

Abstract
Bacillus cytotoxicus is a member of the Bacillus cereus group linked to fatal cases of diarrheal disease. Information on B. cytotoxicus is very limited; in particular comprehensive genomic data is lacking. Thus, we applied a genomic approach to characterize B. cytotoxicus and decipher its population structure. To this end, complete genomes of ten B. cytotoxicus were sequenced and compared to the four publicly available full B. cytotoxicus genomes and genomes of other B. cereus group members. Average nucleotide identity, core genome, and pan genome clustering resulted in clear distinction of B. cytotoxicus strains from other strains of the B. cereus group. Genomic content analyses showed that a hydroxyphenylalanine operon is present in B. cytotoxicus, but absent in all other members of the B. cereus group. It enables degradation of aromatic compounds to succinate and pyruvate and was likely acquired from another Bacillus species. It allows for utilization of tyrosine and might have given a B. cytotoxicus ancestor an evolutionary advantage resulting in species differentiation. Plasmid content showed that B. cytotoxicus is flexible in exchanging genes, allowing for quick adaptation to the environment. Genome-based phylogenetic analyses divided the B. cytotoxicus strains into four clades that also differed in virulence gene content.
Introduction
Bacillus cytotoxicus was first described in 2013 as a thermotolerant member of the Bacillus cereus group1. At that time, only five strains had been detected. Four out of the five strains had been linked to severe foodborne diarrheal outbreaks, which included three fatal cases1. The B. cereus group has been rapidly expanding in recent years1–6 and comprises several genetically closely related species. Its most prominent members are Bacillus anthracis7, Bacillus cereus sensu stricto8, B. cytotoxicus1, Bacillus mycoides9, Bacillus pseudomycoides10, Bacillus thuringiensis11, Bacillus toyonensis12, and Bacillus weihenstephanensis13. A clear phylogenetic separation of B. cereus group species is not possible as the criteria that define species were not phylogenetically based14–16. However, three major clades can be differentiated within the B. cereus group, in which species are intermingled17. The evolution of the B. cereus group was recently suggested to be mainly driven by the adaptation to animal hosts as pathogens, symbionts, or saprophytes18. The enormous variation in pathogenic potential shown by members of this group is often linked to plasmid-encoded virulence factors. While in particular B. anthracis and B. cereus are frequent causes of morbidity and mortality, other B. cereus group species are used as probiotics or biopesticides. Still, the virulence potential of B. cytotoxicus, B. thuringiensis and B. weihenstephanensis may need to be reassessed based on new genomic data as well as data confirming the formation of various toxins19–23.
B. cytotoxicus was first isolated in association with three fatal cases of diarrheal disease in a foodborne outbreak in France in 199824. The implicated causative agent, strain NVH 391–98T, produced cytotoxin K, a novel diarrheic enterotoxin, and was first identified as B. cereus24. However, several years later, multi-locus sequence typing and 16S rRNA sequence comparisons showed that NVH 391-98T belonged to the novel species B. cytotoxicus1. This species characteristically harbors the cytK-1 variant of the gene encoding cytotoxin K, which is highly toxic to human intestinal Caco-2 and Vero cells25. Homologues of CytK-1 with up to 89% amino acid identity were found in other B. cereus group members and are referred to as CytK-225. B. cytotoxicus was described as a novel species in 2013 based on five strains, four of which were linked to foodborne disease1. However, it has been known for some time that cytotoxicity of B. cytotoxicus strains varies, with strain NVH 883-00 having been reported to be non-cytotoxic26. We were recently able to show that cytotoxicity of nine B. cytotoxicus isolates obtained from mashed potato powders varied greatly in a Vero cell assay23. While either no (n = 7) or low cytotoxicity (n = 1) was detected for most isolates, one isolate exceeded the cytotoxitiy of reference strain B. cereus NVH 0075-95, which was linked to foodborne disease23, by more than 3-fold.
The B. cereus group can be divided into three clades. Assignment of strains to clades can be performed using spoIIIAB typing27,28. B. cereus group strains can be further differentiated into seven phylogenetic subtypes29, to which new strains can be assigned by panC typing30. B. cytotoxicus strains are exclusively assigned to panC group VII. B cytotoxicus cannot only be differentiated from other B. cereus group members by its panC sequence, but also by the presence of the cytK-1 gene and its capability to grow at 50 °C1,31. Since the isolation of French outbreak strain NVH 391-98T, a small number of other B. cytotoxicus have been isolated, mainly originating from potato products and in particular mashed potatoes1,23,32.
Nevertheless, little is known about the population structure, ecology, and evolution of B. cytotoxicus and only four complete genome sequences were publicly available. In this study, we aimed to extend the genomic information available for this species. We generated complete genome sequences for ten B. cytotoxicus strains and studied the phylogeny and diversity of B. cytotoxicus strains. In addition, B. cytotoxicus-specific genetic traits were determined by comparison with genomes of other B. cereus group members, and plasmids present in the B. cytotoxicus strains were analyzed. The generated data significantly extends the very limited body of knowledge on B. cytotoxicus, in particular allowing for novel insights into the evolution and differentiation of this species.
Results
Species confirmation and position of B. cytotoxicus within the B. cereus group
B. cytotoxicus is phylogenetically different from other B. cereus group species based on 16S rRNA gene sequence similarity, MLST profile, and panC sequence1,31. The four publicly available genomes as well as the ten newly generated complete genomes (Table 1) were checked for correct species annotation using BTyper31. All 14 strains were placed into panC group VII of the B. cereus group17 based on panC sequence homology (Table 1). This confirms the identification of the strains as B. cytotoxicus.
Table 1.
List of strains.
| Species | Strain | Accession number | panC group |
|---|---|---|---|
| B. cytotoxicus | CH_1 | CP024120 | VII |
| CH_2 | CP024116 | VII | |
| CH_3 | CP024113 | VII | |
| CH_4 | CP024111 | VII | |
| CH_13 | CP024109 | VII | |
| CH_15 | CP024107 | VII | |
| CH_23 | CP024104 | VII | |
| CH_25 | CP024101 | VII | |
| CH_38 | CP024098 | VII | |
| CH_39 | CP024096 | VII | |
| NVH 391-98T | NC_009674 | VII | |
| NVH 883-00 | NZ_FMJN00000000 | VII | |
| CVUAS_2833 | NZ_JYPG01000012 | VII | |
| AFSSA_08CEB44bac | NZ_FMIK00000000 | VII | |
| B. anthracis | Ames_Ancestor | NC_007530 | III |
| Ames | NC_003997 | III | |
| CDC684 | NC_012581 | III | |
| H9401 | CP002091 | III | |
| Sterne | AE017225 | III | |
| B. cereus | AH187 | CP001177 | III |
| ATCC10987 | NC_003909 | III | |
| ATCC14579T | NC_004722 | IV | |
| B4264 | CP001176 | IV | |
| E33L | NC_006274 | III | |
| G9842 | CP001186 | IV | |
| Q1 | NC_011969 | III | |
| B. mycoides | ATCC 6462 | NZ_CP009692 | VI |
| B. pseudomycoides | DSM 12442T | ACMX01000000 | I |
| FSL H8-0534 | MUAQ01000000 | I | |
| B. thuringiensis | BMB171 | NC_014171 | IV |
| YBT 1518 | NC_022873 | IV | |
| ser_konkukian_97_27 | NC_005957 | III | |
| B. weihenstephanensis | KBAB4 | NC_010184 | VI |
| WSBC10204T | NZ_CP009746 | VI | |
| B. toyonensis | BAC3151 BCT 7112T | LDKD02000000 | V |
| CP006863 | V |
To check the whole-genome-sequence-based position of B. cytotoxicus within the B. cereus group, we compared the 14 genomes with selected strains from the B. cereus group. The average nucleotide identity (ANI) between all pairs of B. cytotoxicus strains was at least 99.3%, showing that B. cytotoxicus is a homogeneous species (Fig. 1). B. cytotoxicus has an approximate 85% ANI when compared to other B. cereus group members, with the highest value of 86.9% with B. pseudomycoides. Similar to B. cytotoxicus, B. pseudomycoides had an ANI of approximately 86% when compared to other B. cereus group members. As the ANI among the other members of the B. cereus group was at least 90% (Fig. 1), B. cytotoxicus and B. pseudomycoides are the most distinct members of the B. cereus group. The 14 B. cytotoxicus strains also form distinct clades in trees based on the core and pan genome of the 37 strains (Figs 2 and 3).
