

The ESCRT complex protein Vps4 is rapidly depleted upon nerve injury, causing autophagic impediment and axonal degeneration.
Abstract
Injured axons undergo a controlled, self-destruction process, known as Wallerian degeneration. However, the underlying mechanism remains elusive. Using the Drosophila wing nerve as a model, we identify the ESCRT component Vps4 as a previously unidentified essential gene for axonal integrity. Up-regulation of Vps4 remarkably delays degeneration of injured axons. We further reveal that Vps4 is required and sufficient to promote autophagic flux in axons and mammalian cells. Moreover, using both in vitro and in vivo models, we show that the function of Vps4 in maintaining axonal autophagy and suppressing Wallerian degeneration is conserved in mammals. Last, we uncover that Vps4 protein is rapidly depleted in injured mouse axons, which may underlie the injury-induced autophagic impediment and the subsequent axonal degeneration. Together, Vps4 and ESCRT may represent a novel signal transduction mechanism in axon injury and Wallerian degeneration.
INTRODUCTION
Wallerian degeneration (WD), the progressive self-destruction of the distal segment of injured axons, is an active process that is tightly controlled at molecular and cellular levels (1–4). The current understanding of WD centers on nicotinamide adenine dinucleotide (NAD+) and genes regulating NAD+ metabolism (5, 6). The expression of “Wallerian degeneration slow” [WldS, a chimeric gene containing the NAD+ biosynthetic enzyme nicotinamide mononucleotide adenylyltransferase (Nmnat) 1], overexpression (OE) of Nmnat, and supplement of NAD+ or its precursors can all delay WD (5, 7). Consistently, a rapid turnover of the cytoplasmic isoform Nmnat2 is linked to injury-induced NAD+ depletion (8). Last, sterile alpha and TIR motif–containing 1 (Sarm1) may act as a NAD+ hydrolase to promote axonal degeneration (9–12). However, other than NAD+ and the related regulatory mechanisms, the rest of the complex signaling pathways involved in the initiation of WD remain poorly understood.
The endosomal sorting complexes required for transport (ESCRT) machinery plays a vital role in membrane remodeling and is involved in the regulation of a number of important cellular processes such as cellular abscission, viral budding, and the biogenesis of multivesicular bodies (MVBs) (13, 14). The ESCRT machinery consists of four complexes, namely, ESCRT-0, -I, -II, and -III, as well as the ESCRT accessory proteins such as vacuolar protein sorting 4 (Vps4). The ESCRT-III complex recruits Vps4, which catalyzes the dissociation of ESCRT-III components from the endosomal membranes to recycle them as well as Vps4 itself. The repeated assembly-disassembly of the ESCRT-III and the Vps4 complex is required for maintaining the biogenesis and function of MVBs in cells (13, 15, 16).
Autophagy is a “self-eating” process of the cell that sequesters misfolded proteins and dysfunctional organelles to autophagosomes (APs) and subsequently delivers them to lysosomes for degradation. The process of AP formation is regulated by a series of evolutionarily conserved autophagy-related (Atg) genes. In addition, defects in the fusion of APs and lysosomes are associated with Vps4 mutants in worm, fly, and human cells (17–19), implicating a molecular link between Vps4 and autophagy. Emerging evidence shows that autophagy plays an important role in maintaining the homeostasis in neurons, and mutations in the genes encoding the components of the ESCRT machinery are associated with dysfunctional autophagy in neurodevelopment and chronic neurodegenerative diseases (13, 20, 21). In this study, we have identified the ESCRT protein Vps4 as an important novel player in acute neural injury, which is required for maintaining autophagic flux in axons, and its rapid depletion upon injury may underlie the initiation of WD.
RESULTS
Identification of Vps4 as an anti-degenerative gene in WD using an in vivo nerve injury model
To study the process of axonal degeneration in vivo, we utilized the Drosophila wing nerve model (22, 23) and expressed the membrane-bound mCD8–GFP (green fluorescent protein) using a dpr-Gal4 driver to evaluate the integrity of the wing axons (fig. S1). Using this model, we performed a screen of existing RNA interference (RNAi) transgenic fly lines for genes required for the maintenance of the integrity and function of axons. We found three independent RNAi lines (nos. 105977, 35125, and 35126) of the same gene, Vps4, whose knockdown (KD) led to age-dependent axonal degeneration. The three lines showed similar effects, and two of them are shown in Fig. 1 (A to C). In addition, another gene found in the screen, TSG101, is a component of the ESCRT-I complex (15), and the RNAi-TSG101 flies also caused age-dependent axonal degeneration (fig. S2, A and B), suggesting that the function of the ESCRT machinery was required for axonal integrity. To determine whether up-regulation of the two genes could provide axonal protection, we then generated the transgenic flies to overexpress them in the wing nerve. OE of Vps4 (Fig. 1, D to G) but not TSG101 (fig. S2, C to E) was sufficient to suppress injury-induced axonal degeneration; we therefore focused mainly on investigating the axonal function of Vps4 in this study.
Fig. 1. Vps4 is required for axonal integrity and its OE delays WD.
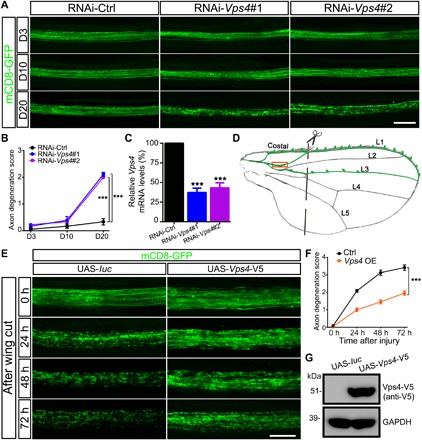
(A and B) Representative images of the wing axons labeled by mCD8-GFP of control (RNAi-Ctrl) or RNAi-Vps4 flies at indicated ages. Axonal degeneration scores are evaluated as described in fig. S1 and quantified in (B). Data shown are means ± SEM; n = 7 to 10 wings per time point per genotype; ***P < 0.001; two-way analysis of variance (ANOVA). D3, day 3; D10, day 10; D20, day 20. (C) The KD efficiency of the RNAi-Vps4 lines is analyzed by quantitative polymerase chain reaction (qPCR) and normalized to actin. Means ± SEM; n = 3; ***P < 0.001; Student’s t test. (D) A schematic drawing of the Drosophila wing, highlighting the neuronal soma and axons in the costal, L1, and L3 wing veins. A pair of scissors indicates the injury site, which completely severed all axons of the L1 nerve, and the boxed area is imaged in (E). (E and F) Representative images (E) and quantification (F) of mCD8-GFP–labeled wing axons of the control (UAS-luc) or Vps4 OE flies (UAS-Vps4-V5) at indicated time points after axotomy. UAS, upstream activation sequence. Means ± SEM; n = 10 to 12 wings per time point per genotype; ***P < 0.001; two-way ANOVA. (G) Western blotting analysis confirming the expression Vps4-V5 in the transgenic flies. Scale bars, 20 μm (A) and 10 μm (E). GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Changes in Vps4 expression critically regulate autophagy levels in axons
Vps4 is a key protein component of the ESCRT machinery, which interacts with the ESCRT-III complex to mediate membrane scission in a variety of cellular processes including MVB biogenesis (13, 15, 16). Since the ESCRT machinery and MVBs play an important role in autophagic clearance, we asked whether the effect of Vps4 KD and OE on axonal integrity and degeneration was due to a function of Vps4 in regulating axonal autophagy. To test this hypothesis, we expressed mCherry-Atg8a in the wing nerve to assess the axonal autophagy levels. Atg8a is the Drosophila homolog of the microtubule-associated protein light chain 3 (LC3), a widely used autophagy marker whose puncta are indications of APs (24). The intact wing axons showed a very low basal level of autophagy, which was markedly increased as early as 3 hours after injury. The destruction of the injured axons began much later, as no significant axonal fragmentation was detected until 12 hours after axotomy (fig. S3).
