

Abstract
Rationale
The ability to memorize threat-associated cues and subsequently react to them, exhibiting escape or avoidance responses, is an essential, often life-saving behavioral mechanism that can be experimentally studied using the fear (threat) conditioning training paradigm. Presently, there is substantial evidence supporting the Synaptic Plasticity-Memory (SPM) hypothesis in relation to the mechanisms underlying the acquisition, retention and extinction of conditioned fear memory.
Objectives
The purpose of this review article is to summarize findings supporting the SPM hypothesis in context of conditioned fear control, applying the set of criteria and tests which were proposed as necessary to causally link lasting changes in synaptic transmission in corresponding neural circuits to fear memory acquisition and extinction with an emphasis on their pharmacological diversity.
Results
The mechanisms of synaptic plasticity in fear circuits exhibit complex pharmacological profiles and satisfy all four SPM criteria: detectability, anterograde alteration, retrograde alteration and mimicry.
Conclusion
The reviewed findings, accumulated over the last two decades, provide support for both necessity and sufficiency of synaptic plasticity in fear circuits for fear memory acquisition and retention, and, in part, for fear extinction, with the latter requiring additional experimental work.
Keywords: Fear Conditioning, Extinction, Synaptic transmission, Synaptic plasticity, Behavior, Animal model
Introduction
The mammalian brain possesses the ability to recognize threats and react to potentially dangerous situations exhibiting characteristic behavioral responses in order to maximize the chances of organismal survival. In both humans and experimental animals, the threat-induced behavioral reactions include those that are inborn (not based on previous life experiences) and those which are learned, thus representing memories of aversive events (Shumyatsky et al. 2005). The latter were extensively studied with classical Pavlovian fear (threat) conditioning – a form of associative learning that results from pairing an initially neutral stimulus which could be of any sensory modality (the conditioned stimulus or CS, such as acoustic tone during auditory fear conditioning) with an aversive stimulus (the unconditioned stimulus US; most commonly, electric footshock) and leads to the formation of a strong CS-US association. In conditioned subjects, CS presentation alone triggers a physiological fear response which is commonly measured as a degree of freezing (LeDoux 2000; Maren 2001). However, if the CS is presented over and over again without aversive reinforcement (i.e. in the absence of the US), the conditioned fear response progressively declines in a process known as fear extinction. In the laboratory, the effectiveness of fear conditioning or fear extinction is commonly measured during a subsequent memory retention test, when the CS is presented without the US either in the new context (probing cued fear memory) or fear extinction context (assaying retention of extinction memory) (Myers and Davis 2007). In such paradigms, animals respond differently to the CS before fear conditioning, after it has been paired with the US, and following extinction of the conditioned response. Thus, the US affects how the CS is perceived by the subject and processed by neuronal circuits of fear control. A major goal in the field is to identify neuronal and synaptic level mechanisms enabling differential coupling of the same stimulus (CS) to specific fear-related behavioral responses (Blair et al. 2001).
Somatosensory inputs carrying the US information and afferent inputs conveying the CS signals converge in the lateral nucleus of the amygdala (LA) where neurons are able to respond to both nociceptive and acoustic stimuli (Fig. 1; Pitkänen et al.1997). Specifically, auditory sensory information is relayed to the LA during auditory fear conditioning via two excitatory glutamatergic projections which carry information about distinct characteristics of the CS (LeDoux, 2000). One pathway, forming the direct thalamic input to the LA, arises in the medial subdivision of the medial geniculate nucleus and the adjacent posterior intralaminar nucleus of the thalamus (MGm/PIN). The second, indirect cortico-amygdala pathway, relays auditory information from auditory thalamus to the LA via auditory cortex (ACx) (LeDoux 2000; Blair et al. 2001; Maren 2001; Maren and Quirk 2004; Dityatev and Bolshakov 2005). Whereas either pathway appears to be sufficient for conditioning to a simple CS (e.g., pure tone), cortical projections may be necessary for the accurate representation of composite auditory stimuli and more complex auditory stimulus processing (e.g., pitch and temporal sound modulation) (reviewed in Armony and LeDoux 1997). In contrast, the US is transmitted to the LA from somatosensory thalamus and somatosensory cortex, as suggested by early studies involving anatomical tracing techniques (Shi and Cassell 1998), lesions (Shi and Davis 1999; Lanuza et al. 2008) and behavioral training combined with electrical stimulation (Cruikshank et al. 1992). The mentioned approaches do not allow, however, the selective manipulation of discrete components within neuronal circuits (e.g., in pathway- and/or cell-type specific manner). The precise nature of the US-transmitting neural circuits is still not completely understood (Brunzell and Kim 2001; Lanuza et al. 2004; Orsini and Maren 2012). Further experiments allowing higher levels of specificity are needed in order to fully uncover nociceptive pathways to the LA contributing to fear conditioning. Among the new technological advances, optogenetic strategies (Boyden et al. 2005; Yizhar et al. 2011), utilizing light-sensitive proteins (opsins), allow accurate spatiotemporal control of neuronal activity in genetically determined neuronal populations, specific brain areas and neuronal pathways (Johansen et al. 2012; Belzung et al. 2014; Lalumiere 2014; Luchkina and Bolshakov 2017). These methodologies have already resulted in important insights into US processing in fear circuits at a cell specific level. For instance, protein kinase Cδ-expressing (PKCδ+) neurons in the lateral central amygdala (CeL) were shown to convey information about the US to LA neurons (Yu et al 2017). About 50% of these neurons exhibited shock-evoked responses during fear conditioning. In a consistent fashion, inhibition of PKCδ+ CeL neurons by DREADD (designer receptor exclusively activated by designer drugs) suppressed US-evoked responses of LA neurons (Yu et al. 2017).
Fig. 1.
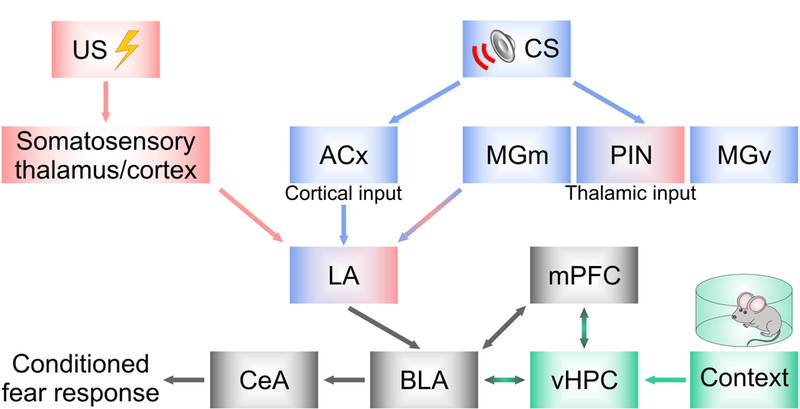
A diagram illustrating brain circuitry of fear-related learning. During fear conditioning, the conditioned stimulus (CS) carrying auditory information (blue) is transmitted to the lateral nucleus of the amygdala (LA) by direct thalamic and indirect cortical pathways (LeDoux 2000; Maren 2001; Dityatev and Bolshakov 2005). Thalamic input to the LA arises in the medial subdivision of the medial geniculate nucleus and the adjacent posterior intralaminar nucleus of the thalamus (MGm/PIN). The auditory thalamus also sends projections to the auditory cortex (ACx). The latter, in turn, projects to the LA, thus forming indirect cortico-amygdala pathway. The LA receives somatosensory signals (red), coding the unconditioned stimulus (US) information from the somatosensory thalamus and cortex (Cruikshank et al. 1992; Shi and Cassell 1998; Shi and Davis 1999; Lanuza et al. 2008). The convergence of CS and US on LA neurons results in lasting synaptic enhancements in auditory CS inputs to the LA, contributing to the encoding of conditioned fear memory (Maren and Quirk, 2004). The signals are then relayed to other components of the learned fear circuits, including BLA, and eventually to the central nucleus of the amygdala (CeA). CeA mediates physiological manifestations of fear through divergent projections to the hypothalamus and brainstem areas (Maren and Quirk 2004). The ventral hippocampus (vHPC) projects to the BLA and is important for the encoding of context-dependency of fear-related behaviors (Herry et al. 2010; Orsini and Maren 2012). Medial prefrontal cortex (mPFC) modulates fear-related behaviors, both fear learning and extinction of fear memory, through its direct projections to the BLA (Milad and Quirk 2002; Likhtik et al. 2005; Quirk et al. 2006).
Overall, the circuits of learned fear are quite complex and more detailed understanding of their anatomical and functional organization may be needed. Fear circuits can be regulated at different levels, including structural and functional modifications associated with learning events. In this review article, we focus on synaptic mechanisms of conditioned fear control when fear is a form of memory.
Synaptic plasticity at inputs to LA as a mechanism of fear learning
The ability of synaptic connections to change their strength in response to stereotyped patterns of neuronal activity, termed activity-dependent synaptic plasticity (i.e., long-term potentiation or LTP and long-term depression or LTD), is widely believed to play a pivotal role in different aspects of learning and memory, providing their physical substrate at the cellular level. According to the popular synaptic plasticity and memory (SPM) hypothesis “activity-dependent synaptic plasticity is induced at appropriate synapses during memory formation, and is both necessary and sufficient for the information storage underlying the type of memory mediated by the brain area in which that plasticity is observed” (Martin et al. 2000). The latter seminal work laid out a set of criteria for determining the necessity and sufficiency of synaptic plasticity in specific brain areas in mechanisms of memory acquisition and storage. Since then, substantial progress has been made (e.g., Takeuchi et al. 2014), especially recently with the introduction of several new methodologies, including multi-electrode array stimulation and recording, optogenetics, chemogenetics, and advanced molecular genetics. Unsurprisingly, strong support for the SPM hypothesis came from studies in the hippocampus, the region in which LTP was discovered (e.g., Whitlock et al. 2006; Garner et al. 2012; Ramirez et al. 2013; Rossetti et al. 2017). However, studies of fear conditioning provided what appears to be the most direct evidence yet that associative LTP at inputs to the LA may constitute a mechanism for encoding the CS-US association and storing fear memories in the course of auditory fear conditioning (Rogan et al. 1997; McKernan and Shinnick-Gallagher 1997; Tsvetkov et al. 2002; Shumyatsky et al. 2002; Rumpel et al. 2005; Shumyatsky et al. 2005; Cho et al. 2012).
Here, we review experimental evidence obtained in studies of fear-related behavior which support the SPM hypothesis, in a manner similar to previous reviews of hippocampus- and cortex-dependent learning (Martin and Morris 2002; Takeuchi et al. 2014). Our intention is to link persistent changes in synaptic transmission in fear circuits (focusing on inputs to the LA) to memory of the CS-US association. Summarizing established ideas and reviewing new findings supporting the SPM hypothesis, we will apply the same set of criteria and tests proposed previously as both necessary and sufficient to causally link synaptic plasticity to memory mechanisms, namely detectability, anterograde and retrograde alterations, and mimicry (Fig. 2) (Martin et al. 2000). It was recently outlined that the experimental findings supporting detectability and mimicry criteria may demonstrate the sufficiency of long-term synaptic plasticity in fear conditioning pathways for encoding and retention of fear memory, whereas the anterograde and retrograde alterations criteria, when met, may provide evidence of necessity (Takeuchi et al. 2014). Below, we describe each of the mentioned criteria in detail in relation to the mechanisms of conditioned fear memory.
Fig. 2.
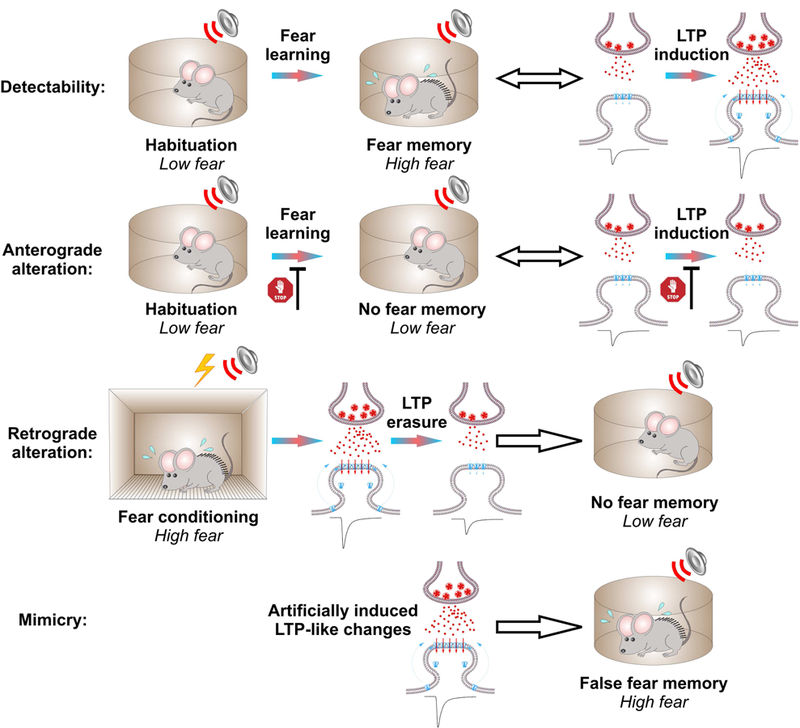
Schematic illustration of the criteria proposed previously to evaluate a causal link between plastic changes in specific brain areas and corresponding brain area-specific memory (Martin et al. 2000). Fear memory and synaptic plasticity at cortical and thalamic projections to the LA are shown here as an example. Similar criteria (i.e., detectability, anterograde and retrograde alterations, and mimicry criteria), can be applied for testing the necessity and sufficiency of plastic changes underlying other amygdala-based forms of learning, such as fear extinction.
