

Abstract
Marijuana use by pregnant women is increasing. To predict developmental risk to the fetus/neonate from such use, in utero fetal exposure to (−)-∆9-tetrahydrocannabinol (THC), the main psychoactive cannabinoid in marijuana and its active psychoactive metabolite, 11-hydroxy-∆9-tetrahydrocannabinol (11-OH-THC), needs to be determined. Since such measurement is not possible, physiologically based pharmacokinetic (PBPK) modeling and simulation can provide an alternative method to estimate fetal exposure to cannabinoids. To do so, pharmacokinetic parameters for the disposition of THC and 11-OH-THC need to be elucidated. Here, we report a first step to estimate these parameters, namely, those related to maternal metabolism of THC/11-OH-THC in human liver microsomes (HLMs) at plasma concentrations observed after smoking marijuana. Using recombinant cytochrome P450 (P450) and UDP-glucuronosyltransferase (UGT) enzymes, CYP1A1, 1A2, 2C9, 2C19, 2D6, 3A4, 3A5, 3A7, and UGT1A9 and UGT2B7 were found to be involved in the disposition of THC/11-OH-THC. Using pooled HLMs, the fraction metabolized (fm) by relevant enzymes was measured using selective enzyme inhibitors, and then adjusted for enzyme cross-inhibition. As previously reported, CYP2C9 was the major enzyme responsible for depletion of THC and formation of 11-OH-THC with fm values of 0.82 ± 0.08 and 0.99 ± 0.10, respectively (mean ± S.D.), while CYP2D6 and CYP2C19 were minor contributors. 11-OH-THC was depleted by UGT and P450 enzymes with fm values of 0.60 ± 0.05 and 0.40 ± 0.05, respectively (mean ± S.D.), with UGT2B7, UGT1A9, CYP2C9, and CYP3A4 as contributors. These mechanistic data represent the first set of drug-dependent parameters necessary to predict maternal-fetal cannabinoid exposure during pregnancy using PBPK modeling.
Introduction
Up to 7.5% of pregnant women in the United States use marijuana at some point during pregnancy (Brown et al., 2017). Furthermore, the prevalence of marijuana use by pregnant women has been increasing (Substance Abuse and Mental Health Services Administration, 2017). The average concentrations of the psychoactive cannabinoid in marijuana, (−)-∆9-tetrahydrocannabinol (THC) have been steadily increasing, with the most recent content (weight per weight) estimated at 12% (ElSohly et al., 2016). To estimate fetal exposure (and therefore risk) to THC and its psychoactive metabolite, 11-hydroxy-∆9-tetrahydrocannabinol (11-OH-THC) (Fig. 1), maternal-fetal exposure to these compounds needs to be measured or predicted. Conducting pharmacokinetic studies in pregnant women is not possible due to ethical and logistical reasons. Maternal exposure to drugs drives drug fetal exposure. Therefore, an alternative is to predict the maternal-fetal exposure to these cannabinoids during pregnancy using physiologically based pharmacokinetic (PBPK) models where gestational age–dependent changes in expression of hepatic drug metabolizing enzymes (DMEs) and transporters can be incorporated (Ke et al., 2012, 2014a; Zhang et al., 2017; Zhang and Unadkat, 2017). The latter is important since activity of many cytochrome P450 (P450) enzymes is either induced or repressed during pregnancy (Anderson, 2005; Ke et al., 2014b).
Fig. 1.

THC metabolic scheme. In vivo, THC is hydroxylated to 11-OH-THC, the primary and psychoactive metabolite. 11-OH-THC is further oxidized to COOH-THC, a nonactive metabolite that is the major circulating cannabinoid metabolite found (as the glucuronide conjugate) in blood and urine. 11-OH-THC is also glucuronidated. Besides the major metabolites shown here, both THC and 11-OH-THC have additional oxidative metabolites.
To predict the disposition of THC/11-OH-THC during pregnancy, among other parameters, one must first identify the enzymes involved in the biotransformation of THC and 11-OH-THC and the fraction metabolized (fm) via these pathways. In vitro studies have identified CYP2C9, 3A4, 2C19, and 2D6 as DMEs responsible for the formation of THC metabolites (Stout and Cimino, 2014). Of these, CYP2C9 and CYP2C19 are the hepatic DMEs responsible for the formation of 11-OH-THC (Bland et al., 2005; Watanabe et al., 2007). Through in vivo studies, CYP2C9 and CYP3A4 have been identified as important DMEs involved in 11-OH-THC clearance (Sachse-Seeboth et al., 2009; Stott et al., 2013). Additionally, UDP-glucuronosyltransferase (UGT) enzymes, UGT1A9 and UGT1A10, are the hepatic DMEs responsible for the glucuronidation of 11-OH-THC in vitro (Mazur et al., 2009).
