the minimum expression value (in number of reads) to be considered, mapq.cutoff for read quality, strand.specific to indicate the sample library strand type (1, 2 and 1.5 for sense, anti-sense and un-stranded libraries), and FLD to specify what fragment length distribution to be used. Two options are available for FLD: normal or empirical, where empirical is recommended when we are not sure about the distribution and let the program to estimate from the data. For most analyses “SKK” is recommend for test.type.
In general, IUTA first estimate transcript abundance and then test for differential usage of isoforms. Upon successful execution, IUTA will output two files: estimates.txt with sample-wise transcript estimates and p_values.txt with results indicating the significance of differential isoform usage.
3.6 Visualize results for a gene with differential isoform usage with intuitive pie chart plot. Below is an example on inspecting the differential usage of gene Gfra1.
> gene<-”Gfra1” > pie_compare(gene, 3, estimates.file = “estimates.txt”, geometry = “Euclidean”, adjust.weight = 1e-300, output.file = paste(“Pieplot_”,gene, “.pdf”, sep = ““), group.name = c(“WT”, “MT”), output.screen=FALSE)
A pie chart is plotted based on isoform abundance estimates from “estimates.txt” output from IUTA function in last step. The pie charts from the command above are shown in Figure 5.
Figure 5. Pie chart showing differential isoform usage for gene Gfra1 between WT and MT samples.

The usage of isoform P10382 is decreased from 80.7% in WT to 65.3 % in MT samples. On the other hand, the usage of P18895 increased in MT samples.
COMMENTARY
Background Information
A typical transcript level analytical pipeline will first align sequence reads to a reference and then estimate the abundance. This approach is highly time consuming and trails far behind accelerating next-generation sequencing rate. For instance, widely used programs like TopHat2 and Cufflinks require 20 hour to map and 14 hour to quantify 20 samples, each with 30 million reads on a 20 core computer (Bray, Pimentel et al. 2016). Despite several streamlined quantification algorithms proposed to speed up the pipeline, alignment overhead cannot be avoided. To address these challenges, ultra-fast lightweight alignment free quantification methods including kallisto and Salmon have been proposed. Kallisto pseudo-aligns reads to a reference transcriptome rather than to a reference genome and is faster than previous methods by two orders of magnitude without compromising on accuracy (Bray, Pimentel et al. 2016, Jin, Wan et al. 2017). Though transcriptome analysis can drive new insights, it is limited in terms of understanding non-coding genome, transcriptional regulation and other non-coding variations (Conesa, Madrigal et al. 2016).
Critical Parameters
Kallisto is an expectation maximization (EM) -based alignment-free quantification method. As in any EM-based algorithm, sample size is the main factor affecting the estimation. In this scenario, the number of reads available for quantification is the sample size. Thus, to avoid any quantification bias, it is important to design experiment such that all the samples have comparable sequencing depth.
To build the Kallisto index, authors of Kallisto recommended a default “kmer” values of 31 (maximum kmer allowed). However, the kmer value should be adjusted according to the sequenced read length, smaller kmer for shorter reads. It is recommended to try out different kmer values and do a saturation check.
Performance of EM-based methods will drop with the increase in number of isoform for a gene. Both IUTA and Kallisto suffer from this issue. Any significant hits with high number of isoforms should be interpreted cautiously.
Fragment length distribution (FLD) used in IUTA is a critical parameter. Since different samples may have different fragment lengths, it is always recommended to set FLD to “empirical” for the distribution to be computed for each sample based on the sample input itself.
Troubleshooting
Processing data from public repositories like GEO (gene expression omnibus) may cause read pair mismatch issues with IUTA. This can be fixed by renaming paired read IDs to be the same.
Advanced Parameters
Similar to advanced parameters section discussed in protocol 1, the strand-specific RNA-seq library should be handled slightly different. Specifically, the quantification step in step 1 of this protocol should be modified as follows:
$ kallisto quant [–-single/--fr-stranded/--rf-stranded] –i index fastq files
Use –-single for single-end data, --fr-stranded for first read forward and --rf-stranded for first read reverse.
