

Abstract
Background:
Protein disulfide isomerase (PDI) serves an essential role in thrombus formation and PDI inhibition is being evaluated clinically as a novel anticoagulant. Yet little is known about the regulation of PDI in the vasculature. Thiols within the catalytic motif of PDI are essential for its role in thrombosis. These same thiols bind nitric oxide (NO), a potent regulator of vessel function. To determine whether regulation of PDI represents a mechanism by which NO controls vascular quiescence, we evaluated the effect of NO on PDI function in endothelial cells, platelets, and thrombus formation in vivo.
Aim:
To assess the effect of S-nitrosylation on the regulation of PDI and other thiol isomerases in the vasculature.
Methods and results:
The role of endogenous NO on PDI activity was evaluated by incubating endothelium with a NO scavenger, which resulted in exposure of free thiols, increased thiol isomerase activity, and enhanced thrombin generation on the cell membrane. Conversely, exposure of endothelium to NO+ carriers or elevation of endogenous NO levels by induction of NO synthesis resulted in S-nitrosylation of PDI and decreased surface thiol reductase activity. S-nitrosylation of platelet PDI inhibited its reductase activity and S-nitrosylated PDI interfered with platelet aggregation, α-granule release, and thrombin generation on platelets. S-nitrosylated PDI also blocked laser-induced thrombus formation when infused into mice. S-nitrosylated ERp5 and ERp57 were found to have similar inhibitory activity.
Conclusions:
These studies identify NO as a critical regulator of vascular PDI and define regulation of PDI function as an important mechanism by which NO maintains vascular quiescence.
Keywords: Nitric Oxide, Protein Disulfide Isomerase, S-nitrosylation, Thrombosis, Platelets
Introduction
Protein disulfide isomerase (PDI) is a multifunctional thiol isomerase that serves a critical role in both protein folding and regulating disulfide-bond formation in nascent proteins synthesized in the endoplasmic reticulum. PDI has an a-b-b’-a’ domain structure. The a and a’ domains contain a CGHC (Cys-Gly-His-Cys) catalytic motif responsible for its oxidoreductase activities, while the b and b’ domains serve as substrate binding domains [1,2]. In addition to its localization at high concentrations in the endoplasmic reticulum, PDI can traffic to extracellular compartments [3,4], including to external membranes of vascular cells [5,6]. There is now compelling evidence that extracellular PDI functions in thrombus formation in vivo [7,8]. Inhibition of PDI by either inhibitory antibodies or small molecules directed at PDI blocks both platelet accumulation and fibrin formation following multiple models of vascular injury [7,9–12]. However, the mechanisms by which PDI contributes to thrombosis and how PDI is regulated in the vasculature are poorly understood.
NO serves a critical role in maintaining vascular patency [13,14]. NO produced by endothelial nitric oxide synthase (eNOS) controls vascular tone and, together with endothelial prostaglandins [15], inhibits platelet-endothelial cell interactions in resting blood vessels [16]. The mechanism of NO inhibition of platelet activity appears to be multifaceted. NO binds to the heme moiety of soluble guanylyl cyclase (sGC), up-regulating its activity, resulting in increased cytoplasmic levels of platelet cGMP. cGMP activates protein kinase G, which phosphorylates multiple substrates and inhibits platelet reactivity [17]. NO also blocks platelet reactivity via a guanylyl cyclase-independent pathway that may involve S-nitrosylation of surface proteins [18–22].
NO can interact with PDI and this interaction can have several consequences. First, PDI can nitrosylate substrates, adding NO to form S-nitrosylated proteins, or denitrosylate substrates, removing NO from S-nitrosylated proteins [23,24]. Second, the reaction of NO with PDI can inhibit its activity [25,26]. Third, PDI on the cell surface can act as a transnitrosylase, delivering NO into cells [27]. Studies in HEL cells showed that knockdown of PDI reduced elevation of cGMP observed upon incubation with exogenous NO [28]. Extracellular vascular PDI participates in thrombosis mainly through disulfide bond reshuffling in target substrates [29,30]. Since vascular cells synthesize NO that can interact with extracellular thiol isomerases, we reasoned that NO might control vascular PDI activity by reacting with its active site cysteines and blocking its oxidoreductase activities.
We now show that endothelial PDI activity is controlled by NO synthesis, which functions in the S-nitrosylation of PDI, and that this reaction inhibits extracellular isomerase activity. Furthermore, NO is capable of inhibiting PDI-dependent thrombin generation on the endothelial cell surface. Platelet PDI is also controlled by S-nitrosylation, which inhibits disulfide cleavage on its cell surface. PDI is capable of transferring NO into platelets and S-nitrosylated PDI (SNO-PDI) interferes with platelet aggregation and granule release. In a mouse model of thrombus formation, S-nitrosylated PDI is antithrombotic, inhibiting both platelet accumulation and fibrin formation induced by vascular injury. These studies demonstrate that S-nitrosylation is an important regulatory mechanism for controlling PDI activity in blood vessels and reveal a previously unrecognized mechanism by which NO maintains vascular quiescence.
Methods
Recombinant protein purification
ERp5 (His-tagged), ERp57, PDI and catalytically inactive (AGHA) PDI (FLAG-tagged on N-terminus and streptavidin-binding protein-tagged on the C-terminus) were cloned into a pET-15b vector and expressed in the BL21 strain of E. Coli and purified as described previously [11].
Di-eosin-GSSG disulfide assay
Redox activity of thiol isomerases was assessed using di-eosin-GSSG as described previously [11,31]. For inhibition studies using NO+ carriers, cPTIO and eNOS manipulators no reducing agent was used.
Preparation of S-nitrosylated thiol isomerases
Nitric oxide carriers were prepared as previously described [32]. For the preparation of S-nitrosylated thiol isomerasees, a 200-fold molar excess of S-nitrosocysteine (SNOC) was incubated with PDI, ERp5 or ERp57 and reacted for 30 minutes at 37°C. For experiments were removal of the NO+ carrier was required, samples were acetone precipitated, resuspended and subjected to size-exclusion. The filtrate containing any decayed NO+ carrier was termed ‘eluate’ and used as a negative control.
Labeling of –SNO and -SH bonds
For S-nitrosylation, thiol isomerases were incubated with a 200-molar excess of SNOC or vehicle in PBS. After 30 minutes at 37°C, HENS buffer (100 mM HEPES (pH 7.3), 1 mM EDTA, 0.1 mM Neocuproine, 1% SDS) containing 50 mM NEM was added to block any remaining free thiols. Subsequently, 6 volumes of ice-cold acetone (−20°C) were added and thiol isomerases were precipitated and resuspended in HENS buffer containing 5 mM L-ascorbic acid and 100 μM 5-fluorescein-maleimide. Similarly, washed platelets were exposed to 20 mM S-nitrosoglutatione (GSNO) or vehicle and subsequently lysed in HENS buffer containing 20 mM NEM. Acetone-precipitated pellets were resuspended in PBS containing 10 mM maleimide-PEG2-biotin (MPB) and 20 mM L-ascorbic acid. Following another acetone precipitation pellets were resuspended in PBS and lysates were loaded on a monomeric avidin column (Thermo Fisher) as per the manufacturer. PDI was detected via western blot using an anti-PDI antibody (RL-90, 1:1000). Confluent HUVECs were incubated for 24 hours with vehicle, L-NAME (1 mM), histamine (100 μM) or L-NAME and histamine. Subsequently, cells were washed thrice with PBS and lysed using HENS buffer containing 50 mM NEM. Acetone-precipitated cell pellets were resuspended in HENS buffer containing 5 mM L-ascorbid acid and 100 μM MPB. After desalting, lysates were subjected to streptavidin-coated magnetic beads and biotinylated proteins were captured. PDI was detected via western blot using an anti-PDI antibody (RL-90, 1:1000). For the measurement of –SH bonds in HUVECS, cells were subjected to 100 μM of cPTIO or vehicle control for 3 hours. Cells were subsequently lysed using HENS buffer and 100 μM 5-fluorescein-maleimide was added to label free thiols. Gels were subjected to Cy2-light emitted by the ImageQuant LS4000.
