

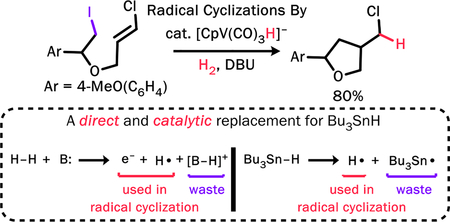
Abstract
Radical cyclizations are most often achieved with Bu3SnH in the presence of a radical initiator, but environmental considerations demand that alternative reagents be developed – ones that can serve as a synthetic equivalent to the hydrogen atom. We have revisited [CpV(CO)3H]−, a known replacement for Bu3SnH, and found that it can be used catalytically under H2 in the presence of a base. We have carried out the tin-free and catalytic radical cyclization of alkyl iodide substrates. The reactions are atom-efficient and the conditions are mild, with broad tolerance for functional groups. We have, for example, achieved the first 5-exoradical cyclization involving attack onto a vinyl chloride. We suggest that the radicals are generated by an initial electron transfer.
Graphical Abstract
Authors are required to submit a graphic entry for the Table of Contents (TOC) that, in conjunction with the manuscript title, should give the reader a representative idea of one of the following: A key structure, reaction, equation, concept, or theorem, etc., that is discussed in the manuscript. Consult the journal’s Instructions for Authors for TOC graphic specifications.
Tin hydrides such as Bu3SnH are frequently used to reduce organic halides R-X to the corresponding hydrocarbons R-H. These reactions occur by the radical chain mechanism in eqs 1–5, and thus require stoichiometric amounts of the tin hydride. Both the resulting Bu3SnX and any leftover Bu3SnH are toxic and difficult to remove.1 The few reactions that use tin catalytically rely on a strong hydride donor to reduce the Bu3SnX back to Bu3SnH, reducing functional-group tolerance.2
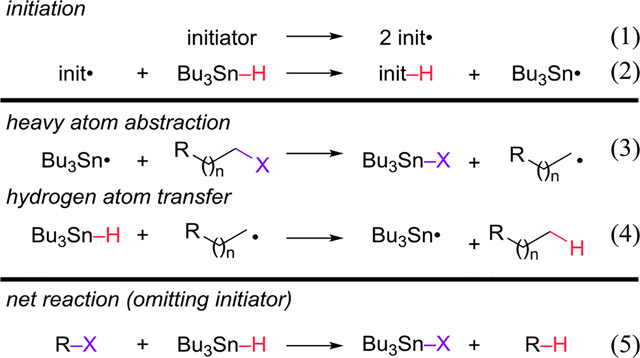 |
Transition-metal hydrides are also potent H• donors. Their M-H bonds are often weaker3 than the Sn-H bond of Bu3SnH (78 kcal/mol),4 allowing them to transfer H• to substrates that do not react with the latter. For example, transition-metal hydrides can generate radicals by H• transfer to activated alkenes and alkynes.5 The resulting M• can be converted back to M-H by H2,6 making these radical-generating reactions catalytic, with near-perfect atom economy.7 H• transfer reactions are finding synthetic applications;8 the Snyder group recently found that H• transfer from a metal hydride was the only practical way of hydrogenating a key enone in the synthesis of exochomine.9
Still needed, however, is a replacement for Bu3SnH in the reduction of R-X. Transition-metal hydrides are known to reduce alkyl halides,10 but in general these reactions are stoichiometric, have limited functional group tolerance, or use harsh reductants to make the reaction catalytic. Hydrides that can be regenerated from H2 are a more atom efficient approach for catalyzing the reduction of R-X.
