

Abstract
A series of carbamate-based inhibitors of glutamate carboxypeptidase II (GCPII) were designed and synthesized using ZJ-43, N-[[[(1S)-1-carboxy-3-methylbutyl]amino]carbonyl]-L-glutamic acid, as a molecular template in order to better understand the impact of replacing one of the two nitrogen atoms in the urea-based GCPII inhibitor with an oxygen atom. Compound 7 containing a C-terminal 2-oxypentanedioic acid was more potent than compound 5 containing a C-terminal glutamic acid (2-aminopentanedioic acid) despite GCPII’s preference for peptides containing an N-terminal glutamate as substrates. Subsequent crystallographic analysis revealed that ZJ-43 and its two carbamate analogs 5 and 7 with the same (S,S)-stereochemical configuration adopt a nearly identical binding mode while (R,S)-carbamate analog 8 containing a D-leucine forms a less extensive hydrogen bonding network. QM and QM/MM calculations have identified no specific interactions in the GCPII active site that would distinguish ZJ-43 from compounds 5 and 7 and attributed the higher potency of ZJ-43 and compound 7 to the free energy changes associated with the transfer of the ligand from bulk solvent to the protein active site as a result of the lower ligand strain energy and solvation/desolvation energy. Our findings underscore a broader range of factors that need to be taken into account in predicting ligand-protein binding affinity. These insights should be of particular importance in future efforts to design and develop GCPII inhibitors for optimal inhibitory potency.
Keywords: glutamate carboxypeptidase II, prostate-specific membrane antigen, crystal structure, metallopeptidase
Graphical Abstract
1. Introduction
Glutamate carboxypeptidase II (GCPII, EC 3.4.17.21) is a zinc metallopeptidase that cleaves the C-terminal glutamate from N-acetylaspartylglutamate (NAAG) producing N-acetylaspartate and glutamate. GCPII is also known as prostate-specific membrane antigen (PSMA), a marker expressed on prostate cancer cells, and as folate hydrolase (FOLH1) for its ability to cleave C-terminal glutamate residues from folate polyglutamate. Therefore, the therapeutic utility of GCPII inhibitors has been investigated in a wide range of diseases, including neurological disorders,1-3 prostate cancer,4 and inflammatory bowel disease (IBD).5 A number of potent GCPII inhibitors have been reported to date. Nearly all of these inhibitors have a general structure consisting of a zinc-binding group (ZBG) linked to a carboxylic acid-containing moiety designed to occupy the S1’ glutamate recognition site of the enzyme.2 Zinc-binding groups exploited in the design of GCPII inhibitors include phosphonate, phosphinate, phosphoramidate, thiol, and hydroxamate groups which were previously utilized in inhibitors of other metallopeptidases. Urea-based GCPII inhibitors first reported in 2001 by Kozikowski et al.6 are distinct from these traditional GCPII inhibitors in that the urea group has rarely been seen in the pharmacophore of other metalloprotease inhibitors. Since the first report, a number of urea-based GCPII inhibitors have been developed and pharmacologically characterized.7-13 Their highly potent GCPII inhibitory activity coupled with their high degree of chemical compatibility with conjugation processes has prompted many researchers to conjugate them to other functional molecules to target PSMA for diagnosis and treatment of prostate cancer.4 For instance, [177Lu]PSMA-617 (LuPSMA),14 a β-particle emitting PSMA ligand containing the urea scaffold (Fig. 1a), is currently being tested in patients with castration-resistant prostate cancer.
Fig. 1.

Chemical structures of [177Lu]PSMA-617, compounds 1-3, and ZJ-43. (a) [177Lu]PSMA-617 contains a component acting as a urea-based GCPII inhibitor that binds to the active site. (b) Compounds 1-3 possess either a urea or carbamate moiety. (c) Chemical structure of ZJ-43.
More recently, in an attempt to achieve more desirable tissue distribution for radiotherapy, Yang et al. developed a new class of GCPII inhibitors (Fig. 1b) in which either one of the two nitrogen atoms in the urea-based inhibitors was replaced by an oxygen atom, forming a carbamate moiety. 15 Comparison of these inhibitors that differ in the orientation of the carbamate moiety have revealed that those containing a C-terminal 2-oxypentanedioic acid are generally more potent than those containing a C-terminal glutamic acid (2-aminopentanedioic acid). For instance, as shown in Fig. 1, a substantial loss of potency was seen when the N-carbamoyl glutamic acid of compound 1 is replaced by an N-carboxy-glutamic acid as seen in compound 2. In contrast, compound 3, in which a carbamate moiety was inserted in the opposite orientation, was found to be substantially more potent than compound 2. This SAR trend is rather unexpected considering that the endogenous GCPII substrates possess a C-terminal glutamic acid and are anticipated to be better mimicked by carbamates containing a C-terminal glutamic acid.
These observations prompted us to conduct systematic SAR studies of carbamate-based GCPII inhibitors and subsequent structural and computational analyses. This is partly driven by our successful application of these combined experimental and theoretical approaches - the latter represented mostly by hybrid quantum and molecular mechanics (QM/MM) calculations - in understanding the GCPII catalytic actions and its inhibition. In particular, QM/MM assisted in revealing the GCPII reaction mechanism, the role of specific mutations in GCPII on the substrate binding16, 17 and last but not least, the molecular basis of the binding of urea-based inhibitors in the internal cavity of GCPII.18
The inhibitory potency of urea-based inhibitors has been shown to be substantially enhanced by expanding towards the substrate entry channel away from the glutamate recognition site 2. These high molecular weight inhibitors may not provide suitable templates for assessing the intrinsic effects of the carbamate moiety replacing the urea group. Consequently, we have chosen ZJ-43 (Fig. 1c),8 one of the smallest urea-based GCPII inhibitors, as a molecular template for our studies as we expected a robust response in inhibitory potency with modifications to the urea group. Subsequently, co-crystal structures were obtained for some of these compounds and further investigated by QM/MM simulations to gain insights into the structural basis for GCPII inhibition by carbamate-based inhibitors.
2. Results and discussion
2.1. Chemistry
ZJ-43 was synthesized using a method similar to that previously reported with minor modification (Fig. 2).8 Specifically, ethyl acetate was used as a solvent in the catalytic hydrogenation process. This modified method was successfully applied to urea- and carbamate-based GCPII inhibitors 4-10 (Table 1) by replacing benzyl leucine and/or dibenzyl glutamate systematically with other benzyl esters, including both enantiomers of dibenzyl 2-hydroxypentanedioate and benzyl 2-hydroxy-4-methylvalerate.
Fig. 2.

Synthesis of ZJ-43. Reagents and conditions: (a) triphosgene, triethylamine, DCM, −78 °C to rt, 16 h, 62% yield; (b) H2, 5% Pd/C, EtOAc, 30 psi, 12 h, quantitative yield.
Table 1:
Inhibition of GCPII by urea- and carbamate-based compounds.
| Cmpd | Structure | Stereochemistry | IC50 (nM) | Cmpd | Structure | Stereochemistry | IC50 (nM) |
|---|---|---|---|---|---|---|---|
| ZJ-43 | 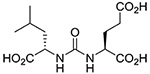 |
(S,S) | 0.50 ± 0.11 | 7 | 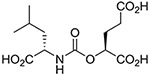 |
(S,S) | 0.65 ± 0.11 |
| 4 | 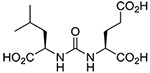 |
(R,S) | 15.9 ± 9.9 | 8 | 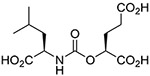 |
(R,S) | 8.9 ± 0.1 |
| 5 | 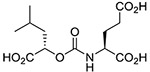 |
(S,S) | 16.9 ± 7.2 | 9 | 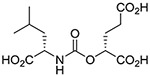 |
(S,R) | 630 ± 8 |
| 6 | 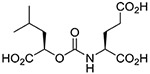 |
(R,S) | 531 ± 111 | 10 | 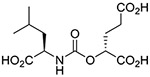 |
(R,R) | 8530 ± 20 |
IC50 values were calculated from the inhibition curves using non-linear analysis and are shown in the format mean ± standard deviation.