Cellular NO detection
Nitric oxide incorporation in washed platelets using the intracellular fluorescent probe 4-amino-5-methylamino-2’,7’-difluorofluorescein diacetate (DAF-FM diacetate) (Ex: 485 nM/Em: 535 nM) was assessed as described previously with minor changes [27]. For endothelial cells, 2 μM DAF-FM diacetate was incubated for 1 hour and subsequently cells were washed thrice with PBS (pH 7.4) and incubated with eNOS manipulators or indicated concentrations of GSNO. DAF-FM fluorescence was then measured. For extracellular NO detection on HUVECs, 2,3-diamino-naphthalene (DAN) fluorescence was used (Ex: 375 nm, Em: 415 nm).
Platelet aggregation
Washed human platelets were generated as previously described [11]. Platelets (2.5 × 108 / mL) were incubated for 10 minutes with SNO-PDI, SNO-ERp5 or SNO-ERp57 or the control eluate obtained after size-exclusion of SNO-PDI and decayed NO+ carrier. Platelets were stimulated with 3 μM of the PAR-1 activating peptide, SFLLRN. Aggregation was measured by light transmission using a Chrono-Log 680 Aggregation System.
FXa and Thrombin Generation Assay
Confluent HUVECs were incubated with either 100 μM of cPTIO, 10 μM N-(2,4-Dimethoxyphenyl)-N-(1-oxo-2-propyn-1-yl)-2-(2-thienyl)glycyl-glycine ethyl ester (PACMA-31), 1 mM SNOC or vehicle for 30 min followed by a wash as indicated. Subsequently cells were incubated with LPS (final concentration 100 ng/mL), sCD14 (final concentration 10 ng/mL) and LBP (final concentration 10 ng/mL) for 3 hours. Cells were washed three times with HBS-BSA assay buffer (20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 150 mM NaCl, 5 mM KCl, 5 mM CaCl2 and 1 mg/mL BSA, pH 7.4). FX (Haematologic Technologies) was added to a final concentration of 100 nM, followed by addition of 200 μM of FXa chromogenic substrate (Biophen). FXa generation was initiated upon addition of 10 nM FVIIa (Haematologic Technologies) and absorbance was measured at 405 nm for 90–120 minutes using the SpectraMax spectrophotometer. For thrombin generation, medium was replaced by a mix of pooled human plasma 5 mM Gly-Pro-Arg-Pro (GPRP) in Hepes Buffered Saline (pH 7.4) in a ratio 80µl/20µl. Thrombin generation was initiated with CaCl2 and measured as previously described [29]. For platelet-dependent thrombin generation, platelet-rich plasma (PRP) was diluted to platelet concentration of 250,000 platelets/μL and thrombin generation was measured as previously described [29].
P-Selectin exposure
Washed human platelets were incubated for 10 minutes with SNO-PDI or the eluate obtained after size-exclusion of SNO-PDI and decayed NO+ carrier. After exposure to 3 µM of SFLLRN, P-selectin exposure was measured using PE-conjugated CD62P (BD Biosciences) using a Gallios Flow Cytometer (Beckman Coulter).
Laser-injury model of the cremaster arteriole
WT C57BL/6JRj mice were from Janvier Labs (Le Genest-Saint-Isle, France) and from Jackson Laboratory (Bar Harbor, ME). Approval from local animal welfare committees was obtained. For the visualization of nitric oxide, DAF-FM diacetate was infused into WT mice pretreated with either vehicle or with the NO scavenger 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide (cPTIO). In a separate experiment SNO-PDI (25 μg) or the eluate after size-exclusion of SNO-PDI and decayed NO+ carrier was infused before laser-induced injury. Intravital microscopy of the cremaster arteriole, visualization of platelet accumulation and fibrin formation was performed as previously described [11,33,34].
For additional information on the methods used, see Data S1.
Results
Endogenous NO inhibits thiol isomerase reductase activity on endothelial cells
Regulation of endothelial thiol isomerases is a potentially important mechanism for controlling the initiation of thrombus formation on endothelial cell surfaces, as occurs in inflammatory states [35]. Little is known, however, regarding the regulation of vascular thiol isomerases. Purified PDI is inhibited by NO+, which binds to its catalytically active cysteines (Fig. S1). NO is essential for maintaining vascular quiescence and could act, in part, by inhibiting PDI on endothelium. To visualize NO accumulation in vivo, mice were infused with diacetate-coupled DAF-FM, which is deacetylated upon cellular uptake and fluoresces upon reaction with NO within the endothelium of cremaster arterioles (Fig. 1A). Pretreatment of mice with 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide (cPTIO), a NO scavenger, abrogates the fluorescent signal. To determine whether endogenous NO production affects thiol isomerase reductase activity on the endothelial cell surface, human umbilical cord endothelial cells (HUVECs) were exposed to cPTIO. Incubation with cPTIO resulted in a dose-dependent decrease in endothelial NO as indicated by DAN fluorescence (Fig. 1B). Reductase activity on the endothelial surface was evaluated by testing the ability of the cells to reduce a probe consisting of GSSG labeled with two molecules of eosin (di-eosin-GSSG) [31,36]. A concomitant increase in di-eosin-GSSG cleavage was observed with cPTIO exposure (Fig. 1C). Scavenging of NO also resulted in increased exposure of free thiols as indicated by an increase in maleimide-PEG2-biotin (MPB) labeling of endothelial proteins before and after cPTIO exposure (Fig. 1D). Incubation of endothelial cells with GSNO, a physiological NO+ carrier involved in transnitrosylation, resulted in decreased free thiols (Fig. 1D) and increased intracellular NO, as monitored using DAF-FM (Fig. 1E). GSNO did not affect di-eosin-GSSG fluorescence in the absence of endothelial cells (Fig. S2). Measurable endothelial reductase activity on the endothelial cell surface was reduced to 24.03 ± 11.55 % of baseline activity when the endothelium was incubated with GSNO for 15 minutes (Fig. 1F-G). Similar results were obtained when a longer GSNO exposure was used (Fig. S3A-B). Di-eosin-GSSG is cell impermeable, and therefore this data indicates that NO controls reductase activity on the endothelial cell surface. The endothelium possesses several proteins with Cys-X-X-Cys motifs and enzymes which are able to react with glutathione [37]. Therefore, the relative contribution of protein disulfide isomerase on di-eosin-GSSG turnover was assessed (Fig. S3C-D). Both quercetin-3-rutinoside (rutin) and PACMA-31, at low concentrations known to be relatively selective for PDI, inhibit about 50% of the di-eosin-GSSG turnover at the endothelial cell surface.
Figure 1.