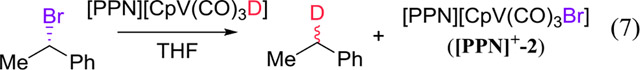
A hydride known to generate R• stoichiometrically from R-X (eq 6) is the [CpV(CO)3 H]− anion (1), reported by Kinney, Jones, and Bergman by 1978.11 The loss of stereochemistry that is observed when 1 reacts with α-bromoethylbenzene (eq 7) is consistent with a radical intermediate.
 |
A series of observations in the literature imply that 1 can be regenerated from [CpV(CO)3Br]− (2) under H2. Dissociation of Br− from 2 should generate the coordinatively unsaturated CpV(CO)3 (3),12 which can be trapped by many two-electron donors to generate complexes like CpV(CO)3(PMe)3 (4a), CpV(CO)3THF (4b),13 and CpV(CO)3MeCN (4c).11a Similarly, 3 should coordinate H2 to generate the dihydrogen complex CpV(CO)3(H2) (5),14 which will regenerate the hydride anion [CpV(CO)3H]− (1) if the H2 ligand were deprotonated. This sequence (Scheme 1) would enable the catalytic hydrodehalogenation of alkyl halides.
Scheme 1.

Reactions Relevant to Catalysis Under H2
H2 must, however, compete with other L (including polar solvents) for the vacant coordination site of 3. We have therefore studied the equilibrium between CpV(CO)3L and CpV(CO)3(H2) (eq 10). The THF adduct of 3 (4b) has been generated by removal of H− from 1,15 and the MeCN adduct 4c has been made by protonating 1 in CH3CN.16 We have, however, found that 4c can be made more easily by oxidizing 1 with Cp*2Fe+ (eq 8).17
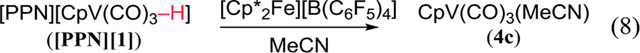 |
Eq 10 must lie far to the left, as the 1H NMR of 4c18 does not change under 1 atm of H2. However, sufficiently strong base pulls 4c through 5 to the hydride anion 1. There is no reaction between 4c and H2 in the presence of Et3N, but DBU gives 1 (by 1H NMR) after several h (eq 9, 10). These experiments imply that the pKa of 5 lies between 18 and 25 in MeCN.
 |
Knowing that each step in Scheme 1 was practical, we tried 119 as a catalyst for the reduction of alkyl bromides (vide infra) and alkyl iodides in the presence of DBU and H2. For alkyl iodide substrate 6, we observed a catalytic hydrodehalogenation20 to generate 7 in MeCN, but the reaction worked better in the less-coordinating solvent THF (Table 1). A higher H2 pressure and an increased concentration of DBU led to a quantitative yield of 7.
Table 1.
Brief optimization of Radical Deiodination Conditions

| entry | cat. loading | H2 | DBU | Solventa | SM:Pdt |
|---|---|---|---|---|---|
| 1 | 10% | 75 psig | 1 eq. | MeCN | 56: 44 |
| 2 | 10% | 75 psig | 1 eq. | THF | 40: 60 |
| 3 | 10% | 75 psig | 1 eq. | Ether | 71 : 29 |
| 4 | 10% | 75 psig | 2 eq. | THF | 30: 70 |
| 5 | 15% | 90 psig | 2 eq. | THF | 05: >95b |
All reactions run overnight at r.t.
Concentration: 50 mM in substrate
isolated yield
Since the hydrodehalogenation reactions of 1 proceed by radical intermediates, appropriate organic substrates should undergo radical cyclization reactions. Indeed, 1 catalyzes the cyclization of 8 with DBU under H2 (Table 2). There is no catalysis if the 1 (entry 1), H2 (entry 2), or base (entry 7) is omitted. A background elimination reaction between 8 and DBU results in 9 or its hydrolysis product 10 (entry 10). Optimizing reaction conditions has allowed us to obtain a quantitative yield of the cyclized product 11 (entry 9).
Table 2.