Detectability
According to the detectability criterion (Martin et al. 2000), if fear memory acquisition is associated with synaptic potentiation (i.e., LTP or long-term potentiation),changes in synaptic efficacy should be detected during or following fear learning in corresponding neural circuits (e.g., in cortico-amygdala and/or thalamo-amygdala pathways).
Consistent with this notion, fear conditioning significantly increases both tone-evoked firing rates of LA neurons, detected with multiple single-unit recordings (Quirk et al. 1995; Repa et al. 2001; Goosens et al. 2003), and auditory CS-evoked field potentials in the LA in the course of training in freely behaving rats (Rogan et al. 1997). Evoked excitatory postsynaptic glutamatergic currents (EPSCs) in LA neurons, elicited by electrical stimulation of afferents from the medial geniculate nucleus (MGN) in vitro, were found to be potentiated in slices from fear-conditioned rats as compared to slices from naïve or unpaired control rats (McKernan and Shinnick-Gallagher 1997). Contextual fear conditioning is associated with a concomitant strengthening of glutamatergic synaptic transmission at cortical inputs to the amygdala (Nonaka et al. 2014). Moreover, fear conditioning occluded LTP in the cortico-LA pathway (Tsvetkov et al. 2002; Cho et al. 2012). This is consistent with the idea that fear learning induces LTP-like synaptic enhancements in vivo that mechanistically resemble electrically-induced cortico-amygdala LTP in brain slices. However, the above-mentioned studies, describing an increased responsiveness of LA neurons to the CS or electric stimulation of auditory pathways during the course or immediately following fear learning, could not evaluate the specificity of observed changes in synaptic strength in the auditory CS pathways (e.g., whether it is restricted to the conditioned tone only). Furthermore, any detected increases in synaptic efficacy could be due to fear-related changes in the auditory cortex and/or auditory thalamus upstream to the LA. Experiments involving discriminative conditioning paradigms and/or optogenetic activation of thalamic or cortical afferents specifically in the LA have successfully addressed these issues (Collins and Paré 2000; Nabavi et al. 2014; Kim and Cho 2017). Discriminative fear conditioning, in which one auditory cue (CS+, e.g., 5 kHz) is paired with the US whilst a second stimulus (CS-, e.g., 10 kHz) does not predict danger, increased auditory-evoked activity specifically to the former (CS+), but not the latter (CS-) (Collins and Paré 2000; Goosens et al. 2003; Ghosh and Chattarji 2015). In particular, using a combination of cutting-edge methodologies, including behaviorally-relevant activity-dependent neuronal labeling techniques together with optogenetics and electrophysiology, LTP was induced preferentially in the auditory CS+ inputs to a subset of LA neurons activated during fear conditioning (approximately 20% of LA cells), but not in randomly selected ACx/MGm to LA pathways (Kim and Cho 2017). Long-lasting changes in synaptic efficacy (phenotypically resembling LTP) were observed in vitro and in vivo at synapses in projections from the auditory thalamus to the lateral amygdala following fear learning. Thus, input-specific LTP in functionally identified pathways in fear circuits that transmit distinct CS information to the amygdala may encode tone-specific fear memory (Kim and Cho 2017).
Other fear-related brain areas and subdivisions of the amygdala also demonstrate fear learning-associated synaptic plasticity. For example, following auditory fear conditioning, associative synaptic plasticity was induced at inputs both to and within the central nucleus of the amygdala (CeA) (Paré et al. 2004; Wilensky et al. 2006; Ciocchi et al. 2010; Duvarci et al. 2011; Li et al. 2013a), at synapses onto interneurons in the LA and basolateral amygdala (BLA) (Mahanty and Sah 1998; Bauer and LeDoux 2004), and the prelimbic cortex-BLA pathway (Arruda-Carvalho and Clem 2014). Furthermore, the auditory thalamus (MGm/PIN) has been alternatively suggested to serve as a possible neuronal substrate of auditory fear learning (not just as a sensory relay) due, in part, to the observed convergence of auditory and nociceptive inputs at single MGm/PIN neurons and to evidence for the induction of MGm/PIN associative synaptic plasticity during fear conditioning (reviewed in Weinberger 2011). Less studied types of synaptic plasticity, at least in relation to the function of fear-controlling circuits, such as spike timing-dependent synaptic plasticity (Shin et al. 2006) and input timing– dependent plasticity in afferent projections to the LA (Cho et al. 2012), may provide further mechanisms of synaptic strengthening during fear learning.
Different induction and expression mechanisms can underlie behaviorally-induced LTP-like synaptic enhancements in fear conditioning pathways. Cellular and molecular mechanisms of LTP at synaptic inputs to the LA have been extensively investigated in in vitro experiments implicating electrophysiological recordings from neurons in amygdalar slices. LTP induction in LA was shown to involve an activation of N-methyl-D-aspartate (NMDA) receptors and/or voltage-gated Ca2+ channels, depending on the induction protocol (Huang and Kandel 1998; Weisskopf et al. 1999; Bauer et al. 2002; Table 1). The resulting elevation of the intracellular Ca2+ concentration may cause further increases in intracellular Ca2+ through the Ca2+-induced Ca2+ release from intracellular stores and result in a subsequent activation of different downstream signaling molecules, such as Ca2+/calmodulin-dependent protein kinase II (CaMKII) and other protein kinases (Dityatev and Bolshakov, 2005). Upon activation, CaMKII translocates from an F-actin-bound state in the cytosol to a postsynaptic density (PSD)-bound form at the synapses (Shen and Meyer 1999) where its synaptic targets are located. Correspondingly, fear conditioning results in an increased amount of the active (autophosphorylated) form of CaMKII in dendritic spines in the LA (Rodrigues et al. 2004). Activated protein kinases, in turn, can alter properties of different synaptic proteins and their interactions by phosphorylation. This leads to persistent changes involving either pre- (an increase in neurotransmitter release (Tsvetkov et al. 2002; Li et al. 2013b; Nonaka et al. 2014) or postsynaptic modifications (quantal size) (Rumpel et al. 2005; Clem and Huganir 2010). The latter manifests as larger postsynaptic responses to the same amount of neurotransmitter released, mediated by an increase in the number of postsynaptic α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionic acid (AMPA) receptors or modulation of existing AMPA receptors (increased ion channel conductance, increased open probability, agonist affinity and/or changes in channel kinetics). For example, phosphorylation of GluA1 AMPA subunit at Ser831 by CaMKII increases conductance of homomeric GluA1 channels (Derkach et al.1999) and insertion of GluA1 into synapses (Hayashi et al. 2000), both leading to an increase in synaptic strength at activated synapse. In support of the contribution of such mechanisms to fear learning, it was found that GluA1 (but not GluA2/3) AMPA receptor subunit expression and trafficking to synaptic sites in the LA is increased following either LTP induction in vitro or fear conditioning (Rumpel et al. 2005; Yeh et al. 2006; Humeau et al. 2007; Clem and Huganir 2010; Nedelescu et al. 2010). Specifically, an enhancement of AMPA receptor-mediated miniature EPSCs in LA neurons as well as a sustained increase in AMPAR to NMDAR (AMPAR/NMDAR) EPSC amplitude ratio in thalamic input to the LA have been observed following auditory fear conditioning in mice, both of which lasting for 7 days (Clem and Huganir 2010). Furthermore, this increase in synaptic strength following fear conditioning was associated with synaptic incorporation of GluA1 subunit-containing (Rumpel et al. 2005), GluA2 subunit-lacking Ca2+-permeable (Clem and Huganir 2010) AMPA glutamate receptors.
Table 1.
Effects of pharmacological agents on neuronal activity and synaptic plasticity implicated in fear conditioning (FC).
| Target, mechanism of action & agent |
Pathway | Effects on neuronal activity and synaptic plasticity |
Site | Time | Effect on FC |
|---|---|---|---|---|---|
| NMDA receptor | |||||
|
Antagonist AP-5 |
CTX-LA THAL-LA |
Blocks tetanus-induced, but not pairing-induced LTP (Huang and Kandel 1998; Weisskopf et al. 1999; Bauer et al. 2002) |
AMY (LA) |
Pre-FC Post-FC |
Blocks the acquisition, but not expression (Miserendino et al. 1990; Campeau et al. 1992) |
|
GluN2B antagonist Ifenprodil |
THAL-LA | Impairs tetanus-induced LTP (Bauer et al. 2002) |
LA | Pre-FC Pre-Test |
Disrupts the acquisition, but not expression (Rodrigues et al. 2001) |
| L-type VGCC | |||||
|
Blocker Verapamil Nifedipine |
THAL-LA | Blocks pairing-induced, but not tetanus-induced LTP (Weisskopf et al. 1999; Bauer et al. 2002) |
LA | Pre-FC | Impairs long-term (24 h) fear memory (Bauer et al. 2002) |
| Group I mGluR | |||||
|
mGluR5 antagonist MPEP |
THAL-LA | Impairs tetanus-induced LTP (Rodrigues et al. 2002) |
LA | Pre-FC Post-FC Pre-Test |
Impairs the acquisition, but not expression or consolidation (Rodrigues et al. 2002) |
|
mGluR1 antagonist CPCCOEt |
LA | Pre-FC | No effect on fear memory acquisition (Kim et al. 2007a) |
||
| CaMKII | |||||
|
Inhibitor KN-62 |
THAL-LA | Impairs tetanus-induced LTP (Rodrigues et al. 2004) |
LA | Pre-FC Pre-Test |
Impairs the acquisition, but not the expression (Rodrigues et al. 2004) |
| PKA & AC | |||||
|
PKA inhibitor Rp-cAMPS KT5720 |
CTX-LA THAL-LA |
Blocks tetanus-induced E-LTP (Huang and Kandel 1998) and L-LTP (Huang et al. 2000) |
LA | Post-FC | (RP)-cAMPS impairs fear memory consolidation (Schafe and LeDoux 2000) |
|
Rp-cAMPS H-89 |
CTX-LA | Blocks heterosynaptic associative presynaptic LTP (Fourcaudot et al. 2008) |
|||
|
AC activator Forskolin |
LA | Enhances tone-induced firing at lower shock intensities (Ghosh and Chattarji 2015) |
LA | Increases freezing to CS−, leads to generalized fear (Ghosh and Chattarji 2015) |
|
| PKA & PKC | |||||
|
Inhibitor H7 |
BLA Not CEA |
Pre-FC | Impairs acquisition of long-term fear memories (Goosens et al. 2000) |
||
| Protein synthesis | |||||
|
Inhibitor Anisomycin |
CTX-LA | Blocks tetanus-induced L-LTP, but not E-LTP (Huang et al. 2000) |
LA | Post-FC | Impairs fear memory consolidation (Schafe and LeDoux 2000) |
| CTX-LA | Blocks slow onset L-LTP induced by LFS in CTX- LA, but not THAL-LA (Huang and Kandel 2007) |
CeA | Post-FC | Impairs fear memory consolidation (Wilensky et al. 2006) |
|
| PI-3 kinase | |||||
|
Inhibitor Wortmannin |
AMY | Blocks FC-induced changes in phosphorylation of CREB (Lin et al. 2003c) |
LA BLA |
Pre-FC | Blocks long-term (24 h) but not short-term memory (1h) (Lin et al. 2001) |
|
Inhibitors Wortmannin LY294002 |
CTX-LA | Blocks tetanus-induced LTP (Lin et al. 2001) |
|||
| GABAA | |||||
|
Agonist Muscimol |
LA BLA |
Silences neuronal activity (Herry et al., 2008; Ghosh and Chattarji 2015) |
LA | Pre-FC | Blocks fear generalization in animals conditioned to a strong US (Ghosh and Chattarji 2015) |
| LA BLA |
Pre-FC Post-FC |
Impairs acquisition, but not expression (Wilensky et al. 2000; Wilensky et al. 2006, but see Sierra-Mercado et al. 2011) |
|||
| CeA | Pre-FC Pre-Test |
Impairs both fear acquisition and fear expression (Wilensky et al. 2006) |
|||
| PL | Post-FC | Affects fear expression (Sierra-Mercado et al. 2011) |
|||
|
Antagonist Picrotoxin |
THAL-LA CTX-LA |
Facilitates in vitro LTP induction (Bissière et al. 2003; Shin et al. 2006; Tully et al. 2007) |
|||
List of abbreviations used:
AC - adenylyl cyclase, AMY – amygdala, BLA – basolateral amygdala, CaMKII - Ca2+/calmodulin-dependent protein kinase II, CeA – central amygdala, CREB - cAMP response element-binding protein, CTX-LA – cortical pathway to the LA, E-LTP – early phase of LTP, FC – fear conditioning, H7– 1-(5’-isoquinolinesulfonyl)-2-methylpiperazine, LA – lateral anygdala, LFS – low-frequency stimulation, L-LTP – late phase of LTP, LTP – long-term potentiation, mGluR - metabotropic glutamate receptor, MPEP - 2-methyl-6-(phenylethynyl)-pyridine, PI-3 - Phosphatidyl inositol 3, PKA – protein kinase A or cAMP-dependent protein kinase, PKC – protein kinase C, PL - prelimbicmedial prefrontal cortex (mPFC), THAL-LA – thalamic input to the LA, VGCC – voltage-gated calcium channel.