2.2. Inhibition of GCPII by urea- and carbamate-based inhibitors
Inhibitory potencies of the urea- and carbamate-based compounds were measured using N-acetyl-L-aspartyl-[3H]-L-glutamate as a substrate and purified human recombinant GCPII. The results are summarized in Table 1. In our assay, ZJ-43 showed subnanomolar inhibitory activity against GCPII with an IC50 value of 0.50 nM. Substitution of D-leucine for L-leucine of ZJ-43 resulted in 32-fold loss of GCPII inhibitory potency as observed with compound 4. Carbamate derivative 5 in which the L-leucine of ZJ-43 was replaced by (S)-2-hydroxy-4-methylvaleric acid (L-leucic acid) also showed a weaker potency as compared to ZJ-43 despite possessing the stereochemical configuration corresponding to that of ZJ-43. Further decline in potency was observed upon introduction of (R)-2-hydroxy-4-methylvaleric acid (D-leucic acid) as seen in compound 6. This finding is consistent with the trend seen in the urea series described earlier. Subsequently, we reversed the orientation of the carbamate moiety by replacing the glutamate portion of ZJ-43 with (S)-2-hydroxypentanedioic acid (L-2-hydroxyglutaric acid). Despite the loss of the C-terminal glutamate moiety recognized in all known endogenous GCPII substrates, carbamate derivative 7 was found to be substantially more potent than compound 5 with an IC50 value of 0.65 nM and nearly equipotent to ZJ-43. The superior potency of 7 over 5 indicates that the trend initially observed by Yang et al. applies to a wider range of carbamate-based GCPII inhibitors regardless of the size and nature of the components expanding towards the substrate entry channel away from the glutamate recognition site. In order to further elaborate on these findings, all remaining three diastereomers 8-10 were also tested in the GCPII assay. Substitution of D-leucine for L-leucine of 7 resulted in 14-fold loss in potency as seen in compound 8. As described earlier, a similar degree of decline in potency was observed when the L-leucine of ZJ-43 was replaced with D-leucine. It should be noted that compound 8 is substantially more potent than compound 6 which shares the same stereoconfiguration but has a carbamate moiety in the opposite orientation. As apparent from compounds 9 and 10, (R)-2-hydroxypentanedioic acid (D-2-hydroxyglutaric acid) is not tolerated by GCPII as a substitute for (S)-2-hydroxypentanedioic acid. This is in a good agreement with the specificity shown by GCPII towards substrates containing a terminal L-glutamate.19
Based on our results, ZJ-43 and its analogs can be classified into four well-established clusters by their inhibitory potency (Fig. 3). The first cluster contains the two subnanomolar inhibitors, ZJ-43 and compound 7. The three low nanomolar inhibitors 4, 5, and 8 constitute the second cluster. The third cluster consists of the two submicromolar inhibitors 6 and 9. Compound 10, the enantiomer (both chiral centers are reversed) of 5, represent the least potent GCPII inhibitors with an IC50 value of 8.5 μM. As expected, the S,S-stereoisomers (ZJ-43, compounds 5 and 7) are preferred by GCPII although the enzyme displays some degree of tolerance to the R-configuration on the leucine side. The superior potency of compound 7 containing a terminal (S)-2-oxyglutarate over compound 5 containing a terminal L-glutamate is consistent with the prior findings.15 Given that the endogenous GCPII substrates possess a C-terminal glutamic acid, this SAR trend is rather unexpected considering that the endogenous GCPII substrates possess a C-terminal glutamic acid and are anticipated and may have an important implication for the design of new GCPII inhibitors.
Fig. 3.

Dose response curves of in vitro GCPII inhibition by ZJ-43 and compounds 4-10 determined by the radioenzymatic assay.
2.3. Co-crystal structures of GCPII in complex with urea- and carbamate-based inhibitors
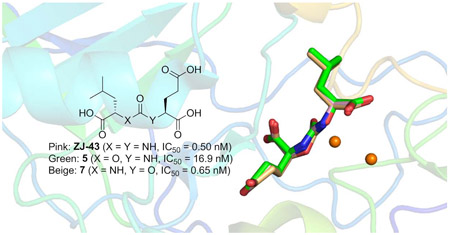
In order to gain further insight into the structural basis for GCPII inhibition by carbamate-based inhibitors, we have determined co-crystal structures of GCPII in complex with compounds ZJ-43, 5, 7 and 8 to 1.52, 1.61, 1.63, and 1.68 Å resolution limits, respectively (Table 2). For all complexes, the interpretable Fo-Fc electron density peaks representing the active site-bound ligands were observed and individual compounds were fitted into the density in the final stages of the refinement (Fig. 4A). Positioning of ZJ-43, 5, and 7 possessing S,S-configuration in the internal cavity of GCPII is virtually indistinguishable (Fig. 4B). The P1’ glutarate moiety occupies the S1’ pocket with the “canonical” binding mode observed previously in other urea-based GCPII inhibitors.20, 21 At the non-prime side, the most extensive contacts are observed between GCPII and the P1 carboxylate function. First, it forms four salt bridges with guanidinium groups of Arg534 (2.8 Å) and Arg536 (2.9 Å and 3.0 Å) of the arginine patch. Furthermore, it is engaged in the water-mediated H-bonds with the side chains of Ser454 (2.8 Å) and Arg534 (2.9 Å) as well as the carbonyl of Ser517 (2.8 Å). The interaction of the glutarate portion of compound 8 possessing R,S-configuration with GCPII appears nearly identical to that of the above three compounds (Fig. 4C), owing to the stereochemistry corresponding to that of L-glutamate at the P1’ site. The decline in the inhibitory potency of compound 8 can be attributed to the less extensive interaction network observed between GCPII and the P1 carboxylate of D-leucine residue (Fig. 4D). Here, the direct interactions involve the guanidinium group of Arg534 (2.9 Å) and the side-chain amide of Asn519 (2.9 Å) and water-mediated H-bonds with side chains of Arg534 (2.9 Å), Arg536 (3.0 Å), and the carbonyl of Ser517 (2.7 Å).
Table 2:
Crystallographic Data Collection and Refinement Statistics.
