

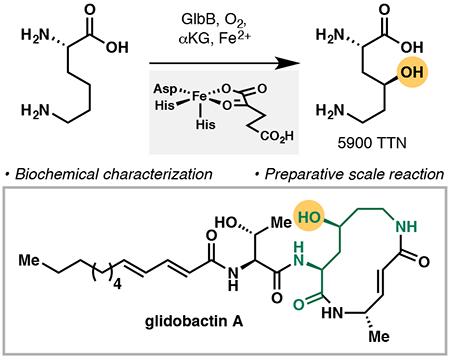
Abstract
We present the functional characterization of GlbB, a lysine 4-hydroxylase from the glidobactin biosynthetic gene cluster. Despite its narrow substrate specificity, GlbB is able to catalyze the hydroxylation of L-lysine with excellent total turnover numbers and complete regio- and diastereoselectivity. The synthetic utility of GlbB is illustrated by its use in the efficient preparation of a key dipeptide fragment of glidobactin.
Graphical Abstract
The syrbactins are a family of hybrid peptide-polyketide natural products that share a common 12-membered unsaturated macrolactam core (Figure 1A).1 Originally isolated as virulence factors in plant-pathogen interactions, the syrbactins were found to boost virulence by targeting the eukaryotic 20S proteasome core particle.2 With reported IC50 values of < 20 nM, two members of this family, glidobactin A (1) and cepafungin I (2) represent some of the most potent natural proteasome inhibitors isolated to date.3 Crystallographic evidence suggests that inhibition occurs primarily by covalent binding of the α,β-unsaturated lactam to the chymotrypsin-like threonine residue in the β5-subunit,2–4 akin to the mode of action of several other proteasome inhibitors such as epoxomicin and salinosporamide A.5 Proteasome inhibitors have recently attracted attention as potential antitumor agents due to their ability to disrupt the regulated degradation of pro-apoptotic factors in malignant cells.6 At present, three proteasome inhibitors, namely bortezomib, carfilzomib, and ixazomib, have been approved by the FDA for the treatment of multiple myeloma, and a few others are undergoing clinical trials. Despite these developments, new proteasome inhibitors still need to be advanced due to the potential emergence of drug resistance and the syrbactins hold significant promise as such candidates. Towards this end, total syntheses efforts and subsequent medicinal chemistry development by Ichikawa, Pirrung, and others have provided initial validation towards the use of this natural product family as antineoplastic agents.4,7
Figure 1.
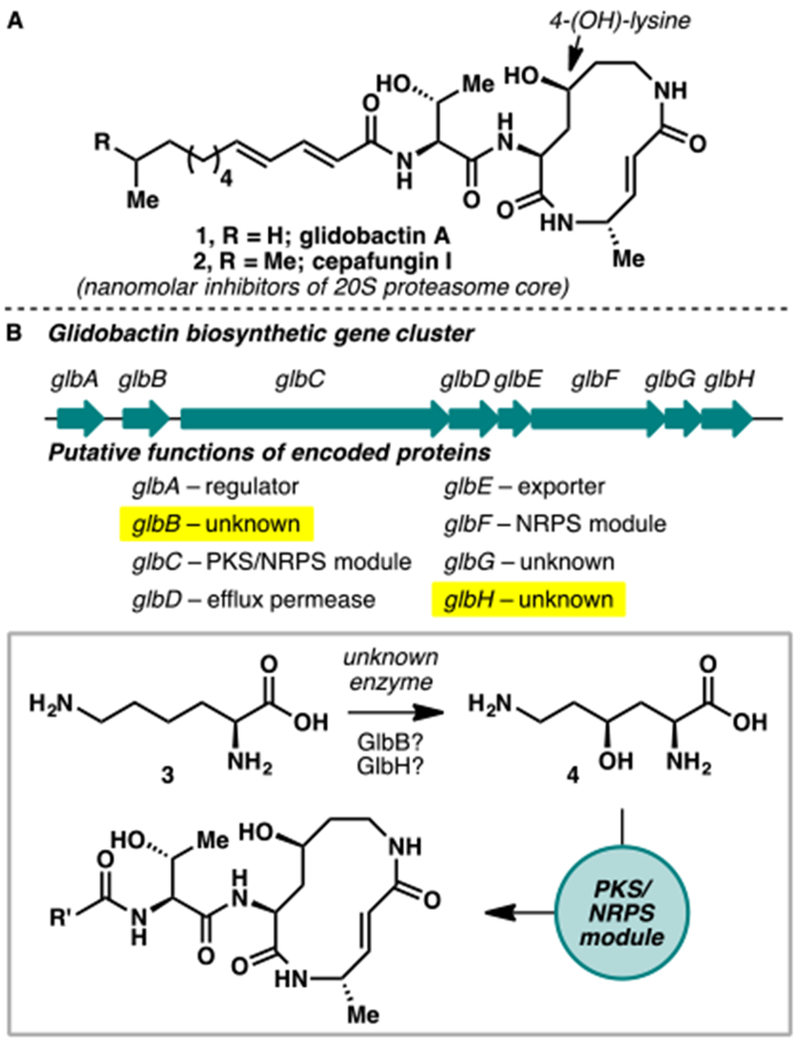
(A) Structures of glidobactin A (1) and cepafungin I (2), members of the syrbactin family of natural products with potent inhibitory activity against the 20S proteasome core. (B) Biosynthetic gene gluster of glidobactin in Polyangium brachysporum and predicted functions of the respective genes in the formation of 1.