Together, these findings provide strong support to the notion that potentiation of neurotransmission in inputs conveying auditory CS information to the amygdala may serve as a cellular mechanism of fear learning.
Anterograde alteration
Simply put, any manipulations perturbing the normal induction or expression of synaptic plasticity in neural circuits underlying learning (e.g., at auditory inputs to the lateral amygdala) should have an effect on learning and memory (fear learning and memory) and vice versa. Interventions preventing changes in synaptic efficacy during fear learning should lead to fear memory impairment (Martin et al. 2000; Takeuchi et al. 2014). Notably, several pharmacological agents capable of perturbing LTP induction, but not baseline excitatory synaptic transmission per se, in amygdala brain slices in vitro also impair auditory fear memory acquisition or expression, when infused into the LA prior or post conditioning training, correspondingly (Table 1). These include antagonists of NMDA receptor (Rodrigues et al. 2001; Bauer et al. 2002), L-type voltage-gated Ca2+ channel (VGCC) blockers (Bauer et al. 2002), mGluR5 subtype of group I metabotropic glutamate receptor (mGluR) antagonists (Rodrigues et al. 2002), as well as Ca2+/calmodulin-dependent protein kinase II (CaMKII) (Rodrigues et al. 2004) and cAMP-dependent protein kinase (protein kinase A or PKA) (Schafe and LeDoux 2000; Fourcaudot et al. 2008) inhibitors (Table 1). Whereas fear learning may require activation of both NMDA receptors and VGCCs, earlier studies indicate differential involvement of NMDA receptors and VGCCs in fear memory acquisition and consolidation, correspondingly (Miserendino et al. 1990; Campeau et al. 1992; Rodrigues et al. 2001; Bauer et al. et al. 2002; Table 1).
Experimental interventions interfering with expression mechanisms of behaviorally-induced synaptic plasticity in CS projections to the LA in fear-conditioned animals also cause fear memory impairments. For instance, HSV vector-mediated expression of GluA1 C-tail in LA neurons was used to disrupt AMPA receptor trafficking associated with fear conditioning (Rumpel et al. 2005). Blockade of GluA1-containing AMPA receptor trafficking in a quarter of LA neurons was sufficient to impair LTP in the amygdala and significantly reduced freezing responses (by approximately 50%) as assessed during memory retention tests 3 h and 24 h following training. Thus, limiting LTP expression in the LA results in a deficit in fear acquisition that is translated into disrupted fear memory (Rumpel et al. 2005).
Studies on genetically modified mice provided evidence that behaviorally-induced synaptic and neuronal modifications in brain circuits of fear learning and memory may be regulated by circuitry-specific gene expression (Shumyatsky et al. 2002; Shumyatsky et al. 2005; Humeau et al. 2007; Riccio et al. 2009; Riccio et al. 2014). For instance, a differential involvement of GluA1 and GluA3 AMPA receptor subunits in expression mechanisms of pathway specific LTP in the LA and auditory fear conditioning was previously analyzed using GluA1 and GluA3 knock-out mice (GluA1−/− and GluA3−/−) (Humeau et al. 2007). GluA1−/− mice exhibited no LTP in thalamic inputs, whereas both GluA1 and GluA3 subunits contributed to LTP in the cortical pathway. However, fear conditioning was selectively impaired only in GluA1−/− animals, suggesting that GluA1-dependent LTP is a dominant form of plasticity underlying fear learning. As another example, genetic ablation of a particular gene, stathmin, enriched in fear conditioning circuits, was associated with deficits in spike timing-dependent LTP at amygdala synapses and decreased ability of mutant mice to learn and remember fear, providing further evidence that LTP in the auditory CS pathways may serve as a mechanism for fear memory formation (Shumyatsky et al. 2005).
These results demonstrate the necessity of synaptic potentiation in the LA for fear memory acquisition and, as such, fulfill the anterograde alteration criterion.
Retrograde alteration
Any interference that changes the spatial distribution of synaptic weights across neurons and their dendrites within the lateral amygdala formed during fear memory acquisition should lead to the alteration of this particular fear memory. In this way, any modification of synaptic changes induced by a specific fear learning paradigm should lead to the modification of this fear memory (including its erasure), detectable at the behavioral level (Martin et al. 2000; Takeuchi et al. 2014).
Even though LTP in the LA is a strong candidate for serving as the underlying mechanism of auditory fear learning, it has been difficult to satisfy the retrograde alteration criterion (particularly in respect to conditioned fear erasure) and provide reliable evidence that this criterion is met during fear conditioning. Newly-developed methodologies enable more selective manipulation of behavior-driving neuronal circuits, including those involved in fear learning and memory, and allow direct attempts to investigate such phenomena. For instance, pairing an electric foot shock with optogenetic stimulation of auditory projections to the LA resulted in a formation of conditioned fear memory (Nabavi et al. 2014). Subsequent delivery of low-frequency photostimulation, comprising an optogenetic depotentiation protocol, to the same inputs resulted in diminished conditioned fear responses during a memory retention test 24 hours later. Therefore, fear memory may be suppressed after depotentiation-inducing photostimulation (Nabavi et al. 2014). Likewise, depotentiation of the CS+-specific ACx/MGm-LA pathway was sufficient to prevent conditioned fear responses to the CS+, whilst low-frequency photostimulation of the CS-pathway had no effect (Kim and Cho 2017). In addition, ablation of LA neurons previously recruited into a fear memory trace (about 15–20% of LA neurons as identified by higher expression levels of the transcription factor CREB; Han et al. 2007; Han et al. 2009) after fear learning blocked fear memory expression (Han et al. 2009). The emergence of data fulfilling the retrograde alteration criterion provides further strong support to the view that long-lasting synaptic plasticity at inputs to the amygdala underlies fear learning.
Mimicry
Mimicry denotes the process whereby artificial fear memories may be experimentally created by replicating the spatial and temporal patterns of synaptic activity (Martin et al. 2000; Takeuchi et al. 2014). The fulfilment of the mimicry criterion may provide the most rigorous test of the STM hypothesis as it potentially has the power to prove a causal link between lasting synaptic changes in the amygdala and fear memory. Early studies demonstrated that pairing local electric stimulation of the MGm (the auditory CS area) with an aversive footshock produced LTP-like enhancements of synaptic transmission at the level of evoked field potentials in the LA which correlated with freezing responses (Kwon and Choi 2009). By replacing electrical stimulation with photostimulation of channelrhodopsin-2 (ChR2)-expressing thalamic axonal terminals in the LA in the previous paradigm, a form of optical fear conditioning can be induced. This is advantageous as it overcomes any putative nonspecific effects of electrical stimulation (e.g., stimulation of MGm fibers not terminating in the LA). During a fear memory retention test conducted 24 hours later, the same optogenetic photostimulation induced robust conditioned responses, suggesting that activation of auditory thalamic projections in the LA temporally paired with the US presentation is sufficient for the induction of associative fear learning (Kwon et al. 2014; Nabavi et al. 2014). Furthermore, in the reversed experimental design, optogenetic activation of ChR2-expressing LA principal neurons (replacing the US) in conjunction with tone presentations also mimicked fear learning and resulted in the formation of artificial fear memory in the absence of an electric shock (Johansen et al. 2010).
Thus, studies of synaptic mechanisms of learned fear appear to satisfy all four proposed criteria namely detectability, anterograde alteration, retrograde alteration and mimicry, thereby supporting the SPM hypothesis (Martin et al. 2000). More generally, these findings provide possibly the best evidence to date for both the necessity and sufficiency of long-term synaptic plasticity in the circuits of learned behavior as a cellular mechanism of learning and memory.
Depotentiation in CS inputs to amygdala as a cellular mechanism underlying extinction
As reviewed above, there is substantial evidence linking LTP in the auditory projections to the LA (cortical and thalamic inputs) to the acquisition and retention of conditioned fear memory. The mechanisms of fear extinction have also been extensively studied behaviorally, as well as at the level of underlying neural circuits and implicated signaling pathways (Myers and Davis 2007). The main components of the neural circuitry of fear extinction are the amygdala, medial prefrontal cortex (mPFC) and hippocampus. Within the amygdala, the BLA (including LA) and intercalated cells (ITC; γ-aminobutyric acid (GABA)-releasing densely packed groups of cells between the BLA and CeA) play key roles in the acquisition of extinction memory and its retention (Paré et al. 2004; Likhtik et al. 2008; Amano et al. 2010). mPFC, specifically its infralimbic (IL) division, is involved in fear inhibition and fear extinction (Milad and Quirk 2002; Maren and Quirk 2004; Santini et al. 2004; Likhtik et al. 2005; Quirk et al. 2006; Sierra-Mercado et al. 2011; Bloodgood et al. 2018; but see Do-Monte et al. 2015). CeA is a major output of the amygdala, which mediates physiological fear responses via its divergent projections to the hypothalamus and brainstem areas (Maren and Quirk 2004). The hippocampus is important for the encoding of context-dependency of fear extinction (Herry et al. 2010; Orsini and Maren 2012). Extinction of fear memory is dependent on the activation of NMDA receptors (Falls et al. 1992; Walker et al. 2002; Ledgerwood et al. 2003; Lin et al. 2003c; Mao et al. 2006; Burgos-Robles et al. 2007; Sotres-Bayon et al. 2007; Sotres-Bayon et al. 2009), metabotropic glutamate receptors (mGluRs) (Kim et al. 2007a; Fontanez-Nuin et al. 2011; Kim et al. 2015), GABAA receptors (Chhatwal et al. 2005; Akirav et al. 2006; Herry et al. 2008; Sierra-Mercado et al. 2011), L-type VGCCs (Cain et al. 2005), mitogen-activated protein kinase (Lu et al. 2001), phosphatidyl inositol 3 (PI-3) kinase (Lin et al. 2003c; Mao et al. 2006), calcineurin (Lin et al. 2003b; Almeida-Corrêa et al. 2015) and new protein synthesis (Lin et al. 2003c), where pharmacological manipulations in the LA, BLA or mPFC prior to extinction training affect extinction memory (summarized in Table 2). Despite this wealth of experimental observations, there is no consensus in the field as to the underlying synaptic mechanisms and contribution of long-term synaptic plasticity to the mechanisms of fear extinction.
Table 2.
Effects of pharmacological agents on neuronal activity and synaptic plasticity implicated in fear extinction (Ext).