| Data collection statistics | ||||
| Inhibitor | ZJ-43 | 5 | 7 | 8 |
| PDB code | 6FE5 | 6EZ9 | 6F5L | 6ETY |
| Space group | I222 | I222 | I222 | I222 |
| Unit-cell parameters a, b, c (Å) | 101.7, 130.7, 159.8 |
101.5, 130.8, 158.6 | 101.1, 130.3, 158.6 | 101.7, 130.7, 158.2 |
| Wavelength (Å) | 0.98 | 0.98 | 0.98 | 0.98 |
| Resolution limits (Å) | 30.00-1.52 (1.61-1.52) |
30.00-1.61 (1.71-1.61) |
30 - 1.63(1.73 - 1.63) | 30.00-1.68 (1.78-1.68) |
| No of unique refl. | 161939 (25398) |
132737 (20491) | 128331 (20423) | 118798 (18462) |
| Redundancy | 5.09 (4.83) | 5.12 (5.03) | 5.22 (5.28) | 5.13 (4.95) |
| Completeness (%) | 99.4 (97.3) | 97.6 (94.2) | 98.7 (98.2) | 98.9 (96.1) |
| I/σ(I) | 15.89 (2.23) | 9.77 (1.99) | 16.13 (2.11) | 14.72 (2.08) |
| Rmerge | 0.048 (0.551) |
0.086 (0.382) | 0.059 (0.906) | 0.062 (0.693) |
| Refinement | ||||
| Resolution limits (Å) | 30.00-1.52 (1.56-1.52) |
30.00 - 1.61 (1.66-1.61) |
30.00 - 1.63 (1.67 -1.63) |
30.00 - 1.68 (1.72 - 1.68) |
| Total nmnber of reflections | 156947 | 128675 (8838) | 124566 (9292) | 115124 (8463) |
| Number of reflections in working set | (11078) | 124617 (8547) | 120817 (9007) | 111551 (8214) |
| Number of reflections in test set | 4964 (375) | 4058 (291) | 3749 (285) | 3573 (249) |
| R/Rfree (%) | 15.9/17.9 (26.2/26.7) |
16.3/18.1 (24.2/26.0) |
15.5/17.4 (27.1/32.1) |
15.6/17.8 (24.7/27.4) |
| Total nmnber of non-H atoms | 6638 | 6579 | 6843 | 6600 |
| Number of non-H protein atoms | 5824 | 5788 | 5974 | 5846 |
| Number of inhibitor molecules | 1 | 1 | 1 | 1 |
| Number of water molecules | 579 | 568 | 628 | 531 |
| Average B-factor (Å2) | 32.44 | 29.42 | 33.72 | 32.92 |
| Protein | 31.01 | 27.97 | 31.86 | 31.58 |
| Water molecules | 40.90 | 38.62 | 43.91 | 41.16 |
| Inhibitor | 25.00 | 26.33 | 26.31 | 27.84 |
| &Ramachandran Plot (%) | ||||
| Most favored | 97 % | 97 % | 97 % | 97 % |
| Additionally allowed | 2 % | 2 % | 2 % | 2 % |
| Disallowed | 1 % (Lys 655 A/B, Ser 656 A/B, Val 382) |
1 % (Lys 655 A/B, Ser 656 A/B, Val 382) | 1 % (Lys 655 A/B, Ser 656 A/B, Gly 335,Val 382) | 1 % (Ser 656 A/B, Gly335, Val 382) |
| R.m.s. deviations: bond lengths (Å) | 0.018 | 0.015 | 0.016 | 0.015 |
| bond angles (°) | 1.79 | 1.68 | 1.75 | 1.64 |
| chiral centers (Å3) | 0.13 | 0.12 | 0.13 | 0.12 |
| missing residues | AA 44-54 | AA 44-54 | AA 44-54 | AA 44-54 |
Values in parentheses are for the highest resolution shells.
Structures were analyzed using the MolProbity package
Fig. 4.

Structural characterization of GCPII-inhibitor complexes. (a) Fo-Fc maps (green) for studied inhibitors are contoured at 3.0 σ and modeled inhibitors are shown in stick representation with atoms colored green (carbon), red (oxygen), and blue (nitrogen). The active-site zinc ions are shown as orange spheres. (b) Superposition of SS diastereomers ZJ-43 (pink), 5 (green), and 7 (light orange) in the internal pocket of GCPII. (c) Superposition of 7 (light orange) and its R,S-counterpart 8 (cyan). (d) Comparison of the interaction network of the P1 carboxylate function with GCPII residues. Inhibitors 7 and 8 are shown in stick representation, GCPII residues as lines and water molecules as red spheres. H-bonds and salt bridges are shown as dashed lines, distances in Ångströms.
2.4. QM/MM calculations
Given that X-ray data at >1.1 Å resolution do not enable direct observation of hydrogen atoms, our structural data cannot provide a mechanistic explanation for the observed difference in inhibitory potency between the two carbamate-based inhibitors 5 and 7. To obtain refined binding modes of the individual compounds and insight into the molecular origin of the inhibitory potency, QM/MM calculations of the three inhibitors ZJ-43, 5, and 7 bound to GCPII were carried out. The details of the computational protocols, including the protein preparation and equilibration are mentioned below (Materials and Methods section). The QM regions (324 atoms, or 325 atoms in the case of urea) of the equilibrium QM/MM structures are depicted in Fig. 5 (coordinates are deposited as supplementary data). As shown in Fig. 5, our QM/MM calculations did not reveal any ‘visible’ differences in the binding of ZJ-43, 5, and 7. The binding modes of all three inhibitors, (a) ZJ-43, (b) compound 5, and (c) compound 7 are essentially identical, resulting in similar hydrogen bonding pattern. A small difference is observed for the interaction of urea based inhibitor ZJ-43 with Gly 518 residue, forming two hydrogen bonds using both NH groups of the central urea unit. Since the hydrogen bonding is equivalent for both compounds 5 and 7, such an interaction cannot be responsible for the lower inhibitory potency of compound 5. Thus, the diverse inhibitory potency, particularly the lower potency of compound 5 as compared to ZJ-43 and 7, does not appear to be originated from any specific ligand-protein interactions.
Fig. 5.

Details of hydrogen-bonding interactions between GCPII residues and (a) ZJ-43, (b) compound 5, and (c) compound 7. H-bonds shorter than 3.5 Å are shown as broken lines.
2.5. Calculations of ligand strain energies and solvation/desolvation energies
Subsequently, we examined whether the experimental data can be attributed to the different strain energy of each ligand, i.e. the alteration of ligand structure from its global energetic minimum in bulk solvent (water) to the bound state in the enzyme pocket and to the solvation/desolvation penalty of the compound (ligand).
To map the conformational space of each compound, the exhaustive DFT-D3/COSMO-RS//MD-LLMOD conformational sampling22 was carried out, followed by the highly-accurate DLPNO-CCSD(T) calculations. This combination enables us to obtain a quantitative insight into the energetics governing GCPII affinity for individual compounds. The energy data are summarized in Table 3 whereas full energetic data are deposited as supplementary data (Table S1). The results suggeset that the lower inhibitory activity of 5 originates in its unfavorable strain energy. In comparison to ZJ-43 and compound 7, Gstrain of compound 5 is higher by 3.2 kcal·mol−1 and 1.4 kcal·mol−1, respectively.
Table 3:
The computed ligand strain energies and solvation/desolvation energies for ZJ-43, 5, and 7.
| Gglobal-local | Glocal-enzyme | Gstrain | ΔGdesolv (referenced to ZJ-43) |
ΔGbind | |
|---|---|---|---|---|---|
| ZJ-43 | 3.1 | 1.1 | 4.2 | 0.0 | 4.2 |
| 5 | 5.3 | 2.1 | 7.4 | −0.9 | 6.5 |
| 7 | 2.7 | 3.2 | 6.0 | −1.8 | 4.2 |
All values are reported in kcal mol−1. Gglobal-local is the difference in Gibbs free energy of a substrate corresponding to reorganization from the equilibrium geometry in solvent to the local energy minimum obtained by the geometry optimization of the enzyme-bound structure; Glocal-enzyme is the change in the Gibbs free energy of a substrate corresponding to reorganization from the above local minimum to the exact enzyme-bound geometry (provided by the QM/MM); Gstrain is the sum of the first two terms; Gdesolv corresponds to the Gibbs free energy change due to transfer from water to 1-octanol (referenced to the ZJ-43); and ΔGbind is the sum of Gstrain and Gdesolv. This can be related to the inhibitory affinity of each substrate.
To further validate the accuracy and completeness of the conformational sampling algorithm, the low–lying minima were compared with the experimental NMR data. Solely for the purpose of the comparison, the conformer’s free energies were also computed in DMSO (used as the solvent in NMR measurements). All three obtained global minima (deposited as full 3-D structural data in the SI) are fully consistent with the J-coupling constants provided by the NMR (Table S2). This gives a validation of the computational strategy used. It can be also mentioned that the order of the energetically low-lying isomers changes between water (target solvent) and DMSO (solvent used for NMR).
We estimated solvation/desolvation energy by the relative differences between the COSMO-RS solvation energies in water and octanol. Octanol represents a “generic” protein environment and therefore, even in the relative scale, we admittedly neglect any specific interactions with the protein (more precisely, interactions that would be different for the three ligands). The validity of such an approach is, of course, justified only for the highly similar ligands, such as those studied herein. As can be seen in Table 3, adding the solvation/desolvation energies to the strain energies for the studied compounds, the quantitative agreement between calculation and experimental binding constants is obtained. This suggests that the differences in binding of the three ligands shall be attributed to the combination of the ligand strain energies and their differences in the octanol/water solvation and not to any specific interaction originating in the protein.