Redical Cyclization Optimization
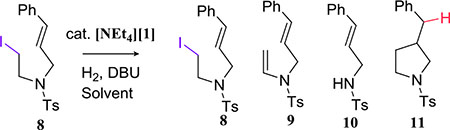
| entry | cat. loading | H2 | DBU | solventa | 8 : 9 : 10 : 11c |
|---|---|---|---|---|---|
| 1 | 0% | 90 psig | 2 eq. | THF | 60 : nd : 40 : nd |
| 2 | 15% | 0 psig | 2 eq. | THF | 60 : 24 : nd : 16 |
| 3 | 15% | 15 psig | 2 eq. | THF | 58 : 13 : nd : 30 |
| 4 | 15% | 40 psig | 2 eq. | THF | 54 : nd : 13 : 34 |
| 5 | 15% | 90 psig | 2 eq. | THF | nd : 10 : nd : 90 |
| 6 | 15% | 90 psig | 1 eq. | THF | 37 : nd : nd : 63 |
| 7 | 15% | 90 psig | 0 eq. | THF | 86 : nd : nd : 14 |
| 8 | 15% | 90 psig | 2 eq. | MeCN | 24 : 42 : nd : 34 |
| 9 | 15% | 90 psig | 2 eq. | THF (dil.)b | nd : nd : nd : 100 |
| 10 | 0% | 0 psig | 2 eq. | THF | 61 : nd : 39 : nd |
All reactions run overnight at r.t.
Concentration: 50 mM in substrate
Concentration: 10 mM in substrate
Given as percentages; all crude yields were within 5% expected mass, ratios determined by 1H NMR analysis
With the optimized conditions from Table 2, we examined substrate scope (Table 3). The first three entries involve substrates for which radical cyclizations have previously been reported; catalysis by 1 with DBU/H2 generates the products with similar yields and selectivity. The yield is lower in the 6-exo case in entry 3, presumably because the cyclization is slow enough that the H• transfer is competitive. The functional group tolerance is quite good: the free alcohol in entry 4 is not a problem. Polyhalogenated substrates react selectively at the iodide (for example, the aryl bromide in entry 5 remains unchanged). Entry 6 is the first 5-exo cyclization of an alkyl radical21 onto a chlorinated double bond; Sharp and Zard have observed cyclizations promoted by Bu3SnH result in the dechlorination of such substrates.22
Table 3.
Radical Cyclization Substrate Scope
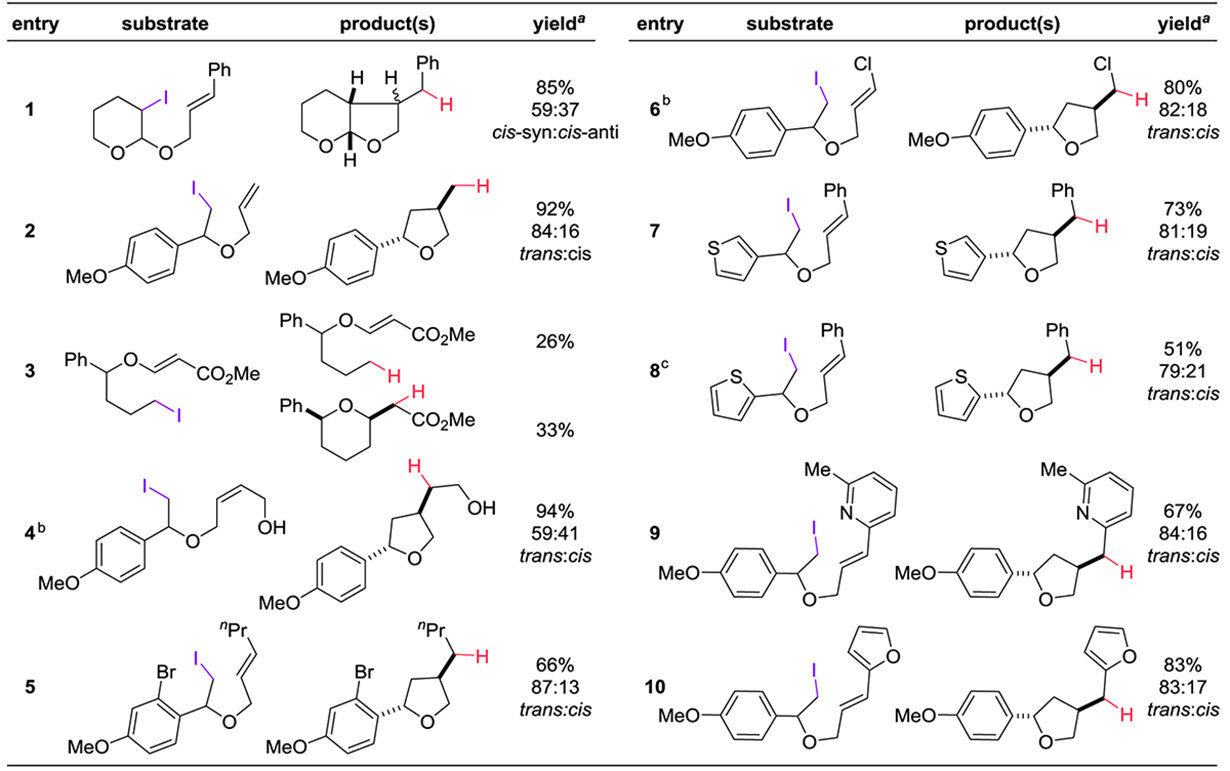
|
Reaction conditions: 1(15 mol%), DBU(2 equiv), 90 psig H2, 10 mM in substrate at r.t. overnight.