Despite the aforementioned data, there are key drug parameters missing that are necessary for PBPK modeling and simulation. In vitro metabolic studies have focused on THC metabolite formation rather than on THC depletion. As such, there may be additional enzymes responsible for the clearance of THC. In addition, the enzymes involved in depletion of 11-OH-THC have not all been identified. Furthermore, the fm via each enzyme, a necessary parameter for PBPK modeling and simulation, is missing. Finally, the majority of in vitro studies have examined the metabolism of supraphysiological cannabinoid concentrations, which may not be representative of enzyme contributions at cannabinoid concentrations observed in vivo. Therefore, the aims of this study were to profile the hepatic metabolic pathways of THC and 11-OH-THC by 1) identifying the relevant recombinant enzymes (both P450s and UGTs) capable of metabolizing these compounds, and 2) using the results from the aforementioned studies as a guide to quantify the fm via the identified enzymes in pooled human liver microsomes (HLMs) at plasma concentrations of THC and 11-OH-THC observed after smoking marijuana. The results from these studies are a first step in generating the maternal mechanistic pharmacokinetic parameters necessary to build a linked THC/11-OH-THC PBPK model that can be used to predict maternal-fetal exposure to THC/11-OH-THC during pregnancy.
Materials and Methods
Chemicals and Reagents.
(−)-∆9-THC (1 mg/ml), (±) 11-OH-THC (0.1 mg/ml), and (±) 11-nor-9-carboxy-∆9-THC (COOH-THC) (0.1 mg/ml) Drug Enforcement Administration–exempt methanol stocks and deuterated internal standards [(−)-∆9-THC-D3, (±) 11-OH-THC-D3, (±) COOH-THC-D3] were purchased from Cerilliant (Round Rock, TX). Low-binding (LB) microcentrifuge tubes (made out of chemical-resistant polypropylene), bovine serum albumin (BSA) (Fraction V, heat-shock treated), acetonitrile, and formic acid [liquid chromatography (LC)/mass spectrometry (MS) grade] were purchased from Fisher Scientific (Hampton, NH). Amber silanized glass vials were purchased from Waters Corporation (Milford, MA). β-NADP+, D-glucose 6-phosphate, glucose-6-phosphate dehydrogenase, uridine 5′-diphosphoglucuronic acid, and all chemical inhibitors and probes were purchased from Sigma-Aldrich (St. Louis, MO). Milli-Q water was used in all preparations. All other chemicals and reagents were obtained at the highest quality available commercially.
Recombinant Enzymes and HLMs.
Recombinant cytochrome P450 (CYP19, 1A1, 1A2, 2A6, 2B6, 2C8, 2C9*1 (Arg144), 2C19, 2D6*1, 2E1, 3A4, 3A5, 3A7) and UDP-glucuronosyltransferase (UGT1A4, 1A9, 1A10, and 2B7) expressed in baculovirus-infected insect cells (Supersomes) and pooled adult HLMs (n = 50, equal mixed gender) were purchased from Corning (Corning, NY).
Cannabinoid Experimental Optimization.
THC has low solubility, high protein, and nonspecific binding to plastic and glassware (Garrett and Hunt, 1974). Due to these reasons, experimental reactions were optimized for cannabinoid adsorption, solubility, and extraction. To limit adsorption, initial studies were performed in amber silanized glass. Due to practical and economic reasons, subsequent studies were performed in LB microcentrifuge tubes. Depletion kinetics of THC was similar in silanized glass versus LB tubes (data now shown). BSA was added to reactions to combat low solubility and limit adsorption. This analytical strategy has been previously suggested for improving radioimmunoassay of THC (Cook et al., 1976). To improve recovery of cannabinoids from reactions, a freeze-liquid technique previously described by Prasad and Singh (2009) was used (further described subsequently).
Reaction Phenotyping Using Recombinant Enzymes.