Vps4 KD in the wing nerve led to a significant increase of axonal mCherry-Atg8a puncta, which was evident at day 10 (D10) and became worse with age (Fig. 2, A and B). The RNAi-Vps4–induced increase of axonal APs occurred much earlier than the breakdown of axonal integrity, which was not detected until D20 (Fig. 1, A and B). Vps4 OE substantially reduced the levels of injury-induced autophagy in the wing axons (Fig. 2, C and D). Unlike Vps4, TSG101 OE did not have the same regulatory impact on autophagy levels in axon injury (fig. S2, D and F), which might underlie the inability of TSG101 OE to protect injured axons (fig. S2, D and E). Although OE of the known neuroprotector Nmnat, a key enzyme in the NAD+ biosynthesis pathway, delayed the breakdown of injured axons, it did not deter the injury-induced autophagy (Fig. 2, E to G). These data suggest that the function of Vps4 in regulating autophagy in axon injury was rather unique and that the increase in autophagy levels was not merely subsequent to injury-induced NAD+ depletion. Instead, the autophagy response and the regulation by Vps4 might represent another important signal transduction pathway in axon injury and WD.
Fig. 2. Up-regulation of Vps4 but not Nmnat alleviates the autophagy response in injured wing axons.
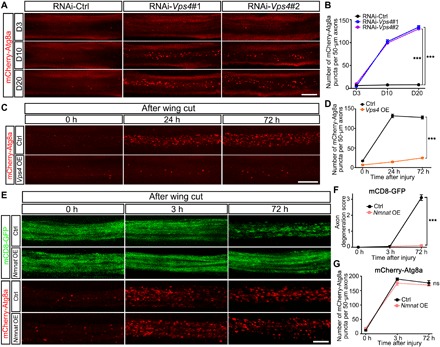
(A to D) Representative images (A and C) and quantifications (B and D) of axonal APs labeled by mCherry-Atg8a in Vps4 KD (A and B) or Vps4 OE (C and D) flies at indicated time points during aging or after injury. RNAi-Ctrl, RNAi-luc; OE Ctrl, UAS-luc; and Vps4 OE, UAS-Vps4-V5. The average numbers of mCherry-Atg8a puncta per 50 μm of axons in (A) and (C) are quantified in (B) and (D), respectively. (E to G) Representative images (E) and quantifications (F and G) of mCD8-GFP–labeled wing axons and mCherry-Atg8a–labeled AP puncta in the control (UAS-lacZ) and Nmnat OE (UAS-Nmnat) flies. Means ± SEM; n = 7 to 12 wings per time point per group; ***P < 0.001; ns, not significant; two-way ANOVA. Scale bars, 20 μm (A), 10 μm (C), and 5 μm (E).
To confirm that the observed mCherry-Atg8a puncta in the wing axons were indeed APs, we conducted RNAi KD of Atg12 or Atg17, two core genes required for AP initiation and expansion, respectively. RNAi-Atg12 or RNAi-Atg17 almost completely blocked the formation of mCherry-Atg8a puncta, and the injury-induced autophagy was suppressed (fig. S4, A and B). Although, as previously reported (25), blocking the initiation of autophagy could delay injury-induced axonal degeneration to some extent (fig. S4, C and D), the protective effect was much weaker than that of Vps4 OE: The axons of the Vps4 OE flies were largely preserved even at 72 hours after injury (Fig. 1, E and F), whereas the protection by RNAi-Atg12 or RNAi-Atg17 was only robust at 24 hours, and the axons were already much degenerated by 72 hours after injury (fig. S4, C and D). Thus, although both cause a significant reduction of injury-induced autophagy, the neuroprotection provided by Vps4 OE was much more potent than simply blocking the formation of axonal APs, suggesting that the mechanism underlying the anti-degenerative function of Vps4 was not merely to suppress the autophagy induction in axon injury.
Vps4 promotes autophagic clearance in mammalian cells
To understand how Vps4 affected autophagy, we extended the study to mammalian systems. We used small interfering RNA (siRNA) to KD Vps4B/SKD1 (the mammalian homolog of Drosophila Vps4) in mouse embryonic fibroblast (MEF) cells and assessed the autophagic flux in the cell using mTagRFP-mWasabi-LC3 (Fig. 3A). The green but not the red fluorescence of mTagRFP-mWasabi-LC3 was bleached in the low pH when APs were fused with lysosomes to form autolysosomes (ALs). In the control cells transfected with scrambled siRNA, the LC3 signal appeared diffused with a few naturally occurring small LC3 puncta, which included both “red and green” APs and “red-only” ALs. In the si-Vps4B group, there was a notable accumulation of large LC3 puncta and aggregates, which were almost all red and green and lacked red-only ALs (Fig. 3A). Moreover, the spectrum analysis showed a significant shift of the Pearson’s correlation coefficient of mTagRFP and mWasabi in the si-Vps4B cells compared to the si-Ctrl group (Fig. 3B), suggesting an impediment of autophagic flux due to Vps4B KD. The result was further supported by Western blotting analysis, as the autophagy markers LC3-II and P62 were accumulated in the cells transfected with si-Vps4B (Fig. 3, C and D). Consistently, Vps4B OE significantly reduced LC3-II and P62 levels in MEF cells (Fig. 3, E and F). In addition, lysosome blockade by chloroquine (CQ) abolished the influence of Vps4B KD or OE on autophagy levels (Fig. 3, C to F). Together, the data indicated that Vps4B functioned by promoting autophagic flux but not autophagy induction.