| Target, mechanism of action & agent |
Pathway | Effects on neuronal activity and synaptic Plasticity |
Site | Time | Effect on fear extinction |
|
|---|---|---|---|---|---|---|
| NMDA receptor | ||||||
|
Antagonist AP-5 |
THAL-LA CTX-LA |
Blocks ex vivo depotentiation induced by LFS in fear-conditioned animals (Kim et al. 2007b; Hong et al. 2009) |
LA BLA |
Pre-Ext | Blocks extinction (Falls et al. 1992; Lin et al. 2003c; Mao et al. 2006) |
|
| CTX-LA | Blocks LFS-induced depotentiation in slices from naïve animals, following LTP induction in vitro (Lin et al. 2003a) |
|||||
| THAL-LA | Blocks LFS and pairing- induced (−50 mV) LTD (Clem and Huganir 2010) |
|||||
| BLA- mITC |
Blocks LTD and LTP (Royer and Paré 2002) |
|||||
|
Antagonist CPP |
Reduces burst firing in mPFC (Burgos-Robles et al. 2007) |
mPFC | Pre-Ext Post-Ext |
Impairs consolidation (Burgos-Robles et al. 2007) |
||
|
GluN2B Antagonist Ifenprodil |
THAL-LA | LA | Pre-Ext Post-Ext |
Impairs acquisition, but not consolidation (Sotres-Bayon et al. 2007; Sotres-Bayon et al. 2009) |
||
| mPFC | Pre-Ext Post-Ext |
Impairs consolidation, but not acquisition (Sotres-Bayon et al. 2009) |
||||
|
Partial agonist DCS (Gly site) |
CTX-LA | Facilitates LFS-induced depotentiation applied 60 min after tetanus stimulation and reduction in surface GluA1 (Mao et al. 2006) |
LA BLA |
Pre-Ext Post-Ext |
Augments consolidation (Walker et al. 2002; Ledgerwood et al. 2003; Mao et al. 2006) |
|
| L-type VGCC | ||||||
|
Blockers Nifedipine Nimodipine |
CTX-LA | Partially/fully blocks LFS- induced depotentiation in slices from naïve animals, following LTP induction in vitro (Lin et al. 2003a) |
Sys | Pre-Ext | Blocks extinction conducted 1 or 3 h post-acquisition (Cain et al. 2005) |
|
| Group I mGluR | ||||||
|
mGluR5 antagonist MPEP |
THAL-LA | No impact on LFS and pairing-induced (−50 mV) LTD (Clem and Huganir 2010) |
IL | Pre-Ext | Impairs extinction recall (Fontanez-Nuin et al. 2011) |
|
|
mGluR5 agonist CHPG |
IL | Increases intrinsic excitability of IL neurons, decreases the slow AHP (Fontanez-Nuin et al. 2011) |
||||
|
mGluR1 antagonist CPCCOEt |
THAL-LA | Blocks ex vivo depotentiation induced by LFS in fear-conditioned animals (Kim et al. 2007b) |
LA | Pre-Ext | Impairs extinction initiated 48 h (but not 2 h) after FC (Kim et al. 2007a) |
|
|
mGluR1 antagonist LY367385 |
THAL-LA | Blocks LFS and pairing- induced (−50 mV) LTD (Clem and Huganir 2010) |
||||
| Group II mGluR | ||||||
|
Antagonist EGLU |
CTX-LA | No effect on LFS-induced depotentiation in slices from naïve animals, following LTP induction in vitro (Lin et al. 2003a) |
||||
|
Antagonist LY341495 |
CTX-LA | Blocks ex vivo depotentiation induced by LFS in fear-conditioned animals (Hong et al. 2009) |
LA, not CeA |
Pre-Ext | Impairs retention, no effect on acquisition (Kim et al. 2015) |
|
|
Agonist DCG-IV |
CTX-LA | Elicits in vitro depotentiation following LTP induction by tetanus stimulation (Lin et al. 2005) |
LA & BLA |
Post-FC | Reduces a conditioned response (Lin et al. 2005) |
|
|
Protein synthesis |
||||||
|
Inhibitor Anisomycin |
LA BLA |
Pre-Ext | Blocks extinction (Lin et al. 2003c) | |||
| mPFC | Pre-Ext | Blocks retention, but not acquisition (Santini et al. 2004) |
||||
| PI-3 kinase | ||||||
|
Inhibitor Wortmannin |
AMY | Blocks extinction-induced changes in phosphorylation of CREB (Lin et al. 2003c) |
LA BLA |
Pre-Ext | Blocks extinction (Lin et al. 2003c; Mao et al. 2006) |
|
| PP2B (CaN) | ||||||
|
Inhibitors Cyclosporin A FK-506 |
CTX-LA | Blocks LFS-induced depotentiation in slices from naïve animals, following LTP induction in vitro (Lin et al. 2003a) |
LA BLA |
Pre-Ext | FK-506 blocks extinction (Lin et al. 2003b) |
|
| GABAA | ||||||
|
Agonist Muscimol |
LA BLA |
Silences neuronal activity (Herry et al., 2008; Ghosh and Chattarji 2015) |
BLA | Pre-Ext | Impairs extinction (Herry et al. 2008; Sierra-Mercado et al. 2011) |
|
| mPFC | Pre-Ext | Long-term enhancement of extinction (Akirav et al. 2006) |
||||
| IL | Pre-Ext | Impairs acquisition of extinction and extinction memory (Sierra-Mercado et al. 2011) |
||||
| LA &BLA |
Ext (after 5 CSs) |
Facilitates the consolidation (Akirav et al. 2006) |
||||
List of abbreviations used:
AC - adenylyl cyclase, AHP – afterhyperpolarization, AMY – amygdala, BLA – basolateral amygdala, CaMKII - Ca2+/calmodulin-dependent protein kinase II, CeA – central amygdala, CHPG – (RS)-2-chloro-5-hydroxyphenylglycine, CREB - cAMP response element-binding protein, CTX-LA – cortical pathway to the LA, DCS - D-cycloserine, E-LTP – early phase of LTP, FC – fear conditioning, Gly site - glycine site of the NMDA receptor, IL - infralimbic division of medial prefrontal cortex, LA – lateral anygdala, LFS – low-frequency stimulation, List of abbreviations used: L-LTP – late phase of LTP, LTP – long-term potentiation, mGluR - metabotropic glutamate receptor, mITC – medial cluster of intercalated cells, MPEP - 2-methyl-6-(phenylethynyl)-pyridine, PI-3 - Phosphatidyl inositol 3, PKA – protein kinase A or cAMP-dependent protein kinase, PL – prelimbic medial prefrontal cortex (mPFC), PP2B (CaN) - serine/threonine-protein phosphatase 2B or calcineurin, Sys – systemic (injection), THAL-LA – thalamic input to the LA, VGCC – voltage-gated calcium channel.
There are different potential mechanisms by which fear responses could be diminished following extinction training. A widely accepted hypothesis suggests that extinction may involve the formation of new associations outside the amygdala (in the mPFC, specifically) competing with the original conditioned fear response and inhibiting fear-promoting circuitry either via plasticity at excitatory inputs to inhibitory interneurons or increased inhibition of principal cells in the BLA (Milad and Quirk 2002; Quirk 2002; Maren and Quirk 2004; Quirk et al. 2006; Santini et al. 2008; Bukalo et al. 2014). This new learning is often less stable than the originally acquired fear memory (Myers and Davis 2007) and extinction-associated plastic changes could possibly dissipate with the passage of time, thus explaining certain behavioral features of fear extinction (e.g., spontaneous recovery). Another possibility is depotentiation of the thalamo-amygdala or cortico-amygdala synapses undergoing LTP after fear conditioning (Kim et al. 2007b; Hong et al. 2009). Consistent with the view of extinction as depotentiation, it was shown previously that certain cellular mechanisms can be more prominently implicated in extinction than in initial fear learning (e.g., activation of L-type VGCCs; Cain et al. 2002). Under this scenario, synaptic enhancements underlying the acquisition of memory of the CS-US association in fear conditioned subjects would be lost, essentially resulting in the erasure of original fear memory. We will review the available evidence in support of different proposed mechanisms of fear extinction, first focusing on the possible link between depotentiation-like synaptic plasticity in the CS pathways and extinction of conditioned fear memory with the emphasis on proposed criteria supporting the SPM hypothesis (Martin et al. 2000).
Detectability, anterograde and retrograde alteration, and mimicry SPM criteria in relation to depotentiation as a mechanism of extinction
As mentioned above, suppression of conditioned fear responses following extinction training may result from a loss of synaptic modifications underlying memory of the CS-US association formed during fear learning via conditioning-induced synaptic plasticity at inputs to the LA. Several earlier studies implicated this mechanism in fear extinction, drawing a parallel between a reduction in synaptic efficacy in slices of the amygdala in response to the depotentiation-inducing stimulation of auditory CS projections to the LA and a decrease in the magnitude of conditioned fear responses following fear extinction training. Depotentiation can be readily induced by low frequency stimulation (with single or paired pulses) at both thalamic (Kim et al. 2007b) and cortical inputs (Hong et al. 2009) to the LA in slices from fear conditioned animals as well as in slices from naïve animals following LTP induction ex vivo (Lin et al. 2003a). The suggested role for depotentiation as an underlying mechanism of fear extinction is supported by various experimental findings. For instance, it was found that extinction may be associated with a return of synaptic efficacy in thalamic and cortical inputs to the LA in slices from fear-conditioned animals to the pre-conditioning baseline level as assayed with synaptic input-output curves for evoked EPSCs (Kim et al. 2007b; Hong et al. 2009). This observation fulfils the detectability criterion in evaluating the depotentiation-extinction link (Martin et al. 2000). These findings, however, are not universally accepted as two independent studies suggested that synapses in the thalamo-LA pathway may remain in a potentiated state following extinction (Clem and Huganir 2010; Kim and Cho 2017), demonstrated by the sustained increase in the AMPAR/N-methyl-D-aspartate (NMDA)R amplitude ratio (AMPAR/NMDAR ratio) of thalamo-LA EPSCs following extinction training. Specifically, excitatory synaptic transmission in the thalamo-LA pathway was enhanced for 7 days following fear conditioning and was not depotentiated after fear extinction (Clem and Huganir 2010). By expressing ChR2 in CS+ activated ACx/MGm neurons, Kim and Cho (2017) demonstrated that synaptic efficacy remained strengthened in the auditory CS+-specific inputs to the LA following extinction of the CS+ in the discriminative fear memory paradigm. When ChR2 was expressed in the auditory cortex and thalamus (the CS specificity was lacking under these conditions), AMPAR/NMDAR EPSC amplitude ratio in CS inputs to the LA was similar following fear conditioning and extinction (Kim and Cho 2017). Moreover, both depotentiation-inducing electric stimulation, delivered to the external capsule in vivo in fear conditioned animals after 24h-retention test (Lin et al. 2003a) and optogenetically delivered LTD protocol (Nabavi et al. 2014) significantly attenuated the expression of fear memory, satisfying both retrograde alteration and mimicry criteria. Notably, optogenetically administered LTP-inducing stimulation was not capable of reactivating extinguished fear memory and reversing extinction (Nabavi et al. 2014).
The finding that extinction occludes ex vivo depotentiation (Kim et al. 2007b; Hong et al. 2009) suggests that extinction and depotentiation may have similar underlying synaptic mechanisms. Notably, ex vivo depotentiation is dependent on activation of NMDA receptors (Lin et al. 2003a; Kim et al. 2007b; Hong et al. 2009), mGluRs (Kim et al. 2007b; Hong et al. 2009), L-type VGCCs and protein phosphatase calcineurin (Lin et al. 2003a), all of which have been implicated in mechanisms of extinction at the behavioral level (Table 2; Falls et al. 1992; Walker et al. 2002; Lin et al. 2003a; Lin et al. 2003b; Cain et al. 2005; Kim et al. 2007a; Sotres-Bayon et al. 2007; Sotres-Bayon et al. 2009). Additionally, GluA2-dependent AMPA receptor endocytosis may contribute to both synaptic depotentation and fear extinction (Kim et al. 2007b; Dalton et al. 2008). Specifically, it was found that a GluR2-derived peptide, capable of blocking regulated AMPAR endocytosis, inhibited depotentiation in thalamic input to the LA, whereas microinjections of a cell-permeable form of the peptide into the LA inhibited extinction (Kim et al. 2007b), fulfilling the anterograde alteration criterion.
Extinction as new learning within and outside amygdala
One consistent observation in studies of fear learning is that associative memory formed as a result of fear conditioning does not disappear completely following extinction training (Fig. 3; Myers and Davis 2007). Extinguished fear can return at extended time intervals following extinction training in a process of spontaneous recovery (Bouton 2002; Rescorla 2004). Furthermore, due to context-specificity of fear extinction, the conditioned fear response can reappear when the context is changed (fear renewal). In addition, conditioned fear can be reinstated following exposure to the unsignalled US. Such features of extinction cannot be readily explained through a relatively simple mechanism involving synaptic depotentiation in CS pathways, which would be expected to manifest itself as a complete erasure of original fear memory. Therefore, it was proposed that fear memory should, at least in part, be retained (but inhibited) through additional neuronal and synaptic processes located elsewhere in the brain, and extinguished fear memory could be reactivated under certain conditions (Myers and Davis 2007; Singewald et al. 2015). For example, spontaneous recovery would occur if newly acquired extinction-associated plasticity is less stable than fear learning-induced synaptic enhancements in CS inputs to the LA.
Fig. 3.
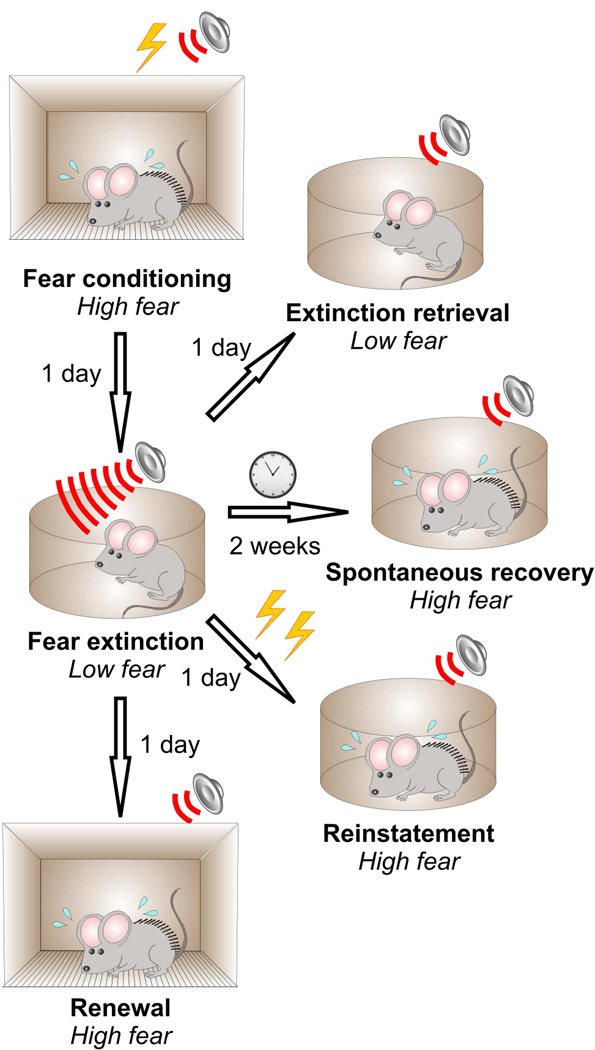
Relapse of fear following fear extinction. Fear is reduced following fear extinction, as assessed during extinction retrieval test the next day after training. However, fear can return by the following mechanisms: spontaneous recovery with the passage of time after fear extinction, reinstatement due to an exposure to the unsignalled US or renewal with exposure to the fear conditioning context that is different from extinction context (Myers and Davis 2007; Singewald et al. 2015).