Yields refer to isolated yields.
In these cases, the remaining mass balance was isolated starting material.
Also isolated was 41% of 2-acetylthiophene (from a background elimination reaction).
We have encountered few difficulties when cyclizing substrates containing heterocyclic moieties. Both 3-substituted and 2-substituted thiophenes (entries 7 and 8) generate the corresponding tetrahydrofuranyl thiophenes. The elimination product in entry 8 (see Table 3, footnote c) is presumably the result of its more acidic C-H bond.23 Pyridyl- and furanyl-substituted alkenes (entries 9 and 10) generate other polycyclic products in good yield.
This reaction (catalytic cyclization by [CpV(CO)3H]−) compares favorably to the corresponding reactions with Bu3SnH. We generate only BH+ ([DBU•HI] precipitates from solution, and can be removed by a filtration through a silica plug); Bu3SnH generates an equivalent of Bu3Sn• waste, which is difficult to remove. Our reaction are done at room temperature, whereas reactions with Bu3SnH must be run at elevated temperatures to initiate a radical chain.
Table 4 shows substrates that [CpV(CO)3H]− does not process effectively. Aryl iodides (entry 1), tertiary alkyl iodides (entry 3), and alkyl bromides (entry 2) only undergo partial conversion under catalytic conditions, although these reactions work when the hydride anion is used stoichiometrically. The catalyst decomposes before the substrate can be completely converted to product.
Table 4.
Substrates That Work Poorly with the Catalytic System
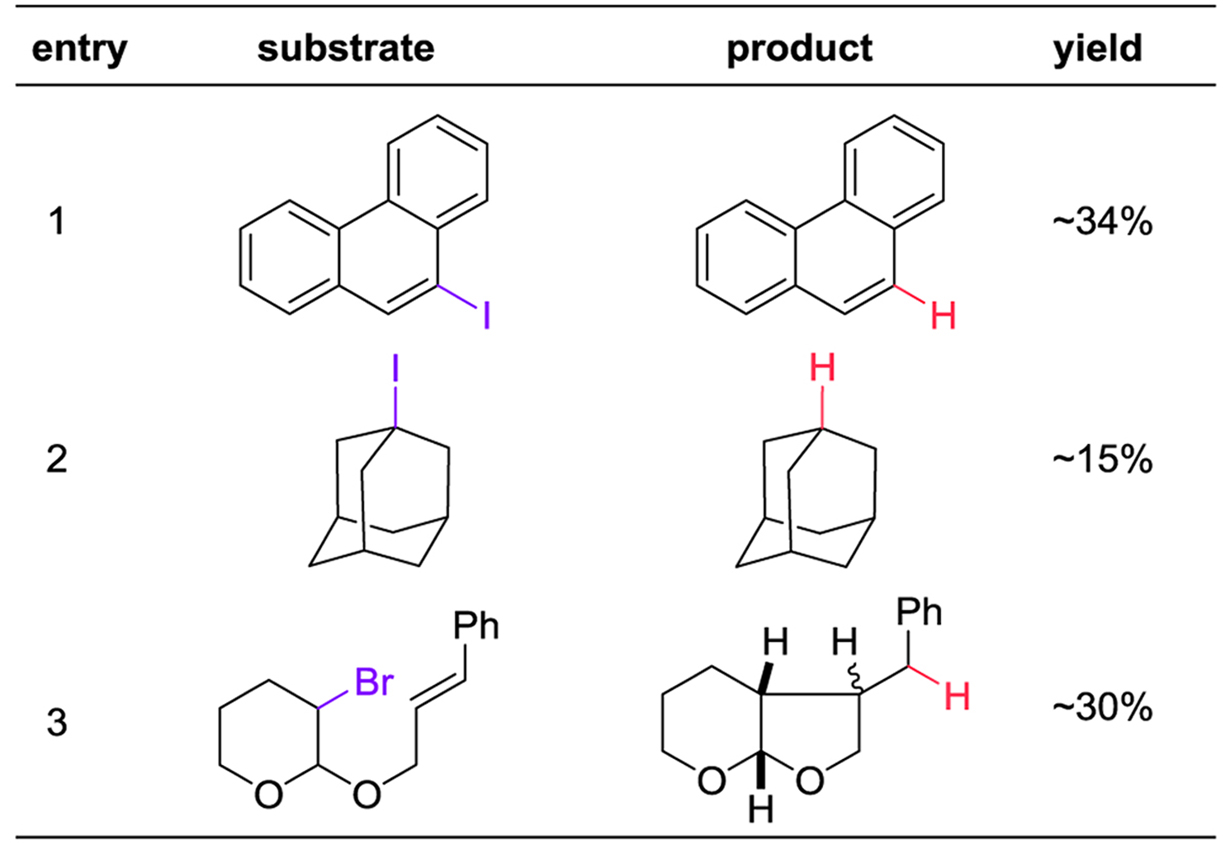
|
Reaction conditions as in table 3. The remaining mass balance was recovered starting material
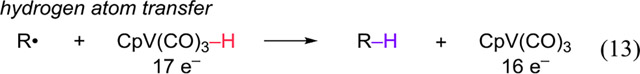
Our observations suggest that the mechanism of the reaction between alkyl halides and [CpV(CO)3H]− should be reexamined. Kinney, Jones, and Bergman believed that the reactions occurred by a radical chain mechanism like that in eqs 1–5, and that the vanadium hydride was a better hydrogen atom donor than Bu3SnH. However, they also considered the possibility