

Abstract
Bromodomain and PHD finger containing protein transcription factor (BPTF) is an epigenetic protein involved in chromatin remodelling and is a potential anticancer target. The BPTF bromodomain has one reported small molecule inhibitor (AU1, rac-1). Here, advances made on the structure-activity relationship of a BPTF bromodomain ligand are reported using a combination of experimental and molecular dynamics simulations leading to the active enatiomer (S)-1. Additionally, a ligand deconstruction analysis was conducted to characterize important pharmacophores for engaging the BPTF bromodomain. These studies have been enabled by a protein-based fluorine NMR approach, highlighting the versatility of the method for selectivity, ligand deconstruction, and ligand binding. To enable future analysis of biological activity, cell activity in a panel of cancer cell lines was carried out using CRISPR-Cas9 and (S)-1 to evaluate cell-based model systems that are sensitive to BPTF inhibition.
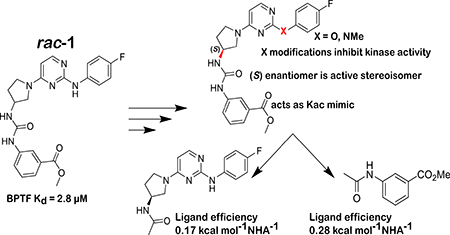
Graphical Abstract
Introduction
BPTF (bromodomain and PHD finger containing transcription factor) is the largest component of the NURF (nucleosome remodelling factor) complex and is essential for transcriptional regulation of many processes in mammalian chromatin biology including early embryo development,1 stem cell pluripotency,2 and T-cell immune function.1 The 3046 amino acid protein was found to contain a bromodomain and two PHD fingers, domains commonly found on histone and DNA binding proteins (Figure 1).3 To facilitate transcriptional regulation, structural studies have shown that BPTF engages the nucleosome in a bivalent manner with its C-terminal PHD finger and bromodomain via binding to trimethylated histone H3K4me3 and acetylated histone H4K16ac respectively.4
Figure 1:

Left) structure of racemic AU1 (rac-1). Right) Ribbon diagrams from the crystal structures of the bromodomain of BPTF (gray) overlaid with the bromodomain of 5FW BPTF (blue) with the fluorine of W2824 labeled in green. Bottom) Domain scheme for BPTF with PHD and bromodomain (BRD) labeled.
Motivating interests for therapeutic development, BPTF associates with the oncoprotein c-MYC and is required for tumorigenesis in high-expressing MYC cells.5 Dysregulation of the protein has been implicated in a number of diseases including: pancreatic cancer,5 melanoma,6 colorectal cancer,7 hepatocellular carcinoma,8 breast cancer,2 bladder cancer,9 and lung cancer.10 Given the relevance of BPTF in disease and its association with NURF, we sought to discover what role the engagement of the bromodomain with acetylated histones on chromatin plays in the context of disease states. Using a newly developed protein-observed fluorine NMR (PrOF NMR) assay for bromodomain ligand discovery,11 we reported the first small molecule inhibitor of the BPTF bromodomain (AU1, rac-1, figure 1), selective for BPTF over BRD4.12 Rac-1 was further shown to be cell active, via reducing transcription in a BPTF-specific reporter assay.12 Additionally, inhibitor treatment of breast epithelial cells decreased c-MYC regulated genes, c-MYC chromatin occupancy, and led to G1 cell cycle arrest.2
Despite the biological importance of BPTF, and the success of chemical probe development for other bromodomains,13,14 outside of rac-1, ligands for the BPTF bromodomain are lacking. Rac-1 has only a moderate potency (Kd = 2.8 μM), and its selectivity profile has not been fully characterized. Here we report our efforts to more thoroughly characterize rac-1 and use PrOF NMR in structure-property studies for improving its metabolic stability and selectivity. We further analyze the mode and efficacy of binding through ligand deconstruction in combination with weighted ensemble molecular dynamics simulations. These studies highlight the versatility of PrOF NMR for bromodomain ligand characterization, and identify areas for improving future BPTF bromodomain inhibitors.