Figure 1.

Whole-genome-based analysis of B. cytotoxicus compared to B. cereus sensu lato. Heat map of the average nucleotide identity (ANI) among the 37 strains. The percentages are listed in the figure and are also available as Supplementary Table S1. BA = Bacillus anthracis, Bcyt = B. cytotoxicus, BM = B. mycoides, Bp = B. pseudomycoides, Bcer = B. cereus, BW = B. weihenstephanensis, Bth = B. thuringiensis, Bto = B. toyonensis.
Figure 2.
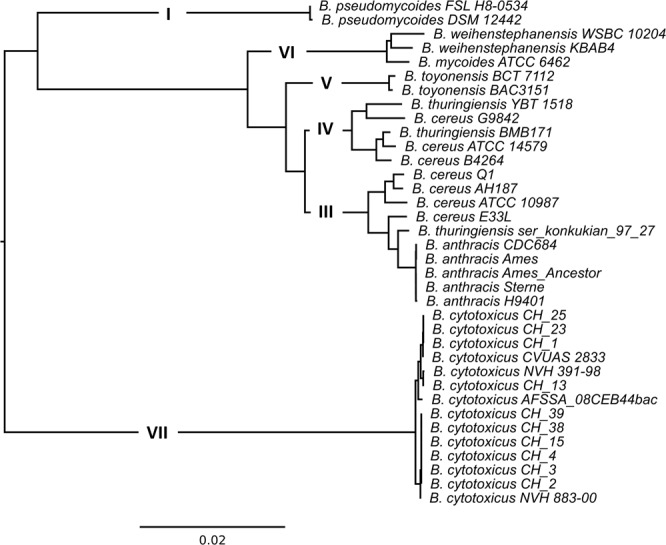
Neighbor-joining mid-point-rooted tree of the concatenated core proteins of the 37 strains. The clades as predicted by panC analysis are displayed in the tree.
Figure 3.
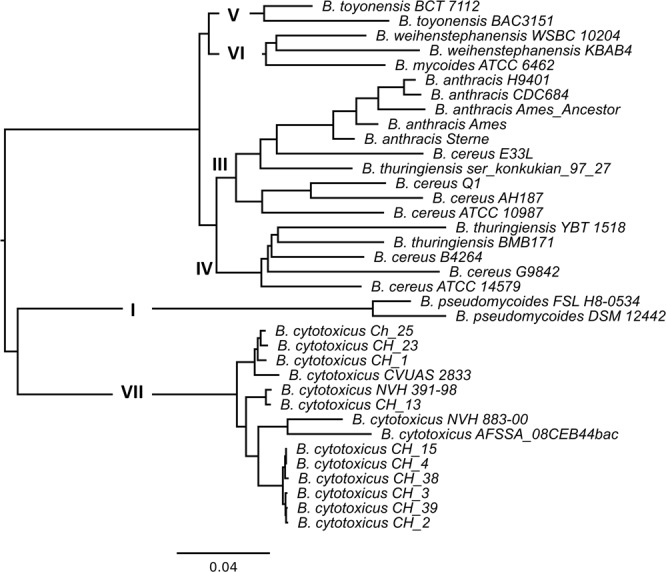
Hierarchical clustered mid-point-rooted tree based on the pan genome of 37 strains from the B. cereus group. The clades as predicted by panC analysis are displayed in the tree.
Phylogenetic analysis of the species B. cytotoxicus
While the species B. cytotoxicus seems homogenous in the core genome analysis, the individual strains appeared to be more divergent from one another in the pan genome analysis. In particular, a shift in position was observed for strain NVH 883-00 (Figs 2 and 3). Therefore, we evaluated the phylogeny of the 14 B. cytotoxicus genomes more closely.
First, we selected the sequences of the seven MLST genes commonly used for B. cereus typing (glp, gmk, ilv, pta, pur, pyc, and tpi)33. The sequences of glp, gmk, and pyc were 100% identical among the 14 B. cytotoxicus strains. Two different alleles occurred for ilv, pur and tpi, and three different alleles for pta. A tree based on the concatenated sequences of the seven genes revealed a close relation between strains CH_1, CH_23, CH_25 and CVUAS 2833, between strain CH_13 and the type strain NVH 391-98, and between strains NVH 883-00, CH_2, CH_3, CH_4, CH_15, CH_38 and CH_39 (Fig. 4). Strain AFSSA_08CEB44bac formed a clade of its own.
Figure 4.
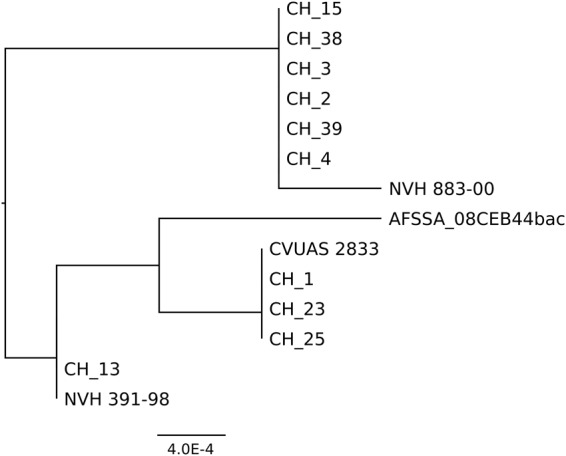
Neighbor-joining tree for strains of the species B. cytotoxicus. The tree is based on the alignment of the concatenated sequences of the MLST genes glp, gmk, ilv, pta, pur, pyc, and tpi.
An ANI matrix of the 14 strains was constructed and revealed four clades, designated A-D, which differ from each other by at least 0.3% (Fig. 5). Strain AFSSA_08CEB44bac is an outlier, differing at least 0.5% from the other strains.
Figure 5.

Heat map of the average nucleotide identity (ANI) for strains of the species B. cytotoxicus.
In a next step, we compared the number of single nucleotide polymorphisms (SNPs) between the core genomes of the strains (Fig. 6). The strains of clade A (CH_13 and type strain NVH391-98T) had 946 SNPs. The strains of clade B (CH_1, CH_23, CH_25, CVUAS 2833) had only ten to 54 SNPs. The strains of clade C either had 12–115 SNPs (CH_2, CH_3, CH_4, CH_38 and CH_39) or 219–227 SNPs (NVH 883–00). Strains in clade A had 7,562 to 9,480 SNPs compared to strains from clade B and 7,426 to 9,806 SNPs compared to strains from clade C (see also Supplementary Table S1). Clade B and C had approximately 10,000 SNPs. Strain AFSSA_08CEB44bac representing clade D had 16,000-20,000 SNPs compared to all other strains. B. cytotoxicus has a genome size of approximately 4.1 Mbp and the SNP rates between the clades are therefore 1.8 to 4.8 SNPs per kbp.
Figure 6.