that the reaction of R-X and 1 began by an electron transfer (eqs 11–13). Rates of electron transfer to R-X vary in the order R-I > R-Br > R-Cl,24 and alkyl halides > aryl halides. These trends match what is observed in Tables 3 and 4, suggesting that the radicals are generated by an electron transfer. A fast electron transfer enables the complete conversion of substrate before complete catalyst decomposition. Electron transfers from low valent transition metal complexes have ample precedent for generating organic radicals,25 and an analogous electron transfer mechanism has been reported for the reaction of 1 with metal carbonyls.11c We thus propose the overall catalytic cycle depicted in Scheme 2. The isolation of the hydrodehalogenation product in Table 2, Entry 3 suggests that the vanadium hydride may not be as effective a hydrogen atom donor as is presently believed.
Scheme 2.

Proposed Catalytic Cycle with [CpV(CO)3H]−
 |
 |
 |
There have been previous reports — some catalytic — of the initiation of radical cyclizations by electron transfer (for example with tetra(dimethylamino)ethylene,26 Ni(0) with Zn,10a or Fe(CO)5 with phenanthroline,10d). The present system is not only catalytic but uses no reducing agent stronger than H The [CpV(CO)3H]− system, with H2 as a chemical fuel, may be seen as an alternative to photo-redox catalysis27 for the generation of reducing intermediates
Supplementary Material
ACKNOWLEDGMENT
We thank Robert G. Bergman and William D. Jones for helpful discussions (and for their prior studies on [CpV(CO)3H]−). Scott A. Snyder, Derek A. Pratt, and Andreas Gansäuer are acknowledged for helpful discussions. J.L.K. acknowledges the National Science Foundation and the Arun Guthikonda Memorial Fellowship for financial support, and J.M.A. acknowledges the Bernice G. Segal Summer Internship and the Barnard College Department of Chemistry for financial support. Research reported in this publication was supported by National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health under award number R01GM124295.