Results and Discussion
PrOF NMR identification of the active enantiomer of rac-1
For chemical probe development, having access to an inactive stereoisomer with similar physicochemical properties can be a useful control. In the original identification of AU1 (rac-1) as a ligand for the BPTF bromodomain, the racemate was tested. Using our previously reported methods, we synthesized both the (R) and (S) enantiomers ((R)-1, and (S)-1) and tested them using PrOF NMR.2
PrOF NMR is a structure-based method for characterizing ligand interactions and protein binding sites. A comparative study has shown PrOF NMR to be comparable in speed and hit identification to ligand-observed NMR, however an advantage is the ability to rapidly quantify affinities of weak binding ligands based on perturbation of fluorine resonances.15 The BPTF bromodomain contains a single tryptophan at position 2824 in the WPF shelf of the protein near the histone binding site (Figure 2A). As described previously, for NMR analysis we labeled the BPTF bromodomain with 5-fluorotryptophan (5FW). We monitored the 19F resonance upon addition of both enantiomers. PrOF NMR binding isotherms can be used to quantify the dissociation constant based on measuring dose-dependent changes in chemical shifts of protein ligands that exhibit fast chemical exchange rates. In the case of moderate affinity ligands such as rac-1, significant resonance broadening is observed as the intermediate chemical exchange rate for binding is longer, but not sufficiently long enough to resolve the bound and unbound state. In the PrOF NMR spectrum for the (S) enantiomer, more significant broadening of the 5FW resonance into baseline than the racemate was observed. (Figure 2B). Conversely, (R)-1 showed no broadening in the NMR assay, indicating that only the (S) enantiomer significantly contributed to the binding affinity originally reported, whereas the (R) enantiomers of future analogs can serve as useful control molecules.
Figure 2:

A) Ribbon structure of the acetylated lysine binding site of the bromodomain of BPTF with 5FW(2824) displayed. The fluorine atom is colored in cyan. N2881 is a key residue for binding of acetylated lysines on histone proteins B) PrOF NMR using the BPTF bromodomain labeled with 5FW at 2824 of rac-1 and both enantiomers against 5FW BPTF. Each ligand is in a two-fold excess of the protein. Both experiments with rac-1 and (S)-1 resulted in significant broadening of the fluorine resonance.
X-ray crystal structure of 5FW BPTF supports a lack of structure perturbation from fluorine for PrOF NMR experiments.
To gain further insight into how (S)-1 interacts with the BPTF bromodomain, a co-crystal structure was highly desirable. Although to date we have been unsuccessful, during these studies we investigated the crystallization of the 5FW-BPTF bromodomain. Previously, we characterized the stability of the fluorine-labeled bromodomain by circular dichroism and determined a negligible change in protein thermal stability from fluorination relative to the unlabelled protein.12 To further verify the native fold is intact, we have now solved the crystal structures of the BPTF bromodomain in both the fluorinated and non-fluorinated form. Alignment of the structures gave a root-mean-square deviation (RMSD) of 0.1548 Å for the protein backbone and a RMSD of 0.3516 Å for W2824 (See Figures 1 and S2-1). Proline 2836, far from the binding site, had the largest RMSD at 0.8093 Å. The similarity in structure of the 5FW and unlabeled BPTF further supports fluorine labeling and PrOF NMR as a viable binding assay. Given the lack of a co-crystal structure of (S)-1), and further verification of the non-perturbing nature of the fluorine in the bromodomain, we thus turned to indirect methods using PrOF NMR to guide medicinal chemistry efforts using structure-activity relationships (SAR) and ligand deconstruction.
(S)-1 Structure-Activity Relationships.
In addition to a modest binding affinity of 2.8 μM, rac-1, and by analogy (S)-1, possess several properties which we wished to improve upon. One significant challenge was the low solubility of rac-1, as observed by an increase in solution turbidity at concentrations above ~100 μM. Additionally, given its cellular activity, for future use of (S)-1 analogs we were concerned about the susceptibility to cellular esterase hydrolysis of the methyl ester. Consistent with our concern regarding metabolic liabilities, pharmacokinetics (PK) studies of (S)-1 in mice, produced a Cmax of 4,540 ng/mL (10 μM) in the plasma, and a rapid half-life at 0.68 h (Figure S2).