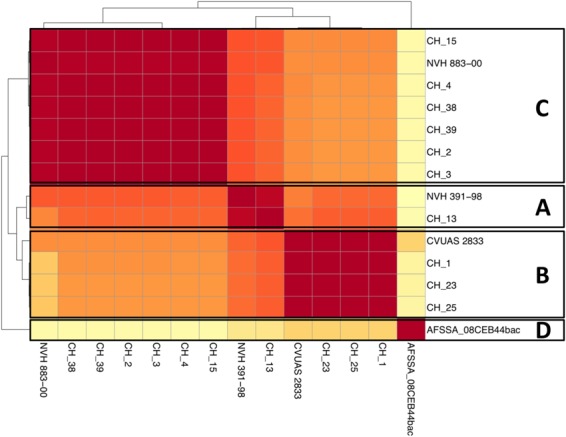
SNP based tree and heat map of the species B. cytotoxicus. The four clades are indicated.
A core genome-based tree further confirmed that B. cytotoxicus can be differentiated into four clades (Fig. 7). In addition, a pan genome tree is useful to distinguish between strains that are hardly distinguishable in core genome analysis34. The pan genome tree of B. cytotoxicus strains confirmed the grouping of strains into four clades. However, AFSSA_08CEB44bac and NVH883-00 are now both outliers (Fig. 8).
Figure 7.
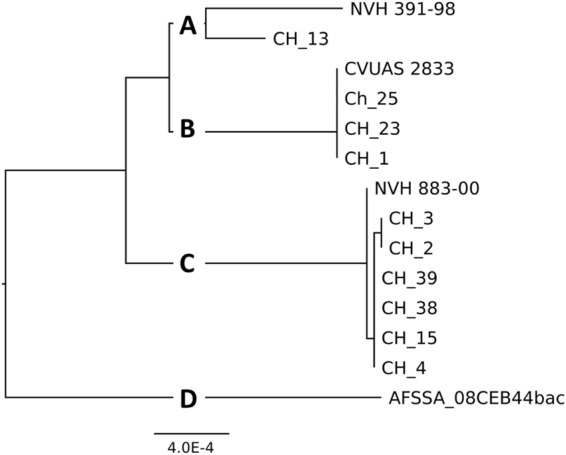
Core genome tree of the species B. cytotoxicus.
Figure 8.
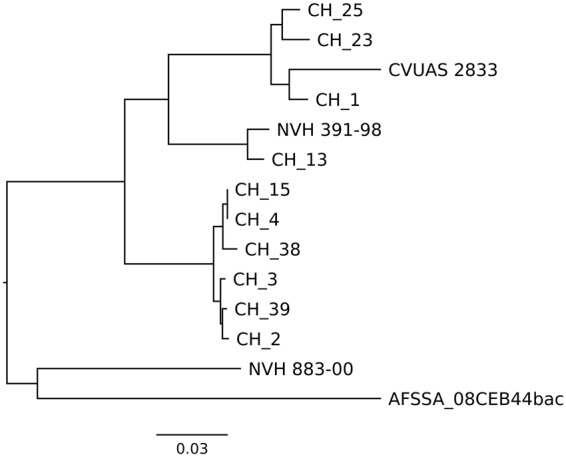
Pan genome tree of the species B. cytotoxicus.
Taken together, MLST, ANI, and SNP and core-genome analyses, resulted in the same grouping of the 14 B. cytotoxicus strains into four clades. Clade A comprised the type strain NVH391-98 and CH_13. Closely related to clade A is clade B, which comprised CH_1, CH_23, CH_25, and CVUAS 2833. Clade C consisted of CH_2, CH_3, CH_4, CH_38, CH_39, and NVH 883-00 and clade D exclusively consisted of AFSSA_08CEB44bac.
Genetic contents of B. cytotoxicus
The core genome of the 14 B. cytotoxicus strains contained 3,151 protein encoding genes. The formula of the fitting curve for the core genome dynamics (Supplemental Fig. 1A) predict a decrease of 45 genes after the addition of a 15th genome and therefore the core-genome is still open. Of the 3,151 core genes, 129 were not found in any of the 23 B. cereus group genomes used in this study and thus comprise potential B. cytotoxicus-specific genes. A functional analysis of B. cytotoxicus-specific genes revealed, among others, genes encoding 77 hypothetical proteins, ten transcriptional regulators, 2 CRIPSR cas associated genes and 2 transposases (Table 2). Interestingly, a putative hydroxyphenylalanine (hpa) operon was exclusively present in all 14 B. cytotoxicus strains. This operon encodes enzymes to degrade the aromatic compound 4-hydroxyphenylacetate to components of the citrate cycle35,36. The operon consists of ten genes and a LysR regulator gene is situated directly upstream in the opposite direction (Fig. 9). This parallels the organization in Escherichia coli, where the hpa regulator is located upstream of the operon37. Further, the regulation of operons by an upstream regulator is frequent in bacteria38 and LysR is therefore a likely candidate to regulate the hpa-operon in B. cytotoxicus. A cytidyltransferase is located in the operon, which is not present in the E. coli operon. The operon has a GC-content of 39.6%, which exceeds the average GC-content of 35.8% for B. cytotoxicus genomes. A search among all B. cereus group genomes revealed that nine of the 11 genes of the operon were present in Bacillus pseudomycoides strain AFS092012, but not in B. pseudomycoides DSM12442, FSL H8-0534 or any of the other 99 B. pseudomycoides genomes in the public database. Furthermore, the operon was not detected in genomes of other members of the B. cereus group.
Table 2.
B. cytotoxicus core genome genes not found in other genomes of the B. cereus group.
| Locus in CH_13a | Gene in CH_13 | Functionb |
|---|---|---|
| CG479_RS05485 | CG479_RS05485 | 2,4-dihydroxyhept-2-ene-1,7-dioic acid aldolase |
| CG479_RS05500 | hpaD | 3,4-dihydroxyphenylacetate 2,3-dioxygenase |
| CG479_RS05515 | CG479_RS05515 | cytidyltransferase |
| CG479_RS05535 | CG479_RS05535 | 4-hydroxyphenylacetate isomerase |
| CG479_RS05530 | CG479_RS05530 | 2-hydroxyhepta-2,4-diene-1,7-dioate isomerase |
| CG479_RS05525 | CG479_RS05525 | 5-carboxymethyl-2-hydroxymuconate isomerase |
| CG479_RS15270 | CG479_RS15270 | glycerophosphodiester phosphodiesterase |
| G479_RS06185 | G479_RS06185 | LTA synthase family protein |
| CG479_RS07295 | CG479_RS07295 | transcriptional regulator |
| CG479_RS07395 | CG479_RS07395 | sporulation protein YjcZ |
| CG479_RS07860 | CG479_RS07860 | TetR/AcrR family transcriptional regulator |
| CG479_RS07990 | CG479_RS07990 | thiamine biosynthesis protein ThiF |
| CG479_RS08090 | CG479_RS08090 | GbsR/MarR family transcriptional regulator |
| CG479_RS08120 | CG479_RS08120 | aspartate aminotransferase family protein |
| CG479_RS09375 | CG479_RS09375 | spore germination protein |
| CG479_RS09435 | CG479_RS09435 | methyl-accepting chemotaxis protein |
| CG479_RS10095 | CG479_RS10095 | transcriptional regulator |
| CG479_RS10100 | CG479_RS10100 | ImmA/IrrE family metallo-endopeptidase |
| CG479_RS10240 | CG479_RS10240 | DMT family transporter |
| CG479_RS10490 | CG479_RS10490 | MarR family transcriptional regulator |
| CG479_RS10975 | cas2 | CRISPR-associated endonuclease Cas2 |
| CG479_RS10980 | cas4 | CRISPR-associated protein Cas4 |
| G479_RS11880 | G479_RS11880 | acyltransferase |
| CG479_RS11890 | CG479_RS11890 | N-acetyltransferase |
| CG479_012025 | CG479_012025 | phosphonate ABC transporter permease |
| CG479_RS12175 | CG479_RS12175 | transcriptional regulator |
| CG479_RS12350 | CG479_RS12350 | IS30 family transposase |
| G479_RS12365 | G479_RS12365 | MATE family efflux transporter |
| CG479_RS12655 | CG479_RS12655 | IS256 family transposase |
| CG479_RS12690 | CG479_RS12690 | ArsR family transcriptional regulator |
| CG479_RS12835 | CG479_RS12835 | enterotoxin |
| G479_RS15125 | G479_RS15125 | transcriptional regulator |
| CG479_RS15995 | CG479_RS15995 | MFS transporter |
| CG479_RS17000 | CG479_RS17000 | non-ribosomal peptide synthetase |
| G479_RS18300 | G479_RS18300 | S-layer protein |
| CG479_RS18315 | CG479_RS18315 | ArsR family transcriptional regulator |
| CG479_RS18510 | CG479_RS18510 | YjcZ family sporulation protein |
| CG479_RS18930 | CG479_RS18930 | NupC/NupG family nucleoside CNT transporter |
| CG479_RS20300 | CG479_RS20300 | NADH-quinone oxidoreductase subunit C |
| CG479_RS20520 | CG479_RS20520 | DNA-directed RNA polymerase subunit delta |
| CG479_RS21035 | CG479_RS21035 | transcriptional antiterminator BglG |
| CG479_RS01030 | CG479_RS01030 | CPBP family intramembrane metalloprotease |
| CG479_RS01060 | CG479_RS01060 | MFS transporter |
| CG479_RS02825 | CG479_RS02825 | histidine kinase |
| CG479_RS02835 | CG479_RS02835 | amidohydrolase family protein |
| CG479_RS02890 | CG479_RS02890 | alpha/beta hydrolase |
| CG479_RS03705 | CG479_RS03705 | histidine kinase |
| CG479_RS04625 | CG479_RS04625 | LytR family transcriptional regulator |
| CG479_RS05280 | CG479_RS05280 | spore coat protein |
astrain CH_13 was used as reference strain, bhypothetical genes were omitted.