Footnotes
Supporting Information
The Supporting Information (synthetic procedures for the substrate/catalyst, a general procedure for the radical cyclization, characterization data (1H- and 13C-NMR, tabulated IR and HRMS data) is available free of charge on the ACS Publications website.
The authors declare no competing financial interests.
REFERENCES
- 1.(a) Breslow R; Light J Tetrahedron Lett. 1990, 31, 2957–2958; [Google Scholar]; (b) Baguley PA; Walton JC Angew. Chem., Int. Ed. Engl 1998, 37, 3072–3082; [DOI] [PubMed] [Google Scholar]; (c) Studer A; Amrein S Synthesis. 2002, 835–849; [Google Scholar]; (d) Clive DLJ; Wang J J. Org. Chem 2002, 67, 1192–1198; [DOI] [PubMed] [Google Scholar]; (e) Stien D; Gastaldi S J. Org. Chem 2004, 69, 4464–4470; [DOI] [PubMed] [Google Scholar]; (f) Le Grognec E; Chrétien J-M; Zammattio F; Quintard J-P Chem. Rev 2015, 115, 10207–10260. [DOI] [PubMed] [Google Scholar]
- 2.(a) Tormo J; Hays DS; Fu GC J. Org. Chem 1998, 63, 5296–5297; [DOI] [PubMed] [Google Scholar]; (b) Hays DS; Fu GC Tetrahedron. 1999, 55, 8815–8832; [Google Scholar]; (c) Corey EJ; Suggs JW J. Org. Chem 1975, 40, 2554–2555; [Google Scholar]; (d) Stork G; Sher PM J. Am. Chem. Soc 1986, 108, 303–304. [Google Scholar]
- 3.(a) Choi J; Pulling ME; Smith DM; Norton JR J. Am. Chem. Soc 2008, 130, 4250–4252; [DOI] [PubMed] [Google Scholar]; (b) Li G; Han A; Pulling ME; Estes DP; Norton JR J. Am. Chem. Soc 2012, 134, 14662–14665; [DOI] [PubMed] [Google Scholar]; (c) Estes DP; Grills DC; Norton JR J. Am. Chem. Soc 2014, 136, 17362–17365. [DOI] [PubMed] [Google Scholar]
- 4.Laarhoven LJJ; Mulder P; Wayner DDM Acc. Chem. Res 1999, 32, 342–349. [Google Scholar]
- 5.(a) Smith DM; Pulling ME; Norton JR J. Am. Chem. Soc 2007, 129, 770–771; [DOI] [PubMed] [Google Scholar]; (b) Kuo JL; Hartung J; Han A; Norton JR J. Am. Chem. Soc 2015, 137, 1036–1039; [DOI] [PubMed] [Google Scholar]; (c) Li G; Kuo JL; Han A; Abuyuan JM; Young LC; Norton JR; Palmer JH J. Am. Chem. Soc 2016, 138, 7698–7704; [DOI] [PubMed] [Google Scholar]; (d) Estes DP; Norton JR; Jockusch S; Sattler W J. Am. Chem. Soc 2012, 134, 15512–15518. [DOI] [PubMed] [Google Scholar]
- 6.Norton JR; Spataru T; Camaoni D; Lee S-J; Li G; Choi J; Franz JA Organometallics. 2014, 33, 2496–2502. [Google Scholar]
- 7.(a) Anastas PT; Warner JC, Green Chemistry: Theory and Practice. Oxford University Press: New York, 1998; [Google Scholar]; (b) Laird T Org. Process Res. Dev 2012, 16, 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Hoffmann RW Chem. Soc. Rev 2016, 45, 577–583; [DOI] [PubMed] [Google Scholar]; (b) Iwasaki K; Wan KK; Oppedisano A; Crossley SWM; Shenvi RA J. Am. Chem. Soc 2014, 136, 1300–1303; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Crossley SW; Barabé F; Shenvi RA J. Am. Chem. Soc 2014, 136, 16788–16791; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Obradors