3. Conclusion
Since the first co-crystal structure was solved for GCPII in complex with an inhibitor in 2006,23 many additional structures have been reported for various GCPII-ligand complexes. Structure-based drug design has consequently played a key role in guiding the design of GCPII inhibitors.24 While these efforts have been primarily focused on the analysis of ligand-protein interactions, our SAR findings coupled with crystallographic analysis and comprehensive computational studies highlight a broader range of factors that need to be taken into account in predicting binding affinity to GCPII. These insights should be of particular importance in future efforts to design and develop GCPII inhibitors for optimal inhibitory potency.
4. Materials and methods
4.1. Chemical Synthesis of urea- and carbamate-based GCPII inhibitors
4.1.1. General
All solvents were reagent grade or HPLC grade. Unless otherwise noted, all materials were obtained from commercial suppliers and used without further purification. Melting points were obtained on a Mel-Temp apparatus and are uncorrected. 1H NMR spectra were recorded at 400 MHz. Chemical shifts are reported in parts per million relative to TMS. The HPLC solvent system consisted of distilled water and acetonitrile, both containing 0.1% formic acid. Analytical HPLC was performed on an Agilent 1200 Series HPLC system equipped with an Agilent G1315D DAD detector (detection at 220 nm) and an Agilent 6120 Quadrupole MS detector. Unless otherwise specified, the analytical HPLC conditions involve a gradient of 5% acetonitrile/95 % water for 0.5 min followed by an increase to 40% acetonitrile/60% water over 1.75 min and continuation of 40% acetonitrile/60% water for 1 min with an Eclipse Plus C18 column (2.1 mm × 50 mm, 3.5 μm) at a flow rate of 1.25 mL/min. All final compounds tested were confirmed to be of >95% purity by the analytical HPLC methods described above unless otherwise noted.
4.1.2. Synthesis of (S)-2-(3-((S)-1-carboxy-3-methylbutyl)ureido)pentanedioic acid (ZJ-43).
4.1.2.1. (S)-Dibenzyl 2-(3-((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yl)ureido)pentanedioate.
A solution of (S)-dibenzyl 2-aminopentanedioate 1.38 g (4.22 mmol, 1.0 eq.) in anhydrous DCM (60 mL) at −78 °C was added triphosgene (426 mg, 1.43 mmol,0.34 eq.) dropwise in DCM (20 mL) and then triethylamine (640 mg, 1.5 eq.). Upon warming to rt, (S)-benzyl 2-amino-4-methylpentanoate (930 mg, 4.22 mmol, 1.0 eq.), and triethylamine (430 mg, 1.0 eq.) were added and the reaction was stirred overnight. The organic phase was washed with water (2 × 50 mL), brine (50 mL), and dried over MgSO4, concentrated. The reside purified by silica gel chromatography (30%-50% EtOAc/hexanes) to give 1.50 g of the product as a white solid (62% yield); 1H NMR (CDCl3) δ 0.91 (dd, J = 6.3, 3.5 Hz, 6 H) 1.20 - 1.35 (m, 3 H) 1.41 - 1.73 (m, 5 H) 1.92 - 2.05 (m, 1 H)2.21 (d, J = 8.1 Hz, 1 H) 2.35 - 2.57 (m, 2 H) 4.14 (q, J = 7.1 Hz, 2 H) 4.45 - 4.65 (m, 2 H) 4.95 (d, J = 8.6 Hz, 1 H) 5.07 - 5.27 (m, 7 H) 7.29 - 7.41 (m, 15 H).
4.1.2.2. (S)-2-(3-((S)-1-Carboxy-3-methylbutyl)ureido)pentanedioic acid (ZJ-43).
A suspension of (S)-dibenzyl 2-(3-((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yl)ureido)pentanedioate (1.20 g, 2.08 mmol) and 5% Pd/C (106 mg, 0.94 mmol) in EtOAc (50 mL) was hydrogenated at 30 psi for 12 hours, filtered through a pad of celite, and then triturated with diethyl ether to give 633 mg of ZJ-43 as a white solid (quantitative yield); mp 64-67 °C; 1H NMR (DMSO-d6) δ 0.87 (dd, J = 12.5, 6.7 Hz, 6 H) 1.33 - 1.55 (m, 2 H) 1.54 - 1.77 (m, 2 H) 1.81 - 2.03 (m, 1 H) 2.15 - 2.37 (m, 2 H) 3.99 - 4.25 (m, 2 H) 6.27 (dd, J = 10.1, 8.6 Hz, 2 H) 12.42 (brs, 3 H). Analytical HPLC; retention time 0.617 min, m/z 303.1 [M - H]−.
4.1.3. Synthesis of (S)-2-(3-((R)-1-carboxy-3-methylbutyl)ureido)pentanedioic acid (4)
4.1.3.1. (S)-Dibenzyl 2-(3-((R)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yl)ureido)pentanedioate.
(S)-Dibenzyl 2-(3-((R)-1-(benzyloxy)-4-methyl-l-oxopentan-2-yl)ureido) was prepared as was described for (S)-dibenzyl 2-(3-((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yl)ureido)pentane-dioate except (R)-benzyl 2-amino-4-methylpentanoate was used in place of (S)-benzyl 2-amino-4-methylpentanoate. The reside purified by silica gel chromatography (1% MeOH/chloroform): white solid (37% yield); 1H NMR (CDCl3) δ 0.90 (d, J = 2.1 Hz, 3H), 0.91 (d, J = 2.0Hz, 6H), 1.46-1.53 (m, 1H), 1.57-1.69 (m, 2H), 1.98-2.04 (m, 1H), 2.18-2.23 (m, 1H), 2.39-2.49 (m, 2H), 4.51-4.57 (m, 2H), 5.04-5.26 (m, 8H), 7.31-7.38 (m, 15).
4.1.3.2. (S)-2-(3-((R)-1-Carboxy-3-methylbutyl)ureido)pentanedioic acid (4).
Compound 4 was prepared as was described for ZJ-43 except (S)-dibenzyl 2-(3-((R)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yl)ureido)pentanedioate was used in place of (S)-dibenzyl 2-(3-((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yl)ureido)pentanedioate and the material was hydrogenated at 1 atm for 60 minutes. Filtration through celite afforded a white solid (80% yield); mp 50-55 °C; 1H NMR (D2O) δ 0.81 (d, J = 5.9 Hz, 3H), 0.85 (d, J = 5.9 Hz, 3H), 1.54 (t, J = 7.2 Hz, 2H), 1.58-1.64 (m, 1H), 1.86-1.93 (m, 1H), 2.09-2.14 (m, 1H), 2.41 (t, J = 7.4 Hz, 2H), 4.14-4.22 (m 2H). Analytical HPLC; retention time 0.74 min, m/z 303.1 [M - H]−.
4.1.4. Synthesis of (S)-2-(((S)-1-carboxy-3-methylbutoxy)carbonylamino)pentanedioic acid (5)
4.1.4.1. (S)-Dibenzyl 2-(((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yloxy)carbonylamino)-pentanedioate.
(S)-Dibenzyl 2-(((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yloxy)carbonylamino)-pentanedioate was prepared as was described for (S)-dibenzyl 2-(3-((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yl)ureido)pentanedioate except (S)-benzyl 2-hydroxy-4-methylpentanoate25 was used in place of (S)-benzyl 2-amino-4-methylpentanoate. The residue purified by silica gel chromatography (25%-40% EtOAc/hexanes), but used in the subsequent step without further purification or characterization (62% yield).
4.1.4.2. (S)-2-(((S)-1-Carboxy-3-methylbutoxy)carbonylamino)pentanedioic acid (5).