In silanized amber glass vials, the following was added (shown as final concentrations) to 0.1 M potassium phosphate buffer (pH 7.4): either 500 nM THC or 100 nM 11-OH-THC, recombinant cytochrome P450 (rCYP) enzyme (2–40 pmol), and BSA. Thus, the final total protein concentration (BSA + rCYP) was 0.2 mg/ml. Mixtures were preincubated in a shaking-block heater for 10 minutes at 37°C and 300 rpm. Reactions were initiated with a NADPH regenerating system (final concentrations: 1.3 mM NADP+, 3.3 mM D-glucose 6-phosphate, 3.3 mM MgCl2, and 0.4 U/ml glucose-6-phosphate dehydrogenase). Reactions using recombinant UGT enzymes were set up as previously described with the following changes: recombinant UGT enzymes (0.05–0.75 mg/ml), 0.2% BSA, 5 mM MgCl2, and 25 μg/ml alamethicin. Reactions were incubated for 15 minutes on ice to allow for pore formation by alamethicin, and then preincubated at 37°C as described previously. Reactions were initiated by adding uridine 5′-diphosphoglucuronic acid (2.5 mM, final concentration). Negative control reactions were also set up as previously described and initiated with buffer instead of cofactors. At designated time points, 50 μl of reaction mixture was terminated by adding to 100 μl ice-cold acetonitrile containing internal standards (THC-D3, 11-OH-THC-D3, and COOH-THC-D3) in LB tubes. Samples were centrifuged at 10,000g for 5 minutes to pellet the protein content. Supernatant was removed and incubated at −20°C for >1 hour to separate the aqueous and organic layers. The top organic layer was removed, placed in LC glass insert vials, and stored at −20°C until analysis by LC–tandem MS (LC-MS/MS). Two independent experiments in duplicate were performed.
Reaction Phenotyping Using HLMs.
In LB tubes, either 1) 500 nM THC and 0.02 mg/ml HLM or 2) 50 nM 11-OH-THC and 0.1 mg/ml HLM was added (showing final concentrations) to 0.1 M potassium phosphate buffer (pH 7.4) containing 0.2% BSA. The cannabinoid concentrations were chosen to represent clinically observed concentrations after smoking marijuana (Hunault et al., 2008). Assay conditions were optimized to keep HLM concentration low (<0.5 mg/ml) and sampling time short (<30 minutes), as previously suggested by Jones and Houston (2004), as well as to keep the maximum substrate depletion at ∼75% to limit errors when fitting models to data (Nath and Atkins, 2006). Negative control reactions were initiated with buffer instead of cofactors. THC or 11-OH-THC was incubated in the presence and absence of selective P450 inhibitors: 10 μM sulfaphenazole (CYP2C9), 2 μM itraconazole (CYP3A), 30 μM omeprazole (CYP2C19), 1 μM quinidine (CYP2D6), and 10 μM furafylline (CYP1A). 11-OH-THC was additionally incubated in the presence and absence of selective UGT inhibitors: 2.5 μM niflumic acid (UGT1A9) and 2 mM fluconazole (UGT2B7). All inhibitors, except for furafylline, were added prior to initiating reaction with cofactor. Since furafylline is a mechanism-based inhibitor, the reaction mixture was preincubated in the presence of the NADPH regenerating system for 15 minutes at 37°C with shaking and the reaction was initiated by the addition of THC. The organic solvent content was <0.75% v/v except for incubations with fluconazole, where the organic content was 3%. Control reactions had matching organic content. Reactions were terminated and processed as described for reactions using recombinant enzymes. Calibration curves for THC, 11-OH-THC, and COOH-THC were prepared in buffer with 0.2% BSA, incubated, and then processed in an identical fashion as the reactions. Three independent experiments, each in duplicate, were performed.
Cross-inhibition of Selective Enzyme Inhibitors.
Each selective P450 inhibitor was incubated in 0.1 M potassium phosphate buffer (pH 7.4) containing either 1) 0.5 mg/ml HLM, 0.2% BSA, and 40 μM S-mephenytoin (CYP2C19 probe) or 2) 0.1 mg/ml HLM, 0.2% BSA, and a cocktail containing the following P450 probes: 5 μM diclofenac (CYP2C9), 10 μM testosterone (CYP3A), and 5 μM dextromethorphan (CYP2D6). Incubations (50 μl, final volume) were initiated with the NADPH regenerating system. Reactions were terminated with addition of 100 μl ice-cold acetonitrile containing tolbutamide as the internal standard. The formation of 4-OH-mephenytoin at 30 minutes, or the formation of 4-OH-diclofenac, 6β-OH-testosterone, and dextrorphan at 20 minutes, was monitored. The P450 probe cocktail was adapted from Chen et al. (2016) and was checked for protein and time linearity. While the time and HLM concentrations for the P450 probe cocktail incubation were chosen to best represent the cannabinoid reaction phenotyping using the HLM setup, the quantification limitation of S-mephenytoin necessitated a higher HLM concentration and longer incubation time. The selective UGT inhibitors were incubated as described in Reaction Phenotyping Using HLMs with a cocktail that (among other UGT probes) included 20 μM propofol (UGT1A9) and 10 μM naloxone hydrochloride (UGT2B7). The UGT cocktail has been previously described by Bhatt et al. (2018). Reactions (50 μl) were terminated by adding 100 μl ice-cold acetonitrile containing androsterone glucuronide as the internal standard. The formation of metabolites propofol-glucuronide and naloxone-3-glucuronide at 30 minutes was monitored. Three independent experiments, each in triplicate, were performed.