C; Martinez RM; Shenvi RA J. Am. Chem. Soc 2016, 138, 4962–4971; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Green SA; Matos JL; Yagi A; Shenvi RA J. Am. Chem. Soc 2016, 138, 12779–12782; [DOI] [PubMed] [Google Scholar]; (f) King SM; Ma X; Herzon SB J. Am. Chem. Soc 2014, 136, 6884–6887; [DOI] [PubMed] [Google Scholar]; (g) Ma X; Herzon SB Chem. Sci 2015, 6, 6250–6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao AX; Hamada T; Snyder SA Angew. Chem. Int. Ed 2016, 55, 10301–10306. [DOI] [PubMed] [Google Scholar]
- 10.(a) Kim H; Lee C Org. Lett 2011, 13, 2050–2053; [DOI] [PubMed] [Google Scholar]; (b) Ekomie A; Lefevre G; Fensterbank L; Lacote E; Malacria M; Ollivier C; Jutand A Angew. Chem., Int. Ed 2012, 51, 6942–6946; [DOI] [PubMed] [Google Scholar]; (c) Rettenmeier C; Wadepohl H; Gade LH Chem. Eur. J 2014, 20, 9657–9665; [DOI] [PubMed] [Google Scholar]; (d) Hwang JY; Baek JH; Shin TI; Shin JH; Oh JW; Kim KP; You Y; Kang EJ Org. Lett 2016, 18, 4900–4903; [DOI] [PubMed] [Google Scholar]; (e) Iwamoto H; Akiyama S; Hayama K; Ito H Org. Lett 2017, 19, 2614–2617; [DOI] [PubMed] [Google Scholar]; (f) Haibach MC; Stoltz BM; Grubbs RH Angew. Chem., Int. Ed 2017, 56, 15123–15126; [DOI] [PubMed] [Google Scholar]; (g) Kyne SH; Clémancey M; Blondin G; Derat E; Fensterbank L; Jutand A; Lefèvre G; Ollivier C Organometallics. 2017, 37, 761–771; [Google Scholar]; (h) Wang Z; Li X; Sun H; Fuhr O; Fenske D Organometallics. 2018, 37, 539–544. [Google Scholar]
- 11.(a) Kinney RJ; Jones WD; Bergman RG J. Am. Chem. Soc 1978, 100, 7902–7915; [Google Scholar]; (b) Kinney RJ; Jones WD; Bergman RG J. Am. Chem. Soc 1978, 100, 635–637; [Google Scholar]; (c) Jones WD; Huggins JM; Bergman RG J. Am. Chem. Soc 1981, 103, 4415–4423. [Google Scholar]
- 12.Jones WD Reactivity Patterns of Transition Metal Hydrides and Alkyls. Ph.D Thesis, California Institute of Technology, Pasadena, CA, 1979. (from the research group of Prof R. G. Bergman) [Google Scholar]
- 13.Hoch M; Rehder DJ Organomet. Chem 1985, 288, C25–C29. [Google Scholar]
- 14.George MW; Haward MT; Hamley PA; Hughes C; Johnson FPA; Popov VK; Poliakoff MJ Am. Chem. Soc 1993, 115, 2286–2299. [Google Scholar]
- 15.In ref. 13, CpV(CO)3(THF) (4b) was generated by mixing [Et4N][CpV(CO)3H] with [Ph3C][BF4] in THF, presumably via the coordinatively unsaturated CpV(CO)3. We tried the analogous reaction in MeCN to generate CpV(CO)3(MeCN) (4c) using MeOTf in place of [Ph3C][BF4], but found that the resulting solutions decompose in a few minutes at room temperature.
- 16.In ref 11a, Bergman showed that [CpV(CO)3H]− (1) can be mixed with TfOH to generate CpV(CO)3MeCN (4c). This presumably first generates the dihydrogen complex CpV(CO)3(H2) (5), which loses H2. We reacted 1 with the more mild acid [Et3NH][BF4] to generate 4c, but observed its decomposition over a few hours at room temperature.