Figure 9.

Genetic organization and functional comparison of the hpa operon in B. cytotoxicus strains. Putative functions are as follows: lysR – regulator of the operon; hpaI - 2,4-dihydroxyhept-2-ene-1,7-dioic acid aldolase; hpaE - 5-carboxymethyl-2-hydroxymuconate semialdehyde dehydrogenase; hpaD - 3,4-dihydroxyphenylacetate 2,3-dioxygenase; Cyt-T – cytidyltransferase; acetate transporter - cation acetate symporter; hpaG - 2-hydroxyhepta-2,4-diene-1,7-dioate isomerase; hpaF - 5-carboxymethyl-2-hydroxymuconate Delta-isomerase; tetratricop. Prot - tetratricopeptide repeat protein. The 330-bp hypothetical gene between Cyt-T and the acetate transporter is not shown.
In addition, the pan genome of the 14 B. cytotoxicus strains consisted of 5,111 genes. Dynamics of the pan genome resulted in a fitting curve with an α ≈ 0.11, indicating an open pan genome. Remarkably, the three draft genomes in the dataset, i.e. AFSSA_08CEB44bac, NVH 883-00, and CVUAS 2833, contained a high number of unique genes: 335, 204, and 64 respectively (Table 3). Moreover, AFSSA_08CEB44bac and NVH 883-00 uniquely shared 40 genes, mainly encoding phage-related or hypothetical functions. All other strains exhibited less than seven unique genes. Remarkably, if the 40 genes shared solely by AFSSA_08CEB44bac and NVH 883-00 are deleted from the pan-genome matrix, the strains do not cluster together anymore in a pan genome tree (data not shown).
Table 3.
Comparison of the characteristics of all B. cytotoxicus strains, which were either full genomes sequenced in the course of this study (marked by an asterisk) or complete genome sequences that were already publicly available.
| Strain | Source | Genes | Plasmids | ||||
|---|---|---|---|---|---|---|---|
| Total | Accessory | Unique | Exclusively absent | Virulence factors | |||
| CH_1* | Mashed potatoes, Switzerland, 2014 | 3866 | 691 | 2 | 7 | 264 | 14 kb, 53 kb, 67 kb |
| CH_2* | Mashed potatoes, Switzerland, 2014 | 3981 | 794 | 0 | 9 | 262 | 53 kb, 83 kb |
| CH_3* | Mashed potatoes, Switzerland, 2014 | 3984 | 796 | 1 | 1 | 263 | 53 kb, 83 kb |
| CH_4* | Mashed potatoes, Switzerland, 2014 | 3943 | 766 | 0 | 0 | 260 | 83 kb |
| CH_13* | Mashed potatoes, Switzerland, 2014 | 3837 | 664 | 2 | 13 | 264 | 79 kb |
| CH_15* | Mashed potatoes, Switzerland, 2014 | 3943 | 766 | 0 | 0 | 260 | 83 kb |
| CH_23* | Mashed potatoes, Switzerland, 2014 | 3888 | 696 | 5 | 37 | 259 | 53 kb, 67 kb |
| CH_25* | Mashed potatoes, Switzerland, 2014 | 3926 | 712 | 4 | 23 | 259 | 53 kb, 67 kb |
| CH_38* | Mashed potatoes, Switzerland, 2014 | 3922 | 742 | 0 | 12 | 262 | 53 kb, 83 kb |
| CH_39* | Mashed potatoes, Switzerland, 2014 | 3989 | 799 | 0 | 5 | 264 | 83 kb |
| AFSSA_08CEB44bac | Cooked semolina, France, 2008, | 4218 | 840 | 335 | 35 | 268 | — |
| CVUAS-2833 | Mashed potatoes linked to foodborne illness, Germany, 2007 | 3782 | 579 | 64 | 93 | 266 | — |
| NVH391-98T | Vegetable puree, France, 1998 | 3844 | 663 | 6 | 6 | 265 | 7 kb |
| NVH 883-00 | Spices, Norway, 2000 | 4254 | 1014 | 222 | 12 | 259 | — |
The low abundance of unique genes in most strains might be caused by high similarity of the strains; thus, we carried out an in-depth analysis of the unique genes detected. Strains NVH 883-00 and CVUAS 2833 were omitted because of their high number of unique genes. Strains NVH 391-98, CH_13, CH_1, CH_23, and CH_25 of clades A and B had 73 genes not found in other clades. Functional analyses revealed 39 hypothetical proteins, whereas other functions were mainly related to phages (see Supplementary Table S2). The strains of clade C (CH_2, CH_3, CH_4, CH_15, CH_38, and CH_39) had 167 clade-specific genes, with 115 representing hypothetical proteins. In addition, a lactoylglutathione lyase and a glutathione transferase were found. Both enzymes might be involved in methylglyoxal detoxification. Further, an operon encoding the functions to convert mannitol to fructose with parallel conversion of mannonate to pyruvate was present.
Genes encoding virulence factors and toxins in B. cytotoxicus
In a next step, the virulence of the B. cytotoxicus strains was assessed via a comparison to the virulence factor database VFDB39. The strains had between 259 and 268 putative virulence factors (Table 2), with 220 factors being found in all 14 strains. Strains NVH 883-00, CH_23, and CH_25 had the fewest virulence factors (n = 259). Strain NVH 883-00 was missing genes present in all other B. cytotoxicus strains that encode three putative virulence factors: a collagen adhesion protein, the motility protein MotA, and a gene involved in non-ribosomal peptide synthesis. Strain AFSSA_08CEB44bac harbored six genes not found in any other strain, including a gene coding for subtilisin.
The cytK-1 gene characteristic for B. cytotoxicus exhibited one amino acid substitution, T257A, between strains from clade A, B, D and clade C. This change is next to the conserved residue Y25624.