Fig. 3. Vps4 is essential and sufficient to promote autophagic flux.
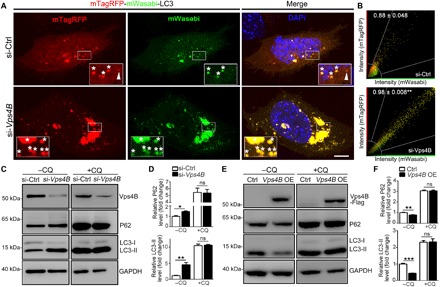
(A) Representative images of mTagRFP-mWasabi-LC3–labeled MEF cells to monitor autophagic flux. si-Ctrl, scrambled siRNA. Zoom-in insets: asterisk, AP; arrowhead, autolysosome. DAPI, 4′,6-diamidino-2-phenylindole. (B) The fluorescence intensity of mTagRFP and mWasabi of each pixel in (A) is measured and plotted. Pearson’s correlation coefficient was used as a measure of colocalization of mTagRFP and mWasabi. The mean correlation coefficient value ± SEM of at least eight cells per group is shown; **P < 0.01; Student’s t test. (C to F) Western blot analysis of P62 and LC3-II in MEF cells with Vps4B KD (C) or Vps4B OE (E) in the absence or presence of CQ (10 μM). All protein levels are normalized to GAPDH, and the ratio in the control group without CQ is set to 1. The relative protein levels of P62 and LC3-II in (C) and (E) are quantified in (D) and (F), respectively. Means ± SEM; n = 3 to 4; *P < 0.05, **P < 0.01, and ***P < 0.001; ns, not significant; Student’s t test. Scale bar, 5 μm.
The neuroprotective function of Vps4 is conserved in mammalian axons in vitro and in vivo
We next wanted to determine whether the axonal function of Vps4 is conserved in mammals. To test this hypothesis, we used the in vitro model of the primary mouse dorsal root ganglion (DRG) neurons. First, we characterized the localization of Vps4B protein in DRG neurons by immunocytochemistry: Vps4B was expressed in both soma and axons (fig. S5A). In addition to the ubiquitously distributed Vps4B signal in the DRG neurites, there were also periodic brighter Vps4B speckles. Vps4 is monomer or dimer in its inactive form in cytosol but assembles to dodecamer once it is recruited to the ESCRT-III complex (26). Thus, the Vps4B speckles along the axons are likely representative of endocytic vesicles where active Vps4B is recruited to the site of action (27).
We then generated lentivirus to overexpress Vps4B in mouse DRG neuron cultures (fig. S5, B and C). Vps4B OE significantly delayed the start of WD of injured DRG neurites: At 12 hours after injury, more than 83% of the DRG neurites in the control group (lenti-Flag) were fragmented, whereas the morphology of the injured DRG neurites in the Vps4B OE group (lenti-Vps4B-Flag) was well maintained and less than 12% of the axons showed degeneration (Fig. 4, A and B). At 24 hours after injury when almost all control DRG neurites had degenerated, the Vps4B OE group just started to degenerate and less than 50% showed minor axonal fragmentation (Fig. 4, A and B). Moreover, we found that Vps4B OE could further delay axonal degeneration in a Sarm1 KD background (Fig. 4, C to E). Loss of Sarm1 was known to potently suppress injury-induced NAD+ decline and delay WD (11). Injured DRG neurites of the sh-Ctrl group showed remarkable axonal fragmentation at 12 hours after injury, which was delayed to 24 hours in the “sh-Sarm1” group and further delayed to 36 hours in the “sh-Sarm1 + Vps4B OE” group. Note that, at the later time points, degenerated DRG neurites were detached from the culture dish and only floating axons or axonal debris were left for imaging. This was observed in all cases of the sh-Ctrl group and in more than half of the “sh-Sarm1” group but not in the “sh-Sarm1 + Vps4B OE” group at 36 hours after injury (Fig. 4C). Together with the finding that Nmnat OE did not suppress injury-induced autophagy (Fig. 2, E to G), the data suggested that the “Vps4-autophagy” pathway might work independently and/or synergistically with the “Nmnat-Sarm1-NAD+” mechanism in triggering WD during axon injury.
Fig. 4. Vps4B OE delays axonal degeneration in the in vitro and in vivo mouse models.
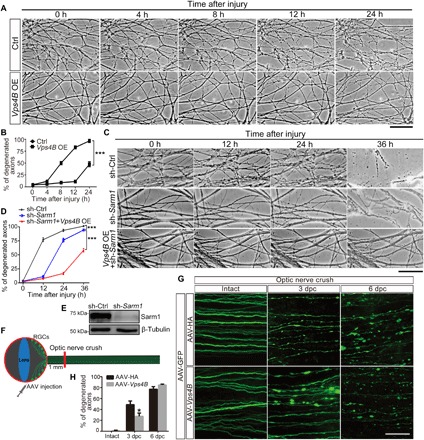
(A to D) Representative phase contrast images (A and C) and quantifications (B and D) of injured neurites of in vitro–cultured DRG neurons. Images are captured live at indicated time points after axotomy. Degenerated axons start to detach from the culture dish from 12 to 24 hours after injury, and in many cases, only floating axons or axonal debris are left at 36 hours. (E) Sarm1 KD is achieved by lentiviral infection of short hairpin RNA (shRNA) and confirmed by Western blotting. Vps4B OE is examined and shown in fig. S5 (B and C). Ctrl, lenti-Flag; Vps4B OE, lenti-Vps4B-Flag; sh-Ctrl, scrambled shRNA; sh-Sarm1, shRNA of mouse Sarm1. Means ± SEM; n = 7 (A and B) and n = 5 to 8 (C and D) DRG cultures per group; ***P < 0.001; two-way ANOVA. (F) A schematic drawing of the mouse retinal ganglion cells (RGCs) and optic nerve injury model. (G) Representative images of GFP-positive optic axons from mice with adeno-associated virus (AAV)–hemagglutinin (HA) (control) or AAV-Vps4B injection at the indicated time after optic nerve crush. The mouse optic nerve was imaged distal to the crush site, at the point about 2 mm proximal to the optic chiasm. Injured axons show swelling, beading, and fragmentation. dpc, days postcrush. (H) Quantification from (G). Means ± SEM; n = 5 mice per group; *P < 0.05; two-way ANOVA. Scale bars, 50 μm (A and C) and 100 μm (G).