Compound 5 was prepared as was described for ZJ-43 except (S)-dibenzyl 2-(((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yloxy)carbonylamino)-pentanedioate was used in place of (S)-dibenzyl 2-(3-((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yl)ureido)pentanedioate. Filtration through celite afforded a white solid (99% yield); mp 38-40 °C; 1H NMR (D2O) δ 0.77-0.81 (m, 6H), 1.49-1.55 (m, 1H), 1.62-1.69 (m, 2H), 1.81-1.87 (m, 1H), 2.07-2.13 (m, 1H), 2.40 (t, J = 7.4 Hz, 2H), 4.08-4.21 (m, 1H), 4.76-4.79 (m, 1H). LCMS (20% acetonitrile/80% H2O for 0.25 minutes followed by an increase to 85% acetonitrile/15% H2O over 1.75 minutes and continuation of 85% acetonitrile/15% H2O until 4 minutes): retention time 0.28 min, m/z 304.1[M - H]−.
4.1.5. Synthesis of (S)-2-(((R)-1-carboxy-3-methylbutoxy)carbonylamino)pentanedioic acid (6)
4.1.5.1. (S)-Dibenzyl 2-(((R)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yloxy)carbonylamino)-pentanedioate.
(S)-Dibenzyl 2-(((R)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yloxy)carbonylamino)-pentanedioate was prepared as was described for (S)-dibenzyl 2-(3-((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yl)ureido)pentanedioate except (R)-benzyl 2-hydroxy-4-methylpentanoate was used in place of (S)-benzyl 2-amino-4-methylpentanoate. The residue purified by silica gel chromatography (20% EtO Ac/hexanes): white solid (40% yield);1H NMR (CDCl3) δ 0.92 (m, 6H), 1.60-1.66 (m, 1H), 1.72-1.78 (m, 2H), 2.22-2.33 (m, 2H), 2.37-2.50 (m, 2H), 4.43-4.46 (m, 1H), 5.01-5.05 (m, 1H), 5.10 (s, 2H), 5.17-5.21 (m, 4H), 5.56 (d, J = 7.7 Hz, 1H), 7.31-7.38 (m, 15H).
4.1.5.2. (S)-2-(((R)-1-Carboxy-3-methylbutoxy)carbonylamino)pentanedioic acid (6).
Compound 6 was prepared as was described for ZJ-43 except (S)-dibenzyl 2-(((R)-1 -(benzyloxy)-4-methyl-1-oxopentan-2-yloxy)carbonylamino)-pentanedioate was used in place of (S)-dibenzyl 2-(3-((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yl)ureido)pentanedioate. Filtration through celite afforded a white solid (99% yield); mp 59-60 °C; 1H NMR (DMSO-d6) δ 0.82-0.92 (m, 6H), 1.46-1.57 (m, 1H), 1.60-1.79 (m, 3H), 1.89-1.99 (m, 1H), 2.24-2.35 (m, 2H), 3.92-3.98 (m, 1H), 4.65 (dd, J = 10.2, 3.2 Hz, 0.2H, minor rotomer, 4.74 (dd, J = 9.2, 4.6 Hz, 0.8H, major rotomer), 7.24 (d, J = 7.9 Hz, 0.2H, minor rotomer), 7.69 (d, J = 7.9 Hz, 0.8H, major rotomer), 12.96 (brs, 3H). Analytical HPLC; retention time 0.422 min, m/z 304.1 [M - H]-. Analytical HPLC; retention time 0.60 min, m/z 304.1 [M - H]−.
4.1.6. Synthesis of (S)-2-((S)-1-carboxy-3-methylbutylcarbamoyloxy)pentanedioic acid (7)
4.1.6.1. (S)-Dibenzyl 2-((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-ylcarbamoyloxy)pentanedioate.
(S)-Dibenzyl 2-((S)-1 -(benzyloxy)-4-methyl-1-oxopentan-2-ylcarbamoyloxy)pentanedioate was prepared in a similar manner to (S)-dibenzyl 2-(3-((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yl)ureido)pentanedioate, but with minor modifications. (S)-dibenzyl 2-hydroxypentanedioate 26 was added to a solution of triphosgene 112 mg, 0.4 mmol, 0.5 eq.) and anhydrous pyridine (66 mg, 0.84 mmol, 1.1 eq.) in anhydrous DCM (30 mL) at 0 °C. The solution was stirred at this temperature for 45 minutes, was washed with water (10 mL), and then the DCM layer was dried over MgSO4, filtered, and concentrated. Without further purification or characterization, to the crude chloroformate (300 mg, 0.76 mmol) in anhydrous THF (20 mL) at 0 °C was added DIEA (250 mg, 1.9 mmol, 2.5 eq.) and then (S)-benzyl 2-amino-4-methylpentanoate hydrochloride (200 mg, 0.76 mmol, 1.0 eq.). The solution was stirred at this temperature for 2h and then concentrated. The residue was dissolved in EtOAc (30 mL) and washed with 10% KHSO4 (25 mL), then sat’d NaHCO3 (25 mL), dried over MgSO4, filtered, and concentrated. The reside was purified by silica gel chromatography (25%-40% EtOAc/hexanes) to give 340 mg of the product as a white solid (78% yield); 1H NMR (CDCl3) δ 0.90 (dd, J = 6.4, 2.2 Hz, 6 H) 1.49-1.56 (m, 1H), 1.60-1.69 (m, 2H), 2.12-2.20 (m, 1H), 2.21-2.30 (m, 1H), 2.40-2.52 (m, 2H), 4.38-4.44 (m, 1H), 5.07-5.25 (m, 8H), 7.33-7.79 (m, 15H).
4.1.6.2. (S)-2-((S)-1-Carboxy-3-methylbutylcarbamoyloxy)pentanedioic acid (7).
Compound 7 was prepared as was described for ZJ-43 except (S)-dibenzyl 2-((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-ylcarbamoyloxy)pentanedioate was used in place of (S)-dibenzyl 2-(3-((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yl)ureido)pentanedioate. Filtration through celite afforded 7 as a white solid (99% yield); mp 37-42 °C; 1H NMR (DMSO-d6) δ 0.84 (d, J = 6.4 Hz, 3H), 0.88 (d, J = 6.4 Hz, 3H), 1.42-1.57 (m, 2H), 1.63-1.69 (m, 1H), 1.85-1.90 (m, 1H), 1.96-2.04 (m, 1H), 2.27-2.42 (m, 2H), 3.90-3.96 (m, 1H), 4.71-4.75 (m, 1H), 7.65 (d, J = 8.3 Hz, 1H), 12.57 (brs, 3H). LCMS (20% acetonitrile/80% H2O for 0.25 minutes followed by an increase to 85% acetonitrile/15% H2O over 1.75 minutes and continuation of 85% acetonitrile/15% H2O until 4 minutes): retention time 0.43 min, m/z 304.1 [M - H]−.
4.1.7. Synthesis of (S)-2-((R)-1-carboxy-3-methylbutylcarbamoyloxy)pentanedioic acid (8).
4.1.7.1. (S)-Dibenzyl 2-((R)-1-(benzyloxy)-4-methyl-1-oxopentan-2-ylcarbamoyloxy)pentanedioate.
(S)-Dibenzyl 2-((R)-1-(benzyloxy)-4-methyl-1-oxopentan-2-ylcarbamoyloxy)pentanedioate was prepared as was described for (S)-dibenzyl 2-(3-((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yl)ureido)pentanedioate except (R)-benzyl 2-amino-4-methylpentanoate hydrochloride was used in place of (S)-benzyl 2-amino-4-methylpentanoate hydrochloride and (S)-dibenzyl 2-hydroxypentanedioate was used in place of (S)-dibenzyl 2-aminopentanedioate . The reside was purified by silica gel chromatography (25%-40% EtOAc/hexanes): white solid (45% yield); 1H NMR (CDCl3) δ 0.92 (m, 6H), 1.27 (m, 1H), 1.53-1.66 (m, 2H), 2.16-2.28 (m, 2H), 2.42 -2.51 (m, 2H), 4.43 (m, 1H), 5.08-5.21 (m, 6H), 5.31 (s, 1H), 7.33-7.36 (m, 15H).
4.1.7.2. (S)-2-((R)-1-Carboxy-3-methylbutylcarbamoyloxy)pentanedioic acid (8).