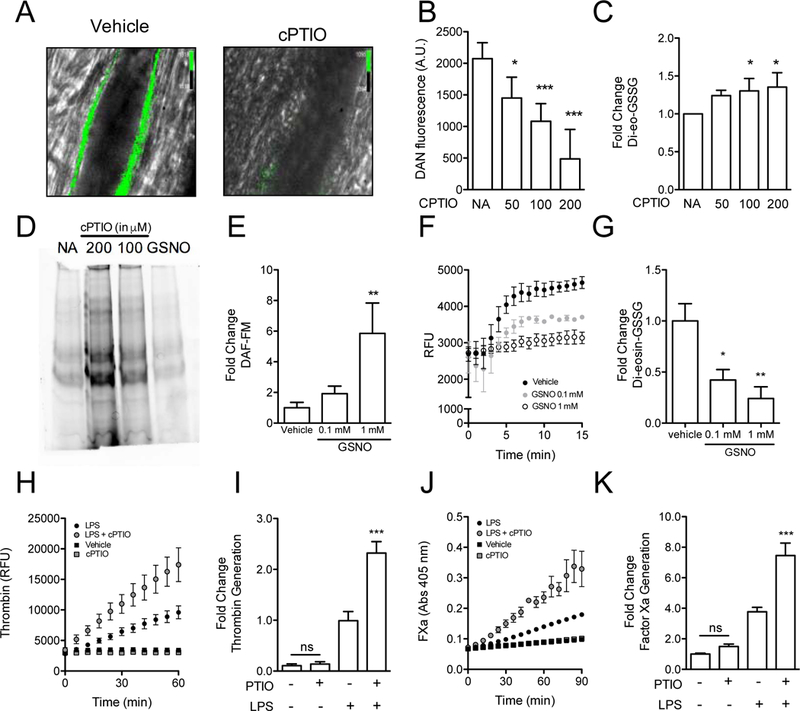
Endogenous nitric oxide controls thiol isomerase activity on the endothelial cell surface. (A) WT C57BL/6RJ mice were infused with the intracellular marker DAF‐FM, to measure intracellular NO generation. Subsequently, mice were exposed to vehicle (left panel) or cPTIO (right panel). Arterioles within the cremaster microvasculature were subsequently visualized using widefield fluorescence microscopy and fluorescence was imaged after 45 minutes (Scale bar: 10 μm). (B) HUVECs were incubated with DAN after 60 minutes exposure to increasing dosage of cPTIO (μM). NO accumulation was assessed by measuring DAN fluorescence (Ex: 375 nm, Em: 450 nm). Bar graphs represent arbitrary units (A.U.) of DAN fluorescence, mean ± SEM (n = 3). (C) Reductase activity was measured in HUVECs after exposure to increasing dosage of cPTIO using a di‐eosin‐GSSG‐based assay. Bar graphs represent fold change of fluorescence as compared to vehicle, mean ± SEM (n = 3). (D) HUVECs were exposed to cPTIO, GSNO or vehicle control (NA) and subsequently free thiols were labelled with 5‐fluorescein maleimide (5‐FM). Cells were subsequently lysed and free thiols visualized using SDS‐polyacrylamide gel electrophoresis followed by fluorescence imaging. (E) HUVECs were incubated with DAF‐FM and then exposed to either vehicle, 0.1 mM or 1 mM of GSNO for 2 hours. Intracellular NO accumulation was assessed by measuring DAF‐FM fluorescence (Ex: 485 nm, Em: 515 nm). Bar graphs represent fold change of fluorescence as compared to vehicle, mean ± SEM (n = 3‐6). (F‐G) Reductase activity was measured in HUVECs exposed to either vehicle, 0.1 mM or 1 mM of GSNO for 15 minutes using a di‐eosin‐GSSG‐based assay. Bar graphs represent fold change of fluorescence as compared to vehicle, mean ± SEM (n = 3). Statistical analysis for bar graphs was performed using one‐way analysis of variance with Bonferroni’s post test (*P < 0.05; **< 0.01). Thrombin (H‐I) and factor Xa (J‐K) generation assays were performed in HUVECs incubated in the presence of 100 μM cPTIO followed by 100 ng/mL LPS. Bar graphs represent fold change in thrombin or FXa generation ± S.E.M. One‐way analysis of variance with Bonferroni’s post test was performed to measure significance (***P < 0.001; ns, non significant). AU, arbitrary units; cPTIO, 2‐phenyl‐4,4,5,5‐tetramethylimidazoline‐1‐oxyl‐3‐oxide; DAF‐FM, 4‐amino‐5‐methylamino‐2′,7′‐difluorofluorescein; DAN, 2,3‐diamino‐naphthalene; FXa, activated factor X; GSNO, S‐nitrosoglutathione; GSSG, glutathione disulfide; LPS, lipopolysaccharide; NA, no addition; NS, not significant; RFU, relative fluorescence units. [Color figure can be viewed at wileyonlinelibrary.com]
Previous studies demonstrating a role for thiol isomerases in thrombin generation on cell surfaces prompted us to evaluate the effect of NO scavenging by cPTIO on thrombin generation on endothelium [38,39]. Incubation with cPTIO enhanced LPS-induced thrombin generation by the endothelium (Fig. 1H-I). To further assess the effect of NO scavenging on the prothrombotic potential of the endothelial surface, we evaluated the effect of cPTIO on endothelial factor Xa generation in the presence of exogenous factors VIIa and X. cPTIO itself showed relatively little enhancement of factor Xa generation on unstimulated endothelium. Significant enhancement by cPTIO was observed, however, in inflamed endothelium exposed to LPS (Fig. 1J-K). These observations indicate that reduction of NO in endothelium enhances both thiol isomerase reductase activity and generation of thrombin.
Endothelial NOS regulation of PDI
NO produced by nitric oxide synthases serves an antithrombotic function in the vasculature. To evaluate the effects of endogenous NO production on endothelial cell thiol isomerase activity, HUVECs were exposed to histamine, which induces NO synthesis [40,41]. Exposure to histamine increased intracellular NO production as evidenced by an increase in DAF-FM fluorescence following exposure (Fig. 2A). Incubation of HUVECs with the NOS inhibitor L N-nitro-L-arginine methyl ester (L-NAME) prior to exposure to histamine blocked the increase in DAF-FM, confirming the activity of NOS isoforms. While informative for detecting NO, DAF-FM can react with dehydroascorbic acid and ascorbic acid, which have been shown to colocalize with nitric oxide synthases [42]. However, we have found no effect of ascorbic acid on DAF-FM fluorescence in our conditions (Fig. S4). Measurement of di-eosin-GSSG cleavage under these same conditions demonstrated that surface reductase activity is inversely proportional to the synthesis of NO; higher levels of NO production correlated with lower levels of reductase activity (Fig. 2B). The inhibition of reductase activity by histamine was mediated through NO synthases as evidenced by the fact that inhibition of NOS by L-NAME blocked the effect of histamine exposure on endothelial cell thiol isomerase activity. The observation that endothelial surface reductase activity is under the control of NO synthases and the fact that PDI is a major endothelial thiol isomerase raised the possibility that endothelial PDI could be regulated by endogenous NO. Specifically, NO produced by NO synthases could bind free thiols within the catalytic motif of PDI and inhibit its reductase activity. To directly assess the effect of NOS activity on the availability of free thiols in PDI, we used an S-nitrosylation switch assay [43]. Following cell lysis and derivatization of free thiols with NEM, S-nitrosylated cysteines were reduced using ascorbate to generate free thiols that were subsequently labeled with MPB. PDI was immunopreciptated and SNO-PDI detected using RL-90, an antibody directed at PDI. This analysis demonstrated S-nitrosylation of PDI following exposure of HUVECs to histamine (Fig. 2C). Histamine-induced S-nitrosylation of PDI was reversed upon preincubation with L-NAME. These data demonstrate a mechanism whereby NO synthesis can regulate endothelial PDI.
Figure 2.

Endothelial nitric oxide synthesis controls PDI on the cell surface of endothelial cells. (A) HUVECs were incubated with either vehicle alone, 1 mM of L‐NAME, 100 μM histamine, or a combination of the latter two for 24 hours. NO levels were then evaluated using DAF‐FM fluorescence. (B) Thiol isomerase activity on the cell surface of HUVECs was assessed after incubation with either vehicle alone, 1 mM of L‐NAME, 100 μM histamine, or a combination of the latter two for 24 hours. (C) HUVECs incubated with either vehicle alone, 1 mM of L‐NAME, 100 μM histamine, or a combination of the latter two were assessed for S‐nitrosylation. Samples were lysed, free thiols were blocked with NEM and then acetone precipitated pellets were resuspended in the presence of 100 μM MPB and 5 mM ascorbic acid. After sample desalting, samples were loaded on streptavidin magnetic beads, washed and the elution was analyzed using SDS‐polyacrylamide gel electrophoresis and western blotting for PDI. Bar graphs represent RFU of DAF‐FM and di‐eosin‐GSSG, mean ± SEM (n = 3‐6). Statistical analysis for bar graphs was performed using one‐way analysis of variance with Bonferroni’s post test (*P < 0.05; **< 0.01). AUC, area under the curve; DAF‐FM, 4‐amino‐5‐methylamino‐2′,7′‐difluorofluorescein; GSSG, glutathione disulfide; l‐NAME, l‐N‐nitro‐l‐arginine methyl ester; PDI, protein disulfide isomerase; RFU, relative fluorescence units.