BASIC PROTOCOL 3
DEEP INTO RNA-SEQ: CRYPTIC SPLICING
With recent advances in deep sequencing technologies, a significant number of novel splice junctions are observed in several eukaryotic datasets (Kapustin, Chan et al. 2011). Such un-annotated cryptic splice cites (CSS) are usually inactive or recognized only at very low levels (Green 1986). Because of their dormant nature, cryptic splice sites are often ignored as noise. However, it has been only recently shown that cryptic sites can be activated if a nearby canonical splice is mutated (Padgett, Grabowski et al. 1986). Cryptic site activation is also linked to a wide range of genetic diseases (Wang and Cooper 2007). Almost 50% of the disease causing mutations disrupt alternative splicing either by mutating canonical splice sites or by activating CSS. Several recent studies have established the role of cryptic site activation in various human diseases, from cancers to neurological disease like amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (Ling, Pletnikova et al. 2015). Owing to their potential roles in disease, it is extremely important to thoroughly understand the mechanism of CSS activation. Identification of target cryptic sites is the first step to decode any CSS-dependent disease mechanism. The following protocol guides through CrypSplice (Tan, Yalamanchili et al. 2016), a novel computational approach to identify cryptic splice sites from RNA-seq data.
Necessary Resources
Hardware: A computer or server with access to Unix command environment.
Software: CrypSplice, Bedtools version 2.17 or higher, R libraries ibb and MASS, Integrative Genomics Viewer (IGV) and samtools. Web resources of respective tools are listed in Table 1.
Input files: Aligned bam files. Files must be sorted and indexed (can use the files from step 3 of protocol 1). Junction bed files also from step 3 of protocol 1. Genome model files: Alternative splicing events, gene model and junction database files (can be downloaded directly from CrypSplice link from above reference).
Step 1. Preparing alignment and junction files.
CrypSplice need alignment files in bam format and junction files in bed format. Refer to steps 1 to 3 in protocol 1 to get bam and bed files from raw fastq reads using tophat2. Create a directory named CrypSpliceAnalysis and copy all the required input files mentioned above into it. By default, alignment files and junction files (from step 3 of protocol 1) are saved as accepted_hits.sorted.bam and junctions.bed respectively. Rename them according to sample names while copying. Then, sort and index all the bam files using samtools. Sort command will output a sorted alignment files based on chromosomal location. The samtools index command will create and index file for the sorted bam file. This index is used by other tools like bedtools to quickly extract the information.
$ mkdir CrypSpliceAnalysis $ cp MT1/Tophat_Out/accepted_hits.sorted.bam CrypSpliceAnalysis/MT1.bam $ cp MT1/Tophat_Out/junctions.bed CrypSpliceAnalysis/MT1.bed $ samtools sort CrypSpliceAnalysis/MT1.bam CrypSpliceAnalysis/MT1.sorted $ samtools index CrypSpliceAnalysis/MT1.sorted.bam
Repeat these commands for other five samples, MT2, MT3, WT1, WT2, WT3 by changing the directory and file names accordingly.
Step 2. Running CrypSplice
Next, run CrypSplice on sorted and indexed bam and junction files with user desired parameters.
# Download genome annotation files from http://www.liuzlab.org/CrypSplice/ to the # directory CrypSpliceAnalysi and uzip the downloaded file. $ cd CrypSpliceAnalysis $ python CrypSplice.py –C WT1.bed,WT2.bed,WT3.bed –T MT1.bed,MT2.bed,MT3.bed –G MM10 –F 10 –M 0.95 –P 6
Input junction files should be indicated in comma-separated list followed by parameters –C and –T for control and treatment samples respectively. Known alternative splicing events, gene model and known junction database files (files for model organisms available from http://www.liuzlab.org/CrypSplice/) should be stored in the genome directory specified by –G. Noise filter –F is used to indicate that any junction less than this threshold is considered noise and will be discarded from analysis. Junction match –M followed by a floating value ranging from 0 to 1 specifies the minimum fractional overlap to filter out known junctions. Parallel processes –P specifies the number of processing core used to run CrypSplice (one core per sample is recommended). More details on how to choose appropriate parameters for CrypSplice will be discussed in the critical parameters section below. In brief, CrypSplice first filters out noisy and all annotated junctions provided in genome model directory from the junction files provided. Then, 5’ exon coverage is computed for every junction. Junction counts and respective exon counts are used to test for statistical significance using a beta binomial model. Finally, every cryptic junction is adjusted for multiple testing and classified into three categories: junction gains, junction losses and differential junctions.