Furthermore, to validate the axonal protective effect of Vps4B in an in vivo mammalian system, we generated adeno-associated virus (AAV) to overexpress Vps4B in mouse neurons. The AAV-Vps4B was injected into the mouse retinal ganglion cells (RGCs), and the optic nerve was then injured by a crush (Fig. 4F). Degeneration of the crushed mouse optic nerve was evident at 3 days postcrush (dpc), displaying remarkable axonal swelling, beading, and fragmentation (Fig. 4G). Vps4B OE markedly delayed the start of the optic nerve degeneration from 3 to 6 dpc (Fig. 4G), as there was a significant reduction of fragmented optic axons in the AAV-Vps4B group compared to the AAV-HA control group at 3 dpc (Fig. 4H). Together, in both in vitro and in vivo mouse models as well as in the Drosophila nervous system, up-regulation of Vps4 significantly delayed WD of injured axons.
The Vps4B protein is rapidly depleted in mouse DRG axons after injury
To investigate how Vps4B responds to axon injury and whether the response underlies its axonal function in WD, we vigorously examined Vps4B levels before and after injury in mouse DRG neurites. We found that Vps4B protein was rapidly depleted from injured DRG axons—the decrease was evident as early as 2 hours and became more robust at 4 hours, at which the Vps4B signals showed a “beading” appearance (Fig. 5A). This was likely reminiscent of the periodic brighter Vps4B dots in intact axons (fig. S5A). By 6 hours after injury, Vps4B signals were largely undetectable (Fig. 5, A and C). The integrity of axonal cytoskeleton of the DRG neurites (Tuj 1 staining) did not show significant fragmentation until 6 to 8 hours after injury (Fig. 5, A and C). In contrast, a remarkable increase of LC3 puncta began at ~4 hours after axotomy (Fig. 5, B and C), which was subsequent to Vps4B reduction but before the start of axonal degeneration (Fig. 5, A and C). Moreover, Western blotting analysis of the protein extracts from the severed DRG neurites confirmed that Vps4B protein levels decreased abruptly at 2 hours, whereas the autophagy marker LC3-II started to increase from 2 hours, continued to accumulate, and was maintained at high levels throughout all time points tested in this study (Fig. 5, D and E).
Fig. 5. The impact of injury on Vps4 protein and autophagy levels in mouse DRG neurites.
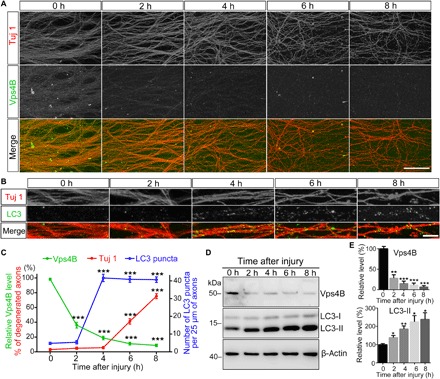
(A to C) Representative confocal images of mouse DRG explants immunostained for β-tubulin III (Tuj 1) and Vps4B in (A) or LC3 in (B) at the indicated time after injury. The relative intensity of Vps4B fluorescence and the percentage of degenerated axons (Tuj 1) in (A) and the average number of LC3 puncta per 25 μm of axons in (B) are quantified in (C). (D and E) The representative Western blot (D) and quantification analysis (E) of Vps4B and LC3-II protein levels in injured DRG neurites at indicated time points. All proteins are normalized to β-actin and shown as relative levels to that of the uninjured axons (0 hours). Means ± SEM; n = 8 (A), n = 10 (B), and n = 3 (D) per group; *P < 0.05, **P < 0.01, and ***P < 0.001; Student’s t test, compared to the level at 0 hours (sham control). Scale bars, 100 μm (A) and 5 μm (B).
DISCUSSION
The temporal relationship of Vps4B decrease, autophagy increase, and axonal degeneration, together with the findings that down-regulation of Vps4 leads to AP accumulation and axon self-destruction whereas up-regulation of Vps4 improves autophagic flux and suppresses WD, implicates that the rapid depletion of Vps4 protein may trigger injury-induced autophagy and axonal degeneration. In addition, the injury-induced depletion may make Vps4B the “bottleneck step” of the ESCRT machinery in the endocytic/autophagic pathway, which might explain why up-regulation of only Vps4 but not of the other ESCRT components such as TSG101 is sufficient to reduce AP accumulation and delay degeneration of injured axons.
Functions of ESCRT in neuronal axons have been largely neglected, and major gaps exist in current knowledge. In this study, we identify ESCRT-Vps4 as a novel anti-degenerative molecule in axon injury. We show that rapid Vps4 depletion impairs autophagic flux in axons that contributes considerably to injury-induced autophagy, which adds substantially to the previous view that autophagy in injury was primarily an “activation” of AP biogenesis (25). Both insufficient and excessive autophagy can lead to axonal degeneration. Our current data indicate that the accumulation of APs per se is likely “toxic” and may serve as a signal to trigger the self-destruction process in injured axons.
The “ESCRT-Vps4-autophagy” is likely one of the wide array of signal transduction pathways that are activated upon axon injury. For example, since the ESCRT machinery is required for membrane protein sorting and turnover (15), the consequence of Vps4 depletion may also involve alteration of membrane receptor signaling pathways. We show that Nmnat OE does not suppress injury-induced autophagy, whereas Vps4B OE shows an additive effect to Sarm1 KD in protecting injured axons. Thus, “ESCRT-Vps4-autophagy” may represent a novel and important signaling pathway that functions in parallel to the Nmnat/Sarm1-NAD+ mechanism in initiating WD (Fig. 6). Future studies are needed to understand how Vps4 protein is down-regulated in injury and to develop intervention strategies against axonal degeneration in neural injury and diseases via modulating ESCRT activity.
Fig. 6. A working model of Vps4 in axon injury and WD.
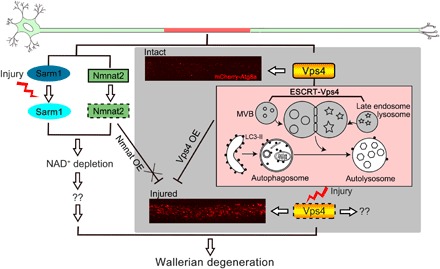
Injury induces rapid depletion of Vps4, which impairs autophagic flux and causes massive AP accumulation, which can lead to axonal degeneration. Meanwhile, Sarm1 is activated and Nmnat2 is degraded upon injury, which cause the NAD+ depletion that can also trigger axonal degeneration. Since Nmnat OE does not suppress injury-induced autophagy, whereas Vps4 OE can further delay WD in the Sarm1 KD background, the two mechanisms likely function independently to transduce the injury signal and synergistically promote the initiation of WD. “??”, unknown or additional downstream targets.