Given the concerns above, we took advantage of our four-step synthesis for SAR studies (see supporting information for synthetic schemes, Figure S5). To test for ways to improve the stability of our compound based on ester hydrolysis concerns, we synthesized new analogs of (S)-1 (2–14, Figure 3). We first tested the would-be product from ester hydrolysis, 12, against 5FW BPTF by PrOF NMR and determined that binding to the bromodomain was eliminated. To improve the stability of the scaffold in the presence of cellular esterases for future cell-based studies, we replaced the ester group of (S)-1 with a series of amides, 9–11. For these analogs, only small amides were tolerated although binding was weakened. The methyl amide (9) showed over an order of magnitude loss in affinity relative to rac-1. Fast chemical exchange allowed for titration to derive a Kd = 30 μM (Figure 4). The ethyl amide (10) also showed fast exchange binding behaviour consistent with a weak interaction, but the affinity was unable to be quantified. A larger amide (11) resulted in a complete loss of binding (Figures S10–11). Unfortunately, upon changing the ester to an amide, we also observed an attenuated selectivity with respect to BRD4(1) by PrOF NMR (Figure S34). Future analogs containing the more stable methyl amide will need to have improved binding activity and selectivity through modifications on alternative sites on the molecule.
Figure 3:

Structures of select rac-1 analogs. PrOF NMR titrations of all compounds can be found in the supporting information
Figure 4:

Sample titration data of 9, a broadening and shifting of the 5FW BPTF resonance is observed. Full titration can be found in the Supporting Information.
Due to the importance of the ester, we also explored isomers of the compound, substituting the methyl ester around the aromatic ring (2 and 3). In these cases, changing the meta substitution of the methyl ester on the aryl urea showed a complete loss of binding in both 2 and 3, indicating meta substituted methyl esters were critical for binding (Figure S8). Conversely, substitutions made on the aniline ring consistently show binding activity through our PrOF NMR assay. Replacing the aniline nitrogen with an oxygen (5) or methylating the nitrogen (6) maintained binding to BPTF but resonance chemical shifts were in the fast exchange regime by PrOF NMR (Figure S11-S12). Substituting the fluorine for a benzyl amine (7) as well as a 3,5-dimethyl substitution pattern (8) show similar characteristics as (S)-1 in an NMR titration, indicating a reduced sensitivity to compound modification (Figure S16). The regioisomer of (S)-1, 13, also showed similar binding characteristics to the original scaffold by PrOF NMR (Figure S21). We conclude, from these PrOF NMR SAR studies, that the amino pyrimidine terminus of the molecule may be more solvent exposed when binding to the protein, whereas the methyl ester is bound more deeply in the bromodomain binding site
(S)-1 has limited solubility in aqueous solution and the broadening of its 19F resonance by NMR indicates a highly associated state. We looked at the urea moiety within the scaffold as a potential source of aggregation and sought to alter this functional group to improve the behaviour of (S)-1 in aqueous solution. The urea was separated by inserting a methylene as seen in compound 14, drastically improving the water solubility to > 1 mM at pH 7.4 as assessed by a lack of solution turbidity. However, we observed a reduction in binding affinity using PrOF NMR titrations (Kd = 39 μM, Figure S22) when the urea was interrupted. We also attempted to reverse the substitution of the pyrrolidine ring with the hopes of interrupting the aggregation potential of the urea by reducing the hydrogen bond capability of one of the nitrogens ((S)-4 and (R)-4). Upon this change, we observed by PrOF NMR in (S)-4 a stoichiometric binding at the concentration of protein that we used, indicating that its binding affinity was at least an order of magnitude below our protein concentration, i.e., 5 μM or lower. We then had this compound tested via AlphaScreen (Reaction Biology) as a competition-based assay between the bromodomain and an acylated histone; however, an IC50 of 70 μM was observed. This difference in binding affinity observed via PrOF NMR and AlphaScreen could potentially be due to the reduced DMSO concentration necessary for the the AlphaScreen assay 0.25% vs. the 1% used in PrOF NMR, this reduction in co-solvent concentration limits the solubility of this analog for the assay, and points to further improvements that are needed for solubility enhancement of our compounds.