Step 3. Filtering and visualizing results
CrypSplice outputs results to three tab-delimited text files. Junction gains: Cryptic junctions observed only in treatment samples. Junction loses: Cryptic junctions observed only in control samples. Differential junctions: Cryptic Junctions observed in both control and treatment samples but with different expression strengths. Statistically significant results can be filtered using an adjusted p-value cutoff. Finally, junctions can be visualized in Integrative Genomics Viewer (IGV) with the following steps: (i) Choose appropriate genome, mm10 for this example. (ii) Load bam files to IGV using “File” load option. (iii) Paste junction coordinates of interest to visualize junction reads (Fig. 6a). (iv) Right click on any track to open a menu and choose sashimi plot. Sashimi plot is an intuitive junction visualization graph showing both exon coverage and junction connections. Example sashimi plots from the demo data are shown in Fig. 6b.
Figure 6: Visualizing splicing events in IGV:

(a) Screen shot showing exon coverage and (b) Sashimi plots with black arrows pointing to junction gains and red arrows pointing to junction losses.
COMMENTARY
Background Information
Recently, more studies had shown the important connections of cryptic splice sites to various human diseases, ranging from cancer to neurological disease such as amyotrophic lateral sclerosis. Despite of huge interest from the transcriptomic communities, we remains lack of a robust computational approach to identify cryptic splice sites from RNA-seq data for the past decade. CrypSplice is the first computational tool focusing exclusively on identifying cryptic splice sites from RNA-seq data. It uses a beta-binomial model to effectively handle both inter- and intra-sample variance. Its merit is apparent from the large number of TDP-43-dependent splicing defects identified that were not previously discovered (Tan, Yalamanchili et al. 2016). Supporting experimental validations also conforms the practical usability of the method.
Critical Parameters
Noise threshold (-F): Noisy alignments are unavoidable in any high-throughput sequencing data. However, these noisy alignments should not be recognized as cryptic events. CrypSplice uses a user defined noise threshold to filter out these noisy alignments. By default, a cutoff of 10 reads is used to filter out noisy junctions. However, it is recommended to choose this cutoff as a function of sequencing depth, with large cutoff for higher coverage and vice versa.
Junction Match (-M): CrypSplice filters out all known junction from that are observed to detect cryptic sites. Any minor mapping errors could result in putative cryptic sites. To account for such mapping shifts, CrypSplice allows a junction match threshold. Any two junctions falling within the cutoff are collapsed to one. However, if the threshold is weak, the potential novel junction could be collapsed. It is recommended to choose this cutoff as a function of anchor length used to map junction reads (0.95 is used in this protocol). Longer anchor length should use a higher cutoff.
Troubleshooting
CrypSplice is mainly dependent on bedtools and ibb package. Any changes or upgrades in these packages can potentially cause problems. In case of any issues, downgrading to original reported versions of bedtools (V2.17) and ibb package (13.06) can help. Alternatively, one can edit bedtools intersect and bedtools coverage commands accordingly in the file “CrypSplice.py”.
GUIDELINES FOR UNDERSTANDING RESULTS
Upon successful execution of the steps described in the protocols above, one would obtain a list of candidates that are dysregulated at the gene, isoform and junction levels. With the candidate list, users could further investigate the regulatory mechanisms that are disturbed due to the experiment, for example, from a functional analysis on the candidate genes, gene set enrichment analysis, etc (Pathan, Keerthikumar et al. 2015).
Although the pipeline was successfully executed, problems with the analysis could still exist. A few common red flags could be checked while interpreting the results:
Poor quality of raw sequencing reads could be detected from the FastQC step (Step 1 of the basic protocol 1). If the FastQC reports failures on many metrics, the quality of the library being sequenced might have been compromised.
Poor overall mappability of the samples. Overall mappability of a typical pair-end RNA-Seq data is 80% or higher. Quality of the sample and library will lead to variation in mappability. For example, degraded sample such as frozen tissue, degraded RNA, library with high GC contents will pose a challenge in mapping and lead to lower mappability.
Discordant mappability of 10% or higher. This hints that a large number of the pair-end reads were not mapped according to the expected pair-reads assumptions, such as the insertion size and paring of the input reads. In this case, users should make sure the input files are in correct order and the reads in the files are in pair at the same order. In addition, users could tune the insertion size parameter (-r parameter of tophat2) to obtain better mappability.