The endothelial cell membrane has been increasingly recognized as an important surface for thrombin activation [44]. PDI was the first thiol isomerase shown to function in thrombosis and the endothelium is an essential source of PDI during thrombus formation in vivo [45]. However, the regulation of endothelial PDI is not well understood. We evaluated the role of PDI in thrombin generation on the endothelial cell surface and determined whether NO controlled this process. Incubation of endothelial cells with PACMA-31 blocked lipopolysaccharide (LPS)-induced thrombin and factor Xa generation on HUVECs (Fig. 3A-D). Incubation of HUVECs with SNOC also inhibited thrombin and factor Xa generation in this system. Neither SNOC nor PACMA-31 interfered with expression of TF (Fig. S5), which has previously been shown to be upregulated after 3 hour LPS stimulation [46]. These studies demonstrate the importance of PDI and regulation of NO levels on thrombin generation on endothelial cell surfaces.
Figure 3.

Endothelial cell thrombin generation is controlled by NO and PDI. Thrombin (A‐B) and factor Xa (C‐D) generation assays were performed using HUVECs. Cells were incubated with PACMA‐31 (10 μM) for 15 minutes followed by washing with PBS, and subsequently stimulated with 100 ng/mL LPS as indicated. In different wells, vehicle or SNOC (1 mM) were simultaneously added with 100 ng/mL LPS to cells. Graphs show representative tracings of thrombin and FXa generation. Bar graphs represent fold change in thrombin or FXa generation ± S.E.M. One‐way analysis of variance with Bonferroni’s post test was performed to measure significance (***P < 0.001; ns, non significant). FXa, activated factor X; LPS, lipopolysaccharide; NS, not significant; PACMA‐31, N‐(2,4‐dimethoxyphenyl)‐N‐(1‐oxo‐2‐propyn‐1‐yl)‐2‐(2‐thienyl)glycyl‐glycine ethyl ester; RFU, relative fluorescence units; SNOC, S‐nitrosocysteine.
S-nitrosylated PDI inhibits platelet function
NO produced by the endothelium serves an important function in preventing platelets from adhering to intact endothelium. Inhibition of platelet thiol isomerase activity and, specifically, S-nitrosylation of PDI could be a mechanism of controlling platelet activation. To evaluate this possibility, we incubated platelets with the NO+ carrier GSNO to determine the effect of GSNO exposure on NO incorporation into platelets. Exposure to GSNO resulted in a dose-dependent incorporation of NO into platelets as measured by the intracellular NO sensor DAF-FM (Fig. 4A). GSNO exposure also inhibited platelet surface reductase activity in a dose-dependent manner (Fig. 4B), with an IC50 of ~15 µM. To assess whether GSNO exposure resulted in S-nitrosylation of platelet PDI, we evaluated the formation of SNO-PDI as detected using the S-nitrosylation switch assay following incubation with GSNO (Fig. 4C). This analysis confirmed GSNO-mediated S-nitrosylation of PDI. Evaluation of platelet releasates, showed a significant increase in SNO labelling PDI following thrombin stimulation compared with platelets treated with vehicle alone (fold increase of 1.65 ± 0.26, mean ± SEM, p:0.02) (Fig. S6). Using mass spectrometry analysis, we detected S-nitrosylation in the active cysteines of PDI, Cys53, Cys56, Cys397 and Cys400. Upon treatment with GSNO, there was a marked increase of Cys-SNO in Cys397 and Cys400 in the a’ domain (Fig. S7 & Table S1).
Figure 4.

Cell surface PDI on platelets is controlled by NO. (A) Platelet incorporation of NO following incubation with increasing doses of GSNO was assessed using DAF‐FM. Values represent DAF‐FM relative fluorescent units (RFU, mean ± SEM (n = 3‐4). (B) Di‐eosin‐GSSG cleavage on the cell surface of platelets was measured following incubation with increasing doses of GSNO. Values represent percentage cleavage of di‐eosin‐GSSG as compared to vehicle alone, mean ± SEM (n = 3‐4). (C) Platelets were incubated with GSNO (20 mM), lysed, and subjected to a MPB‐switch assay. MPB‐labeled platelet lysates were loaded on a monoavidin column, washed, and the elution was subsequently analyzed using SDS‐polyacrylamide gel electrophoresis and immunoblotting for PDI. Samples were analyzed on the same gel. Quantification of densitometry was performed to assess SNO‐PDI generation on platelets. Mann‐Whitney U test was used to determine significance, *P < 0.05) (D) Platelet‐dependent thrombin generation was performed after samples were incubated with either vehicle, 0.1 mM or 1 mM SNOC, 100 μM PACMA‐31 and activated with the PAR‐1 activating peptide, SFLLRN. Bar graphs represent thrombin generation/min ± S.E.M. (n = 6), one way analysis of variance was performed using Bonferroni’s post test (***P < 0.001). DAF‐FM, 4‐amino‐5‐methylamino‐2′,7′‐difluorofluorescein; GNSO, S‐nitrosoglutathione; GSSG, glutathione disulfide; PACMA‐31, N‐(2,4‐dimethoxyphenyl)‐N‐(1‐oxo‐2‐propyn‐1‐yl)‐2‐(2‐thienyl)glycyl‐glycine ethyl ester; PDI, protein disulfide isomerase; RFU, relative fluorescence units.
PDI has previously been shown to participate in activation-induced platelet-dependent thrombin generation [29,39]. We reasoned that inhibition of surface PDI by S-nitrosylation would inhibit activation-induced platelet-dependent thrombin generation. Incubation of platelets with PACMA-31 blocked platelet-dependent thrombin generation induced by the PAR1 agonist, SFLLRN (Fig. 4D). Incubation of platelets with SNOC also inhibited thrombin generation. These results raise the possibility that NO blocks platelet-dependent thrombin generation at least in part by inhibiting platelet PDI activity.
To evaluate the effect of SNO-PDI on platelet function, we generated SNO-PDI by incubation of PDI with a 200-fold molar excess of SNOC. SNOC was used instead of GSNO because it has a substantially shorter half-life and is more easily removed, avoiding contamination with unreacted NO+ carrier [47]. Removal of SNOC was achieved by acetone precipitation and subsequent filtration by size-exclusion. The filtrate, containing decayed SNOC, was used as a negative control and termed ‘eluate’ to assess for free NO+ carrier. Before examining effects of SNO-PDI on platelet function, we first confirmed that the SNOC treatment and acetone precipitation did not irreversibly inactivate PDI. Thus, PDI and SNO-PDI were assayed for their ability to cleave di-eosin-GSSG after acetone precipitation (Fig. S8A). After SNOC treatment, SNO-PDI lost its reductase activity, whereas the untreated acetone precipitated PDI retained activity. The treatment of SNO-PDI with DTT resulted in reversal of the observed inhibition (Fig. S8B), indicating that the S-nitrosylation of PDI is reversible. To assess effects of SNO-PDI on platelet function, platelet aggregation was performed in the presence of SNO-PDI, untreated PDI, and control eluate (Fig. 5A,B). SNO-PDI inhibited SFLLRN-induced platelet aggregation. In contrast, we found that neither control eluate nor untreated PDI significantly affected platelet aggregation under the conditions of our assay. SNO-PDI also inhibited SFLLRN-induced P-selectin expression, indicating inhibition of α-granule release (Fig. 5C). These results indicate that S-nitrosylated PDI inhibits platelet activation, as measured by aggregation and α-granule release.
Figure 5.
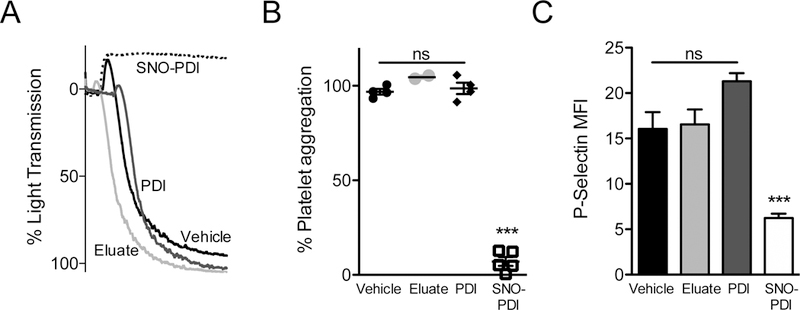
SNO‐PDI inhibits platelet aggregation and granule release. (A) Washed human platelets were stimulated with 3 μM SFLLRN after incubation with either vehicle (black), eluate (light gray), 285 nM PDI (dark gray), or 285 nM SNO‐PDI (dashed) and assessed for platelet aggregation. (B) Quantification of maximal aggregation was performed. Bar graphs represent maximal aggregation ± S.E.M. One‐way analysis of variance with Bonferroni’s post test was performed for significance (***P < 0.001). (C) Washed human platelets were stimulated with 3 μM SFLLRN after incubation with either vehicle (black), eluate (light gray), PDI (dark gray), or SNO‐PDI (white) and assessed for P‐selectin exposure. One‐way analysis of variance with Bonferroni’s post test was performed for significance (***P < 0.001). NS, not significant; MFI, mean fluorescence intensity; PDI, protein disulfide isomerase; SNO‐PDI, S‐nitrosylated protein disulfide isomerase.