Plasmids of B. cytotoxicus
The 11 completely assembled genomes of B. cytotoxicus each contain one or more plasmids. No plasmid-like sequences were identified in the draft genomes of strains AFSSA_08CEB44bac, NVH 883-00, and CVUAS 2833. In total, 18 plasmids with sizes from 7 to 83 kb were identified (Table 2). The plasmids can be divided into four groups that exhibit no sequence similarity (Fig. 10). The first group contains ten plasmids of 83 kb, 79 kb, and 67 kb, the second group contains six plasmids of 53 kb, and the final two groups each contain one plasmid of 14 kb and 7 kb, respectively. The first group of plasmids was found throughout the ten strains sequenced in this study, i.e. CH_1 to CH_39 (Table 2). Remarkably, the plasmid size was 79 kb in clade A, 67 kb in clade B and 83 kb in clade C. Additional genes on the 83 kb plasmids encoded one conjugational protein and 12 hypothetical proteins. The 83 kb plasmid group had an average nucleotide identity of >99.8% with a coverage of virtually 100%. The maximum nucleotide identity with other Bacillus plasmids was 79%, but with a maximum coverage of 56% as revealed by a blastN search against the NCBI nucleotide database. The 53 kb plasmids had an average nucleotide identity of >99.7% with a coverage of virtually 100%. The plasmids were similar to the plasmid pBCM1301 from Bacillus cereus M13 with 97% identity and a coverage of 86%.
Figure 10.
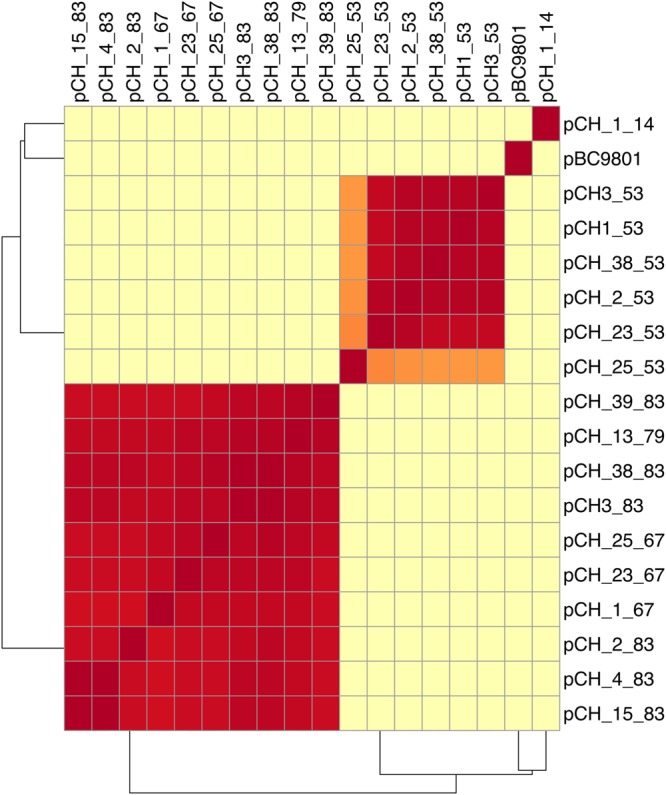
ANI analyses of plasmids in B. cytotoxicus. The plasmids are indicated with the respective strain identifier followed by the size in kb.
The 18 plasmids encoded 1,258 identified genes encoding 52 different functions. The complete list can be found online as Supplementary Table S3. The 67–83 kb plasmids carried genes coding for proteins with conjugational functions, stress proteins, transposons, a peptide transporter and a sulfite exporter. A toxin-antitoxin system of the type III ToxN/AbiQ family toxin-antitoxin system was also present on these plasmids. Homologs of this system with up to 94.2% amino acid identity were found in several plasmids of the B. cereus group. The 53 kb plasmid-encoded genes associated with stress-related proteins and transposons. Genes conferring resistance to antibiotic agents or encoding virulence factors were not identified on any of the plasmids.
Discussion
B. cytotoxicus is a recently described species that causes severe foodborne illness1. This study presents the phylogeny and genome content of B. cytotoxicus and provides novel clues on the evolution of the species. Complete genome-based phylogeny revealed that B. cytotoxicus is a distinct species within the B. cereus group, consistent with results obtained for the genome of type strain NVH 391-9817. The ANI and core genome-based trees of the B. cereus group indicate that B. cytotoxicus is – like B. anthracis – a relatively homogenous species, which is in stark contrast to other members of the B. cereus group such as B. cereus and B. thuringiensis, which exhibit a rather heterogeneous population structure40,41. While the pan genome-based tree showed a higher degree of evolutionary divergence (Fig. 1), this is in agreement with the generally higher divergence exhibited by pan genome-based trees compared to core genome-based trees34,42. The core genome-based tree containing only B. cytotoxicus strains also depicted the species as less homogenous, possibly because it was based on a higher number of genes than the B. cereus group core genome tree.
The heterogeneity was reflected in clustering in four clades. The clades were already visible in MLST analysis but based on only one or two SNPs. The genome-based trees in this study are far more reliable, as the number of SNPs between the clades is at least 7,428. Further, the SNP rate is 300-fold higher than the sequencing error rate for Pacbio SMRT sequencing (https://www.pacb.com/uncategorized/a-closer-look-at-accuracy-in-pacbio/) and complete genome trees therefore represent a valid hypothesis for evolution of B. cytotoxicus. In addition, as leading evolutionary biologist Ernst Mayr pointed out, evolution occurs in a complete organism and not on a single gene (https://www.edge.org/conversation/ernst_mayr-what-evolution-is) and hence complete genomes are preferred for phylogenetic analyses.
The SNP based analysis has the highest resolving power as it is based on DNA sequences and takes into consideration intergenic regions. The core-genome-based analysis is based on protein sequences and ignores intergenic regions and silent mutations. Nevertheless, the four clades also appeared in the core-genomes tree, showing that their distinction is robust.
The occurrence of four clades in core, pan, and SNP-based trees (Fig. 2) indicates that the clades are results of true evolutionary events. Strain NVH_883-00 can be found in clade C in the MLST, SNP and core genome trees, but clusters close to AFSSA_08CEB44bac in the pan genome tree (Fig. 2). Such differences can be considered as alternative evolutionary hypotheses42. NVH 883-00 shared many phage genes with AFSSA_08CEB44bac and their close clustering in the pan genome tree, which was based on a presence-absence matrix, was due to the high amount of uniquely shared genes. In addition, the presence of these genes in the two strains suggests that horizontal gene transfer mediated via phages occurred between the two strains.
The species B. cytotoxicus harbors a complete hydroxyphenylalanine operon that is absent in all other B. cereus group members and encodes the machinery to degrade aromatic compounds to succinate and pyruvate35. The pathway is normally associated with soil bacteria and is also known to contribute to para-cresol production out of tyrosine in the human large intestine43,44. Lignin-derived aromatic compounds are a major carbon source in soil and the capability to degrade these compounds is present in many soil bacteria36. In addition, free amino acids are released in the human gut due to the activity of peptidases and proteases from human and bacterial origin45. The presence of the hpa-operon suggests that B. cytotoxicus can utilize aromatic compounds, including tyrosine as carbon source. Furthermore, Gram-negative bacteria are susceptible to para-cresol and production of para-cresol in the intestine leads to reduced numbers of Gram-negatives46. The hpa-operon thus provides a clear evolutionary advantage for B. cytotoxicus through access to an additional carbon source and potentially also through production of the inhibiting agent para-cresol. However, the activity of the hpa-operon and its role in the metabolism of B. cytotoxicus remains to be elucidated. The presence of the operon points towards soil as the ecological niche of B. cytotoxicus, which is consistent with isolation of the organism almost exclusively from potato products23,32. Soil may represent the source of contamination for mashed potatoes after detecting B. cytotoxicus on a raw potato32.
The substantially higher GC content of the operon compared to the rest of the chromosome suggests that it was acquired from a higher GC% organism, likely another soil bacterium. The operon might have given a B. cytotoxicus ancestor an evolutionary advantage through utilization of tyrosine. This evolutionary advantage eventually resulted in species differentiation. The presence of a cytidyltransferase even suggests that energy can be gained in this pathway via CTP dependent substrate-level phosphorylation.