Extinction-related plastic changes in mPFC
Numerous studies demonstrated previously that mPFC projections to amygdala contribute to the mechanisms of extinction, inhibiting the expression of conditioned fear response in extinguished animals. Thus, it was suggested that enhanced neuronal activity in the mPFC and, in particular, its infralimbic division, during recall of fear memory in extinguished subjects may suppress signal flow within amygdala and thereby diminish fear responses (Milad and Quirk 2002; Milad et al. 2004; Likhtik et al. 2005; Quirk et al. 2006). Accordingly, increased neuronal firing in IL negatively correlated with freezing levels and the rate of spontaneous recovery in fear extinguished rats (Milad and Quirk 2002). The role of mPFC as a locus of long-term extinction memory storage was further supported by the results of pharmacological studies within the mPFC using NMDA receptor antagonists (Burgos-Robles et al. 2007; Sotres-Bayon et al. 2009), group I metabotropic glutamate receptor blockers (Fontanez-Nuin et al. 2011) and protein synthesis inhibitors (Santini et al. 2004). These pharmacological manipulations resulted in impaired consolidation of extinction memory, whereas having no effect on its acquisition. Inactivation of IL, but not of the prelimbic division of the mPFC (PL), with the GABAA receptor agonist muscimol impaired both the acquisition of extinction and extinction memory (Sierra-Mercado et al. 2011, but see Akirav et al. 2006; Table 2).
At the cellular level, extinction leads to an increase in intrinsic membrane excitability and enhanced firing of IL neurons (Santini et al. 2008). It appears that the latter is required for consolidation of extinction memory (Burgos-Robles et al. 2007). Neuronal burst firing may promote synaptic plasticity in downstream regions (e.g., at mPFC inputs to principal neurons and/or interneurons in the amygdala or other targets of prefrontal projections) by conveying information more reliably, and, perhaps, in upstream areas as well through backpropagation of action potentials (e.g., Buzsáki et al. 2002). Electrical microstimulation of the IL, paired with conditioned stimuli designed to mimic neuronal tone responses, significantly decreased freezing responses in non-extinguished animals, mirroring extinction memory to a certain extent (Milad and Quirk 2002; Milad et al. 2004). In contrast to the IL, microstimulation of PL led to the enhanced expression of conditioned fear and prevented fear extinction (Vidal-Gonzalez et al. 2006). Electrical stimulation of projections from the mediodorsal thalamus (MD) to prefrontal cortex also affected extinction of fear memory (Herry et al. 1999; Herry and Garcia 2002). Specifically, the induction of long-term depression (LTD) in the mPFC by low-frequency stimulation of the mediodorsal thalamic nucleus during extinction training was associated with a resistance to express extinction (Herry and Garcia 2002). On the other hand, LTP in the mPFC, induced by thalamic nucleus high frequency stimulation (HFS) prior to extinction training, facilitated long-term maintenance of extinction memory when assessed 1 week later (Herry and Garcia 2002), providing further support for the role of plastic changes in the mPFC in extinction.
The delineation of specific functional roles in fear mechanisms of two mPFC subdivisions, IL and PL, with their proposed antagonistic contributions to fear control (Vidal-Gonzalez et al. 2006; Sierra-Mercado et al. 2011), became technically feasible with the development of novel methodologies, allowing more precise time- and region-specific manipulations of behavior-controlling neural circuits (e.g., reviewed in Bukalo et al. 2014; Riga et al. 2014; Luchkina and Bolshakov 2017). Thus, optogenetic activation of ChR2-expressing and photo-inhibition of halorhodopsin (eNpHR) or archaerhodopsin-3 (ArchT) expressing IL neurons or their projections to the amygdala during extinction training promoted and impaired extinction memory formation, respectively (Bukalo et al. 2015; Do-Monte et al. 2015). Consistent with these findings, chemogenetic inhibition of IL-BLA projections during extinction acquisition impaired extinction memory recall (Bloodgood et al. 2018). The role of IL activity in extinction memory is further supported by the results of a recent study providing evidence that feed-forward inhibition in the IL, driven by excitatory projections from the ventral hippocampus to parvalbumin-expressing IL interneurons, may mediate relapse of extinguished fear (Marek et al. 2018a). However, photoinhibition of IL principal neurons or their inputs to the amygdala in experiments using neuron-specific CaMKIIα promoter for opsin targeting during retrieval of extinction memory did not alter freezing responses (Bukalo et al. 2015; Do-Monte et al. 2015; Kim et al. 2016). Finally, silencing of both excitatory and inhibitory cells (neurons and interneurons, respectively; with only ~24% of infected cells being GABAergic interneurons) in the IL with eNpHR under control of the pan-neuronal human synapsin promoter resulted in impaired expression of fear extinction memory at the time of retrieval (Kim et al. 2016), suggesting a role for IL inhibitory interneurons in extinction memory retrieval. Interestingly, activation of glutamatergic IL neurons by light at the time of extinction retrieval resulted in enhanced expression of extinction in some studies (Do-Monte et al. 2015; Kim et al. 2016), but had no effect in others (Bukalo et al. 2015). Together, these findings provide evidence for the functional role of IL-amygdala projections in extinction memory formation, whereas their role in extinction retrieval may still need further investigation. It is possible that excitatory IL projections to other brain regions besides the amygdala (Bukalo et al. 2015) or GABAergic IL cells and their projections (Kim et al. 2016) may be implicated in retrieval of extinction memory.
In contrast to earlier neuroanatomical studies (reviewed in Pape and Pare 2010), more recent experiments with optogenetic tools, which allow both neuroanatomical tracing and testing of functional connectivity in the same experiment, demonstrated that mPFC projection patterns to the amygdala and excitatory and inhibitory synaptic responses in mPFC-amygdala pathway are not significantly different between IL and PL, suggesting a possibility of the functional overlap between the two mPFC subdivisions (Cho et al. 2013; Arruda-Carvalho and Clem 2014; Hübner et al. 2014). Moreover, it has been recently demonstrated that neurons in layer 5/6 of the PL send excitatory projections to the IL, and activation of these connections enhances fear extinction, indicating a role for PL-IL interactions in extinction mechanisms (Marek et al. 2018b). Additionally, different neural circuits, different cell types and connections within and between mPFC subdivisions may mediate distinct behavioral effects. For example, optogenetic activation of IL principal neurons in vivo did not affect IL interneurons, but inhibited PL neurons, suggesting that IL may possibly diminish the conditioned fear response by controlling PL output (Ji and Neugebauer 2012). Parvalbumin (PV)-expressing interneurons in dorsomedial PFC (PL and Cg1 area of the anterior cingulate cortex) inhibit fear expression, as their optogenetic silencing coupled with CS+ presentations following fear extinction resulted in reinstatement of extinguished fear responses (Courtin et al. 2014). However, much work remains to be done in order to fully characterize the role of different mPFC areas and their specific projections and neuronal types in fear extinction-related behaviors.
Notably, the role of extra-amygdala brain structures in fear-related behaviors is further supported by a recent study showing that neurons in the parabrachial nucleus (PBn), expressing calcitonin gene-related peptide (CGRP), are activated by the auditory cues which were previously paired with electric footshocks (Campos et al. 2018). Silencing of these neurons in the PBn attenuated conditioned fear responses. This opens up an interesting possibility, which could be tested experimentally, that CGRPPBN cells (involved in encoding of danger), and synaptic plasticity in circuits where they are located, may contribute to the mechanisms of extinction as well.
Amygdala as a part of extinction circuitry
mPFC may effectively inhibit fear responses following extinction via several mechanisms. The mPFC sends robust projections to the amygdala as well as other brain areas receiving inputs from the amygdala, such as hypothalamus and brainstem (periaqueductal grey, specifically) (Quirk et al. 2006; Franklin et al. 2017). The IL division of mPFC may inhibit fear responses via direct or indirect BLA-mediated activation of the medial ITC (mITC) group, located between CeA and BLA, which in turn, leads to feed-forward inhibition of the CeA neurons (Royer et al. 1999; Berretta et al. 2005; Likhtik et al. 2005; Likhtik et al. 2008; Amir et al. 2011; Strobel et al. 2015).Selective lesioning of the ITCs following extinction training caused a deficit in extinction retrieval (Likhtik et al. 2008). The ITCs receive excitatory and inhibitory inputs from many brain areas, including LA, BLA and mPFC, as well as from the auditory thalamus and cortex, all of which could undergo plastic changes thus contributing to mechanisms of extinction.
In the experiments using ex vivo slice electrophysiology following behavioral training, extinction was associated with the enhancement of synaptic efficacy at BLA-to-mITC synapses, with ITCs inhibiting CeM and, therefore, leading to the reduction of conditioned fear responses (Amano et al. 2010), satisfying the detectability SMP criterion. In addition, IL neuronal activity was shown to drive this extinction-related synaptic plasticity in the amygdala as demonstrated in muscimol-mediated inactivation experiments (Amano et al., 2010). Moreover, it was found that neuropeptide S facilitates fear extinction when locally infused into the amygdala by increasing glutamatergic transmission from the LA to mITCs via corresponding presynaptic receptors on connected principal neurons, without affecting other amygdala cell types or other inputs to ITCs (Jüngling et al. 2008). In contrast, anothe r study showed that BLA inputs to mITCs undergo potentiation following fear learning, which was reversed upon extinction training (Huang et al. 2014). These conflicting findings may be at least partially explained by heterogeneity of mITCs in their synaptic properties (e.g., neurotransmitter release probabilities) and differential connectivity (Geracitano et al. 2007; Amir et al. 2011; Busti et al. 2011; Mańko et al. 2011; Duvarci and Pare 2014). Thus, dorsal mITCs, which mostly receive their inputs from the LA and project to the CeL, are preferentially activated during fear expression, whereas ventral mITCs receive inputs from the BLA and project to the CeM and are activated during extinction training and extinction retrieval (Amir et al. 2011; Busti et al. 2011; Duvarci and Pare 2014). However, this model does not account for the existence of alternative inputs to ITCs as well as their projections to other nuclei of the amygdala outside of the CeA and the functional connectivity between separate ITC clusters (Royer et al. 1999; Royer et al. 2000; Amir et al. 2011; Busti et al. 2011; Asede et al. 2015). Both fear conditioning and extinction have been shown to induce plasticity in thalamic and cortical inputs to mITCs (Asede et al. 2015), whereas neither fear conditioning nor fear extinction had an effect on synaptic transmission at mPFC-mITC connections (Cho et al. 2013).
Additionally, other pathway- and cell type-specific synaptic changes have been identified following extinction training. For instance, a recent study, using ex vivo electrophysiology and optogenetics in slices from behaviorally trained mice, showed reduced synaptic efficacy of excitatory glutamatergic transmission at mPFC-BLA synapses following extinction training. In contrast, feedforward GABAergic inhibition in these projections remained unchanged, thereby shifting the overall excitatory/inhibitory balance in the mPFC-BLA pathways towards a greater functional efficiency of inhibition (Cho et al. 2013), also fulfilling the detectability SMP criterion. Moreover, synaptic transmission in mPFC projections to mITCs remained intact following extinction, leading to further suppression of fear responses during memory retrieval in fear-extinguished mice. Notably, BLA neurons, including “extinction” and “fear” neurons, demonstrate differential activity patterns during distinct behavioral states (low and high fear, correspondingly) as well as specificity in their afferent and efferent projections (Herry et al. 2008; Senn et al. 2014), providing additional routes of efficient fear control. mPFC may affect CeA neuronal activity leading to diminished fear responses via recruitment of “extinction” neurons, which, in turn, can drive mITCs or inhibitory CeL neurons as well as inhibit BLA “fear” neurons. Thus, recent functional studies revealed the existence of complex networks, distributed plasticity and parallel processing in neuronal circuits outside and within the amygdala underlying fear extinction. Further studies, specifically manipulating distinct clusters of ITCs as well as identified projections to and from ITCs in vivo, may be needed to provide evidence supporting both the necessity and sufficiency of plastic modifications at the cellular and network levels for extinction-related behaviors.
Merging depotentiation-like and inhibitory mechanisms of extinction
There is substantial evidence in support of both depotentiation and inhibitory mechanisms underlying fear extinction at the synaptic level. This is consistent with the notion that both inhibition (new learning) and depotentiation (unlearning or erasure of previously formed associations) may coexist and co-function within the corresponding neural circuits and are not mutually exclusive in relation to fear extinction (Clem and Schiller 2016). Thus, a recently developed biologically realistic network model of the LA activity, incorporating experimentally validated biophysical single-cell models, connectivity, and synaptic plasticity mechanisms, predicted that extinction training may lead to both depotentiation of conditioned synapses onto principal neurons and potentiation of inputs to local interneurons, resulting in suppressed CS-induced responses of pyramidal cells (Li et al. 2009). Based on optimal LA network dynamics, both depotentiation and increased inhibition mechanisms may be required for the acquisition and maintenance of fear extinction memory (Li et al. 2009).