Ligand deconstruction of (S)-1 by PrOF NMR
Although PrOF NMR SAR experiments provided insights for improving solubility and stability of the rac-1 scaffold and the identification of the active enantiomer, (S)-1, none of the changes led to a significant increase in affinity. These results led us to explore a ligand deconstruction analysis of (S)-1 to evaluate the contribution to binding of specific pharmacophores of the parent scaffold.16,17 PrOF NMR has mainly been used for hit identification, and only a preliminary ligand deconstruction study was reported before.18 However, NMR is well-suited for deconstruction analysis due to its ability to quantify the affinities of weak binding fragments. We synthesized fragments F1-F6 of the parent scaffold, which could be used to test the relative contributions of each portion of the molecule by PrOF NMR. In the original study which identified rac-1 as a BPTF bromodomain ligand, apparent important pharmacophores for binders of BPTF were the aryl urea and the meta carbonyl group (ester or ketone).19 In light of this and the SAR results described above, we predicted a stronger interaction from this portion of the molecule when taken alone. Fragment F1 which contains the amino-pyrimidine portion of (S)-1 did not bind BPTF despite being a motif found in other bromodomain inhibitors. 13,14 Alternatively, fragment F4 containing the methyl ester displayed a Kd as determined by PrOF NMR of 1.3 mM with the highest ligand efficiency of 0.28. This can be compared with rac-1 with a ligand efficiency of 0.22. We tested additional fragments, F3, F5, and F6 with results summarized in figure 5. In these cases, a similar ligand efficiency was maintained (0.22–0.23) of fragments tested. Extending fragment F1 to F2 to include the amino-pyrrolidine and the carbonyl group of the urea also bound with a Kd of 1.3 mM, albeit with a reduced ligand efficiency of 0.17. We note, that fragments can bind in multiple orientations if not anchored by a strong non-covalent interaction.20 Despite this possibility, these data support that the binding of (S)-1 to the bromodomain of BPTF is the result of an additive effect of several weak contacts throughout the (S)-1 scaffold.
Figure 5:

Ligand deconstruction study of (S)-1, with the exception of F1, all fragments bind 5FW BPTF with similar ligand efficiencies, indicating binding contacts are dispersed throughout the molecule. Using PrOF NMR, a binding isotherm can be generated to quantify weak binding ligands. F6 is shown as a representative example. NB = No observable binding. Rac-1 ligand efficiency = 0.22
Molecular dynamic simulations of (S)-1 with BPTF
We have not solved a co-crystal structure of (S)-1 to validate a binding mode to support our SAR and ligand deconstruction analysis, necessitating alternative methods to provide higher resolution information on binding. We therefore turned towards molecular dynamics (MD) simulations of (S)-1 binding to BPTF using a recently reported weighted ensemble approach21 that has been used to investigate bromodomain-ligand interactions.22 The inclusion of full protein motion in explicit solvent in our MD simulation versus our prior docking studies was anticipated to improve our predicted binding pose. Starting from multiple docked initial structures, a network model of ligand poses was constructed, and the weights of each state were used to predict an ensemble of probable poses (Figure 6A). These poses support that the methyl ester serves as an acetylated lysine mimic as it can hydrogen bond to the conserved N2881 of BPTF (Figures 2, 6B, and S4). The importance of the methyl ester in the acetyl lysine binding site is consistent with the high ligand efficiency of F4 and is consistent our SAR study that alterations of the methyl ester such as substitutions around the aromatic ring obliterate binding, as well as the intolerance for large amide groups at this position. MD simulations show the attached aryl ring also interacts with Phe2887 (via a stacking interaction, while the urea is involved in H-bonding interactions with D2834. Finally, the p-fluoroaniline is exposed to solvent near W2824 in the WPF shelf, consistent with our findings that other groups can be installed at this position (e.g., 7, 8) and that replacement of the p-fluoroaniline with a chlorine maintains modest affinity (F3, Kd = 35 μM). In addition to helping explain our SAR, this docking pose will be used to guide future analog designs in the absence of a co-crystal structure.
Figure 6:

A) Conformation space network describing all of the binding poses found and their interconnectivity. Each node represents a ligand pose and nodes are colored according to the solvent accessible surface area of the ligand (blue/black = unbound, green/yellow = fully bound). The two starting poses, as well as the predicted most probable pose are labeled. B) Views of the most probable pose. A density isosurface was computed using 10 random ligand structures taken from most probable state (shown in transparent gray). Interacting residues are labeled.