Biogenetically, the macrolactam core of 1 and 2 arises from sequential condensations of 4-hydroxylysine, L-alanine, and malonyl-CoA catalyzed by modules of nonribosomal peptide synthetases. In 2007,8 Dudler and co-workers identified the biosynthetic gene cluster responsible for the production of glidobactin from a soil bacterium belonging to Burkholderiales. Eight genes, named glbA–H, were found to be crucial for the biosynthesis of glidobactin (Figure 1B). Two of the genes, glbC and glbF, were assigned to encode for the NRPS modules responsible for the formation of the macrolactam core structure. Of the remaining six, glbA, glbD, and glbE are thought to encode for proteins involved in the export and regulation of the metabolite production. GlbH shares homology with FAD-dependent nitrodioxygenases and GlbB contains a conserved domain within the DUF2257 family, hitherto a family of uncharacterized conserved bacterial proteins. Though it is likely that either of the latter two genes is responsible for the production of 4-(OH)-Lys, the exact biosynthetic origin of this motif was never elucidated on a molecular level.
In surveying the literature, several lines of evidence seem to point to the role of GlbB in the production of 4-(OH)-Lys. Firstly, deletion of glbH from the producer strain by Dudler led to only reduction and not total abolition of glidobactin production.8 A glbB disruption mutant, however, was found to be completely incapable of producing glidobactin. A homologous syrbactin synthetase from Photorhabdus luminescens, despite containing only homologs of glbB–G, is capable of producing glidobactin A when expressed in Pseudomonas putida.9 Finally, while the function of DUF2257 (now known as PF10014) was unknown at the time of Dudler’s work, subsequent investigation by Ogawa and co-workers revealed that it belongs to the iron- and α-ketoglutarate-dependent (Fe/αKG) superfamily of enzymes and that its members are capable of hydroxylating free-standing aliphatic amino acids.10 Indeed, a homology model of GlbB, built using a solved crystal structure of a member of PF10014 from M. petroleiphilum as a template,11 revealed the conserved His1-X-Asp-Xn-His2 metal binding triad consisting of His188, Asp190, and His251. On the opposite side of the pocket, an arginine residue (Arg267) is likely involved in the binding of αKG via salt bridge formation. The homology model also suggests a hydrophilic pocket formed by Asp73, Arg82, Gln115, and Arg128, which likely forms key polar contacts with the amino acid substrate (Figure S1). Based on these observations, we postulate that GlbB might be responsible for the C4 hydroxylation of L-lysine in the biosynthesis of glidobactin. Recent results from our laboratory have argued for the general utility of Fe/αKG enzymes as enabling tools in the production of valuable building blocks for the synthesis of complex bioactive molecules.12 Hypothesizing that selective C4 hydroxylation of L-Lys can be leveraged for the chemoenzymatic synthesis of syrbactins and related analogues, we set out to functionally characterize GlbB.