The clade-specific amino acid substitution in CytK-1 next to a conserved residue might lead to differences in the activity of CytK-1 and hence virulence. Type strain NVH 391-98T that caused fatal cases of foodborne disease in an outbreak in France and CVUAS 2833 linked to foodborne disease in Germany clustered in clade A and B, respectively. This supports the hypothesis that strains from these clades could be more virulent. Strain NVH 883-00 is non-cytotoxic26 and belongs to clade C. This is consistent with the hypothesis that strains from clade C exhibit low toxicity. Furthermore, virulence factor analyses revealed that NVH 883-00 uniquely lacks three virulence factors, the flagellar motor MotA, a collagen adhesion protein, and a gene involved in non-ribosomal peptide synthesis. The low virulence of this strain may therefore also be attributed to the deficiency in these three factors. Cytotoxicity and virulence studies are needed to provide comprehensive data supporting or dismissing a link between the virulence potential and the different clades.
Clade C possesses unique genes encoding two functions, a lactoylglutathione system and a mannitol conversion system. Both may give clade C strains an advantage; the first to survive methylglyoxal, the second to regenerate NAD+. The latter reaction is useful if redox regeneration is limited, and is used by fermentative bacteria47,48.
The distribution of plasmids in strains from the different clades suggests horizontal gene transfer in B. cytotoxicus, at least between strains from the same geographical region (Switzerland). The 53 kb plasmids were solely found in clade C. Further, the 53 kb plasmids had higher similarity to plasmids of the B. cereus group than to the 83 kb plasmids. This higher similarity strongly suggests that the 53 kb plasmids were acquired by clade C strains after the 83 kb plasmids were acquired and after differentiation into clades occurred. The encoded stress proteins may handle stress linked to conjugation, rather than providing the host with increased tolerance when faced with environmental stress49. The toxin-antitoxin system is one commonly found on B. cereus plasmids and ensures plasmid maintenance during germination and sporulation37.
Conclusion
We were able to show that, in contrast to some other B. cereus group species, B. cytotoxicus strains are phylogenetically distinct. Genomic content analysis revealed that a hydroxyphenylalanine (hpa) operon and its putative regulator are present in B. cytotoxicus, but absent in all other members of the B. cereus group. This operon codes for the machinery to degrade aromatic compounds to succinate and pyruvate and was likely acquired from another Bacillus species. It allows for utilization of tyrosine and might have given a B. cytotoxicus ancestor an evolutionary advantage that eventually resulted in species differentiation. Plasmid content of the strains showed that B. cytotoxicus is flexible in exchanging genes, allowing for quick adaptation to the environment. Genome-based phylogenetic analysis divided the investigated B. cytotoxicus strains into four clades that also differed in virulence gene content. In conclusion, our results provide novel insights essential to extend the currently very limited understanding of B. cytotoxicus virulence, ecology, and evolution.
Methods
Bacterial strains
Ten B. cytotoxicus strains (CH_1, CH_2, CH_3, CH_4, CH_13, CH_15, CH_23, CH_25, CH_38, CH_39) were isolated from mashed potatoes, which had been collected from regiment kitchens of the Swiss Army in 2014 and 2015. For bacterial isolation, ten-fold dilution series of the samples in 0.85% NaCl were streaked on MYP agar (Oxoid) and incubated at 37 °C over night. Plates were subsequently checked for colonies exhibiting a mannitol-negative and egg-yolk lecithinase-positive phenotype and a colony morphology consistent with the strains of the B. cereus group.
In addition, complete genome sequences of various reference strains were downloaded from the NCBI database in January-May 2018. A comprehensive list of all strains used in this study is provided as Table 1.
Complete genome sequencing
Genomic DNA was extracted and purified from B. cytotoxicus strains CH_1, CH_2, CH_3, CH_4, CH_13, CH_15, CH_23, CH_25, CH_38, CH_39 using the GenElute Bacterial Genomic DNA Kit (Sigma, Buchs, Switzerland). Genomes were sequenced on a PacBio RS II sequencer (Pacific Biosciences, Menlo Park, USA) using the single-molecule real-time sequencing technology (SMRT) chemistry at the Functional Genomics Centre Zurich. Sequencing each sample on two SMRTcells with P6/C4 chemistry and 180-minute movies generated 102,934 to 170,329 sequence reads with a mean read length of 10,120 to 18,569 bp, corresponding to approximately 200 to 600-fold genome coverage, depending on the sample. PacBio sequences were assembled de novo using the SMRT Analysis v2.3.0 software and the Hierarchical Genome Assembly Process (HGAP_3) workflow. Annotation of the genomes was carried out using the NCBI Prokaryotic Genome Automatic Annotation Pipeline (PGAAP; http://www.ncbi.nlm.nih.gov/genome/annotation_prok/)50.
General genome analyses
Core and pan genomes were identified using the USEARCH algorithm51 at a 75% sequence identity cut-off within the Bacterial Pan Genome Analysis (BPGA) software package. The dynamics of the core and pan genome were plotted and a fitting curve calculation was performed using a power fit for the pan genome and an exponential fit for the core genome, both embedded in BPGA (supplemental Fig. 1). If the dynamics of the pan genome result in a fitting curve with an α < 1, the pan genome is considered open52.
Average nucleotide identity (ANI) was calculated using the perl script “get_homologous”42 and calculated using the following settings: -E < 1e-05 for BLAST searches and -C 75% minimum alignment coverage. The options “-A” to produce a tab-separated file with relative average sequence identity and the option “-a “CDS” “ to run BlastN on the CDS nucleotide sequences were triggered to obtain an ANI matrix file. The ANI matrix was visualized in a heat map using clustvis53, with a Manhattan distance calculation and a complete linkage for rows and columns.
The ANI for plasmids was calculated using the python script average_nucleotide_identity.py in the pyani suite available at, https://github.com/widdowquinn/pyani. It calculates the ANI according to Richter54. In short, it aligns sequences using nucmer in Mummer55 and uses TETRA56 to calculate nucleotide frequencies. Nucleotide fasta files were used as input.
Phylogeny methodology
Core and pan genomes were identified using USEARCH51 at a 75% sequence identity cut-off within the Bacterial Pan Genome Analysis tool (BPGA) software package57. Maximum likelihood trees of the core genomes were constructed based on MUSCLE58 alignments of the concatenated core proteins and performed in BPGA. Pan genome trees were constructed from a presence-absence matrix (1/0) from orthologous clusters using standard settings in BPGA. The resulting Newick files were visualized as midpoint rooted trees in Figtree 1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/).
MLST-based trees were obtained by aligning the concatenated nucleotide sequences using MAFFT59. The alignment was converted to a Newick format using the nearest neighbor joining method based on the Jukes-Cantor substitution model and a midpoint-rooted tree was visualized in FigTree 1.4.3. Phylip format tree files are available in Supplementary Material S4.
Single nucleotide polymorphisms analysis
Single nucleotide polymorphisms (SNPs) were identified using Parsnp and Gingr in the Harvest suite60. SNPs were first identified with Parsnp using genomic nucleotide fasta files as input data. All 14 B. cytotoxicus genomes were compared to each B. cytotoxicus genome as reference genome. Output data files were converted to variant calling files using Gingr in the harvest suite60. The number of SNPs was calculated as being the sum of the variants compared to the reference strain. A matrix was produced with the SNPs per strain. The matrix was hierarchically clustered using complete linkage and Manhattan distance in R (R-project.org) using an in-house R-script. A heatmap was produced using Clustvis53.