MATERIALS AND METHODS
Statement of animal care and surgical procedures
All surgical procedures in mice were performed in compliance with the institutional guidelines on the scientific use of living animals at the Interdisciplinary Research Center on Biology and Chemistry, Chinese Academy of Sciences (CAS), or with the protocols approved by the Animal and Plant Care Facility at the Hong Kong University of Science and Technology. In this study, “Principles of Laboratory Animal Care” [National Institutes of Health (NIH) publication no. 86-23, revised 1985] were followed. Animal distress and conditions requiring euthanasia were addressed, and the number of animals used was minimized.
Drosophila stocks
The UAS-Dcr2 (24651), UAS-RNAi-luciferase (31603), UAS-RNAi-mCherry (35785), UAS-RNAi-Atg12 (34675), and UAS-RNAi-Atg17 (36918) flies were from the Bloomington Drosophila Stock Center (BDSC); the three UAS-RNAi-Vps4 lines (nos. 105977, 35125, and 35126) were from the Vienna Drosophila Resource Center (VDRC); and the UAS-RNAi-TSG101 (9712R-1) flies were from the Fly Stocks of National Institute of Genetics (NIG-FLY). The UAS-mCherry-Atg8a fly line was previously described (28). For neuronal expression of long hairpin RNAi lines, RNAi-luciferase was used as a control and a copy of UAS-Dcr2 was coexpressed to boost the KD efficiency (29); for V20 RNAi lines that express short hairpin RNA (shRNA), no additional UAS-Dcr2 was needed and RNAi-mCherry was used as a control.
For in vivo imaging of axon membrane and axonal APs or to simultaneously manipulate gene expression, the following stable fly lines carrying multiple transgenes were generated by chromosomal recombination:
yw;dpr-Gal4,UAS-mCD8-GFP,UAS-mCherry-Atg8a/CyO
yw;dpr-Gal4,UAS-mCD8-GFP,UAS-mCherry-Atg8a/CyO;UAS-Dcr2/TM3.Ser
yw;dpr-Gal4,UAS-GAL4,UAS-mCD8-CherryRFP/CyO;UAS-Dcr2/TM3.Ser
All flies tested in this study were raised on standard cornmeal media and maintained at 25°C and 60% relative humidity.
Generation of transgenic fly strains
To generate pBID-UASC-Vps4-V5 and pBID-UASC-TSG101-V5 plasmids, DNA fragments encoding Drosophila Vps4 and TSG101 were amplified from complementary DNA (cDNA) [the Drosophila Genomics Resource Center (DGRC), GH02678, and RE26756, respectively], subcloned into pIZ-V5-6xHis vector using the Eco RI/Sac II sites, and then subcloned with a V5 tag into the pBID-UASC vector (30) using the Eco RI/Eag I sites. The polymerase chain reaction (PCR) primers used are listed below:
Vps4 F1: 5′-CCGAATTCTAAATCACCATGGCAGCCG-3′
Vps4 R1: 5′-TTCCGCGGGCCCTCCTGCCCAAAGTC-3′
Vps4 F2: 5′-CCGAATTCTAAATCACCATGGCAGCCG-3′
Vps4 R2: 5′-GACGGCCGCTCAATGGTGATGGTGATGATG-3′
TSG101 F1: 5′-CCGAATTCAGCACAATGCCTGCGGTT-3′
TSG101 R1: 5′-TTCCGCGGTGCTTTGATGTGCTCCTCG-3′
TSG101 F2: 5′-CCGAATTCAGCACAATGCCTGCGGTT-3′
TSG101 R2: 5′-GACGGCCGCTCAATGGTGATGGTGATGATG-3′
The transgenic fly strains of UAS-Vps4-V5 and UAS-TSG101-V5 were generated by ΦC31 integrase-mediated, site-specific integration into the fly genome, which allowed uniform transgene expression across different lines. The attP landing site stock used in this study was UAS-phi2b2a;VK5 (75B1) and a transgenic pBID-UASC-luciferase fly strain generated using the same method, and the same landing site (31) was used as a control.
Drosophila wing nerve injury and imaging procedure
The Drosophila wing nerve was injured and visualized as previously described (22, 23). Briefly, to sever the wing axons, flies were anesthetized using CO2 gas and the wings were cut using handheld spring scissors (Fine Science Tools, 15000-08). The integrity and autophagy levels of the wing axons were indicated by mCD8-GFP and mCherry-Atg8a, respectively. For confocal images, wings were detached from fly bodies, briefly washed with PBST (0.2% Triton X-100/PBS), and then mounted directly in VECTASHIELD Mounting Medium (Vector Laboratories, H-1000). Images were taken with a Leica TCS SP8 confocal microscope using a 63× oil objective [numerical aperture (NA) = 1.4]. Images were processed and analyzed in LAS X and ImageJ software and assembled into figures using Photoshop CS6 (Adobe Systems Inc.) with similar settings. The axonal degeneration score and the number of axonal APs were quantified as described in fig. S1.