Codon optimization and over-expression of GlbB as an N-His-tagged protein was found to provide good yield of soluble enzyme (Figure S3). In vitro assays with recombinant GlbB were next performed to test its ability to hydroxylate L-Lys (Figure 2). In the presence of FeSO4, α-ketoglutarate (αKG), and ascorbic acid, reaction of L-Lys with GlbB was observed to generate a new product with m/z = 163, corresponding to one hydroxylation event. Omission of either FeSO4 or αKG from the reaction led to abolition of catalytic activity, confirming the dependency of the enzyme on these components for activity. Subsequent NMR characterization verified the identity of the reaction product as 4, thus establishing the role of GlbB as lysine 4-hydroxylase in the biosynthesis of glidobactin.
Figure 2.
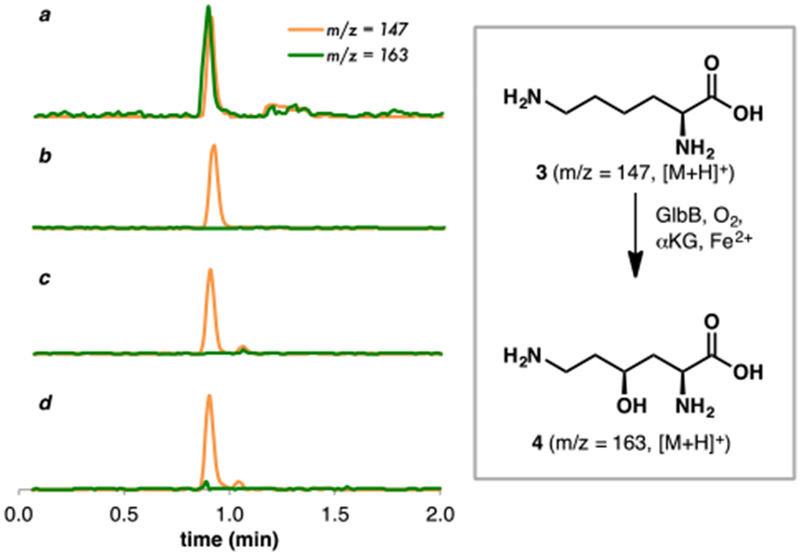
LCMS profile of L-Lys hydroxylation (a) in the presence of GlbB, FeSO4, αKG, and ascorbic acid; (b) in the absence of GlbB; (c) in the absence of FeSO4; (d) in the absence of αKG.
To investigate the utility of GlbB as a hydroxylation biocatalyst, we tested its activity against a suite of free amino acid substrates. Among the 20 substrates tested (Figure 3 and Figure S4), only L-Lys, L-Leu and L-Met yielded the corresponding hydroxylation products. Hydroxylation of L-Lys proceeded with complete diastereoselectivity and high total turnover number (5900 TTN). Similar to several other members of the PF10014 family, GlbB can catalyze the C4 hydroxylation of L-Leu to 5, albeit with only moderate TTN. Productive oxidation could also be observed with L-Met to provide the corresponding sulfoxide product 6 with 330 TTN and ca. 10:1 dr. Additionally, steady-state kinetic parameters were obtained for hydroxylation of L-Lys, L-Leu, and L-Met (Figure S5). Consistent with the observed TTNs, the catalytic efficiency of L-Lys hydroxylation of L-Lys was found to be a few orders of magnitude higher than that of L-Leu or L-Met, arising from significantly higher kcat and much lower KM. With the exception of PolL—an Fe/αKG involved in the biosynthesis of carbamoylpolyoxamic acid13—previously characterized members of PF10014 have mostly been limited to Fe/αKGs accepting only amino acid substrates with aliphatic side-chains.10 To the best of our knowledge, GlbB is the first member of this family capable of acting on both polar and aliphatic amino acids. Prior to our work, the two Fe/αKG lysine hydroxylases characterized thus far, KDO1 and KDO2,14 are classified as clavaminate-synthase-like Fe/αKG dioxygenases (IPR014503 family), with minimal sequence similarity to GlbB. In fact, despite catalyzing the hydroxylation of the same carbon of L-Lys, KDO2 and GlbB share only 11% sequence identity. The identification of lysine hydroxylation activity within both PF10014 and IPR014503 families suggests convergent evolution of this function from highly dissimilar ancestral enzymes.