- 17.In ref 11a, Bergman reported that mixing 1 with AgCl generates the solvate complex 4c. We speculated that this occurs by an initial electron transfer, and so we tried the more mild single-electron oxidant [Cp*2Fe][B(C6F5)4].
- 18.It has been said that 4b and 4c are thermally unstable at room temperature (ref 13). However, we have found that solutions of CpV(CO)4 in MeCN undergo conversion to CpV(CO)3MeCN (4c) in ambient light (31% conversion over 2977 minutes), there is no measurable decomposition of 4c at room temperature on this time scale. We suggest 4b and 4c are, in fact, stable at room temperature. Solutions of 4c generated from [Cp*2Fe][B(C6F5)4] are stable for many hours, and suitable for studying the equilibrium with H2.
- 19.During this time, we developed an improved protocol for the synthesis of [NEt4][CpV(CO)3H] (see SI). There were no differences between the reactions of the [NEt4]+ salt and the [PPN]+ salt, so the tetraethylammonium salt was used subsequently.
- 20.The importantance of green hydrodehalogenation methods is discussed further in ref 10f and 27a.
- 21.(a) Patel VF; Andis SL; Enkema JK; Johnson DA; Kennedy JH; Mohamadi F; Schultz RM; Soose DJ; Spees MM J. Org. Chem 1997, 62, 8868–8874; [Google Scholar]; (b) Boger DL; Boyce CW; Garbaccio RM; Searcey M Tetrahedron Lett. 1998, 39, 2227–2230; [Google Scholar]; (c) Stephenson MJ; Howell LA; O’Connell MA; Fox KR; Adcock C; Kingston J; Sheldrake H; Pors K; Collingwood SP; Searcey MJ Org. Chem 2015, 80, 9454–9467. [DOI] [PubMed] [Google Scholar]
- 22.Sharp LA; Zard SZ Org. Lett 2006, 8, 831–834. [DOI] [PubMed] [Google Scholar]
- 23.(a) Catalan J. Am. Chem. Soc 1984, 106, 421–422; [Google Scholar]; (b) Seino J. Am. Chem. Soc 1995, 117, 12181–12193. [Google Scholar]
- 24.(a) Ashby EC; DePriest R; Goel A; Wenderoth B; Pham TJ Org. Chem 1984, 49, 3545–3556; [Google Scholar]; (b) Ashby EC; Argyropoulos JN J. Org. Chem 1985, 50, 3274–3283; [Google Scholar]; (c) Ashby EC; Welder CO; Doctorovich F Tetrahedron Lett. 1993, 34, 7235–7238; [Google Scholar]; (d) Ashby EC; Deshpande AK J. Org. Chem 1994, 59, 3798–3805; [Google Scholar]; (e) Welder CO; Ashby EC J. Org. Chem 1997, 62, 4829–4833; [Google Scholar]; (f) Ashby EC; Welder CO J. Org. Chem 1997, 62, 3542–3551. [Google Scholar]
- 25.(a) Torii S; Inokuchi T; Yukawa TJ Org. Chem 1985, 50, 5875–5877; [Google Scholar]; (b) Pattenden G Chem. Soc. Rev 1988, 17, 361–382; [Google Scholar]; (c) Gansäuer A; Fan C-A; Piestert FJ Am. Chem. Soc 2008, 130, 6916–6917; [DOI] [PubMed] [Google Scholar]; (d) Alonso F; Beletskaya IP; Yus M Chem. Rev 2002, 102, 4009–4091. [DOI] [PubMed] [Google Scholar]
- 26.Murphy JA; Khan TA; Zhou SZ; Thomson DW; Mahesh M Angew. Chem., Int. Ed 2005, 44, 1356–1360. [DOI] [PubMed] [Google Scholar]
- 27.(a) Nguyen JD; D’Amato EM; Narayanam JMR; Stephenson CR J. Nature Chem 2012, 4, 854–859; [DOI] [PubMed] [Google Scholar]; (b) Kim H; Lee C Angew. Chem., Int. Ed 2012, 51, 12303–12306; [DOI] [PubMed] [Google Scholar]; (c) Shen YY; Cornella J; Julia-Hernandez F; Martin R Acs Catal. 2017, 7, 409–412. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.