Bromodomain selectivity analysis of (S)-1
As (S)-1 remained our best compound, we next conducted a preliminary analysis of bromodomain selectivity for (S)-1. Rac-1 was originally shown to be selective for BPTF over the N-terminal BRD4 bromodomain, BRD4(1), which also contains a WPF shelf and is a member of the bromodomain and extra-terminal domain (BET) family of proteins. In addition to 5FW-labeled BRD4(1), we tested three other WPF shelf-containing proteins: a second BET bromodomain, 5FW BRDT(1), PCAF, which is on the same branch of the phylogenetic tree as BPTF,23 and a highly homologous bromodomain from Plasmodium falciparum, PfGCN524 (Figure 7). Although PfGCN5 is not a human bromodomain, acetylated peptides have been shown to bind to both the PfGCN5 bromodomain and BPTF with similar affinity.25 Additionally, PCAF inhibitors also inhibit PfGCN5 suggesting high similarity in binding sites of these structurally related bromodomains.26 In the PrOF NMR spectra, (S)-1 did not perturb resonances for either BRD4(1) or BRDT(1). In the case of the PCAF bromodomain, although PCAF and BPTF are very close on the phylogenetic tree, only a small chemical shift was observed with slight broadening, without a significant dose dependence (see Supporting Information). These results indicate a moderate level of selectivity is maintained for BPTF over the structurally similar PCAF bromodomain. Finally, in the case of PfGCN5, a significant broadening of the most downfield resonance was observed indicating an interaction similar to BPTF. We have tentatively assigned this resonance to the tryptophan in the WPF shelf based on prior binding studies with pan-inhibitor bromosporine.27 Interestingly rac-1 has shown antimalarial activity,28 consistent with bromodomain targeting, although target identification for rac-1 has not been carried out against the parasite. When potency is increased, a more complete bromodomain profiling will be conducted.
Figure 7:

Selectivity of (S)-1 toward four different bromodomains, BRD4(1), BRDT(1), PCAF, and PfGCN5. All experiments were carried out with 50 μM protein in the absence (black spectrum) or presence of 100 μM (S)-1 (red spectrum).
Kinase selectivity analysis of rac-1 stereoisomers
Rac-1 was originally discovered using the published kinase inhibitor set library.12 Therefore, we also tested for kinase activity of (S)-1. In a recent report evaluating inhibition against the kinome, rac-1 showed 82% inhibition of two kinases, CDKL2 and TRKC, when tested at 1 μM.29 Four additional kinases (CDK11, TRKB, HPK1, p38-δ) were inhibited between 51–68%. Based on this data, we utilized the KINOMEscan service from DiscoverX to test (R)-1 and (S)-1 against CDKL2 and TRKC. (S)-1 showed dissociation constants of 260 nM and 200 nM against CDKL2 and TRKC respectively, whereas (R)-1 had dissociation constants of 1200 nM and 400 nM against the two kinases (Table 1). While there is a nearly five-fold difference in affinity toward CDKL2, there is only a two-fold difference toward TRKC, indicating that the stereocenter is not critical to the binding of these targets, in particular TRKC. These kinases need to be considered in future cell-based studies.
Table 1:
Binding affinities of (S)-1 and analogs against known kinase targets.
| Compound Number | TRKC Kd | CDKL2 Kd |
|---|---|---|
| (S)-1 | 200 nM | 260 nM |
| (R)-1 | 400 nM | 1,200 nM |
| 5 | NB | NB |
| 6 | 2,600 nM | 19,000 nM |
NB= No significant binding
We looked to make changes to limit the kinase binding of the molecule, thereby reducing potential off-target effects in vivo. The amino-pyrimidine pharmacophore found in (S)-1 is a known kinase-binding functionality which recognizes the hinge region of the ATP binding pocket in kinases. Two of the simplest changes that we envisioned were replacing the protic nitrogen with an oxygen (5), and the alkylation of the protic nitrogen (6). N-methylation of the aniline nitrogen yielded stoichiometric binding again in the PrOF NMR assay but a lack of activity was observed in AlphaScreen, potentially for the same solubility reason previously discussed. Upon exchanging the nitrogen for an oxygen, binding was maintained, albeit weakened relative to (S)-1, as the ligand approached fast-intermediate exchange in the PrOF NMR assay. Encouragingly, after testing these two compounds against CDKL2 and TRKC we observed a drastic reduction in kinase binding in the N-methyl case (6), and a complete loss of binding with compound 5 (Table 1). The phenoxypyrimidine motif will be a useful design element for future ligands to remove kinase binding.