It is possible that the extent to which depotentiation or inhibition mechanisms contribute to the behavioral manifestations of extinction may depend on the age of animals and specific behavioral training paradigms. Specifically, it has been shown that rats, when fear-extinguished during early development (P16–17), do not exhibit reinstatement (Kim and Richardson 2007a) or renewal (Kim and Richardson 2007b), and their extinction memory acquisition is independent of NMDA receptors (Langton et al. 2007) and/or GABAA receptors activation (Kim and Richardson 2007b). Therefore, NMDA receptor independent mGluR-dependent depotentiation (Lin et al. 2005) and unlearning of the original CS-US association could possibly contribute to extinction mechanisms during early development. Another study suggested that the time interval between fear conditioning and extinction training may define mechanisms of fear extinction (new learning vs. unlearning). As extinction 24–72 h following fear acquisition was associated with subsequent reinstatement, renewal, and spontaneous recovery, it could be largely mediated by inhibition mechanisms. On the other hand, depotentiation-mediated mechanisms, not associated with reinstatement, renewal, or spontaneous recovery, were suggested to predominate during extinction training performed at short intervals (10 min to 1 h) after fear acquisition (Myers et al. 2006). Accordingly, extinction training applied 1 h, but not 24 h post-acquisition reversed the increase in surface expression of GluA1 induced by fear conditioning in LA and BLA (Mao et al. 2006).However, this time-dependence of mechanisms of extinction was not universally observed (Maren and Chang 2006; Alvarez et al. 2007; Schiller et al. 2008; Woods and Bouton 2008; Chang and Maren 2009). It was proposed that early (single-session extinction) and late phases (multiple-session extinction training) of extinction may engage inhibition and depotentiation mechanisms, correspondingly (An et al. 2017). It was suggested also that within-session and between-session extinction may differ from each other mechanistically (Plendl and Wotjak 2010; Almeida-Corrêa et al. 2015). Other studies showed that manipulations of the mPFC (low-frequency electric stimulation or lesions to mimic the inhibitory mechanism) do not affect the magnitude of extinction within a single extinction session (Quirk et al. 2000; Herry and Garcia 2002). The synaptic removal of calcium-permeable AMPARs at synapses in the LA can, possibly, underlie the diminished recovery from extinction during reconsolidation update (a modified behavioral paradigm where extinction session is presented after an isolated retrieval trial; Monfils et al. 2009), but not classical extinction without a preceding single retrieval trial (Clem and Huganir 2010).
Furthermore, synaptic connections to different populations of neurons in the LA and BLA may display plastic changes of opposite directions. Indeed, both the LA and BLA incorporate populations of cells specifically active during states of high (fear conditioning), low fear states (extinction) or both (extinction resistant neurons), identified using in vivo single unit recordings (Repa et al. 2001; Herry et al. 2008; An et al. 2012). One population of cells (i.e. extinction-resistant neurons) demonstrated persistent neuronal activity throughout the behavioral training (during both post-fear conditioning and extinction sessions, independently of freezing levels) (Repa et al. 2001; Herry et al. 2008; An et al. 2012). Therefore, these cells may be responsible for long-term memory of the CS-US association (Repa et al. 2001; An et al. 2012). Following extinction, their outputs could be controlled by GABAergic inhibitory mechanisms. In contrast, another cell subpopulation (i.e. extinction-susceptible fear neurons) showed a decrease in CS responses upon extinction training (Repa et al. 2001; Herry et al. 2008; An et al. 2012), which could be mediated by depotentiation mechanisms. Furthermore, these cells showed strong potentiation after reconditioning, suggesting that subpopulation of extinction-susceptible fear neurons may also encode the updated CS–US association strength (An et al. 2012).
Conclusions
Long-term synaptic plasticity at specific projections in fear circuits of the brain is both necessary and sufficient for retaining memory of aversive events, as well as for extinction of conditioned fear memory, which was demonstrated in the experiments using fear conditioning training paradigm. Both depotentiation-like decreases of synaptic efficacy in the conditioned stimulus projections to the amygdala, potentiated by fear learning, and inhibitory mechanisms implicating plasticity outside the amygdala (e.g., in mPFC) at the synaptic level were observed following extinction training. This is in agreement with the view that new learning and depotentiation (erasure of earlier established CS-US association) can co-function in mediating extinction of conditioned fear memory. Notably, the necessity and sufficiency SPM criteria (detectability, anterograde and retrograde alteration, and mimicry), validating the synaptic plasticity-memory connection (Martin et al. 2000), were shown to be met in relation to synaptic depotentiation as a mechanism of extinction. However, further studies will be required to provide experimental evidence for the validity of SPM hypothesis for the mechanisms of extinction implicating new inhibitory learning. The neural circuits of the latter are more diffuse, compared to the circuits of fear conditioning, and the applicability of SPM criteria would need to be tested at different components of the extinction circuitry undergoing plastic changes. Overall, recently-developed and emerging experimental methodologies may give new insights into the function of behavior-driving neural circuits, including circuits of fear control. Combined with established techniques for pharmacological interventions, the ever-expanding experimental toolbox can help us to elucidate the detailed mechanisms of fear-related behaviors, and potentially result in developing treatments of mental illnesses implicating dysfunctions of brain mechanisms responsible for fear-related behavioral responses.
Acknowledgments:
We thank Vernon Clarke and members of the laboratory for help and constructive discussions. This work was supported by grants R01MH108665 and R01MH105851 from NIMH (to V.Y.B.) and The Phyllis & Jerome Lyle Rappaport Foundation (to N.V.L.).
Footnotes
Conflict of interest statement: On behalf of all authors, the corresponding author (Dr. Bolshakov) states that there is no conflict of interest.
References
- Akirav I, Raizel H, Maroun M (2006) Enhancement of conditioned fear extinction by infusion of the GABA(A) agonist muscimol into the rat prefrontal cortex and amygdala. Eur J Neurosci 23:758–764 [DOI] [PubMed] [Google Scholar]
- Almeida-Corrêa S, Moulin TC, Carneiro CFD, Gonçalves MMC, Junqueira LS, Amaral OB (2015) Calcineurin inhibition blocks within-, but not between-session fear extinction in mice. Learn Mem 22:159–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez RP, Johnson L, Grillon C (2007) Contextual-specificity of short-delay extinction in humans: renewal of fear-potentiated startle in a virtual environment. Learn Mem 14:247–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano T, Unal CT, Paré D (2010) Synaptic correlates of fear extinction in the amygdala. Nat Neurosci 13:489–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir A, Amano T, Pare D (2011) Physiological identification and infralimbic responsiveness of rat intercalated amygdala neurons. J Neurophysiol 105:3054–3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An B, Hong I, Choi S (2012) Long-term neural correlates of reversible fear learning in the lateral amygdala. J Neurosci 32:16845–16856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An B, Kim J, Park K, Lee S, Song S, Choi S (2017) Amount of fear extinction changes its underlying mechanisms. eLife 6:e25224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armony JL, LeDoux JE (1997) How the brain processes emotional information. Ann N Y Acad Sci 821:259–270 [DOI] [PubMed] [Google Scholar]
- Arruda-Carvalho M, Clem RL (2014) Pathway-selective adjustment of prefrontal-amygdala transmission during fear encoding. J Neurosci 34:15601–15609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asede D, Bosch D, Lüthi A, Ferraguti F, Ehrlich I ( 2015) Sensory inputs to intercalated cells provide fear-learning modulated inhibition to the basolateral amygdala. Neuron 86:541–554 [DOI] [PubMed] [Google Scholar]
- Bauer EP, LeDoux JE (2004) Heterosynaptic long-term potentiation of inhibitory interneurons in the lateral amygdala. J Neurosci 24:9507–9512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer EP, Schafe GE, LeDoux JE (2002) NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci 22:5239–5249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgärtel K, Genoux D, Welzl H, Tweedie-Cullen RY,Koshibu K, Livingstone-Zatchej M, Mamie C, Mansuy IM (2008) Control of the establishment of aversive memory by calcineurin and Zif268. Nat Neurosci 11:572–578 [DOI] [PubMed] [Google Scholar]
- Belzung C, Turiault M, Griebel G (2014) Optogenetics to study the circuits of fear- and depression-like behaviors: a critical analysis. Pharmacol Biochem Behav 122:144–157 [DOI] [PubMed] [Google Scholar]
- Berretta S, Pantazopoulos H, Caldera M, Pantazopoulos P, Paré D (2005) Infralimbic cortex activation increases c-Fos expression in intercalated neurons of the amygdala. Neuroscience 132:943–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissière S, Humeau Y, Lüthi A (2003) Dopamine gates LTP induction in lateral amygdala by suppressing feedforward inhibition. Nat Neurosci 6:587–592 [DOI] [PubMed] [Google Scholar]
- Blair HT, Schafe GE, Bauer EP, Rodrigues SM, LeDoux JE (2001) Synaptic plasticity in the lateral amygdala: a cellular hypothesis of fear conditioning. Learn Mem 8:229–242 [DOI] [PubMed] [Google Scholar]
- Bloodgood DW, Sugam JA, Holmes A, Kash TL (2018) Fear extinction requires infralimbic cortex projections to the basolateral amygdala. Transl Psychiatry 8:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K (2005) Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci 8:1263–1268 [DOI] [PubMed] [Google Scholar]
- Bouton ME (2002) Context, ambiguity, and unlearning: sources of relapse after behavioral extinction. Biol Psychiatry 52:976–986 [DOI] [PubMed] [Google Scholar]
- Brunzell DH, Kim JJ (2001) Fear conditioning to tone, but not to context, is attenuated by lesions of the insular cortex and posterior extension of the intralaminar complex in rats. Behav Neurosci 115:365–375 [PubMed] [Google Scholar]
- Bukalo O, Pinard CR, Holmes A (2014) Mechanisms to medicines: elucidating neural and molecular substrates of fear extinction to identify novel treatments for anxiety disorders. Br J Pharmacol 171:4690–4718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukalo O, Pinard CR, Silverstein S, Brehm C, Hartley ND, Whittle N, et al. (2015) Prefrontal inputs to the amygdala instruct fear extinction memory formation. Sci Adv 1(6):e1500251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos-Robles A, Vidal-Gonzalez I, Santini E, Quirk GJ (2007) Consolidation of fear extinction requires NMDA receptor-dependent bursting in the ventromedial prefrontal cortex. Neuron 53:871–880 [DOI] [PubMed] [Google Scholar]
- Busti D, Geracitano R, Whittle N, Dalezios Y, Mańko M, Kaufmann W, et al. (2011) Different fear states engage distinct networks within the intercalated cell clusters of the amygdala. J Neurosci 31:5131–5144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G, Csicsvari J, Dragoi G, Harris K, Henze D, Hirase H (2002) Homeostatic maintenance of neuronal excitability by burst discharges in vivo. Cereb Cortex 12:893–899 [DOI] [PubMed] [Google Scholar]
- Cain CK, Blouin AM, Barad M (2002) L-type voltage-gated calcium channels are required for extinction, but not for acquisition or expression, of conditional fear in mice. J Neurosci 22:9113–9121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain CK, Godsil BP, Jami S, Barad M (2005) The L-type calcium channel blocker nifedipine impairs extinction, but not reduced contingency effects, in mice. Learn Mem 12:277–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau S, Miserendino MJ, Davis M (1992) Intra-amygdala infusion of the N-methyl-D-aspartate receptor antagonist AP5 blocks acquisition but not expression of fear-potentiated startle to an auditory conditioned stimulus. Behav Neurosci 106:569–574 [DOI] [PubMed] [Google Scholar]
- Campos CA, Bowen AJ, Roman CW, Palmiter RD (2018) Encoding of danger by parabrachial CGRP neurons. Nature 555:617–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch Chang, Maren S (2009) Early extinction after fear conditioning yields a context-independent and short-term suppression of conditional freezing in rats. Learn Mem 16:62–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhatwal JP, Myers KM, Ressler KJ, Davis M (2005) Regulation of gephyrin and GABAA receptor binding within the amygdala after fear acquisition and extinction. J Neurosci 25:502–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JH, Bayazitov IT, Meloni EG, Myers KM, Carlezon WA, Zakharenko SS, et al. (2012) Coactivation of thalamic and cortical pathways induces input timing-dependent plasticity in amygdala. Nat Neurosci 15:113–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JH, Deisseroth K, Bolshakov VY (2013) Synaptic encoding of fear extinction in mPFC-amygdala circuits. Neuron 80:1491–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciocchi S, Herry C, Grenier F, Wolff SBE, Letzkus JJ, Vlachos I, et al. (2010) Encoding of conditioned fear in central amygdala inhibitory circuits. Nature 468:277–282 [DOI] [PubMed] [Google Scholar]