Genetic content analyses
Pan genome gene presence-absence matrices were produced using get_homologous42: First, a cluster of orthologous groups (COG) and an orthologous Markov clustering (OMCL)-based pan genome was determined using the following settings: -E < 1e-05 for blast searches, -C 75% minimum alignment coverage, and -t 0 for obtaining all clusters. Subsequently, a pan genome matrix was produced using the script compare_clusters.pl using the OMCL and COG-based pan genomes as input and the option -m to obtain a matrix42. This method is more stringent than the BPGA method, as it builds a matrix based on both OMCL and COG and thus reduces the number of false positives in the gene content analysis. The matrix was converted to a presence-absence matrix (1/0) in Microsoft’s Excel (Microsoft, Redmond, WA, USA) to determine gene content.
Identification of genes encoding putative virulence factors
Virulence factors were identified by comparing the predicted proteome of each strains to the large protein set B of the virulence factor database VFDB, which contains 26,594 protein sequences, including 397 from the B. cereus group39, downloaded April 3rd, 2018. A bidirectional best-hit approach using blastp61 was applied to minimize the number of false positive hits. Blast settings were E < 1e-05 and an additional cut-off of 75% minimum alignment was used.
Accession numbers
Sequence and annotation data of the complete genomes of B. cytotoxicus strains CH_1, CH_2, CH_3, CH_4, CH_13, CH_15, CH_23, CH_25, CH_38, and CH_39 are deposited in the GenBank database under the accession numbers listed in Table 1.
Electronic supplementary material
Acknowledgements
We thank Katrin Zurfluh for laboratory work. We are grateful for financial support to JK provided by the Laboratory of Food Microbiology, Institute of Food, Nutrition and Health, ETH Zurich.
Author Contributions
Conceptual design: S.J., M.E.S., M.J.A.S. and R.S. Data analysis: M.J.A.S., T.T., J.K. Interpretation of results and preparation of the manuscript: M.J.A.S., S.J., M.E.S. All authors read and approved the final manuscript.
Data Availability
The datasets supporting the conclusions of this article are available in the GenBank repository. For accession numbers see Table 1.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-36254-x.
References
- 1.Guinebretière MH, et al. Bacillus cytotoxicus sp. nov. is a novel thermotolerant species of the Bacillus cereus group occasionally associated with food poisoning. Int. J. Syst. Evol. Microbiol. 2013;63:31–40. doi: 10.1099/ijs.0.030627-0. [DOI] [PubMed] [Google Scholar]
- 2.Liu B, et al. Bacillus bingmayongensis sp. nov., isolated from the pit soil of Emperor Qin’s Terra-cotta warriors in China. Antonie Van Leeuwenhoek. 2014;105:501–510. doi: 10.1007/s10482-013-0102-3. [DOI] [PubMed] [Google Scholar]
- 3.Jung M-Y, et al. Bacillus gaemokensis sp. nov., isolated from foreshore tidal flat sediment from the Yellow Sea. J. Microbiol. 2010;48:867–871. doi: 10.1007/s12275-010-0148-0. [DOI] [PubMed] [Google Scholar]
- 4.Miller RA, et al. Bacillus wiedmannii sp. nov., a psychrotolerant and cytotoxic Bacillus cereus group species isolated from dairy foods and dairy environments. Int. J. Syst. Evol. Microbiol. 2016;66:4744–4753. doi: 10.1099/ijsem.0.001421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jung MY, et al. Bacillus manliponensis sp. nov., a new member of the Bacillus cereus group isolated from foreshore tidal flat sediment. J. Microbiol. 2011;49:1027–1032. doi: 10.1007/s12275-011-1049-6. [DOI] [PubMed] [Google Scholar]
- 6.Liu Y, et al. Proposal of nine novel species of the Bacillus cereus group. Int. J. Syst. Evol. Microbiol. 2017;67:2499–2508. doi: 10.1099/ijsem.0.001821. [DOI] [PubMed] [Google Scholar]
- 7.Cohn F. Untersuchungen über Bacterien. Beiträge zur Biol. der Pflanz. 1872;1:127–224. [Google Scholar]
- 8.Frankland GC, Frankland P. Studies on some new microorganisms obtained from air. Philos.Trans. R. Soc. Lond. B. 1887;178:257–287. doi: 10.1098/rstb.1887.0011. [DOI] [Google Scholar]
- 9.Flügge, C. Die Mikroorganismen: mit besonderer Berücksichtigung der Infektionskrankheiten. (F. C. W. Vogel, 1886).
- 10.Nakamura L. Bacillus pseudomycoides sp. nov. Int. J. Syst. Bacteriol. 1998;48:1031–1035. doi: 10.1099/00207713-48-3-1031. [DOI] [PubMed] [Google Scholar]
- 11.Berliner E. Über die Schlaffsucht der Mehlmottenraupe (Ephestia kühniella Zell.) und ihren Erreger Bacillus thuringiensis n. sp. Zeitschrift für Angew. Entomol. Berlin. 1915;2:29–56. [Google Scholar]
- 12.Jiménez G, et al. Description of Bacillus toyonensis sp. nov., a novel species of the Bacillus cereus group, and pairwise genome comparisons of the species of the group by means of ANI calculations. Syst. Appl. Microbiol. 2013;36:383–391. doi: 10.1016/j.syapm.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 13.Lechner S, et al. Bacillus weihenstephanensis sp. nov. is a new psychrotolerant species of the Bacillus cereus group. Int. J. Syst. Bacteriol. 1998;48:1373–1382. doi: 10.1099/00207713-48-4-1373. [DOI] [PubMed] [Google Scholar]
- 14.Ehling-Schulz M, Messelhäusser U. Bacillus ‘next generation’ diagnostics: Moving from detection toward subtyping and risk-related strain profiling. Front. Microbiol. 2013;4:1–8. doi: 10.3389/fmicb.2013.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Helgason E, et al. Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis − one species on the basis of genetic evidence. Appl. Environ. Microbiol. 2000;66:2627–2630. doi: 10.1128/AEM.66.6.2627-2630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, et al. Genomic insights into the taxonomic status of the Bacillus cereus group. Sci. Rep. 2015;5:14082. doi: 10.1038/srep14082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bazinet AL. Pan-genome and phylogeny of Bacillus cereus sensu lato. BMC Evol. Biol. 2017;17:1–16. doi: 10.1186/s12862-017-1020-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rasigade JP, Hollandt F, Wirth T. Genes under positive selection in the core genome of pathogenic Bacillus cereus group members. Infect. Genet. Evol. 2018;65:55–64. doi: 10.1016/j.meegid.2018.07.009. [DOI] [PubMed] [Google Scholar]
- 19.Böhm ME, Huptas C, Krey VM, Scherer S. Massive horizontal gene transfer, strictly vertical inheritance and ancient duplications differentially shape the evolution of Bacillus cereus enterotoxin operons hbl, cytK and nhe. BMC Evol. Biol. 2015;15:246. doi: 10.1186/s12862-015-0529-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johler S, et al. Enterotoxin production of Bacillus thuringiensis isolates from biopesticides, foods, and outbreaks. Front. Microbiol. 2018;9:1915. doi: 10.3389/fmicb.2018.01915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guérin A, et al. Cereulide production by Bacillus weihenstephanensis strains during growth at different pH values and temperatures. Food Microbiol. 2017;65:130–135. doi: 10.1016/j.fm.2017.02.006. [DOI] [PubMed] [Google Scholar]
- 22.Thorsen L, Budde BB, Henrichsen L, Martinussen T, Jakobsen M. Cereulide formation by Bacillus weihenstephanensis and mesophilic emetic Bacillus cereus at temperature abuse depends on pre-incubation conditions. Int. J. Food Microbiol. 2009;134:133–139. doi: 10.1016/j.ijfoodmicro.2009.03.023. [DOI] [PubMed] [Google Scholar]
- 23.Heini N, Stephan R, Ehling-Schulz M, Johler S. Characterization of Bacillus cereus group isolates from powdered food products. Int. J. Food Microbiol. 2018;283:59–64. doi: 10.1016/j.ijfoodmicro.2018.06.019. [DOI] [PubMed] [Google Scholar]