S-nitrosylated PDI inhibits thrombus formation
The observation that SNO-PDI inhibits platelet function was striking, since it suggests that PDI functions to transfer NO into platelets. The fact that endogenous PDI demonstrates this property is evidenced by the fact that inhibition of PDI by either antibodies directed at PDI or small molecule PDI inhibitors block NO incorporation into platelets (Fig. S9). Inhibition of PDI by either an inhibitory antibody or small molecules blocks thrombus formation in several in vivo models. Given the ability of SNO-PDI to transfer NO to vascular substrates, we evaluated the effects of SNO-PDI on thrombosis in vivo. SNO-PDI was infused into mice and its effect on thrombus formation in cremaster arterioles following laser-induced injury was evaluated using intravital microscopy (Fig. 6A-G). Infusion of SNO-PDI resulted in a significant reduction of maximal platelet accumulation (Fig. 6B) and maximal fibrin formation (Fig. 6E) at sites of vascular injury compared with eluate controls. Similarly, total fibrin formation as assessed by area under the curve (AUC) measurements was significantly reduced (Fig. 6F), whereas a trend towards reduced platelet accumulation was observed (Fig. 6C). These results demonstrate that SNO-PDI is active in vivo and inhibits thrombus formation.
Figure 6.
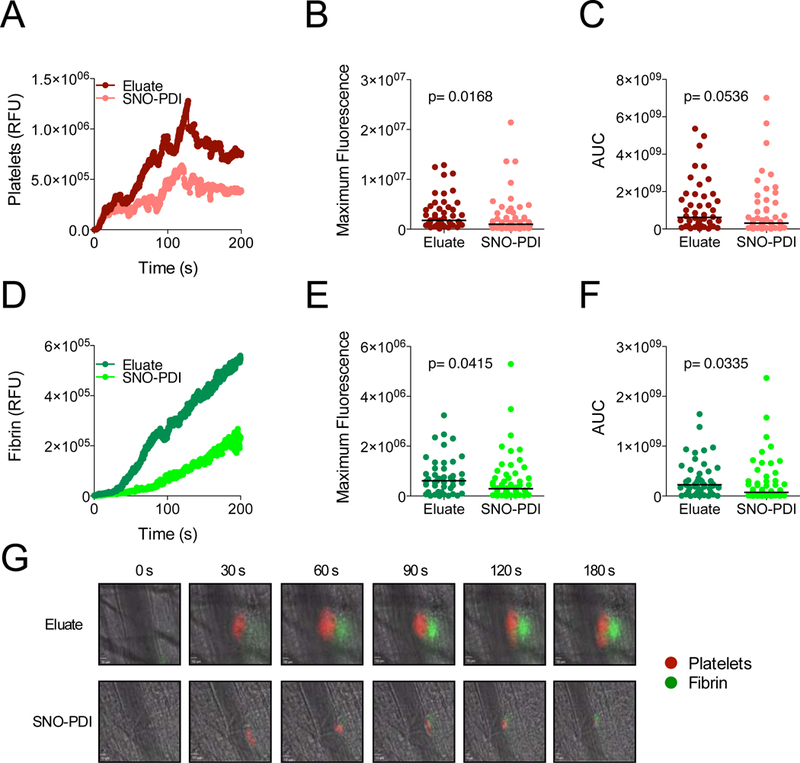
SNO‐PDI inhibits thrombus formation following laser‐induced injury. Both DyLight‐649 conjugated platelet‐specific (anti‐GPIbα) and DyLight‐488 fibrin‐specific (anti‐fibrin) antibodies were infused into mice (0.1 μg/g of mouse weight). Subsequently, mice were infused with either SNO‐PDI (25 μg) or the eluate containing the remaining NO‐donor after size‐exclusion. Thrombi were induced by laser injury of cremaster arterioles after infusion of either SNO‐PDI (n = 58) or the eluate (n = 46). Kinetics of thrombus formation were assessed for 240 seconds after the initial injury. Median‐integrated fluorescent intensities of SNO‐PDI (light red/light green) and eluate (dark red/dark green) after infusion of (A) anti‐GPIbα (platelets) and (D) antifibrin (fibrin) are plotted over time. Analyzed data using either the (B and E) maximum fluorescence or (C and F) area under the curve (AUC) is shown, statistical analysis was performed using a Mann‐Whitney U test. (G) Representative images of intravital microscopy are shown for each condition. (Scale bar: 10 μm). AUC, area under the curve; RFU, relative fluorescence units; SNO‐PDI, S‐nitrosylated protein disulfide isomerase. [Color figure can be viewed at wileyonlinelibrary.com]
Inhibition of other vascular thiol isomerases by NO
Thrombus formation requires the activity of additional vascular thiol isomerases that are distinct from PDI. Inhibition and/or genetic deletion of either ERp5 or ERp57 inhibits injury induced thrombus formation [36,48]. Both ERp5 and ERp57 possess CGHC active site motifs. We therefore determined whether ERp5 and ERp57 are also subject to regulation via S-nitrosylation and whether their S-nitrosylated forms impair platelet activation. Incubation of either ERp5 (Fig. 7A) or ERp57 (Fig. 7B) with the NO+ carrier SNOC inhibited their oxidoreductase activity with an IC50 of ~10 μM. ERp5 and ERp57 incubated with SNOC demonstrated S-nitrosylation as detected by ascorbate reduction and 5-fluorescein-maleimide labeling (Fig. 7C). SNO-ERp5 and SNO-ERp57 inhibited platelet aggregation in a manner similar to SNO-PDI (Fig. 7D-F). These data show that other vascular thiol isomerases known to function in thrombus formation are S-nitrosylated and that their S-nitrosylated forms inhibit platelet function.
Figure 7.

Vascular thiol isomerases are controlled by S‐nitrosylation. The effect of increasing doses of SNOC on the reductase activity of (A) ERp5 and (B) Erp57 was evaluated. Values represent percentage cleavage of di‐eosin‐GSSG as compared to vehicle alone, mean ± SEM (n = 3‐4). (C) Recombinant ERp5 and ERp57 were incubated with the NO donor SNOC, followed by acetone precipitation and free thiol blocking with NEM. Fluorescent labeling of –SNO bonds was performed. Images are representative of 3 independent experiments. (D‐E) Vehicle (black), eluate (light gray), 380 nM SNO‐ERp5 (dashed), 285 nM SNO‐ERp57 (dashed), 380 nM ERp5 (dark gray) and 285 nM ERp57 (dark gray) (i.e., 5 ug of each thiol isomerase) were incubated with washed platelets, stimulated with 5 μM SFLLRN, and assessed for platelet aggregation. Representative tracings are shown for each condition. (F) Quantification of % maximal aggregation was performed. Figures represent maximal aggregation ± S.E.M. One‐way analysis of variance with Bonferroni’s post test was performed for significance (***P < 0.001). 5‐FM, 5‐fluorescein maleimide; NS, not significant; SNOC, S‐nitrosocysteine; SNO‐ERp5, S‐nitrosylated ERp5; SNO‐ERp57, S‐nitrosylated ERp57.