Compound 8 was prepared as was described for ZJ-43 except (S)-dibenzyl 2-((R)-1-(benzyloxy)-4-methyl-1-oxopentan-2-ylcarbamoyloxy)pentanedioate was used in place of (S)-dibenzyl 2-(3-((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yl)ureido)pentanedioate. Filtration through celite afforded a white solid (99% yield); mp 48-50 °C;1H NMR (D2o) δ 0.83 (d, J = 6.3 Hz, 3H), 0.86 (d, J – 6.3 Hz, 3H), 1.53-1.67 (m, 3H), 2.01-2.17 (m, 2H), 2.41-2.51 (m, 2H), 4.10 (m, 0.8H, major rotomer), 4.22 (m, 0.2H, minor rotomer), 4.86-4.88 (m, 1H). LCMS (20% acetonitrile/80% H2O for 0.25 minutes followed by an increase to 85% acetonitrile/15% H2O over 1.75 minutes and continuation of 85% acetonitrile/15% H2O until 4 minutes): retention time 0.44 min, m/z 304.1 [M-H]−.
4.1.8. Synthesis of (R)-2-((S)-1-carboxy-3-methylbutylcarbamoyloxy)pentanedioic acid (9).
4.1.8.1. (R)-Dibenzyl 2-((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-ylcarbamoyloxy)pentanedioate.
(R)-Dibenzyl 2-((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-ylcarbamoyloxy)pentanedioate was prepared as was described for (S)-dibenzyl 2-(3-((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yl)ureido)pentanedioate except (R)-dibenzyl 2-hydroxypentanedioate26 was used in place of (S)-dibenzyl 2-aminopentanedioate. The reside purified by silica gel chromatography (25%-40% EtOAc/hexanes): white solid (76% yield); 1H NMR (CDCl3) δ 0.90-0.93 (m, 6H), 1.51-1.68 (m, 3H), 2.15-2.26 (m, 2H), 2.40-2.51 (m, 2H), 4.40-4.41 (m, 1H), 5.06-5.21 (m, 6H), 5.31 (d, J = 8.2 Hz, 1H), 7.33-7.36 (m, 15H).
4.1.8.2. (R)-2-((S)-1-Carboxy-3-methylbutylcarbamoyloxy)pentanedioic acid (9).
Compound 9 was prepared as was described for ZJ-43 except (R)-dibenzyl 2-((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-ylcarbamoyloxy)pentanedioate was used in place of (S)-dibenzyl 2-(3-((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yl)ureido)pentanedioate. Filtration through celite afforded a white solid (99% yield); mp 37-40 °C; 1H NMR (D2O) δ 0.83 (d, J = 6.3 Hz, 3H), 0.86 (d, J = 6.3 Hz, 3H), 1.50-1.57 (m, 3H), 1.97-2.09 (m, 2H), 2.37-2.48 (m, 2H), 4.09-4.13 (m, 0.7H, major rotomer), 4.20-4.24 (m, 0.3H, minor rotomer), 4.80-4.87 (m, 1H). LCMS (20% acetonitrile/80% H2O for 0.25 minutes followed by an increase to 85% acetonitrile/15% H2O over 1.75 minutes and continuation of 85% acetonitrile/15% H2O until 4 minutes): retention time 0.43 min, m/z 304.1 [M - H]−.
4.1.9. Synthesis of (R)-2-((R)-1-carboxy-3-methylbutylcarbamoyloxy)pentanedioic acid (10).
4.1.9.2. (R)-Dibenzyl 2-((R)-1-(benzyloxy)-4-methyl-1-oxopentan-2-ylcarbamoyloxy)pentanedioate.
(R)-Dibenzyl 2-((R)-1-(benzyloxy)-4-methyl-1-oxopentan-2-ylcarbamoyloxy)pentanedioate was prepared as was described for (S)-dibenzyl 2-(3-((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yl)ureido)pentanedioate except (R)-dibenzyl 2-hydroxypentanedioate was used in place of (S)-dibenzyl 2-aminopentanedioate and (R)-benzyl 2-amino-4-methylpentanoate was used in place of (S)-benzyl 2-amino-4-methylpentanoate. The residue purified by silica gel chromatography (25%-40% EtOAc/hexanes), but used in the subsequent step without further purification (40% yield).
4.1.9.2. (R)-2-((R)-1-Carboxy-3-methylbutylcarbamoyloxy)pentanedioic acid (10).
Compound 10, was prepared as was described for ZJ-43 except (R)-dibenzyl 2-((R)-1-(benzyloxy)-4-methyl-1-oxopcntan-2-ylcarbamoyloxy)pentanedioate was used in place of (S)-dibenzyl 2-(3-((S)-1-(benzyloxy)-4-methyl-1-oxopentan-2-yl)ureido)pentanedioate. Filtration through celite afforded a white solid (99% yield); mp 40-43 °C; 1H NMR (D2O) δ 0.82 (d, J = 6.3 Hz, 3H), 0.86 (d, J = 6.3 Hz, 3H) 1.56-1.68 (m, 3H), 2.02-2.19 (m, 2H), 2.45-2.50 (m, 2H), 4.03-4.13 (m, 1H), 4.83-4.87 (m, 1H). LCMS (20% acetonitrile/80% H2O for 0.25 minutes followed by an increase to 85% acetonitrile/15% H2O over 1.75 minutes and continuation of 85% acetonitrile/15% H2O until 4 minutes): retention time 0.42 min, m/z 304.1 [M - H]−.
4.2. GCPII Assay
4.2.1. GCPII expression and purification
Expression and purification of the extracellular domain of human GCPII (amino acids 44 – 750) were carried out as described previously.27 N-terminally tagged GCPII was overexpressed in Schneider’s S2 cells and concentrated to 1/10 of the original volume using tangential flow filtration TFF (Millipore Mosheim, France). Concentrated medium was dialyzed against 50 mM Tris-HCl, 150 mM NaCl, pH 8.0, at 4°C overnight and affinity purified on the Streptactin Sepharose column (IBA, Germany). The N-terminal Strep tag was cleaved by the TEV protease (1:50 molar ratio) at 4°C overnight and the final purification step on Superdex 200 16/60 column (GE Healthcare Bio-Sciences, Uppsala, Sweden) in 20 mM Tris-HCl, 150 mM NaCl, pH 8.0, yielded GCPII with >98% purity. The protein was concentrated to 10 mg/mL and kept at −80°C until further use.
4.2.2. IC50 determination
Inhibition constants of studied inhibitors were determined using the radioenzymatic assay with 3H-NAAG (radiolabeled at the terminal glutamate) as a substrate.28 Briefly, rhGCPII (30 ng/ml) was preincubated in the presence of increasing concentrations of inhibitors in 20 mM Tris, 150 mM NaCl, pH 8.0, for 15 min at 37°C in the total volume of 80 μl. The reaction was initiated by addition of 40 μl of 0.31 μM NAAG (Sigma) and 15 nM 3H-NAAG (50 Ci/mmol, Perkin Elmer) mixture (120 μl total reaction volume). The reaction was terminated after 20 mins by 120 μl of 200 mM potassium phosphate, 50 mM EDTA, 2 mM β-mercaptoethanol, pH 7.4. The released glutamate was separated from the reaction mixture by ion-exchange chromatography and quantified by liquid scintillation. Duplicate reactions were carried out for each experimental point. The data were fitted using the GraphPad Prism software (GraphPad Software, San Diego, CA, USA) and IC50 values were calculated from the inhibition curves of three independent experiments using a non-linear analysis protocol.