- Clem RL, Huganir RL (2010) Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science 330:1108–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem RL, Schiller D (2016) New Learning and Unlearning: Strangers or Accomplices in Threat Memory Attenuation? Trends Neurosci 39:340–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins DR, Paré D (2000) Differential fear conditioning induces reciprocal changes in the sensory responses of lateral amygdala neurons to the CS(+) and CS(−). Learn Mem 7:97–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtin J, Chaudun F, Rozeske RR, Karalis N, Gonzalez-Campo C, Wurtz H, et al. (2014) Prefrontal parvalbumin interneurons shape neuronal activity to drive fear expression. Nature 505:92–96 [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Edeline JM, Weinberger NM (1992) Stimulation at a site of auditory-somatosensory convergence in the medial geniculate nucleus is an effective unconditioned stimulus for fear conditioning. Behav Neurosci 106:471–483 [DOI] [PubMed] [Google Scholar]
- Dalton GL, Wang YT, Floresco SB, Phillips AG (2008) Disruption of AMPA receptor endocytosis impairs the extinction, but not acquisition of learned fear. Neuropsychopharmacology 33:2416–2426 [DOI] [PubMed] [Google Scholar]
- Derkach V, Barria A, Soderling TR (1999) Ca2+/calmodulin-kinase II enhances channel conductance of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci U S A 96:3269–3274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dityatev AE, Bolshakov VY (2005) Amygdala, long-term potentiation, and fear conditioning. Neuroscientist 11:75–88 [DOI] [PubMed] [Google Scholar]
- Do-Monte FH, Manzano-Nieves G, Quiñones-Laracuente K, Ramos-Medina L, Quirk GJ (2015) Revisiting the role of infralimbic cortex in fear extinction with optogenetics. J Neurosci 35:3607–3615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvarci S, Pare D (2014) Amygdala microcircuits controlling learned fear. Neuron 82:966–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvarci S, Popa D, Paré D (2011) Central amygdala activity during fear conditioning. J Neurosci 31:289–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falls WA, Miserendino MJ, Davis M (1992) Extinction of fear-potentiated startle: blockade by infusion of an NMDA antagonist into the amygdala. J Neurosci 12:854–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanez-Nuin DE, Santini E, Quirk GJ, Porter JT (2011) Memory for fear extinction requires mGluR5-mediated activation of infralimbic neurons. Cereb Cortex 21:727–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourcaudot E, Gambino F, Humeau Y, Casassus G, Shaban H, Poulain B, et al. (2008) cAMP/PKA signaling and RIM1α mediate presynaptic LTP in the lateral amygdala. Proc Natl Acad Sci U S A 105:15130–15135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin TB, Silva BA, Perova Z, Marrone L, Masferrer ME, Zhan Y, et al. (2017) Prefrontal cortical control of a brainstem social behavior circuit. Nat Neurosci 20:260–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner AR, Rowland DC, Hwang SY, Baumgaertel K, Roth BL, Kentros C, et al. (2012) Generation of a synthetic memory trace. Science 335:1513–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geracitano R, Kaufmann WA, Szabo G, Ferraguti F, Capogna M (2007) Synaptic heterogeneity between mouse paracapsular intercalated neurons of the amygdala. J Physiol 585(Pt 1):117–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Chattarji S (2015)Neuronal encoding of the switch from specific to generalized fear. Nat Neurosci 18:112–120 [DOI] [PubMed] [Google Scholar]
- Goosens KA, Hobin JA, Maren S (2003) Auditory-evoked spike firing in the lateral amygdala and Pavlovian fear conditioning: mnemonic code or fear bias? Neuron 40:1013–1022 [DOI] [PubMed] [Google Scholar]
- Goosens KA, Holt W, Maren S (2000) A role for amygdaloid PKA and PKC in the acquisition of long-term conditional fear memories in rats. Behav Brain Res 114:145–152 [DOI] [PubMed] [Google Scholar]
- Han JH, Kushner SA, Yiu AP, Cole CJ, Matynia A, Brown RA, et al. (2007) Neuronal competition and selection during memory formation. Science 316:457–460 [DOI] [PubMed] [Google Scholar]
- Han JH, Kushner SA, Yiu AP, Hsiang HLL, Buch T, Waisman A, et al. (2009) Selective erasure of a fear memory. Science 323:1492–1496 [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R (2000) Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science 287:2262–2267 [DOI] [PubMed] [Google Scholar]
- Herry C, Ciocchi S, Senn V, Demmou L, Müller C, Lüt hi A (2008) Switching on and off fear by distinct neuronal circuits. Nature 454:600–606 [DOI] [PubMed] [Google Scholar]
- Herry C, Ferraguti F, Singewald N, Letzkus JJ, Ehrlich I, Lüthi A (2010) Neuronal circuits of fear extinction. Eur J Neurosci 31:599–612 [DOI] [PubMed] [Google Scholar]
- Herry C, Garcia R (2002) Prefrontal cortex long-term potentiation, but not long-term depression, is associated with the maintenance of extinction of learned fear in mice. J Neurosci 22:577–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Vouimba RM, Garcia R (1999) Plasticity in the mediodorsal thalamo-prefrontal cortical transmission in behaving mice. J Neurophysiol 82:2827–2832 [DOI] [PubMed] [Google Scholar]
- Hong I, Song B, Lee S, Kim J, Kim J, Choi S (2009) Extinction of cued fear memory involves a distinct form of depotentiation at cortical input synapses onto the lateral amygdala. Eur J Neurosci 30:2089–2099 [DOI] [PubMed] [Google Scholar]
- Huang CC, Chen CC, Liang YC, Hsu KS (2014) Long-term potentiation at excitatory synaptic inputs to the intercalated cell masses of the amygdala. Int J Neuropsychopharmacol 17:1233–1242 [DOI] [PubMed] [Google Scholar]
- Huang YY, Kandel ER (2007) Low-frequency stimulation induces a pathway-specific late phase of LTP in the amygdala that is mediated by PKA and dependent on protein synthesis. Learn Mem 14:497–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Kandel ER (1998) Postsynaptic induction and PKA-dependent expression of LTP in the lateral amygdala. Neuron 21:169–178 [DOI] [PubMed] [Google Scholar]
- Huang YY, Martin KC, Kandel ER (2000) Both protein kinase A and mitogen-activated protein kinase are required in the amygdala for the macromolecular synthesis-dependent late phase of long-term potentiation. J Neurosci 20:6317–6325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübner C, Bosch D, Gall A, Lüthi A, Ehrlich I (2014. ) Ex vivo dissection of optogenetically activated mPFC and hippocampal inputs to neurons in the basolateral amygdala: implications for fear and emotional memory. Front Behav Neurosci 8:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Reisel D, Johnson AW, Borchardt T, Jensen V, Gebhardt C, et al. (2007) A pathway-specific function for different AMPA receptor subunits in amygdala long-term potentiation and fear conditioning. J Neurosci 27:10947–10956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Shaban H, Bissière S, Lüthi A (2003) Presynaptic induction of heterosynaptic associative plasticity in the mammalian brain. Nature 426:841–845 [DOI] [PubMed] [Google Scholar]
- Ji G, Neugebauer V (2012) Modulation of medial prefrontal cortical activity using in vivo recordings and optogenetics. Mol Brain 5:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen JP, Hamanaka H, Monfils MH, Behnia R, Deisseroth K, Blair HT, et al. (2010) Optical activation of lateral amygdala pyramidal cells instructs associative fear learning. Proc Natl Acad Sci U S A 107:12692–12697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen JP, Wolff SBE, Lüthi A, LeDoux JE (2012) C ontrolling the elements: an optogenetic approach to understanding the neural circuits of fear. Biol Psychiatry 71:1053–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jüngling K, Seidenbecher T, Sosulina L, Lesting J, Sangha S, Clark SD, et al. (2008) Neuropeptide S-mediated control of fear expression and extinction: role of intercalated GABAergic neurons in the amygdala. Neuron 59:298–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Cho HY, Augustine GJ, Han JH (2016) Selective Control of Fear Expression by Optogenetic Manipulation of Infralimbic Cortex after Extinction. Neuropsychopharmacology 41: 1261–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, An B, Kim J, Park S, Park S, Hong I, et al. (2015) mGluR2/3 in the lateral amygdala is required for fear extinction: cortical input synapses onto the lateral amygdala as a target site of the mGluR2/3 Action. Neuropsychopharmacology 40:2916–2928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee S, Park H, Song B, Hong I, Geum D, et al. (2007a) Blockade of amygdala metabotropic glutamate receptor subtype 1 impairs fear extinction. Biochem Biophys Res Commun 355:188–193 [DOI] [PubMed] [Google Scholar]
- Kim J, Lee S, Park K, Hong I, Song B, Son G, et al. (2007b) Amygdala depotentiation and fear extinction. Proc Natl Acad Sci U S A 104:20955–20960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Richardson R (2007a) A developmental dissociation in reinstatement of an extinguished fear response in rats. Neurobiol Learn Mem 88:48–57 [DOI] [PubMed] [Google Scholar]
- Kim JH, Richardson R (2007b) A developmental dissociation of context and GABA effects on extinguished fear in rats. Behav Neurosci 121:131–139 [DOI] [PubMed] [Google Scholar]
- Kim WB, Cho JH (2017) Encoding of discriminative fear memory by input-specific LTP in the amygdala. Neuron 95:1129–1146 [DOI] [PubMed] [Google Scholar]
- Kwon JT, Choi JS (2009) Cornering the fear engram: long-term synaptic changes in the lateral nucleus of the amygdala after fear conditioning. J Neurosci 29:9700–9703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon JT, Nakajima R, Kim HS, Jeong Y, Augustine GJ, Han JH (2014) Optogenetic activation of presynaptic inputs in lateral amygdala forms associative fear memory. Learn Mem 21:627–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalumiere R (2014) Optogenetic dissection of amygdala functioning. Front Behav Neurosci 8:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langton JM, Kim JH, Nicholas J, Richardson R (2007) The effect of the NMDA receptor antagonist MK-801 on the acquisition and extinction of learned fear in the developing rat. Learn Mem 14:665–668 [DOI] [PubMed] [Google Scholar]
- Lanuza E, Moncho-Bogani J, Ledoux JE (2008) Unconditioned stimulus pathways to the amygdala: effects of lesions of the posterior intralaminar thalamus on foot-shock-induced c-Fos expression in the subdivisions of the lateral amygdala. Neuroscience 155:959–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanuza E, Nader K, Ledoux JE (2004) Unconditioned stimulus pathways to the amygdala: effects of posterior thalamic and cortical lesions on fear conditioning. Neuroscience 125:305–315 [DOI] [PubMed] [Google Scholar]
- Ledgerwood L, Richardson R, Cranney J (2003) Effects of D-cycloserine on extinction of conditioned freezing. Behav Neurosci 117:341–349 [DOI] [PubMed] [Google Scholar]
- LeDoux JE (2000) Emotion circuits in the brain. Annu Rev Neurosci 23:155–184 [DOI] [PubMed] [Google Scholar]
- Li G, Nair SS, Quirk GJ (2009) A biologically realistic network model of acquisition and extinction of conditioned fear associations in lateral amygdala neurons. J Neurophysiol 101: 1629–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Penzo MA, Taniguchi H, Kopec CD, Huang ZJ, Li B (2013a) Experience-dependent modification of a central amygdala fear circuit. Nat Neurosci 16:332–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Meloni EG, Carlezon WA, Milad MR, Pitman RK, Nader K, et al. (2013b) Learning and reconsolidation implicate different synaptic mechanisms. Proc Natl Acad Sci U S A 110:4798–4803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likhtik E, Pelletier JG, Paz R, Paré D (2005) Prefrontal control of the amygdala. J Neurosci 25:7429–7437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likhtik E, Popa D, Apergis-Schoute J, Fidacaro GA, Paré D (2008) Amygdala intercalated neurons are required for expression of fear extinction. Nature 454:642–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Lee CC, Gean PW (2003a) Involvement of a calcineurin cascade in amygdala depotentiation and quenching of fear memory. Mol Pharmacol 63:44–52 [DOI] [PubMed] [Google Scholar]
- Lin CH, Lee CC, Huang YC, Wang SJ, Gean PW (2005) Activation of group II metabotropic glutamate receptors induces depotentiation in amygdala slices and reduces fear-potentiated startle in rats. Learn Mem 12:130–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Yeh SH, Leu TH, Chang WC, Wang ST, Gean PW (2003b) Identification of calcineurin as a key signal in the extinction of fear memory. J Neurosci 23:1574–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Yeh SH, Lin CH, Lu KT, Leu TH, Chang WC, et al. (2001) A role for the PI-3 kinase signaling pathway in fear conditioning and synaptic plasticity in the amygdala. Neuron 31: 841–851 [DOI] [PubMed] [Google Scholar]
- Lin CH, Yeh SH, Lu HY, Gean PW (2003c) The similarities and diversities of signal pathways leading to consolidation of conditioning and consolidation of extinction of fear memory. J Neurosci 23:8310–8317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu KT, Walker DL, Davis M (2001) Mitogen-activated protein kinase cascade in the basolateral nucleus of amygdala is involved in extinction of fear-potentiated startle. J Neurosci 21: RC162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchkina NV, Bolshakov VY (2017) Diminishing fear: Optogenetic approach toward understanding neural circuits of fear control. Pharmacol Biochem Behav 10.1016/j.pbb.2017.05.005 [DOI] [PMC free article] [PubMed]
- Mahanty NK, Sah P (1998) Calcium-permeable AMPA receptors mediate long-term potentiation in interneurons in the amygdala. Nature 394:683–687 [DOI] [PubMed] [Google Scholar]