- 24.Lund T, De Buyser ML, Granum PE. A new cytotoxin from Bacillus cereus that may cause necrotic enteritis. Mol. Microbiol. 2000;38:254–261. doi: 10.1046/j.1365-2958.2000.02147.x. [DOI] [PubMed] [Google Scholar]
- 25.Fagerlund A, Ween O, Lund T, Hardy SP, Granum PE. Genetic and functional analysis of the cytK family of genes in Bacillus cereus. Microbiology. 2004;150:2689–2697. doi: 10.1099/mic.0.26975-0. [DOI] [PubMed] [Google Scholar]
- 26.Fagerlund A, Brillard J, Fürst R, Guinebretière M-H, Granum PE. Toxin production in a rare and genetically remote cluster of strains of the Bacillus cereus group. BMC Microbiol. 2007;7:43. doi: 10.1186/1471-2180-7-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ehling-Schulz M, et al. Emetic toxin formation of Bacillus cereus is restricted to a single evolutionary lineage of closely related strains. Microbiology. 2005;151:183–197. doi: 10.1099/mic.0.27607-0. [DOI] [PubMed] [Google Scholar]
- 28.Fricker M, Ågren J, Segerman B, Knutsson R, Ehling-Schulz M. Evaluation of Bacillus strains as model systems for the work on Bacillus anthracis spores. Int. J. Food Microbiol. 2011;145:129–136. doi: 10.1016/j.ijfoodmicro.2010.07.036. [DOI] [PubMed] [Google Scholar]
- 29.Guinebretière MH, et al. Ecological diversification in the Bacillus cereus Group. Environ. Microbiol. 2008;10:851–865. doi: 10.1111/j.1462-2920.2007.01495.x. [DOI] [PubMed] [Google Scholar]
- 30.Guinebretière MH, et al. Ability of Bacillus cereus group strains to cause food poisoning varies according to phylogenetic affiliation (groups I to VII) rather than species affiliation. J. Clin. Microbiol. 2010;48:3388–3391. doi: 10.1128/JCM.00921-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carroll LM, Kovac J, Miller RA, Wiedmann M. Rapid, high-throughput identification of anthrax-causing and emetic Bacillus cereus group genome assemblies via BTyper, a computational tool for virulencebased classification of Bacillus cereus group isolates by using nucleotide sequencing data. Appl. Environ. Microbiol. 2017;83:1–19. doi: 10.1128/AEM.01096-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Contzen M, Hailer M, Rau J. Isolation of Bacillus cytotoxicus from various commercial potato products. Int. J. Food Microbiol. 2014;174:19–22. doi: 10.1016/j.ijfoodmicro.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 33.Jolley KA, Maiden MC. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. 2010;11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Snipen L, Ussery DW. Standard operating procedure for computing pangenome trees. Stand. Genomic Sci. 2010;2:135–141. doi: 10.4056/sigs.38923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prieto MA, Díaz E, García J. Molecular characterization of the 4-hydroxyphenylacetate catabolic pathway of Escherichia coli W: Engineering a mobile aromatic degradative cluster. J. Bacteriol. 1996;178:111–120. doi: 10.1128/jb.178.1.111-120.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Díaz E, Ferrández A, Prieto MA, García JL. Biodegradation of aromatic compounds by Escherichia coli. Microbiol. Mol. Biol. Rev. 2001;65:523–569. doi: 10.1128/MMBR.65.4.523-569.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Short FL, Monson RE, Salmond GPC. A type III protein-RNA toxin-antitoxin system from Bacillus thuringiensis promotes plasmid retention during spore development. RNA Biol. 2015;12:933–937. doi: 10.1080/15476286.2015.1073438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Korbel JO, Jensen LJ, von Mering C, Bork P. Analysis of genomic context: prediction of functional associations from conserved bidirectionally transcribed gene pairs. Nat. Biotechnol. 2004;22:911. doi: 10.1038/nbt988. [DOI] [PubMed] [Google Scholar]
- 39.Chen L, et al. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005;33:325–328. doi: 10.1093/nar/gki008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ehling-Schulz, M., Knutsson, R. & Scherer, S. In Genomes of Foodborne and Waterborne Pathogens (Eds Karhariou, S., Fratamico, P. & Liu, Y.) 147–164 (2011).
- 41.Kolstø A-B, Tourasse NJ, Økstad OA. What sets Bacillus anthracis apart from other Bacillus species? Annu. Rev. Microbiol. 2009;63:451–476. doi: 10.1146/annurev.micro.091208.073255. [DOI] [PubMed] [Google Scholar]
- 42.Contreras-Moreira B, Vinuesa P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 2013;79:7696–7701. doi: 10.1128/AEM.02411-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harwood CS, Parales RE. The β-ketoadipate pathway and the biology of self-Identity. Annu. Rev. Microbiol. 1996;50:553–590. doi: 10.1146/annurev.micro.50.1.553. [DOI] [PubMed] [Google Scholar]
- 44.Dawson, L. F. et al. The analysis of para-cresol production and tolerance in Clostridium difficile 027 and 012 strains. BMC Microbiol. 11, (2011). [DOI] [PMC free article] [PubMed]
- 45.Smith, E. A. & Macfarlane, G. T. Enumeration of human colonic bacteria producing phenolic and indolic compounds: effects of pH, carbohydrate availability and retention time on dissimilatory aromatic amino acid metabolism. J. Appl. Bacteriol. 81, 288–302 (1996). [DOI] [PubMed]
- 46.Passmore, I. J. et al. Para-cresol production by Clostridium difficile affects microbial diversity and membrane integrity of Gram-negative bacteria. PLOS Pathogens14 (2018). [DOI] [PMC free article] [PubMed]
- 47.Richter H, Hamann I, Unden G. Use of the mannitol pathway in fructose fermentation of Oenococcus oeni due to limiting redox regeneration capacity of the ethanol pathway. Arch. Microbiol. 2003;179:227–233. doi: 10.1007/s00203-003-0519-6. [DOI] [PubMed] [Google Scholar]
- 48.Årsköld E, et al. Phosphoketolase pathway dominates in Lactobacillus reuteri ATCC 55730 containing dual pathways for glycolysis. J. Bacteriol. 2008;190:206–212. doi: 10.1128/JB.01227-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baltrus DA. Exploring the costs of horizontal gene transfer. Trends Ecol. Evol. 2013;28:489–495. doi: 10.1016/j.tree.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 50.Angiuoli SV, et al. Toward an online repository of standard operating procedures (SOPs) for (meta)genomic annotation. Omi. A J. Integr. Biol. 2008;12:137–141. doi: 10.1089/omi.2008.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 52.Tettlin, H., Riley, D., Cattuto, C. & Medini, D. Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 12, 472–477 (2008). [DOI] [PubMed]
- 53.Metsalu T, Vilo J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015;43:W566–W570. doi: 10.1093/nar/gkv468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Richter M, Rosselló-Móra R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. 2009;106:19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Teeling H, Waldmann J, Lombardot T, Bauer M, Glöckner FO. TETRA: A web-service and a stand-alone program for the analysis and comparison of tetranucleotide usage patterns in DNA sequences. BMC Bioinformatics. 2004;5:1–7. doi: 10.1186/1471-2105-5-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chaudhari NM, Gupta VK, Dutta C. BPGA-an ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016;6:1–10. doi: 10.1038/srep24373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Katoh, K., Rozewicki, J. & Yamada, K. D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 1–7 (2017). [DOI] [PMC free article] [PubMed]
- 60.Treangen TJ, Ondov BD, Koren S, Phillippy AM. The harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014;15:1–15. doi: 10.1186/s13059-014-0524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the conclusions of this article are available in the GenBank repository. For accession numbers see Table 1.