Cancer cell line sensitivity to BPTF gene deletion and rac-1 stereoisomers
Prior studies of rac-1, supported BPTF target engagement and cellular activity.12 Here, our results led to the identification of the active enantiomer, (S)-1. However, additional kinase off-targets have now also been characterized. Prior to further mechanistic studies, as a preliminary analysis of more global cellular effects of (S)-1, we tested the sensitivity of cells to both (S)-1 and (R)-1 treatment, to identify cell lines that were less susceptible to off target effects shared by (S)-1 and (R)-1, and were sensitive to BPTF inhibition. We first screened for cell lines with increased BPTF gene expression levels, and identified HepG2 (Liver hepatocellular carcinoma cell line), K562 (Chronic myelogenous leukemia cell line), and MCF-7 cells (Hormone positive breast cancer cell lines) amongst the cancer cell lines with high levels of BPTF mRNA (Figure S1 A). Next, we utilized an all-in-one CRISPR-Cas9-GFP lentiviral vector and single stranded guide RNA sequences (sgRNAs) specific to targeting BPTF to define the effects of BPTF deletion on cellular growth, as a readout for growth dependency (Figure S1 A-C). The percentage of GFP positive cells (GFP+), a mark for lentiviral infection, was monitored for several weeks using flow cytometry, as a proxy for the effects of BPTF deletion of growth of HepG2, K562 and MCF-7 cell lines. Control sgRNA for the essential gene RNA Polymerase 3 (RPA3, cell depletion effect), and vectors lacking specific sgRNAs (Empty vector) were used for comparison. HepG2 liver cancer cells were the least sensitive to BPTF depletion through the experimental time course (Figure 8A). Conversely, the percentage of GFP+ cells gradually decreased over time in MCF-7 cells and K562 expressing BPTF sgRNAs, similar to the RPA3 sgRNA depletion control experiment. K562 cells were the most sensitive to BPTF depletion, with the percentage of GFP+ cells dropping ~2-fold over time. Together, these results demonstrate that K562 and MCF-7 cells are dependent on BPTF function for growth, providing useful cell line models to define epigenetic mechanisms associated with BPTF dependency, and for testing the efficacy of BPTF bromodomain inhibitors.
Figure 8:

Sensitivity of cancer cell lines to (S)-1 and (R)-1. A) CRISPR-Cas9 BPTF-targeting. HepG2, MCF-7 and K562 cell lines were infected with all in one CRISPR-Cas9-GFP lentiviral particles, expressing sgRNAs targeting RPA3 gene (depletion control) and the BPTF gene. Fold change was calculated by comparing final measure of GFP to initial infection GFP. n=2 technical replicates. B) Cell viability analysis in cancer cell lines treated with 5 μM of either (S)-1 or (R)-1 for 72 h, following incubation with AlamarBlue for 4 h. Error bars demonstrate standard deviation across biological replicates.
Given that additional loss of protein function beyond bromodomain effects can result from gene deletion studies, we tested the effect of (S)-1 or (R)-1 on the growth of BPTF-dependent (K562 and MCF-7) and independent (HepG2) cancer cell lines. In doing so, cancer cell lines were treated with either (S)-1, or (R)-1 at 5 μM for 72 hours, following quantification of cellular viability/growth using an AlamarBlue viability assay. Our results show that HepG2 cells are not sensitive to treatment with either (S)-1 or (R)-1, consistent with BPTF deletion experiments above (Figure 8). Alternatively, K562 and MCF-7 cells were sensitive to treatment with (S)-1, demonstrated by a marked decrease on cell viability. MCF-7 cells were also sensitive to the inactive bromodomain inhibitor (R)-1, suggesting that for MCF-7 cells, off-target effects in addition to those of BPTF inhibition may be controlling cell growth in response to small molecule treatment (Figure 8). Alternative, K562 cells were only affected by (S)-1. These results support K562 cells as being a useful model cell line for further evaluation of the effects of (S)-1 and for defining the epigenetic mechanisms associated with BPTF dependency with improved BPTF bromodomain inhibitors.