- Mańko M, Geracitano R, Capogna M (2011) Functional connectivity of the main intercalated nucleus of the mouse amygdala. J Physiol 589(Pt 8):1911–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao SC, Hsiao YH, Gean PW (2006) Extinction training in conjunction with a partial agonist of the glycine site on the NMDA receptor erases memory trace. J Neurosci 26:8892–8899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek R, Jin J, Goode TD, Giustino TF, Wang Q, Acca GM, Holehonnur R, Ploski JE, Fitzgerald PJ, Lynagh T, Lynch JW, Maren S, Sah P (2018a) Hippocampus-driven feed-forward inhibition of the prefrontal cortex mediates relapse of extinguished fear. Nat Neurosci 21:384–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek R, Xu L, Sullivan RKP, Sah P (2018b) Excitatory connections between the prelimbic and infralimbic medial prefrontal cortex show a role for the prelimbic cortex in fear extinction. Nat Neurosci 2018 21:654–658 [DOI] [PubMed] [Google Scholar]
- Maren S, Chang Ch (2006) Recent fear is resistant to extinction. Proc Natl Acad Sci U S A 103: 18020–18025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Quirk GJ (2004) Neuronal signalling of fear memory. Nat Rev Neurosci 5:844–852 [DOI] [PubMed] [Google Scholar]
- Maren S (2001)Neurobiology of Pavlovian fear conditioning. Annu Rev Neurosci 24:897–931 [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG (2000) Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci 23:649–711 [DOI] [PubMed] [Google Scholar]
- Martin SJ, Morris RGM (2002) New life in an old idea: the synaptic plasticity and memory hypothesis revisited. Hippocampus 12:609–636 [DOI] [PubMed] [Google Scholar]
- McKernan MG, Shinnick-Gallagher P (1997) Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 390:607–611 [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ (2002) Neurons in medial prefrontal cortex signal memory for fear extinction. Nature 420:70–74 [DOI] [PubMed] [Google Scholar]
- Milad MR, Vidal-Gonzalez I, Quirk GJ (2004) Electrical stimulation of medial prefrontal cortex reduces conditioned fear in a temporally specific manner. Behav Neurosci 118:389–394 [DOI] [PubMed] [Google Scholar]
- Miserendino MJ, Sananes CB, Melia KR, Davis M (1990) Blocking of acquisition but not expression of conditioned fear-potentiated startle by NMDA antagonists in the amygdala. Nature 345:716–718 [DOI] [PubMed] [Google Scholar]
- Monfils MH, Cowansage KK, Klann E, LeDoux JE (2009) Extinction-reconsolidation boundaries: key to persistent attenuation of fear memories. Science 324:951–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers KM, Davis M (2007) Mechanisms of fear extinction. Mol Psychiatry 12:120–150 [DOI] [PubMed] [Google Scholar]
- Myers KM, Ressler KJ, Davis M (2006) Different mechanisms of fear extinction dependent on length of time since fear acquisition. Learn Mem 13:216–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabavi S, Fox R, Proulx CD, Lin JY, Tsien RY, Malinow R (2014) Engineering a memory with LTD and LTP. Nature 511:348–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka A, Toyoda T, Miura Y, Hitora-Imamura N, Naka M, Eguchi M, et al. (2014) Synaptic plasticity associated with a memory engram in the basolateral amygdala. J Neurosci 34:9305–9309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsini CA, Maren S (2012) Neural and cellular mechanisms of fear and extinction memory formation. Neurosci Biobehav Rev 36:1773–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC, Pare D (2010) Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev 90:419–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paré D, Quirk GJ, Ledoux JE (2004) New vistas on amygdala networks in conditioned fear. J Neurophysiol 92:1–9 [DOI] [PubMed] [Google Scholar]
- Pitkänen A, Savander V, LeDoux JE (1997) Organization of intra-amygdaloid circuitries in the rat: an emerging framework for understanding functions of the amygdala. Trends Neurosci 20:517–523 [DOI] [PubMed] [Google Scholar]
- Plendl W, Wotjak CT (2010) Dissociation of within- and between-session extinction of conditioned fear. J Neurosci 30:4990–4998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Garcia R, González-Lima F (2006) Prefront al mechanisms in extinction of conditioned fear. Biol Psychiatry 60:337–343 [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Repa C, LeDoux JE (1995) Fear conditioning enhances short-latency auditory responses of lateral amygdala neurons: parallel recordings in the freely behaving rat. Neuron 15:1029–1039 [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Russo GK, Barron JL, Lebron K (2000) The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci 20:6225–6231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ (2002) Memory for extinction of conditioned fear is long-lasting and persists following spontaneous recovery. Learn Mem 9:402–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez S, Liu X, Lin PA, Suh J, Pignatelli M, Redondo RL, et al. (2013) Creating a false memory in the hippocampus. Science 341:387–391 [DOI] [PubMed] [Google Scholar]
- Repa JC, Muller J, Apergis J, Desrochers TM, Zhou Y, LeDoux JE (2001) Two different lateral amygdala cell populations contribute to the initiation and storage of memory. Nat Neurosci 4: 724–731 [DOI] [PubMed] [Google Scholar]
- Rescorla RA (2004) Spontaneous recovery varies inversely with the training-extinction interval. Learn Behav 32:401–408 [DOI] [PubMed] [Google Scholar]
- Riccio A, Li Y, Moon J, Kim KS, Smith KS, Rudolph U, et al. (2009) Essential role for TRPC5 in amygdala function and fear-related behavior. Cell 137:761–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio A, Li Y, Tsvetkov E, Gapon S, Yao GL, Smith KS, et al. (2014) Decreased anxiety-like behavior and Gαq/11-dependent responses in the amygdala of mice lacking TRPC4 channels. J Neurosci 34:3653–3667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riga D, Matos MR, Glas A, Smit AB, Spijker S, Van den Oever MC (2014) Optogenetic dissection of medial prefrontal cortex circuitry. Front Syst Neurosci 8:230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues SM, Bauer EP, Farb CR, Schafe GE, LeDoux JE (2002) The group I metabotropic glutamate receptor mGluR5 is required for fear memory formation and long-term potentiation in the lateral amygdala. J Neurosci 22:5219–5229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues SM, Farb CR, Bauer EP, LeDoux JE, Schafe GE (2004) Pavlovian fear conditioning regulates Thr286 autophosphorylation of Ca2+/calmodulin-dependent protein kinase II at lateral amygdala synapses. J Neurosci 24:3281–3288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues SM, Schafe GE, LeDoux JE (2001) Intra-amygdala blockade of the NR2B subunit of the NMDA receptor disrupts the acquisition but not the expression of fear conditioning. J Neurosci 21:6889–6896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogan MT, Stäubli UV, LeDoux JE (1997) Fear conditioning induces associative long-term potentiation in the amygdala. Nature 390:604–607 [DOI] [PubMed] [Google Scholar]
- Rossetti T, Banerjee S, Kim C, Leubner M, Lamar C, Gupta P, et al. (2017) Memory Erasure Experiments Indicate a Critical Role of CaMKII in Memory Storage. Neuron 96:207–216.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer S, Martina M, Paré D (1999) An inhibitory interface gates impulse traffic between the input and output stations of the amygdala. J Neurosci 19:10575–10583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer S, Martina M, Paré D (2000) Polarized synaptic interactions between intercalated neurons of the amygdala. J Neurophysiol 83:3509–3518 [DOI] [PubMed] [Google Scholar]
- Royer S, Paré D (2002) Bidirectional synaptic plasticity in intercalated amygdala neurons and the extinction of conditioned fear responses. Neuroscience 115:455–462 [DOI] [PubMed] [Google Scholar]
- Rumpel S, LeDoux J, Zador A, Malinow R (2005) Postsynaptic receptor trafficking underlying a form of associative learning. Science 308:83–88 [DOI] [PubMed] [Google Scholar]
- Santini E, Ge H, Ren K, Peña de Ortiz S, Quirk GJ (2004) Consolidation of fear extinction requires protein synthesis in the medial prefrontal cortex. J Neurosci 24:5704–5710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Quirk GJ, Porter JT (2008) Fear conditioning and extinction differentially modify the intrinsic excitability of infralimbic neurons. J Neurosci 28:4028–4036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, LeDoux JE (2000) Memory consolidation of auditory pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci 20:RC96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller D, Cain CK, Curley NG, Schwartz JS, Stern SA, Ledoux JE, et al. (2008) Evidence for recovery of fear following immediate extinction in rats and humans. Learn Mem 15:394–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senn V, Wolff SBE, Herry C, Grenier F, Ehrlich I, Gründemann J, et al. (2014) Long-range connectivity defines behavioral specificity of amygdala neurons. Neuron 81:428–437 [DOI] [PubMed] [Google Scholar]
- Shen K, Meyer T (1999) Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science 284:162–166 [DOI] [PubMed] [Google Scholar]
- Shi C, Davis M (1999) Pain pathways involved in fear conditioning measured with fear-potentiated startle: lesion studies. J Neurosci 19:420–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi CJ, Cassell MD (1998) Cascade projections from somatosensory cortex to the rat basolateral amygdala via the parietal insular cortex. J Comp Neurol 399:469–491 [DOI] [PubMed] [Google Scholar]
- Shin RM, Tsvetkov E, Bolshakov VY (2006) Spatiotemporal asymmetry of associative synaptic plasticity in fear conditioning pathways. Neuron 52:883–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumyatsky GP, Malleret G, Shin RM, Takizawa S, Tully K, Tsvetkov E, et al. (2005) stathmin, a gene enriched in the amygdala, controls both learned and innate fear. Cell 123:697–709 [DOI] [PubMed] [Google Scholar]
- Shumyatsky GP, Tsvetkov E, Malleret G, Vronskaya S, Hatton M, Hampton L, et al. (2002) Identification of a signaling network in lateral nucleus of amygdala important for inhibiting memory specifically related to learned fear. Cell 111:905–918 [DOI] [PubMed] [Google Scholar]
- Sierra-Mercado D, Padilla-Coreano N, Quirk GJ (2011) Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology 36:529–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singewald N, Schmuckermair C, Whittle N, Holmes A, Ressler KJ (2015) Pharmacology of cognitive enhancers for exposure-based therapy of fear, anxiety and trauma-related disorders. Pharmacol Ther 149:150–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotres-Bayon F, Bush DEA, LeDoux JE (2007) Acquisition of fear extinction requires activation of NR2B-containing NMDA receptors in the lateral amygdala. Neuropsychopharmacology 32: 1929–1940 [DOI] [PubMed] [Google Scholar]
- Sotres-Bayon F, Diaz-Mataix L, Bush DEA, LeDoux JE (2009) Dissociable roles for the ventromedial prefrontal cortex and amygdala in fear extinction: NR2B contribution. Cereb Cortex 19:474–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobel C, Marek R, Gooch HM, Sullivan RKP, Sah P (2015) Prefrontal and Auditory Input to Intercalated Neurons of the Amygdala. Cell Rep 10:1435–1442 [DOI] [PubMed] [Google Scholar]
- Takeuchi T, Duszkiewicz AJ, Morris RGM (2014) The synaptic plasticity and memory hypothesis: encoding, storage and persistence. Philos Trans R Soc Lond B Biol Sci 369: 20130288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetkov E, Carlezon WA, Benes FM, Kandel ER, Bolshakov VY (2002) Fear conditioning occludes LTP-induced presynaptic enhancement of synaptic transmission in the cortical pathway to the lateral amygdala. Neuron 34:289–300 [DOI] [PubMed] [Google Scholar]
- Tully K, Li Y, Tsvetkov E, Bolshakov VY (2007) Norepinephrine enables the induction of associative long-term potentiation at thalamo-amygdala synapses. Proc Natl Acad Sci U S A 104:14146–14150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal-Gonzalez I, Vidal-Gonzalez B, Rauch SL, Quirk GJ (2006) Microstimulation reveals opposing influences of prelimbic and infralimbic cortex on the expression of conditioned fear. Learn Mem 13:728–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DL, Ressler KJ, Lu KT, Davis M (2002) Facilitation of conditioned fear extinction by systemic administration or intra-amygdala infusions of D-cycloserine as assessed with fear-potentiated startle in rats. J Neurosci 22:2343–2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger NM (2011) The medial geniculate, not the amygdala, as the root of auditory fear conditioning. Hearing research 274:61–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisskopf MG, Bauer EP, LeDoux JE (1999) L-type voltage-gated calcium channels mediate NMDA-independent associative long-term potentiation at thalamic input synapses to the amygdala. J Neurosci 19:10512–10519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF (2006) Learning induces long-term potentiation in the hippocampus. Science 313:1093–1097 [DOI] [PubMed] [Google Scholar]
- Wilensky AE, Schafe GE, Kristensen MP, LeDoux JE (2006) Rethinking the fear circuit: the central nucleus of the amygdala is required for the acquisition, consolidation, and expression of Pavlovian fear conditioning. J Neurosci 26:12387–12396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilensky AE, Schafe GE, LeDoux JE (2000)The amygdala modulates memory consolidation of fear-motivated inhibitory avoidance learning but not classical fear conditioning. J Neurosci 20:7059–7066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods AM, Bouton ME (2008) Immediate extinction causes a less durable loss of performance than delayed extinction following either fear or appetitive conditioning. Learn Mem 15:909–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Davidson TJ, Mogri M, Deisseroth K (2011) Optogenetics in neural systems. Neuron 71:9–34 [DOI] [PubMed] [Google Scholar]
- Yu K, Ahrens S, Zhang X, Schiff H, Ramakrishnan C, Fenno L, et al. (2017) The central amygdala controls learning in the lateral amygdala. Nat Neurosci 20:1680–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]