4.3. Structural biology
4.3.1. Crystallization and data collection
Diffracting crystals of GCPII/inhibitor complexes were obtained using procedures described previously 20. Briefly, GCPII (10 mg/ml) was mixed with a 20 mM stock solution of a given inhibitor in water (neutralized with NaOH) at the 20:1 molar ratio and the GCPII/inhibitor solution was then combined with an equivalent volume of the reservoir solution (33% pentaerythritol propoxylate, (Sigma), 2.0% polyethylene glycol 3350 (Sigma), and 100 mM Tris–HCl, pH 8.0). Diffraction quality crystals grew in hanging-drops at 293 K for 14 days and then were harvested and flash frozen in liquid nitrogen directly from crystallization droplets. Diffraction intensities for each complex were collected at 100 K using synchrotron radiation at MX P13 beamline (EMBL Hamburg at the PETRA III storage ring, DESY, Hamburg, Germany; 0.98 Å) equipped with the Dectris PILATUS 3 6M (Dectris AG, Swizerland). The complete dataset for each complex was collected from a single crystal and data were processed using XDS 29. The final data collection statistics are shown in Table 2.
4.3.2. Structure determination, refinement, and analysis
Difference Fourier methods were used to determine structures of GCPII/inhibitor complexes with the 3D7H structure used as a starting model. Calculations were performed using Refmac 5.5.30 and the refinement protocol was interspersed with manual corrections to the model employing the program Coot 0.6.31 The PRODRG server was used to generate restrains library and coordinate files for individual inhibitors.32 Inhibitors were fitted into the positive electron density map in the final stages of the refinement. Approximately 1,000 of the randomly selected reflections were kept aside for cross-validation (Rfree) during the refinement process. The quality of the final models was evaluated using the MOLPROBITY software33 and relevant statistics are summarized in Table 2.
4.4. Computational methods
4.4.1. QM/MM calculations
The computational protocols and the general QM/MM setup that was previously successfully applied in mechanistic and structural studies of GCPII16, 18, 34 has been employed in this work. This also concerns the initial protein structure (further denoted St1) used in the modelling. The St1 is the fully equilibrated all-atom QM/MM model originating in the 1.71 Å resolution X-ray structure of the E424A mutant with the N-Ac-Asp-Glu ligand (PDB code 3BXM). This included a few missing residues in the 3BXM crystal structure, added solvation sphere of ~12 700 water molecules (radius of 50 Å from the center of the protein), and two Na+ ions to neutralize the overall charge of the protein. Apart from utilizing the previous (non-trivial) protein preparation and equilibration,34 we believe that the adopted approach also warrants unbiased structure of the ligand binding. The full St1 structure (in PDB format) is deposited as the supporting information. The ligands were then positioned into the St1 by its superimposition with particular ligand-protein crystal structure reported in the experimental part of this work. The quantum system consisted of 324 atoms for 7 and 5 ligand structures, and of 325 atoms for urea-based ZJ-43, whereas the part of the protein that was allowed to relax in the QM/MM calculations comprised 29 residues and 10 water molecules in the vicinity of the QM system (R= 2.0 Å) – the same for all three studied ligand-protein complexes.
4.4.2. Calculations of ligand strain energies and solvation/desolvation energies
The Gibbs free energy of the equilibrium structure found by the DFT-D3/COSMO-RS//MD-LLMOD methodology was calculated as: G = Eel + Gsolv + EZPVE + pV − RT lnQ, where i) Eel term was obtained using the DLPNO-CCSD(T)/aug-cc-pVDZ method35 as implemented in ORCA 4.0 program;36 ii) EZPVE + pV − RTlnQ term was obtained from frequency calculations with the rigid rotor/harmonic oscillator approximation (for p = 1 bar); and iii) the Gsolv was calculated using the conductor-like screening model for realistic solvation (COSMO-RS) with the radii-based isosurface cavity and the COSMOtherm parameter set BP_TZVPD_FINE_C30_1701.ctd as available in COSMOtherm17 37, 38. The COSMO-RS calculations were carried out following the recommended protocol: RI-BP86-D3/def2-TZVPD/cosmo (ε = ∞) ≡ ECOSMO,∞ and RI-BP86-D3/def2-TZVPD/in vacuo ≡ Ein vacuo single-point calculations were used to calculate Gsolv = (ECOSMO,∞ − Ein vacuo) + μCOSMO-RS(T), where μ(T) is the temperature dependent COSMO-RS chemical potential, as defined in 37. From the practical reasons, we further decompose the strain energy into two parts: i) from equilibrium conformation to the local minimum obtained by geometry optimization of the enzyme-bound geometry (Eglobal-local), and ii) from local minimum to the geometry in enzyme (Elocal-enzyme). The first can be calculated using all contributions to the Gibbs free energy, while the second neglects the thermal contributions to the Gibbs free energy, originating from the frequency calculations (the EZPVE + pV − RTlnQ term), as the enzyme-bound conformation is in constrained geometry invoked by the enzyme environment. The geometry optimizations of the enzyme-bound structures were employed using the RI-TPSS-D3/def2-SV(P)/cosmo(ε = 80) method 39-42 with the Turbomole 7.2.1 program (http://www.turbomole.com). The level of theory is consistent with that of the QM/MM calculations. The desolvation energy (Gdesolv) is estimated from the difference in Gsolv calculated for the enzyme-bound geometry for water and for 1-octanol, mimicking the enzyme environment. This is referenced to the urea-based inhibitor ZJ-43, for which the ΔGsolv was set to zero.
Supplementary Material
Table S1 Primary energy data: molecular energies in gas-phase, E(el), solvation energies, G(solv) and thermal corrections.
Table S2 1H and 13C NMR data of compounds ZJ-43, 5, and 7
Table S3 1H spectrra of compounds 4-10.
Coordinates.zip Equilibrium geometries of the QM systems and one all-atom PDB structure (denoted St1 above)
Acknowledgment:
We acknowledge Iva Jelinkova, Barbora Havlinova, and Petra Baranova for the excellent technical assistance and DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the use of experimental facilities. Parts of this research were carried out at the PETRA III storage ring and we thank Isabel Bento for assistance in using the P13 beamline. This work was in part supported by the CAS (RVO: 86652036), MEYS CR (LM2015043 CIISB), and projects CIISB4HEALTH (CZ.02.1.01/0.0/0.0/16_013/0001776) and BIOCEV (CZ.1.05/1.1.00/02.0109) from the ERDF and by NIH grants R01CA161056 and P30MH075673 awarded to BSS. LR acknowledges the support from Grant Agency of the Czech Republic (grant 17-24155S) and computer time at IT4I infrastructure (MEYS grant LM2015070, Large Infrastructures for Research, Experimental Development and Innovations: IT4Innovations National Supercomputing Center).