Figure 3.
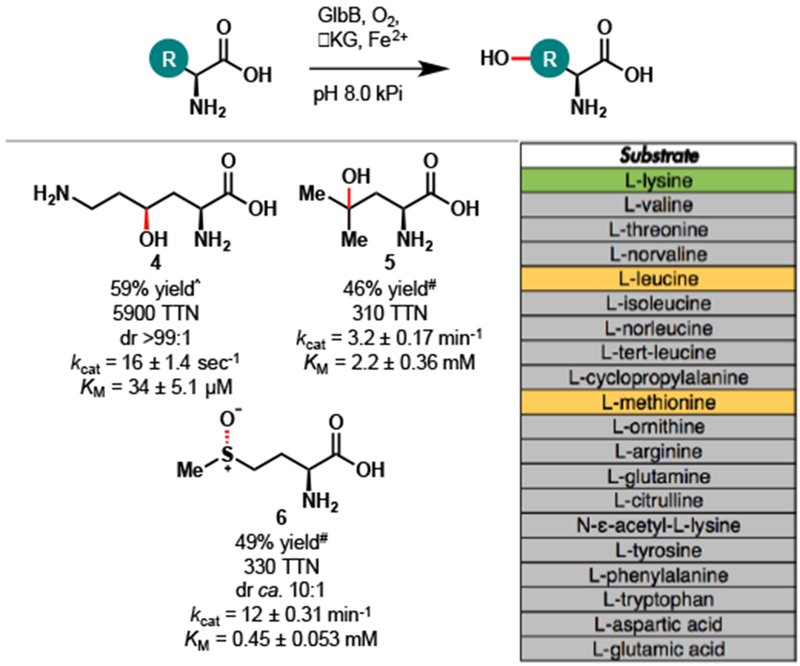
Substrate scope of amino acid hydroxylation with GlbB. Conditions for total turnover number (TTN) measurement: free amino acid (20 mM, 1 equiv), αKG (50 mM, 2.5 equiv), L-ascorbic acid (10 mM, 0.5 equiv), FeSO4 (1 mM, 0.05 equiv), GlbB (0.01–0.15 mol% enzyme loading), kPi buffer (pH = 8.0, 50 mM, 3 mL total volume), 12 h at 20 °C. Yields and TTNs were determined by NMR analysis calibrating against an internal standard of known concentration. Color legend for substrate table: green – excellent substrate, orange – moderate substrate, grey – nonsubstrate. ^Reaction was carried out with 0.01 mol% GlbB. #Reaction was carried out with 0.15 mol% GlbB.