Discussion
NO is an important mediator of vascular patency. In addition to its vasodilatory effects, it prevents vascular occlusion by inhibiting platelet function and endothelial cell activation [49]. We use a pharmacological approach to show that vascular thiol isomerases are targets of NO in the maintenance of vascular quiescence. A limitation of the pharmacological approach is that we cannot identify with certainty the relatively importance of the specific NO synthases or thiol isomerases involved in NO signaling to thiol isomerases. Nonetheless, these approaches demonstrate that NO interactions with thiol isomerases affect vascular quiescence by two general mechanisms. First, production of NO by NO synthases leads to S-nitrosylation of thiol isomerases, which inhibits their reductase activity by binding to catalytic cysteines (Fig. S1, Fig. 2). As thiol isomerases serve an essential role in thrombus formation, inhibition of thiol isomerase activity by NO is antithrombotic. Second, following the reaction with NO, thiol isomerases can act as transnitrosylases and S-nitrosylases, transferring NO into platelets and potentially to substrate molecules (Fig. 4).
Our studies demonstrate that endothelial derived NO controls prothrombotic PDI function on the cell surface. Scavenging of NO results in increased exposure of free thiols on endothelial cells and enhanced reductase activity as evidenced by increased di-eosin-GSSG cleavage. NO scavenging is also associated with increased endothelial thrombin and factor Xa generation in response to LPS (Fig. 1). In contrast, exposure to a NO+ carrier markedly inhibits endothelial di-eosin-GSSG cleavage and activation of NO synthesis results in both increased PDI S-nitrosylation and reduction of surface thiol isomerase activity (Fig. 2). Previous studies using proteomic analyses followed by mass-spectrometry have shown increased levels of S-nitrosylated PDI upon eNOS upregulation [50,51]. These observations together with our studies support a model whereby thiol isomerase activity at the endothelial cells surface is regulated by NO synthesis and that down-regulation of NO synthesis can enhance prothrombotic thiol isomerase activity. Alternative mechanisms of forming S-nitrothiols such as interactions of nitrite with protein metal centers could also have downstream influences on thiol isomerase activity [52]. That thrombosis is sensitive to NO synthesis is evidenced by studies showing that L-NAME augments thrombosis in glomeruli in the context of LPS exposure, induces thrombotic stroke in hypertensive rats, enhances thrombosis in an arterial inversion graft model, and increases platelet aggregation following infusion into mice [53–57]. Conversely, NO donors such as NONOate and LA419 are, antithrombotic [58–60]. While the thrombotic and hemostatic phenotypes following genetic knockouts of NOS isoforms have not consistently been prothrombotic, the interpretation of these studies is complicated by the fact that eNOS deficiency increases fibrinolysis secondary to increased secretion of plasminogen activator [61,62]. Our data support the premise that modulation of endothelial PDI activity by NO could contribute to the antithrombotic effect of NO.
Our studies also show that platelet PDI is regulated by S-nitrosylation. PDI is S-nitrosylated following exposure to NO+ carriers (Fig. 4). PDI is essential for transporting NO from carriers such as GSNO into the platelet cytosol (Fig. S9; Fig. 5) [27]. The neutralization of cell surface platelet thiol isomerase activity observed following S-nitrosylation of PDI by NO+ carriers blocks the prothrombotic function of platelets, since platelet-dependent thrombin generation is dependent on PDI (Figure 4) [29,39]. These observations support a model whereby NO generated by endothelium is transferred into platelets via PDI. According to this model the constitutive production of NO from endothelium both neutralizes the oxidoreductase activity of PDI exposed to the circulation and facilitates the delivery of NO from the endothelium to the platelet cytosol (Fig. S10).
That PDI is a critical mediator of thrombus formation in vivo has been demonstrated in multiple animal models of thrombosis using several PDI inhibitors and a platelet-specific PDI knockout mouse [7,9–11,63–67]. Inhibition of fibrin formation by SNO-PDI in our thrombus formation model (Fig. 6) is consistent with the in vitro observation that thiol isomerase activity and PDI-dependent thrombin generation at the endothelial cell surface is controlled by NO (Fig. 3A-B). We show that both platelet- and endothelial-mediated thrombin generation are dependent on PDI and sensitive to NO. However, the involvement of PDI may vary by cell type. While platelet-dependent thrombin generation is dependent on factor V, which is a substrate of PDI, it is independent of TF [29]. In contrast, PDI has been shown to affect TF activity [38,68–72]. Additional mechanisms by which PDI and NO influence platelet-mediated thrombin generation remains a question of ongoing studies.
Extracellular thiol isomerases are critical mediators of thrombus formation. Control of thiol isomerase activity by S-nitrosylation and denitrosylation provides a rapid means to modulate the activity of these enzymes (Fig. S10). Under conditions of decreased NO generation, extracellular thiol isomerases could become denitrosylated. Different redox environments could promote S-nitrosylation of the catalytic cysteines of PDI or promote the transfer of NO by PDI intracellularly or on the cell surface. Understanding the regulation of thiol isomerases in vascular quiescence and thrombosis will be important in the effective use of thiol isomerase inhibitors as they are tested clinically in the setting of thrombotic disease.
Supplementary Material
Essentials.
Nitric oxide synthesis controls protein disulfide isomerase (PDI) function
Nitric oxide (NO) modulation of PDI controls endothelial thrombogenecity
S-nitrosylated PDI inhibits platelet function and thrombosis
NO maintains vascular quiescence in part through inhibition of PDI
Acknowledgments
R.H. Bekendam has received funding from A*MIDEX – Academie d’Excellence (Aix-Marseille University) and by a National Institute of Health grant (U54 HL112302). D. Iyu has received funding from subprograma estatal de movilidad del Plan Estatal de Investigación Científica, Técnica y de Innovación 2013–2016 del Ministerio de Educación, Cultura y Deportes, Spain. K. De Ceunynck, O. Muse, and J.D. Stopa received funding by a National Institute of Health grant (T32 HL007917). R. Flaumenhaft has received support from the National Heart, Lung and Blood Institute (R01 HL125275 and R35 HL135775). P.K. Bendapudi was supported by a Mentored Research Award from the Hemostasis and Thrombosis Research Society (HTRS), which was funded by an independent medical educational grant from Shire, and a career development grant from the National Institutes of Health (1K08 HL136840–01). P.J. Hogg was supported by the National Health and Medical Research Council of Australia. J. Chiu was supported by the Helen and Robert Ellis Postdoctoral Fellowship and Tony Basten Postdoctoral Fellowship from the Sydney Medical School Foundation.
Footnotes
Disclosure of Conflict of Interest
The authors state that they have no conflict of interest.