Conclusions
In conclusion, we have studied the SAR of rac-1, the first reported inhibitor of the bromodomain of BPTF to show selectivity over that of BRD4(1). We identified that the (S)-enantiomer is the dominant stereoisomer for binding to the bromodomain. In addition, substitution of the amino-pyrimidine moiety led to the diminished binding of the substrate to CDKL2 and TRKC kinases. These results also showcase PrOF NMR as a useful protein-ligand characterization tool for quantifying weak binding ligands during ligand deconstruction, SAR analysis, and testing selectivity against related bromodomains. Finally, cell-based experiments including BPTF depletion, and chemical inhibition, support a BPTF dependence in K562 cells, a chronic myelogenous leukemia cell line. Richart and co-workers, identified cancer cell lines that could be sensitive to BPTF inhibition with high c-Myc levels.5 Chronic myelogenous leukemia cells, were identified to have some of the highest c-Myc levels, supporting our BPTF sensitivity studies. Together these studies provide a framework for the design of inhibitors for the BPTF bromodomain, an important epigenetic effector domain, lacking potent ligands.
Supplementary Material
Acknowledgements
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes under the award numbers (R01GM121414, W.C.K.P, R01GM130794, A.D., R35GM118047 H.A.) the National Cancer Institute (R01CA215755 M.K) and Rita Allen Scholar grant (C.O.D.S.) and Pershing Square Sohn Prize (C.O.D.S) J.A.J. was supported by a National Institutes of Health Biotechnology training grant 5T32GM008347–23 A.K.U. was supported by a UMN Doctoral Dissertation Fellowship. Cold Spring Harbor Labs shared resources were supported through the Cancer Center 5P30CA045508 and the cell lines were distributed through the Cold Spring Harbor Tissue Culture Facility. We would like to thank Emily Sherman for early synthetic efforts on rac-1 analogs.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Landry J, Sharov AA, Piao Y, Sharova LV, Xiao H, Southon E, Matta J, Tessarollo L, Zhang YE, Ko MS, Kuehn MR, Yamaguchi TP and Wu C, PLoS Genet, 2008, 4, e1000241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frey WD, Chaudhry A, Slepicka PF, Ouellette AM, Kirberger SE, Pomerantz WCK, Hannon GJ and dos Santos CO, Stem Cell Reports, 2017, 9, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones MH, Hamana N and Shimane M, Genomics, 2000, 63, 35–39. [DOI] [PubMed] [Google Scholar]
- 4.Ruthenburg AJ, Li H, Milne TA, Dewell S, McGinty RK, Yuen M, Ueberheide B, Dou Y, Muir TW, Patel DJ and Allis CD, Cell, 2011, 145, 692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richart L, Pau E. Carrillo-de Santa, Rio-Machin A, de Andres MP, Cigudosa JC, Lobo VJ and Real FX, Nat Commun, 2016, 7, 10153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dar AA, Nosrati M, Bezrookove V, de Semir D, Majid S, Thummala S, Sun V, Tong S, Leong SP, Minor D, Billings PR, Soroceanu L, Debs R, Miller JR 3rd, Sagebiel RW and Kashani-Sabet M, J.Natl. Cancer. Inst,, DOI: 10.1093/jnci/djv034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiao S, Liu L, Lu X, Long J, Zhou X and Fang M, J Cancer Res Clin Oncol, 2015, 141, 1465–1474. [DOI] [PubMed] [Google Scholar]
- 8.Xiao S, Liu L, Fang M, Zhou X, Peng X, Long J and Lu X, Dig Dis Sci, 2015, 60, 910–918. [DOI] [PubMed] [Google Scholar]