RNA extraction and real-time quantitative PCR
For quantitative PCR (qPCR), total RNA was isolated from the heads of 5-day-old adult flies using TRIzol (Invitrogen). Approximately 30 to 35 fly heads were collected for each repeat, and three biological repeats were performed for the statistical analysis. Deoxyribonuclease (Promega) was applied to remove genomic DNA. Reverse transcription (RT) reactions were then performed using the High-Capacity cDNA Reverse Transcription kit with random primers (Applied Biosystems). The cDNA was then used for qPCR with SYBR Select Master Mix (Applied Biosystems) via the QuantStudio 6 Flex Real-Time PCR System (Life Technologies). Actin was used as an internal reference. The following related fly qPCR primers were used:
actin F: 5′-GAGCGCGGTTACTCTTTCAC-3′
actin R: 5′-GCCATCTCCTGCTCAAAGTC-3′
Atg12 F: 5′-GAGCACTTCCTCCTCCACAC-3′
Atg12 R: 5′-CTACGGTCCAGGTTCGCTTT-3′
Atg17 F: 5′-CACCCAAGAGACATACCAGC-3′
Atg17 R: 5′-AGATCGTAGGCAGAGCTTCT-3′
Vps4 F: 5′-ACTGCGTCTGTATGAGCACG-3′
Vps4 R: 5′-CTCCTTTAGCTTCTCGGCCC-3′
Plasmids and constructs
To generate the pCMV-Vps4B-3xFlag plasmid, the human Vps4B cDNA was amplified from human umbilical vein endothelial cells by RT-PCR and inserted into the pCMV-3Tag-3B expression vector (SnapGene) between Bam HI and Hind III sites using the ClonExpress MultiS One Step Cloning Kit (Vazyme). The PCR primers used are listed below:
Vps4B F1: 5′-GAGCGCGGTTACTCTTTCAC-3′
Vps4B R1: 5′-GCCATCTCCTGCTCAAAGTC-3′
Vps4B F2: 5′-CGCTCTAGCCCGGGCGGATCCGCCACCATGTCATCCACTTCGCCCAA-3′
Vps4B R2: 5′-GTCGACGGTATCGATAAGTCCTTGCCTTCTTGACCAAAATCTTCTG-3′
All constructs were verified by sequencing to ensure the integrity of the cloned open reading frames. The pcDNA3.1-mTagRFP-mWasabi-LC3 plasmid was previously described (32). The Vps4B siRNA oligo was GAUGGAUGUUCCUGGAGAUTT (GenePharma).
Cell cultures and transfection
Mouse MEF cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco) with 10% (v/v) fetal bovine serum (Gibco) and 1% penicillin/streptomycin at 37°C in 5% CO2. Transient transfection was performed using Lipofectamine 3000 (Invitrogen) for DNA and Lipofectamine RNAiMAX (Invitrogen) for siRNA in reduced serum Opti-MEM medium (Gibco) according to the manufacturer’s protocol. For lysosome blockade experiments, MEF cells were treated with 10 μM CQ (Sigma) for 8 hours before harvest.
Lentiviral vectors and infection
To generate the pLenti-hSyn-Vps4B-Flag plasmid, DNA fragments were amplified from pCMV-Vps4B-3xFlag by PCR and subcloned into a pLenti-hSyn vector (33) for neuron-specific expression using the Bam HI/Eco RI sites by homologous recombination as above. The PCR primers are listed below:
Lenti-Vps4B F1: 5′-GCCACCATGTCATCCACTTC-3′
Lenti-Vps4B R1: 5′-CCGGGCCCTATTTATCGT-3′
Lenti-Vps4B F2: 5′-AGAGCGCAGTCGAGAGGATCCGCCACCATGTCATCCACTTC-3′
Lenti-Vps4B R2: 5′-GATAAGCTTGATATCGAATTCCCGGGCCCTATTTATCGT-3′
To generate the pLKO-shRNA-Sarm1 plasmid, the shRNA of the mouse Sarm1 was cloned into the pLKO.1 vector. The oligo primers used were as follows: sh-Sarm1 F, 5′-CCGGCCACATTATCAGAGAGCCAAACTGCAGTTTGGCTCTCTGATAATGTGGTTTTTG-3′; sh-Sarm1 R, 5′-AATTCAAAAACCACATTATCAGAGAGCCAAACTGCAGTTTGGCTCTCTGATAATGTGG-3′. The negative control vector containing scrambled shRNA was obtained from Addgene (no. 1864), and the sequence of the hairpin was CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG.
Lentivirus to express Vps4B-Flag or sh-Sarm1 was prepared according to the established protocols. Briefly, 293T cells were cotransfected with pLenti-hSyn-Vps4B-Flag or pLKO.1-sh-Sarm1, vesicular stomatitis virus glycoprotein, and delta 8.9 with a ratio of 1:1.5:2 in Opti-MEM I medium using PolyJet reagent (SignaGen). Lenti-hSyn-Flag was packaged similarly and used as a control. Culture supernatant was collected at 48 hours after transfection and passed through a 0.45-μm filter. Fresh medium containing viral particles was used or stored at 4°C for less than 1 week before DRG culture infection.
DRG neuron culture, in vitro axotomy, and quantification
Dissection of C57BL/6 mouse embryonic DRG was carried out as previously described (34). For DRG explants, individual DRGs were seeded in the middle of the insert of the Transwell culture system (Corning, 35302) as described before (35). On 7 days in vitro (DIV), axotomy was performed by scraping the upper side of the insert that contained the cell bodies of the DRG explants using a cell scraper. To culture dissociated DRG neurons (for Vps4B viral infection), dissected DRGs were digested with collagenase and TrypLE Express Enzyme (Gibco) and the suspended DRG neurons were placed as a drop (5 to 10 μl) in the center of each well for 15 to 20 min. After cells were attached, 1 ml of culture medium was gently added to each well of a 12-well plate. The DRG neurons were then infected with pLenti-hSyn-Vps4B-Flag on 7 DIV, and axotomy was performed under the microscope using a micro surgical blade (WHB, C21) on 14 DIV.
Bright-field images of in vitro–cultured DRG neurites were taken live at indicated time points after axotomy using a Leica DMi8 inverted microscope. The severity of neurite degeneration was quantified as previously described (36). Briefly, the degeneration index was calculated as the ratio of the degenerated neurite pixel number to the total neurite pixel number. Numbers of random, nonoverlapping images of different DRG explants of each group from pooled results of at least three independent repeats are indicated in each figure.
AAV injection and mouse optic nerve injury
We used AAV serotype 2/2 to overexpress Vps4B, GFP, and 2×HA tag. RGCs of B6 mice were transduced with AAV-GFP virus to do sparse labeling and with AAV2-hSynapsin-2×HA or AAV2-hSynapsin-VPS4B virus to achieve saturated infection. Specifically, 2 μl of AAV2-hSynapsin-GFP virus with a titer of ~108 GC/ml was injected into the vitreous bodies of 5- to 6-week-old B6 mice to achieve RGCs sparse labeling. Four weeks later, mice were injected with AAV2-hSyanpsin-2×HA (control) or AAV2-hSynapsin-Vps4B with a titer of about 1013 GC/ml. After 4 weeks, optic nerve crush was performed with jeweler’s forceps (Dumont #5; Fine Science Tools) for 2 s at the point about 1 mm behind the optic disk. The mice were kept alive for 3 or 6 days before they were euthanized for imaging analysis of the optic nerve.
Imaging and quantification of axonal degeneration in mouse optic nerve
GFP-labeled mouse optic nerve was imaged by confocal microscopy at the point about 2 mm proximal to the optic chiasm. For quantification, around 100 GFP-positive RGC axons of each nerve sample were examined. The axons with fragmentation or breaks were scored as degenerated. Degeneration index was quantified as the percentage of all axons scored as degenerated. Five mice were included in each group.