Short Version: Nitric Oxide controls vascular PDI through S-Nitrosylation
References
- 1.Hatahet F, Ruddock LW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal 2009; 11: 2807–50. [DOI] [PubMed] [Google Scholar]
- 2.Ellgaard L, Ruddock LW. The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep 2005; 6: 28–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crescente M, Pluthero FG, Li L, Lo RW, Walsh TG, Schenk MP, Holbrook LM, Louriero S, Ali MS, Vaiyapuri S, Falet H, Jones IM, Poole AW, Kahr WHA, Gibbins JM. Intracellular Trafficking, Localization, and Mobilization of Platelet-Borne Thiol Isomerases. Arterioscler Thromb Vasc Biol 2016; 36: 1164–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Araujo TLS, Fernandes CG, Laurindo FRM. Golgi-independent routes support protein disulfide isomerase externalization in vascular smooth muscle cells. Redox Biol 2017; 12: 1004–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hahm E, Li J, Kim K, Huh S, Rogelj S, Cho J. Extracellular protein disulfide isomerase regulates ligand-binding activity of M 2 integrin and neutrophil recruitment during vascular inflammation. Blood 2013; 121: 3789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Subramaniam S, Jurk K, Hobohm L, Jäckel S, Saffarzadeh M, Schwierczek K, Wenzel P, Langer F, Reinhardt C, Ruf W. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood 2017; 129: 2291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reinhardt C, Von Brühl ML, Manukyan D, Grahl L, Lorenz M, Altmann B, Dlugai S, Hess S, Konrad I, Orschiedt L, Mackman N, Ruddock L, Massberg S, Engelmann B. Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation. J Clin Invest 2008; 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho J, Kennedy DR, Lin L, Huang M, Merrill-Skoloff G, Furie BC, Furie B. Protein disulfide isomerase capture during thrombus formation in vivo depends on the presence of ??3 integrins. Blood 2012; 120: 647–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho J, Furie BC, Coughlin SR, Furie B. A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. J Clin Invest 2008; 118: 1123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jasuja R, Passam FH, Kennedy DR, Kim SH, Van Hessem L, Lin L, Bowley SR, Joshi SS, Dilks JR, Furie B, Furie BC, Flaumenhaft R. Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. J Clin Invest 2012; 122: 2104–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bekendam RH, Bendapudi PK, Lin L, Nag PP, Pu J, Kennedy DR, Feldenzer A, Chiu J, Cook KM, Furie B, Huang M, Hogg PJ, Flaumenhaft R. A substrate-driven allosteric switch that enhances PDI catalytic activity. Nat Commun 2016; 7: 12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schulman S, Bendapudi P, Sharda A, Chen V, Bellido-Martin L, Jasuja R, Furie BC, Flaumenhaft R, Furie B. Extracellular Thiol Isomerases and Their Role in Thrombus Formation. Antioxid Redox Signal 2015; 24: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vallance P, Collier J, Moncada S. Effects of Endothelium-Derived Nitric Oxide on Peripheral Arteriolar Tone in Man. Lancet 1989; 334: 997–1000. [DOI] [PubMed] [Google Scholar]
- 14.Griffith TM, Lewis MJ, Newby AC, Henderson AH. Endothelium-derived relaxing factor. J Am Coll Cardiol 1988; 12: 797–806. [DOI] [PubMed] [Google Scholar]
- 15.Ruggeri ZM. Von Willebrand factor, platelets and endothelial cell interactions. J Thromb Haemost 2003; 1: 1335–42. [DOI] [PubMed] [Google Scholar]
- 16.Versteeg HH, Heemskerk JWM, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev 2013; 93: 327–58. [DOI] [PubMed] [Google Scholar]
- 17.Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev 2010; 62: 525–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gambaryan S, Kobsar A, Hartmann S, Birschmann I, Kuhlencordt PJ, Müller-Esterl W, Lohmann SM, Walter U. NO-synthase-/NO-independent regulation of human and murine platelet soluble guanylyl cyclase activity. J Thromb Haemost 2008; 6: 1376–84. [DOI] [PubMed] [Google Scholar]
- 19.Akhter S, Vignini A, Wen Z, English A, Wang PG, Mutus B. Evidence for S-nitrosothiol-dependent changes in fibrinogen that do not involve transnitrosation or thiolation. Proc Natl Acad Sci U S A 2002; 99: 9172–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Priora R, Margaritis A, Frosali S, Coppo L, Summa D, Di Giuseppe D, Aldinucci C, Pessina G, Di Stefano A, Di Simplicio P. In vitro inhibition of human and rat platelets by NO donors, nitrosoglutathione, sodium nitroprusside and SIN-1, through activation of cGMP-independent pathways. Pharmacol Res 2011; 64: 289–97. [DOI] [PubMed] [Google Scholar]
- 21.Kokkola T, Savinainen JR, Mönkkönen KS, Retamal M, Laitinen JT. S-nitrosothiols modulate G protein-coupled receptor signaling in a reversible and highly receptor-specific manner. BMC Cell Biol 2005; 6: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oberprieler NG, Roberts W, Graham AM, Homer-Vanniasinkam S, Naseem KM. cGMP-independent inhibition of integrin alphaIIBbeta3-mediated platelet adhesion and outside-in signalling by nitric oxide. FEBS Lett 2007; 581: 1529–34. [DOI] [PubMed] [Google Scholar]
- 23.Ramachandran N, Root P, Jiang X-M, Hogg PJ, Mutus B. Mechanism of transfer of NO from extracellular S-nitrosothiols into the cytosol by cell-surface protein disulfide isomerase. Proc Natl Acad Sci 2001; 98: 9539–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang L-M, St Croix C, Cao R, Wasserloos K, Watkins SC, Stevens T, Li S, Tyurin V, Kagan VE, Pitt BR. Cell-surface protein disulfide isomerase is required for transnitrosation of metallothionein by S-nitroso-albumin in intact rat pulmonary vascular endothelial cells. Exp Biol Med 2006; 231: 1507–15. [DOI] [PubMed] [Google Scholar]
- 25.Sliskovic I, Raturi A, Mutus B. Characterization of the S-denitrosation activity of protein disulfide isomerase. J Biol Chem 2005; 280: 8733–41. [DOI] [PubMed] [Google Scholar]
- 26.Root P, Sliskovic I, Mutus B. Platelet cell-surface protein disulphide-isomerase mediated S -nitrosoglutathione consumption. Biochem J 2004; 382: 575–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bell SE, Shah CM, Gordge MP. Protein disulfide-isomerase mediates delivery of nitric oxide redox derivatives into platelets. Biochem J 2007. p. 283–8. [DOI] [PMC free article] [PubMed]
- 28.Zai A, Rudd MA, Scribner AW, Loscalzo J. Cell-surface protein disulfide isomerase catalyzes transnitrosation and regulates intracellular transfer of nitric oxide. J Clin Invest 1999; 103: 393–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stopa JD, Neuberg D, Puligandla M, Furie B, Flaumenhaft R, Zwicker JI. Protein disulfide isomerase inhibition blocks thrombin generation in humans by interfering with platelet factor V activation. JCI Insight 2017; 2: 351–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bowley SR, Fang C, Merrill-Skoloff G, Furie BC, Furie B. Protein disulfide isomerase secretion following vascular injury initiates a regulatory pathway for thrombus formation. Nat Commun 2017; 8: 14151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raturi A, Mutus B. Characterization of redox state and reductase activity of protein disulfide isomerase under different redox environments using a sensitive fluorescent assay. Free Radic Biol Med 2007; 43: 62–70. [DOI] [PubMed] [Google Scholar]
- 32.Lei SZ, Pan Z-H, Aggarwal SK, Chen H-SV, Hartman J, Sucher NJ, Lipton SA. Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron 1992; 8: 1087–99. [DOI] [PubMed] [Google Scholar]
- 33.Falati S, Gross P, Merrill-Skoloff G, Furie BC, Furie B. Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nat Med 2002; 8: 1175–81. [DOI] [PubMed] [Google Scholar]
- 34.Dubois C, Panicot-Dubois L, Gainor JF, Furie BC, Furie B. Thrombin-initiated platelet activation in vivo is vWF independent during thrombus formation in a laser injury model. J Clin Invest 2007; 117: 953–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cho J Protein disulfide isomerase in thrombosis and vascular inflammation. J Thromb Haemost 2013; : 2084–91. [DOI] [PMC free article] [PubMed]