Outlier sample(s) observed from PCA plot and correlation heatmap. If a sample was not clustered to the other samples of the same genotype from both plots, it could be considered as outlier. Users could remove the sample(s), re-normalized the counts, and plot the PCA and correlation heatmaps again to assess the sample clusters. Due to limited number of samples in most experiments (less than five samples per group), the outlier detection with PCA and heatmap should be taken cautiously. In addition, users should assess the sample clustering on other factors such as batch, library preparation, gender of the mice, types of tissues, and others to check if the samples cluster per other factor instead of the designed treatment.
Differentially expressed genes and isoforms can be filtered and ranked based on adjusted P-value and fold change filters respectively. Adjusted P-value ≤ 0.05 and fold change ≥ 1.5 (at least 50% change) should suffice majority of the studies. Fold change cutoff can be adjusted according to the impact of treatment, smaller value to capture subtle changes and vice versa.
Huge differences in isoform usage are attractive. However, it is strongly recommended to cross check the estimated isoform proportions with actual mapping using IGV. The EM (expectation maximization) procedure of isoform quantification may introduce some bias.
Cryptic junction changes are classified into junction gains, junction losses and differential junctions as described in step 3 of protocol 3. Two consecutive junction gains between any two consecutive exons indicate cryptic exon inclusion. On the other hand, a loss of junction suggests an intron retention. A single junction gain corresponds to an alternative start/stop according to the strand orientation. Though junctions can be ranked based on relative strength (Junction score) always the absolute baseline should also be considered in selecting top hits. High relative junction strength (Junction score) need not guarantee high impact/cryptic activity.
Supplementary Material
ACKNOWLEDGEMENT
This work is partially supported by NSF [DMS 1263932], CPRIT [RP170387], NIH [ R01 GM120033], Houston Endowment, Huffington Foundation, Belfer Foundation. Y.W is also supported by NIH [5R01HD079442-02, and 1RF1AG054111-01]
LITERATURE CITED
- Anders S, Pyl PT and Huber W (2015). “HTSeq--a Python framework to work with high-throughput sequencing data.” Bioinformatics 31(2): 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P and Pachter L (2016). “Near-optimal probabilistic RNA-seq quantification.” Nat Biotechnol 34(5): 525–527. [DOI] [PubMed] [Google Scholar]
- Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, Szczesniak MW, Gaffney DJ, Elo LL, Zhang X and Mortazavi A (2016). “A survey of best practices for RNA-seq data analysis.” Genome Biol 17: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou T, Xu J, Gao Y, Gu J, Ji C, Xie Y and Zhou Y (2010). “Evolution of peroxisome proliferator-activated receptor gamma alternative splicing.” Front Biosci (Elite Ed) 2: 1334–1343. [DOI] [PubMed] [Google Scholar]
- Drăghici S (2012). Statistics and data analysis for microarrays using R and Bioconductor Boca Raton, FL, CRC Press. [Google Scholar]
- Garcia-Blanco MA, Baraniak AP and Lasda EL (2004). “Alternative splicing in disease and therapy.” Nat Biotechnol 22(5): 535–546. [DOI] [PubMed] [Google Scholar]
- Gene Ontology C (2015). “Gene Ontology Consortium: going forward.” Nucleic Acids Res 43(Database issue): D1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MR (1986). “Pre-mRNA splicing.” Annu Rev Genet 20: 671–708. [DOI] [PubMed] [Google Scholar]
- Jin H, Wan YW and Liu Z (2017). “Comprehensive evaluation of RNA-seq quantification methods for linearity.” BMC Bioinformatics 18(Suppl 4): 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapustin Y, Chan E, Sarkar R, Wong F, Vorechovsky I, Winston RM, Tatusova T and Dibb NJ (2011). “Cryptic splice sites and split genes.” Nucleic Acids Res 39(14): 5837–5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R and Salzberg SL (2013). “TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions.” Genome Biol 14(4): R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin JZ, Yassour M, Adiconis X, Nusbaum C, Thompson DA, Friedman N, Gnirke A and Regev A (2010). “Comprehensive comparative analysis of strand-specific RNA sequencing methods.” Nat Methods 7(9): 709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R