In agreement with GlbB’s unique substrate specificity, a global survey of the PF10014 family, generated using the sequence similarity network (SSN) approach, shows that GlbB belongs to a distinct cluster separate from previously characterized members of PF10014 (Figure 4). Notably, this cluster also contains an Fe/αKG encoded by plu1881 (Uniprot accession code: Q7N5R2, 48% sequence identity with GlbB) from the glidobactin synthetase of P. luminescens.9 Assuming that enzymes within this cluster exhibit similar substrate preference, our analysis would predict that Plu1881 is also capable of catalyzing C4 hydroxylation of L-Lys. More importantly, the SSN of PF10014 reveals the existence of several clusters with no functionally characterized members. For example, we identify a cluster containing HrmJ, an Fe/αKG that had been implicated in the formation of 3-(trans-2’-nitrocyclopropyl)alanine from L-Lys in the biosynthesis of hormaomycin.15 We anticipate that functional characterization of enzymes belonging to these lesser-known clusters will expand the breadth of chemical reaction space spanned by the Fe/αKG dioxygenase superfamily.
Figure 4.

Sequence similarity network of PF10014 (100% identical sequences are conflated, alignment score 60). Orange nodes represent previously characterized Fe/αKG belonging to this family.
With an eye towards a chemoenzymatic total synthesis of the syrbactins, we next assessed the utility of GlbB in preparative scale hydroxylation of L-Lys (Scheme 1). At 0.6 mmol scale, full conversion of 3 to 4 could be obtained by using just 0.02 mol% of purified GlbB. Similar to other C4 and C5 hydroxylated amino acids,12,14 4 was prone to undergo intramolecular cyclization to gamma lactone 7 during protection of the free amines. Further stirring of the crude material from the amine protection in the presence of EDC allowed the lactonization to proceed to full conversion, providing 7 in 60% isolated yield. Gratifyingly, 7 was found to undergo facile amide bond formation with L-alaninol (8) at room temperature to afford dipeptide 9 in 92% yield.16 This sequence illustrates the synthetic utility of GlbB as a key dipeptide fragment of glidobactin could be obtained in just four simple steps.
Scheme 1.
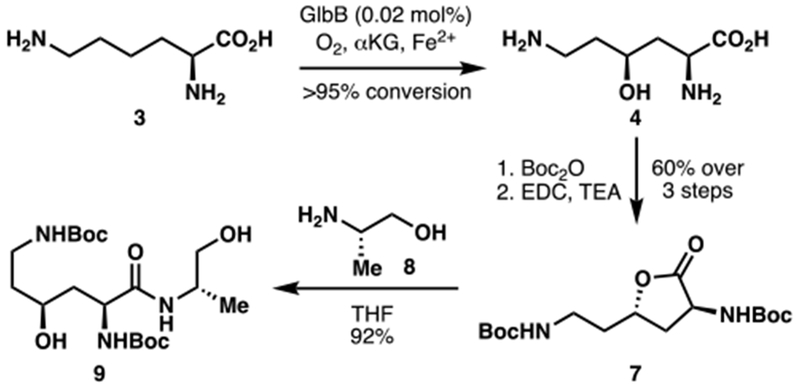
Application of L-Lys hydroxylation with GlbB in the synthesis of a key dipeptide fragment (9) of glidobactin.
Conclusions
Through a series of in vitro experiments, we have functionally characterized a new Fe/αKG dioxygenase that is capable of catalyzing selective C4 hydroxylation of L-Lys. These studies not only shed light on the origin of the 4-(OH)-Lys motif in glidobactin, but also lay the groundwork for the development of a chemoenzymatic strategy to access the syrbactin family of natural products and related structural analogues.
Supplementary Material
Acknowledgements
Financial support for this work was generously provided by The Scripps Research Institute and the National Institutes of Health (Grant R35 GM128895). We thank the Shen lab, the Kodadek lab, and the Roush lab for generous access to their instrumentations.