Abbreviations Used:
- GCPII
glutamate carboxypeptidase II
- PSMA
prostate-specific membrane antigen
- SAR
structure-activity relationship
- NAAG
N-acetyl-aspartyl-glutamate
- ZBG
zinc-binding group
- QM/MM
quantum mechanics/molecular mechanics
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Zhou J, Neale JH, Pomper MG, et al. , NAAG peptidase inhibitors and their potential for diagnosis and therapy. Nat. Rev. Drug Discov 2005;4:1015–1026. [DOI] [PubMed] [Google Scholar]
- [2].Ferraris DV, Shukla K, Tsukamoto T, Structure-activity relationships of glutamate carboxypeptidase II (GCPII) inhibitors. Curr. Med. Chem 2012;19:1282–1294. [DOI] [PubMed] [Google Scholar]
- [3].Vornov JJ, Hollinger KR, Jackson PF, et al. , Still NAAG'ing After All These Years: The Continuing Pursuit of GCPII Inhibitors. Adv Pharmacol. 2016;76:215–255. [DOI] [PubMed] [Google Scholar]
- [4].Eiber M, Fendler WP, Rowe SP, et al. , Prostate-Specific Membrane Antigen Ligands for Imaging and Therapy. J Nucl Med. 2017;58:67S–76S. [DOI] [PubMed] [Google Scholar]
- [5].Rais R, Jiang W, Zhai H, et al. , FOLH1/GCPII is elevated in IBD patients, and its inhibition ameliorates murine IBD abnormalities. JCI Insight. 2016;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kozikowski AP, Nan F, Conti P, et al. , Design of remarkably simple, yet potent urea-based inhibitors of glutamate carboxypeptidase II (NAALADase). J. Med. Chem 2001;44:298–301. [DOI] [PubMed] [Google Scholar]
- [7].Kozikowski AP, Zhang J, Nan F, et al. , Synthesis of urea-based inhibitors as active site probes of glutamate carboxypeptidase II: efficacy as analgesic agents. J. Med. Chem 2004;47:1729–1738. [DOI] [PubMed] [Google Scholar]
- [8].Olszewski RT, Bukhari N, Zhou J, et al. , NAAG peptidase inhibition reduces locomotor activity and some stereotypes in the PCP model of schizophrenia via group II mGluR. J Neurochem. 2004;89:876–885. [DOI] [PubMed] [Google Scholar]
- [9].Mease RC, Dusich CL, Foss CA, et al. , N-[N-[(S)-1,3-Dicarboxypropyl]carbamoyl]-4-[18F]fluorobenzyl-L-cysteine, [18F]DCFBC: a new imaging probe for prostate cancer. Clin Cancer Res. 2008;14:3036–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Olszewski RT, Wegorzewska MM, Monteiro AC, et al. , Phencyclidine and dizocilpine induced behaviors reduced by N-acetylaspartylglutamate peptidase inhibition via metabotropic glutamate receptors. Biol Psychiatry. 2008;63:86–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yamamoto T, Kozikowski A, Zhou J, et al. , Intracerebroventricular administration of N-acetylaspartylglutamate (NAAG) peptidase inhibitors is analgesic in inflammatory pain. Molecular pain. 2008;4:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhang AX, Murelli RP, Barinka C, et al. , A remote arene-binding site on prostate specific membrane antigen revealed by antibody-recruiting small molecules. J Am Chem Soc. 2010;132:12711–12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Feng JF, Van KC, Gurkoff GG, et al. , Post-injury administration of NAAG peptidase inhibitor prodrug, PGI-02776, in experimental TBI. Brain Res. 2011;1395:62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kratochwil C, Giesel FL, Eder M, et al. , [(1)(7)(7)Lu]Lutetium-labelled PSMA ligand-induced remission in a patient with metastatic prostate cancer. Eur J Nucl Med Mol Imaging. 2015;42:987–988. [DOI] [PubMed] [Google Scholar]
- [15].Yang X, Mease RC, Pullambhatla M, et al. , [(18)F]Fluorobenzoyllysinepentanedioic Acid Carbamates: New Scaffolds for Positron Emission Tomography (PET) Imaging of Prostate-Specific Membrane Antigen (PSMA). J Med Chem. 2016;59:206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Klusak V, Barinka C, Plechanovova A, et al. , Reaction mechanism of glutamate carboxypeptidase II revealed by mutagenesis, X-ray crystallography, and computational methods. Biochemistry. 2009;48:4126–4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mlcochova P, Plechanovova A, Barinka C, et al. , Mapping of the active site of glutamate carboxypeptidase II by site-directed mutagenesis. FEBS J. 2007;274:4731–4741. [DOI] [PubMed] [Google Scholar]
- [18].Plechanovova A, Byun Y, Alquicer G, et al. , Novel substrate-based inhibitors of human glutamate carboxypeptidase II with enhanced lipophilicity. J Med Chem. 2011;54:7535–7546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Slusher BS, Rojas C, Coyle JT, Chapter 367 - Glutamate Carboxypeptidase II A2 - Rawlings, Neil D, in: Salvesen G (Ed.) Handbook of Proteolytic Enzymes, Academic Press; 2013, pp. 1620–1627. [Google Scholar]
- [20].Barinka C, Byun Y, Dusich CL, et al. , Interactions between human glutamate carboxypeptidase II and urea-based inhibitors: structural characterization. J Med Chem. 2008;51:7737–7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kopka K, Benesova M, Barinka C, et al. , Glu-Ureido-Based Inhibitors of Prostate-Specific Membrane Antigen: Lessons Learned During the Development of a Novel Class of Low-Molecular-Weight Theranostic Radiotracers. J Nucl Med. 2017;58:17S–26S. [DOI] [PubMed] [Google Scholar]
- [22].Gutten O, Bim D, Rezac J, et al. , Macrocycle Conformational Sampling by DFT-D3/COSMO-RS Methodology. J Chem Inf Model. 2018;58:48–60. [DOI] [PubMed] [Google Scholar]
- [23].Mesters JR, Barinka C, Li W, et al. , Structure of glutamate carboxypeptidase II, a drug target in neuronal damage and prostate cancer. EMBO J. 2006;22:1375–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pavlicek J, Ptacek J, Barinka C, Glutamate carboxypeptidase II: an overview of structural studies and their importance for structure-based drug design and deciphering the reaction mechanism of the enzyme. Curr Med Chem. 2012;19:1300–1309. [DOI] [PubMed] [Google Scholar]
- [25].Buck SB, Huff JK, Himes RH, et al. , Total synthesis and anti-tubulin activity of epi-c3 analogues of cryptophycin-24. J Med Chem. 2004;47:3697–3699. [DOI] [PubMed] [Google Scholar]
- [26].Wiesner M, Neff B, Method for the production of bicyclic aromatic amino acids and intermediate products thereof, US 7,371,854, 2008. [Google Scholar]
- [27].Conway RE, Rojas C, Alt J, et al. , Prostate-specific membrane antigen (PSMA)-mediated laminin proteolysis generates a pro-angiogenic peptide. Angiogenesis. 2016;19:487–500. [DOI] [PubMed] [Google Scholar]
- [28].Hlouchova K, Barinka C, Konvalinka J, et al. , Structural insight into the evolutionary and pharmacologic homology of glutamate carboxypeptidases II and III. FEBS J. 2009;276:4448–4462. [DOI] [PubMed] [Google Scholar]
- [29].Kabsch W, Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Murshudov GN, Skubak P, Lebedev AA, et al. , REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Emsley P, Lohkamp B, Scott WG, et al. , Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Schuttelkopf AW, van Aalten DM, PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. [DOI] [PubMed] [Google Scholar]
- [33].Chen VB, Arendall WB 3rd, Headd JJ, et al. , MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Navratil M, Tykvart J, Schimer J, et al. , Comparison of human glutamate carboxypeptidases II and III reveals their divergent substrate specificities. FEBS J. 2016;283:2528–2545. [DOI] [PubMed] [Google Scholar]
- [35].Sparta M, Neese F, Chemical applications carried out by local pair natural orbital based coupled-cluster methods. Chem Soc Rev. 2014;43:5032–5041. [DOI] [PubMed] [Google Scholar]
- [36].Neese F, The ORCA program system. WIREs Comput Mol Sci. 2012;2:73–78. [Google Scholar]
- [37].Klamt A, Conductor-like Screening Model for Real Solvents: A New Approach to the Quantitative Calculation of Solvation Phenomena. J. Phys. Chem 1995;99:2224–2235. [Google Scholar]
- [38].Klamt A, Jonas V, Börger T, et al. , Refinement and Parametrization of COSMO-RS. J. Phys. Chem 1998;102:5074–5085. [Google Scholar]
- [39].Eichkorn K, Treutler O, Öhm H, et al. , Auxiliary basis sets to approximate Coulomb potentials. Chem. Phys. Lett 1995;240:283–290. [Google Scholar]
- [40].Grimme S, Antony J, Ehrlich S, et al. , A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys. 2010;132:154104. [DOI] [PubMed] [Google Scholar]
- [41].Klamt A, Schüürmann G, COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2 1993:799–805. [Google Scholar]
- [42].Tao J, Perdew JP, Staroverov VN, et al. , Climbing the density functional ladder: nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys Rev Lett. 2003;91:146401. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Primary energy data: molecular energies in gas-phase, E(el), solvation energies, G(solv) and thermal corrections.
Table S2 1H and 13C NMR data of compounds ZJ-43, 5, and 7
Table S3 1H spectrra of compounds 4-10.
Coordinates.zip Equilibrium geometries of the QM systems and one all-atom PDB structure (denoted St1 above)