Antibodies
The following primary antibodies were used for Western blotting and immunostaining in this study: mouse anti-Flag (Sigma-Aldrich, F3165), rabbit anti-Flag (Proteintech, 20543-1-AP), rabbit anti-Vps4B (Proteintech, 17673-1-AP), mouse anti-P62 (Abcam, ab56416), rabbit anti-LC3B (Abcam, ab48394), mouse anti-V5 (Proteintech, 66007-1-Ig), mouse anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Proteintech, 60004-1-Ig), mouse anti–β-tubulin class III (Tuj 1; Abcam, ab78078), rabbit anti–Tuj 1 (Sigma-Aldrich, T2200), and mouse anti–β-actin (Cell Signaling Technology, 3700). The horseradish peroxidase–conjugated secondary antibodies were as follows: goat anti-mouse (Sigma-Aldrich, A4416) and goat anti-rabbit (Sigma-Aldrich, A9169). The fluorophore-conjugated secondary antibodies were as follows: goat anti-rabbit Alexa Fluor 488 (Life Technologies, A11036), goat anti-mouse Alexa Fluor 488 (Life Technologies, A11029), goat anti-mouse Alexa Fluor 568 (Life Technologies, A11031), goat anti-rabbit Cy5 (Life Technologies, A10523), and goat anti-mouse Cy5 (Life Technologies, A105234).
Immunocytochemistry
MEF cells were grown on chamber slides (Lab-Tek) and transfected with the indicated plasmids for 48 hours. Cells were fixed with fresh paraformaldehyde (Sangon, A500684) in PBS for 15 min at room temperature, permeabilized with 0.5% Triton X-100 (Sigma-Aldrich, T8787) in PBS for 15 min, and then blocked with 3% goat serum in PBST (1× PBS + 0.1% Triton X-100) for 60 min at room temperature. Cells were then incubated with the indicated antibodies in blocking buffer at 4°C overnight, followed by incubation of appropriate secondary antibodies at room temperature for 1.5 to 2 hours. The chamber slides were then disassembled according to the manufacturer’s instruction and mounted in VECTASHIELD Mounting Medium with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, H-1200) with cover glass. For DRG neurites, immunostaining was performed similarly except that the filters of the Transwell inserts were taken off, processed, and mounted on a glass slide in VECTASHIELD Mounting Medium (Vector Laboratories, H-1000) with coverslip for confocal imaging.
Protein extraction and Western blotting
Cells were lysed in the cell extraction buffer (Invitrogen, FNN0011) with the protease inhibitor cocktail (Roche), sonicated on ice, and then centrifuged at 13,000g for 20 min at 4°C. Equivalent amounts of lysates were resolved by electrophoresis through a 12% bis-tris SDS–polyacrylamide gel electrophoresis gel (Invitrogen) and probed with the primary and secondary antibodies listed above. Detection was performed using the High-sig ECL Western Blotting Substrate (Tanon). Images were captured using an Amersham Imager 600, and densitometry was measured using the ImageQuant TL software (GE Healthcare) and ImageJ (NIH).
Statistical analysis
Statistical significance in this study was determined by two-way analysis of variance (ANOVA) with Bonferroni’s post hoc test or unpaired, two-tailed Student’s t test at *P < 0.05, **P < 0.01, and ***P < 0.001, as indicated in the figures. Error bars represent SEM.
Supplementary Material
Acknowledgments
We thank the BDSC, the NIG-FLY, and the VDRC for providing fly strains; J. Lin, X. Zhang, Y. Chen, and J. Yuan for reagents; A. Du, S. Qiu, and K. Tian for technical support; and J. Yuan, Y. Chen, members of the Fang laboratory, and colleagues in the field for advice and criticism. Funding: The study was supported by grants from the NKRD Program of China (no. 2016YFA0501902 to Y.F.), the National Science Foundation of China (no. 31471017 and no. 81671254 to Y.F., no. 81671214 to K.L., and no. 81501171 to A.L.), the 863 program (no. 2014AA020526 to Y.F.), the UGC/RGC grants (AoE/M-09/12, AoE/M-604/16, C6004-17G, 16103315, and 16149316 to K.L.), the ITC grant of Hong Kong SAR (ITCPD/17-9 to K.L.), and the SKIP grants (JCYJ20160428145818099 and JCYJ20160427185601855 to K.L.). Author contributions: H.W., K.L., and Y.F. conceived the study and designed the experiments. H.W. and Y.F. performed the fly screen. H.W., X.W., K.Z., X.C., Z.W., and A.L. contributed important new reagents. H.W., Z.W., and X.C. generated new fly strains. H.W. performed fly wing injury experiments and imaging. H.W. and K.Z. performed MEF cell experiments. H.W., Q.W., and S.Z. performed DRG neuron injury experiments, immunohistochemistry, Western blot, and confocal imaging analyses. X.W. performed the mouse optical nerve surgical procedures and data collection. H.W., X.W., K.Z., K.L., and Y.F. analyzed the data, and H.W., A.L., K.L., and Y.F. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Correspondence and requests for materials should be addressed to Y.F. (fangys@sioc.ac.cn). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/2/eaav4971/DC1
Fig. S1. The evaluation system of wing axonal degeneration scores.
Fig. S2. The ESCRT-I component TSG101 is required but not sufficient to maintain axonal integrity.
Fig. S3. Axon injury induces a rapid autophagy response followed by axonal destruction.
Fig. S4. Inhibition of autophagy induction by RNAi-Atg12 or RNAi-Atg17 provides moderate axonal protection.
Fig. S5. Examination of endogenous Vps4B protein and lentiviral OE of Vps4B in mouse DRG neurons.