- 36.Passam FH, Lin L, Gopal S, Stopa JD, Bellido-Martin L, Huang M, Furie BC, Furie B. Both platelet- and endothelial cell-derived ERp5 support thrombus formation in a laser-induced mouse model of thrombosis. Blood 2015; 125: 2276–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cohen RA, Murdoch CE, Watanabe Y, Bolotina VM, Evangelista AM, Haeussler DJ, Smith MD, Mei Y, Tong X, Han J, Behring JB, Bachschmid MM, Matsui R. Endothelial Cell Redox Regulation of Ischemic Angiogenesis. J Cardiovasc Pharmacol 2016; 67: 458–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahamed J, Versteeg HH, Kerver M, Chen VM, Mueller BM, Hogg PJ, Ruf W. Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc Natl Acad Sci 2006; 103: 13932–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jurk K, Lahav J, Van Aken H, Brodde MF, Nofer JR, Kehrel BE. Extracellular protein disulfide isomerase regulates feedback activation of platelet thrombin generation via modulation of coagulation factor binding. J Thromb Haemost 2011; 9: 2278–90. [DOI] [PubMed] [Google Scholar]
- 40.Li H, Burkhardt C, Heinrich U-R, Brausch I, Xia N, Förstermann U. Histamine upregulates gene expression of endothelial nitric oxide synthase in human vascular endothelial cells. Circulation 2003; 107: 2348–54. [DOI] [PubMed] [Google Scholar]
- 41.Warboy CM, Chen N, Zhang Q, Shaifta Y, Vanderslott G, Passacquale G, Hu Y, Xu Q, Ward JPT, Ferro A. Bidirectional cross-regulation between the endothelial nitric oxide synthase and beta-catenin signalling pathways. Cardiovasc Res 2014; 104: 116–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang X, Kim W-S, Hatcher N, Potgieter K, Moroz LL, Gillette R, Sweedler JV. Interfering with nitric oxide measurements. 4,5-diaminofluorescein reacts with dehydroascorbic acid and ascorbic acid. J Biol Chem 2002; 277: 48472–8. [DOI] [PubMed] [Google Scholar]
- 43.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE 2001; 2001: pl1. [DOI] [PubMed] [Google Scholar]
- 44.Yau JW, Teoh H, Verma S. Endothelial cell control of thrombosis. BMC Cardiovasc Disord 2015; 15: 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jasuja R, Furie B, Furie BC. Endothelium-derived but not platelet-derived protein disulfide isomerase is required for thrombus formation in vivo. Blood 2010; 116: 4665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Higgins SJ, De Ceunynck K, Kellum JA, Chen X, Gu X, Chaudhry SA, Schulman S, Libermann TA, Lu S, Shapiro NI, Christiani DC, Flaumenhaft R, Parikh SM. Tie2 protects the vasculature against thrombus formation in systemic inflammation. J Clin Invest 2018; 128: 1471–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mancuso C, Bonsignore A, Di Stasio E, Mordente A, Motterlini R. Bilirubin and S-nitrosothiols interaction: evidence for a possible role of bilirubin as a scavenger of nitric oxide. Biochem Pharmacol 2003; 66: 2355–63. [DOI] [PubMed] [Google Scholar]
- 48.Wu Y, Ahmad SS, Zhou J, Wang L, Cully MP, Essex DW. The disulfide isomerase ERp57 mediates platelet aggregation, hemostasis, and thrombosis. Blood 2012; 119: 1737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zampolli A, Basta G, Lazzerini G, Feelisch M, De Caterina R. Inhibition of endothelial cell activation by nitric oxide donors. J Pharmacol Exp Ther 2000; 295: 818–23. [PubMed] [Google Scholar]
- 50.Huang B, Chen SC, Wang DL. Shear flow increases S-nitrosylation of proteins in endothelial cells. Cardiovasc Res 2009; 83: 536–46. [DOI] [PubMed] [Google Scholar]
- 51.Huang B, Li FA, Wu CH, Wang DL. The role of nitric oxide on rosuvastatin-mediated S-nitrosylation and translational proteomes in human umbilical vein endothelial cells. Proteome Science 2012. [DOI] [PMC free article] [PubMed]
- 52.Broniowska KA, Diers AR, Hogg N. S-nitrosoglutathione. Biochim Biophys Acta 2013; 1830: 3173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shultz PJ, Raij L. Endogenously synthesized nitric oxide prevents endotoxin-induced glomerular thrombosis. J Clin Invest 1992; 90: 1718–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sasaki Y, Noguchi T, Seki J, Giddings JC, Yamamoto J. Protective effects of imidapril on He-Ne laser-induced thrombosis in cerebral blood vessels of stroke-prone spontaneously hypertensive rats. Thromb Haemost 2000; 83: 722–7. [PubMed] [Google Scholar]
- 55.Azizzadeh B, Buga GM, Berke GS, Larian B, Ignarro LJ, Blackwell KE. Inhibitors of nitric oxide promote microvascular thrombosis. Arch Facial Plast Surg 5: 31–5. [DOI] [PubMed] [Google Scholar]
- 56.Provost P, Merhi Y. Endogenous nitric oxide release modulates mural platelet thrombosis and neutrophil-endothelium interactions under low and high shear conditions. Thromb Res 1997; 85: 315–26. [DOI] [PubMed] [Google Scholar]
- 57.Freedman JE, Loscalzo J. Nitric oxide and its relationship to thrombotic disorders. Journal of Thrombosis and Haemostasis 2003. p. 1183–8. [DOI] [PubMed]
- 58.Özüyaman B, Ebner P, Niesler U, Ziemann J, Kleinbongard P, Jax T, Gödecke A, Kelm M, Kalka C. Nitric oxide differentially regulates proliferation and mobilization of endothelial progenitor cells but not of hematopoietic stem cells. Thromb Haemost 2005; 94: 770–2. [DOI] [PubMed] [Google Scholar]
- 59.Chiang S, Azizzadeh B, Buga G, Ignarro L, Calcaterra T, Blackwell K. Local administration of nitric oxide donor significantly impacts microvascular thrombosis. Laryngoscope 2003; 113: 406–9. [DOI] [PubMed] [Google Scholar]
- 60.Vilahur G, Segalés E, Casaní L, Badimon L. A novel anti-ischemic nitric oxide donor inhibits thrombosis without modifying haemodynamic parameters. Thromb Haemost 2004; 91: 1035–43. [DOI] [PubMed] [Google Scholar]
- 61.Iafrati MD, Vitseva O, Tanriverdi K, Blair P, Rex S, Chakrabarti S, Varghese S, Freedman JE. Compensatory mechanisms influence hemostasis in setting of eNOS deficiency. Am J Physiol Heart Circ Physiol 2005; 288: H1627–32. [DOI] [PubMed] [Google Scholar]
- 62.Upmacis RK, Shen H, Benguigui LES, Lamon BD, Deeb RS, Hajjar KA, Hajjar DP. Inducible nitric oxide synthase provides protection against injury-induced thrombosis in female mice. Am J Physiol Heart Circ Physiol 2011; 301: H617–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou J, Wu Y, Wang L, Rauova L, Hayes VM, Poncz M, Essex DW. The C-terminal CGHC motif of protein disulfide isomerase supports thrombosis. J Clin Invest 2015; 125: 4391–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim K, Hahm E, Li J, Holbrook L-M, Sasikumar P, Stanley RG, Ushio-Fukai M, Gibbins JM, Cho J. Platelet protein disulfide isomerase is required for thrombus formation but not for hemostasis in mice. Blood 2013; 122: 1052–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lin L, Gopal S, Sharda A, Passam F, Bowley SR, Stopa J, Xue G, Yuan C, Furie BC, Flaumenhaft R, Huang M, Furie B. Quercetin-3-rutinoside Inhibits Protein Disulfide Isomerase by Binding to Its b’x Domain. J Biol Chem 2015; 290: 23543–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sousa HR, Gaspar RS, Sena EML, da Silva SA, Fontelles JL, AraUjo TLS, Mastrogiovanni M, Fries DM, Azevedo-Santos APS, Laurindo FRM, Trostchansky A, Paes AM. Novel antiplatelet role for a protein disulfide isomerase-targeted peptide: evidence of covalent binding to the C-terminal CGHC redox motif. J Thromb Haemost 2017; 15: 774–84. [DOI] [PubMed] [Google Scholar]
- 67.Zhu S, Welsh JD, Brass LF, Diamond SL. Platelet-targeting thiol reduction sensor detects thiol isomerase activity on activated platelets in mouse and human blood under flow. J Thromb Haemost 2016; 14: 1070–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Versteeg HH, Ruf W. Tissue factor coagulant function is enhanced by protein-disulfide isomerase independent of oxidoreductase activity. J Biol Chem 2007; 282: 25416–24. [DOI] [PubMed] [Google Scholar]
- 69.Chen VM, Ahamed J, Versteeg HH, Berndt MC, Ruf W, Hogg PJ. Evidence for activation of tissue factor by an allosteric disulfide bond. Biochemistry 2006; 45: 12020–8. [DOI] [PubMed] [Google Scholar]
- 70.Raturi A Ruf W Effect of protein disulfide isomerase chaperone activity inhibition on tissue factor activity. J Thromb Haemost 2010; 8: 1863–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Langer F, Ruf W. Synergies of phosphatidylserine and protein disulfide isomerase in tissue factor activation. Thromb Haemost 2014; 111: 590–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen F, Zhao Z, Zhou J, Lu Y, Essex DW, Wu Y. Protein disulfide isomerase enhances tissue factor-dependent thrombin generation. Biochem Biophys Res Commun 2018; 501: 172–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.