- 9.Kim K, Punj V, Choi J, Heo K, Kim JM, Laird PW and An W, Epigenetics and Chromatin, 2013, 6, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dai M, Lu J-J, Guo W, Yu W, Wang Q, Tang R, Tang Z, Xiao Y, Li Z, Sun W, Sun X, Qin Y, Huang W, Deng W and Wu T, Oncotarget, 2015, 6, 33878–33892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mishra NK, Urick AK, Ember SWJ, Scho E and Pomerantz WC, ACS Chem. Biol, 2014, 9, 2755–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urick AK, Hawk LML, Cassel MK, Mishra NK, Liu S, Adhikari N, Zhang W, Dos Santos CO, Hall JL and Pomerantz WCK, ACS Chem. Biol, 2015, 10, 2246–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang G, Smith SG and Zhou M-M, Chem. Rev, 2015, 115, 11625–11668. [DOI] [PubMed] [Google Scholar]
- 14.Brand M, Measures AM, Wilson BG, Cortopassi WA, Alexander R, Höss M, Hewings DS, Rooney TPC, Paton RS and Conway SJ, ACS Chem. Biol, 2015, 10, 22–39. [DOI] [PubMed] [Google Scholar]
- 15.Urick AK, Calle LP, Espinosa JF, Hu H and Pomerantz WCK, ACS Chem. Biol, 2016, 11, 3154–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Babaoglu K and Shoichet BK, Nat. Chem. Biol, 2006, 2, 720–723. [DOI] [PubMed] [Google Scholar]
- 17.Ciulli A, Williams G, Smith AG, Blundell TL and Abell C, J. Med. Chem, 2006, 49, 4992–5000. [DOI] [PubMed] [Google Scholar]
- 18.Ayoub AM, Hawk LML, Herzig RJ, Jiang J, Wisniewski AJ, Gee CT, Zhao P, Zhu JY, Berndt N, Offei-Addo NK, Scott TG, Qi J, Bradner JE, Ward TR, Schönbrunn E, Georg GI and Pomerantz WCK, J. Med. Chem, 2017, 60, 4805–4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Urick AK, Hawk LML, Cassel MK, Mishra NK, Liu S, Adhikari N, Zhang W, dos Santos CO, Hall JL and Pomerantz WCK, ACS Chem. Biol, 2015, 10, 2246–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kozakov D, Hall DR, Jehle S, Luo L, Ochiana SO, Jones EV, Pollastri M, Allen KN, Whitty A and Vajda S, Proc. Natl. Acad. Sci, 2015, 112, E2585–E2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dickson A and Brooks CL, J. Phys. Chem. B, 2014, 118, 3532–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dickson A, Biophys. J, 2018, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Müller S, Pawson T, Gingras AC, Arrowsmith CH and Knapp S, Cell, 2012, 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moustakim M, Clark PGK, Trulli L, de Arriba A. L. Fuentes, Ehebauer MT, Chaikuad A, Murphy EJ, Mendez-Johnson J, Daniels D, Hou CFD, Lin YH, Walker JR, Hui R, Yang H, Dorrell L, Rogers CM, Monteiro OP, Fedorov O, Huber KVM, Knapp S, Heer J, Dixon DJ and Brennan PE, Angew. Chemie - Int. Ed, 2017, 56, 827–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perell GT, Mishra NK, Sudhamalla B, Ycas PD, Islam K and Pomerantz WCK, Biochemistry, 2017, 56, 4607–4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Humphreys PG, Bamborough P, Chung C, Craggs PD, Gordon L, Grandi P, Hayhow TG, Hussain J, Jones KL, Lindon M, Michon A-M, Renaux JF, Suckling CJ, Tough DF and Prinjha RK, J. Med. Chem, 2017, 60, 695–709. [DOI] [PubMed] [Google Scholar]
- 27.Perell GT, Mishra NK, Sudhamalla B, Ycas PD, Islam K and Pomerantz WCK, Biochemistry, 2017, 56, 4607–4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Almela MJ, Lozano S, Lelièvre J, Colmenarejo G, Coterón JM, Rodrigues J, Gonzalez C and Herreros E, PLoS One, 2015, 10, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elkins JM, Fedele V, Szklarz M, Azeez K. R. Abdul, Salah E, Mikolajczyk J, Romanov S, Sepetov N, Huang XP, Roth BL, Zen A. Al Haj, Fourches D, Muratov E, Tropsha A, Morris J, Teicher BA, Kunkel M, Polley E, Lackey KE, Atkinson FL, Overington JP, Bamborough P, Müller S, Price DJ, Willson TM, Drewry DH, Knapp S and Zuercher WJ, Nat. Biotechnol, 2016, 34, 95–103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.