REFERENCES AND NOTES
- 1.Coleman M. P., Freeman M. R., Wallerian degeneration, Wlds, and Nmnat. Annu. Rev. Neurosci. 33, 245–267 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang J. T., Medress Z. A., Barres B. A., Axon degeneration: Molecular mechanisms of a self-destruction pathway. J. Cell Biol. 196, 7–18 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fang Y., Bonini N. M., Axon degeneration and regeneration: Insights from Drosophila models of nerve injury. Annu. Rev. Cell Dev. Biol. 28, 575–597 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Conforti L., Gilley J., Coleman M. P., Wallerian degeneration: An emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci. 15, 394–409 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Ali Y. O., Li-Kroeger D., Bellen H. J., Zhai R. G., Lu H.-C., NMNATs, evolutionarily conserved neuronal maintenance factors. Trends Neurosci. 36, 632–640 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerdts J., Summers D. W., Milbrandt J., DiAntonio A., Axon self-destruction: New links among SARM1, MAPKs, and NAD+ metabolism. Neuron 89, 449–460 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neukomm L. J., Freeman M. R., Diverse cellular and molecular modes of axon degeneration. Trends Cell Biol. 24, 515–523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilley J., Coleman M. P., Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLOS Biol. 1, e1000300 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Osterloh J. M., Yang J., Rooney T. M., Fox A. N., Adalbert R., Powell E. H., Sheehan A. E., Avery M. A., Hackett R., Logan M. A., MacDonald J. M., Ziegenfuss J. S., Milde S., Hou Y.-J., Nathan C., Ding A., Brown R. H. Jr., Conforti L., Coleman M., Tessier-Lavigne M., Züchner S., Freeman M. R., dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337, 481–484 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerdts J., Summers D. W., Sasaki Y., DiAntonio A., Milbrandt J., Sarm1-mediated axon degeneration requires both SAM and TIR interactions. J. Neurosci. 33, 13569–13580 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerdts J., Brace E. J., Sasaki Y., DiAntonio A., Milbrandt J., SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science 348, 453–457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Essuman K., Summers D. W., Sasaki Y., Mao X., DiAntonio A., Milbrandt J., The SARM1 toll/interleukin-1 receptor domain possesses intrinsic NAD+ cleavage activity that promotes pathological axonal degeneration. Neuron 93, 1334–1343.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tooze S. A., Schiavo G., Liaisons dangereuses: Autophagy, neuronal survival and neurodegeneration. Curr. Opin. Neurobiol. 18, 504–515 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Rogov V., Dötsch V., Johansen T., Kirkin V., Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 53, 167–178 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Schmidt O., Teis D., The ESCRT machinery. Curr. Biol. 22, R116–R120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Christ L., Raiborg C., Wenzel E. M., Campsteijn C., Stenmark H., Cellular functions and molecular mechanisms of the ESCRT membrane-scission machinery. Trends Biochem. Sci. 42, 42–56 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Nara A., Mizushima N., Yamamoto A., Kabeya Y., Ohsumi Y., Yoshimori T., SKD1 AAA ATPase-dependent endosomal transport is involved in autolysosome formation. Cell Struct. Funct. 27, 29–37 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Rusten T. E., Vaccari T., Lindmo K., Rodahl L. M. W., Nezis I. P., Sem-Jacobsen C., Wendler F., Vincent J.-P., Brech A., Bilder D., Stenmark H., ESCRTs and Fab1 regulate distinct steps of autophagy. Curr. Biol. 17, 1817–1825 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Djeddi A., Michelet X., Culetto E., Alberti A., Barois N., Legouis R., Induction of autophagy in ESCRT mutants is an adaptive response for cell survival in C. elegans. J. Cell Sci. 125, 685–694 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Yang Y., Coleman M., Zhang L., Zheng X., Yue Z., Autophagy in axonal and dendritic degeneration. Trends Neurosci. 36, 418–428 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y., Song M., Song F., Neuronal autophagy and axon degeneration. Cell. Mol. Life Sci. 75, 2389–2406 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fang Y., Soares L., Teng X., Geary M., Bonini N. M., A novel Drosophila model of nerve injury reveals an essential role of Nmnat in maintaining axonal integrity. Curr. Biol. 22, 590–595 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fang Y., Soares L., Bonini N. M., Design and implementation of in vivo imaging of neural injury responses in the adult Drosophila wing. Nat. Protoc. 8, 810–819 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T., LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wakatsuki S., Tokunaga S., Shibata M., Araki T., GSK3B-mediated phosphorylation of MCL1 regulates axonal autophagy to promote Wallerian degeneration. J. Cell Biol. 216, 477–493 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu Z., Gonciarz M. D., Sundquist W. I., Hill C. P., Jensen G. J., Cryo-EM structure of dodecameric Vps4p and its 2:1 complex with Vta1p. J. Mol. Biol. 377, 364–377 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferguson S. M., Axonal transport and maturation of lysosomes. Curr. Opin. Neurobiol. 51, 45–51 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang Y.-Y., Neufeld T. P., An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol. Biol. Cell 20, 2004–2014 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ni J.-Q., Markstein M., Binari R., Pfeiffer B., Liu L.-P., Villalta C., Booker M., Perkins L., Perrimon N., Vector and parameters for targeted transgenic RNA interference in Drosophila melanogaster. Nat. Methods 5, 49–51 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J.-W., Beck E. S., McCabe B. D., A modular toolset for recombination transgenesis and neurogenetic analysis of Drosophila. PLOS ONE 7, e42102 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao X., Wang H., Wang Z., Wang Q., Zhang S., Deng Y., Fang Y., In vivo imaging reveals mitophagy independence in the maintenance of axonal mitochondria during normal aging. Aging Cell 16, 1180–1190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou C., Zhong W., Zhou J., Sheng F., Fang Z., Wei Y., Chen Y., Deng X., Xia B., Lin J., Monitoring autophagic flux by an improved tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3) reveals that high-dose rapamycin impairs autophagic flux in cancer cells. Autophagy 8, 1215–1226 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Chen Y., Akin O., Nern A., Tsui C. Y. K., Pecot M. Y., Zipursky S. L., Cell-type-specific labeling of synapses in vivo through synaptic tagging with recombination. Neuron 81, 280–293 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L., Marquardt T., Direct live monitoring of heterotypic axon-axon interactions in vitro. Nat. Protoc. 7, 351–363 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Unsain N., Heard K. N., Higgins J. M., Barker P. A., Production and isolation of axons from sensory neurons for biochemical analysis using porous filters. J. Vis. Exp. 89, e51795 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wakatsuki S., Saitoh F., Araki T., ZNRF1 promotes Wallerian degeneration by degrading AKT to induce GSK3B-dependent CRMP2 phosphorylation. Nat. Cell Biol. 13, 1415–1423 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/2/eaav4971/DC1
Fig. S1. The evaluation system of wing axonal degeneration scores.
Fig. S2. The ESCRT-I component TSG101 is required but not sufficient to maintain axonal integrity.
Fig. S3. Axon injury induces a rapid autophagy response followed by axonal destruction.
Fig. S4. Inhibition of autophagy induction by RNAi-Atg12 or RNAi-Atg17 provides moderate axonal protection.
Fig. S5. Examination of endogenous Vps4B protein and lentiviral OE of Vps4B in mouse DRG neurons.