Footnotes
Footnotes relating to the title and/or authors should appear here.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Terui Y, Nishikawa J, Hinoo H, Kato T, Shoji J, J. Antibiot 1990, 43, 788; [DOI] [PubMed] [Google Scholar]; Wäspi U, Hassa P, Staempfli A, Molleyres L-P, Winkler T, Dudler R, Microbiol. Res 1999, 154, 1. [Google Scholar]
- 2.Groll M, Schellenberg B, Bachmann AS, Archer CR, Huber R, Powell TK, Lindow S, Kaiser M, Dudler R, Nature 2008, 452, 755. [DOI] [PubMed] [Google Scholar]
- 3.Stein ML, Beck P, Kaiser M, Dudler R, Becker CFW, Groll M, Proc. Natl. Acad. Sci. USA 2012, 109, 18367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clerc J, Groll M, Illich DJ, Bachmann AS, Huber R, Schellenberg B, Dudler R, Kaiser M, Proc. Natl. Acad. Sci. USA 2009, 106, 6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rentsch A, Landsberg D, Broadmann T, Bülow L, Girbig A-K, Kalesse M, Angew. Chem. Int. Ed 2013, 52, 5450; [DOI] [PubMed] [Google Scholar]; Schneekloth JS Jr., Crews CM, Curr. Drug Targets 2011, 12, 1581; [DOI] [PubMed] [Google Scholar]; Tsukamoto S, Yokosawa H, Planta Med 2010, 76, 1064. [DOI] [PubMed] [Google Scholar]
- 6.Cromm PM, Crews CM, ACS Cent. Sci 2017, 3, 830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmidt U, Kleefeldt A, Mangold R, J. Chem. Soc., Chem. Commun 1992, 1687; [Google Scholar]; Chiba T, Hosono H, Nakagawa K, Asaka M, Takeda H, Matsuda A, Ichikawa S, Angew. Chem. Int. Ed 2014, 53, 4836; [DOI] [PubMed] [Google Scholar]; Pirrung MC, Biswas G, Ibarra-Rivera TR, Org. Lett 2010, 12, 2402; [DOI] [PubMed] [Google Scholar]; Kitahata S, Chiba T, Yoshida T, Ri M, Iida S, Matsuda A, Ichikawa S, Org. Lett 2016, 18, 2312; [DOI] [PubMed] [Google Scholar]; Kitahata S, Yakushiji F, Ichikawa S, Chem. Sci 2017, 8, 6959; [DOI] [PMC free article] [PubMed] [Google Scholar]; Opoku-Ansah J, Ibarra-Rivera TR, Pirrung MC, Bachmann AS, Pharm. Biol 2012, 50, 25. [DOI] [PubMed] [Google Scholar]
- 8.Schellenberg B, Bigler L, Dudler R, Environ. Microbiol 2007, 9, 1640. [DOI] [PubMed] [Google Scholar]
- 9.Dudnik A, Bigler L, Dudler R, Microbiol. Res 2013, 168, 73. [DOI] [PubMed] [Google Scholar]
- 10.Kodera T, Smirnov SV, Samsonova NN, Kozlov YI, Koyama R, Hibi M, Ogawa J, Yokozeki K, Shimizu S, Biochem. Biophys. Res. Commun. 2009, 390, 506; [DOI] [PubMed] [Google Scholar]; Hibi M, Ogawa J, Appl. Microbiol. Biotechnol 2014, 98, 3869. [DOI] [PubMed] [Google Scholar]
- 11.Xu Q, et al. , Proteins 2014, 82, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zwick CR III, Renata H, J. Am. Chem. Soc 2018, 140, 1165; [DOI] [PubMed] [Google Scholar]; Zhang X, King-Smith E, Renata H, Angew. Chem. Int. Ed 2018, 57, 5037. [DOI] [PubMed] [Google Scholar]
- 13.Qi J, Wan D, Ma H, Liu Y, Gong R, Qu X, Sun Y, Deng Z, Chen W, Cell Chem. Biol 2016, 23, 935. [DOI] [PubMed] [Google Scholar]
- 14.Baud D, et al. , ChemCatChem 2014, 6, 3012. [Google Scholar]
- 15.Höfer I, Crüsemann M, Radzom M, Geers B, Flachshaar D, Cai X, Zeeck A, Piel J, Chem. Biol 2011, 18, 381. [DOI] [PubMed] [Google Scholar]
- 16.Marin J, Briand J-P, Guichard G, Eur. J. Org. Chem 2008,1005. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.