and Genome S Project Data Processing (2009). “The Sequence Alignment/Map format and SAMtools.” Bioinformatics 25(16): 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling JP, Pletnikova O, Troncoso JC and Wong PC (2015). “TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD.” Science 349(6248): 650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, O’Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH and Ecker JR (2008). “Highly integrated single-base resolution maps of the epigenome in Arabidopsis.” Cell 133(3): 523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo YH, Chung E, Li Z, Wan YW, Mahe MM, Chen MS, Noah TK, Bell KN, Yalamanchili HK, Klisch TJ, Liu Z, Park JS and Shroyer NF (2017). “Transcriptional Regulation by ATOH1 and its Target SPDEF in the Intestine.” Cell Mol Gastroenterol Hepatol 3(1): 51–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W and Anders S (2014). “Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2.” Genome Biol 15(12): 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon R and Omenn GS (2010). “Proteomic characterization of novel alternative splice variant proteins in human epidermal growth factor receptor 2/neu-induced breast cancers.” Cancer Res 70(9): 3440–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L and Wold B (2008). “Mapping and quantifying mammalian transcriptomes by RNA-Seq.” Nat Methods 5(7): 621–628. [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M and Snyder M (2008). “The transcriptional landscape of the yeast genome defined by RNA sequencing.” Science 320(5881): 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu L, Huang W, Umbach DM and Li L (2014). “IUTA: a tool for effectively detecting differential isoform usage from RNA-Seq data.” BMC Genomics 15: 862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padgett RA, Grabowski PJ, Konarska MM, Seiler S and Sharp PA (1986). “Splicing of messenger RNA precursors.” Annu Rev Biochem 55: 1119–1150. [DOI] [PubMed] [Google Scholar]
- Pathan M, Keerthikumar S, Ang CS, Gangoda L, Quek CY, Williamson NA, Mouradov D, Sieber OM, Simpson RJ, Salim A, Bacic A, Hill AF, Stroud DA, Ryan MT, Agbinya JI, Mariadason JM, Burgess AW and Mathivanan S (2015). “FunRich: An open access standalone functional enrichment and interaction network analysis tool.” Proteomics 15(15): 2597–2601. [DOI] [PubMed] [Google Scholar]
- Pham TV, Piersma SR, Warmoes M and Jimenez CR (2010). “On the beta-binomial model for analysis of spectral count data in label-free tandem mass spectrometry-based proteomics.” Bioinformatics 26(3): 363–369. [DOI] [PubMed] [Google Scholar]
- Quinlan AR and Hall IM (2010). “BEDTools: a flexible suite of utilities for comparing genomic features.” Bioinformatics 26(6): 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztainberg Y, Chen HM, Swann JW, Hao S, Tang B, Wu Z, Tang J, Wan YW, Liu Z, Rigo F and Zoghbi HY (2015). “Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligonucleotides.” Nature 528(7580): 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Q, Yalamanchili HK, Park J, De Maio A, Lu HC, Wan YW, White JJ, Bondar VV, Sayegh LS, Liu X, Gao Y, Sillitoe RV, Orr HT, Liu Z and Zoghbi HY (2016). “Extensive cryptic splicing upon loss of RBM17 and TDP43 in neurodegeneration models.” Hum Mol Genet [DOI] [PMC free article] [PubMed]
- Thorvaldsdottir H, Robinson JT and Mesirov JP (2013). “Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration.” Brief Bioinform 14(2): 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GS and Cooper TA (2007). “Splicing in disease: disruption of the splicing code and the decoding machinery.” Nat Rev Genet 8(10): 749–761. [DOI] [PubMed] [Google Scholar]
- Yalamanchili HK, Li Z, Wang P, Wong MP, Yao J and Wang J (2014). “SpliceNet: recovering splicing isoform-specific differential gene networks from RNA-Seq data of normal and diseased samples.” Nucleic Acids Res 42(15): e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalamanchili HK, Xiao QW and Wang J (2012). “A novel neural response algorithm for protein function prediction.” BMC Syst Biol 6 Suppl 1: S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalamanchili HK, Yan B, Li MJ, Qin J, Zhao Z, Chin FY and Wang J (2014). “DDGni: Dynamic delay gene-network inference from high-temporal data using gapped local alignment.” Bioinformatics 30(3): 377–383. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.