LC-MS/MS Analysis.
Samples were analyzed with an ACQUITY ultra-performance LC system (Waters Corporation) coupled to an AB SCIEX Triple Quad 6500 (SCIEX, Framingham, MA) using an ACQUITY UPLC BEH C18 column (1.7 μM 2.1 × 50 mm) with an attached C18 × 2 mm guard column. The LC-MS/MS method for quantification of cannabinoids, P450, and UGT probes was adapted from Hudson et al. (2013), Chen et al. (2016), and Bhatt et al. (2019), respectively. The LC flow gradient and multiple reactions monitoring parameters are listed in Supplemental Tables 1 and 2.
Analysis of Kinetic Parameters.
The depletion rate-constant (kdep) value was obtained by fitting the THC or 11-OH-THC concentration-time profile with a first-order monoexponential decay equation (eq. 1), where Ct is the concentration of substrate remaining at a given time point (t) and C0 is the substrate concentration at t = 0
| (1) |
The 11-OH-THC formation rate-constant (kform) value was obtained by fitting the 11-OH-THC concentration-time profile using eq. 2, where Mt and Mmax are the metabolite concentrations at a given time point (t) and maximum metabolite formed, respectively. The Mmax value was bound to not exceed the input THC concentrations of 500 nM. Of note, there was no significant depletion of 11-OH-THC at 0.02 mg/ml HLM (data not shown), and as such eq. 2 includes only the formation and not the depletion rate-constant values of 11-OH-THC. Furthermore, while COOH-THC was monitored in all reactions, there was no observed formation of COOH-THC (see Results); therefore, no formation kinetics was established for COOH-THC
| (2) |
Depletion clearance (CLdep) of THC or 11-OH-THC and formation clearance (CLform) of 11-OH-THC were calculated as shown in eqs. 3 and 4, where [HLM] and [rCYP] are the protein and recombinant enzyme concentrations (milligrams per milliliter or picomole per milliliters), respectively
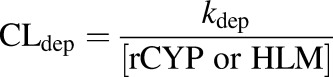 |
(3) |
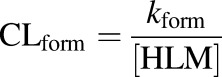 |
(4) |
The fraction metabolized was calculated from kdep or kform in the presence and absence of inhibitor (i) as shown in eq. 5
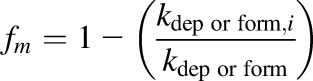 |
(5) |
Adjusting the Fraction Metabolized for Inhibitor Cross-inhibition.
The cannabinoid fm values were adjusted for P450 and UGT inhibitor cross-inhibition (eq. 6) as previously described (Njuguna et al., 2016)
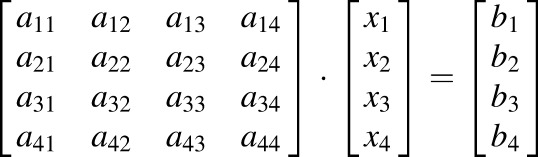 |
(6) |
For example, eq. 6 solves x = A−1*b, where x is the adjusted P450 fm matrix (see Table 4), A is the P450 cross-inhibition matrix (see Table 2), and b is the experimentally observed P450 fm (nonadjusted) matrix (see Table 1). To adjust for the cross-inhibition of UGT inhibitors a 2 × 2 cross-inhibition (a11–a22) (see Table 3) and 2 × 1 experimental fm (b1 to b2) matrix was used. A stochastic simulation approach for error propagation of the adjusted fm values was used. Briefly, a truncated normal distribution (with bounds 0 < μ <1) using the mean and S.D. of each measured cross-inhibition (e.g., a11–a44) or experimental fm (e.g., b1–b4) parameter was simulated and eq. 6 was solved by choosing randomly from the normal distributions. The mean and S.D. from 1000 iterations are reported in Table 4. If the solution for the adjusted fm values was negative, then the fm value was labeled as negligible.
TABLE 4.
Cannabinoid fm values in HLMs adjusted for inhibitor cross-inhibition
Data shown are mean ± S.D. of the fm values from Table 1 adjusted for inhibitor cross-inhibition (P450 enzymes from Table 2 and UGT enzymes from Table 3) using eq. 6.
| Enzyme | THC Depletion | 11-OH-THC |
|
|---|---|---|---|
| Formation | Depletion | ||
| CYP2C9 | 0.82 ± 0.08 | 0.99 ± 0.10 | 0.09 ± 0.05 |
| CYP3A | Negligiblea | Negligiblea | 0.20 ± 0.08 |
| CYP2C19 | Negligiblea | 0.07 ± 0.18 | Negligibleb |
| CYP2D6 | 0.17 ± 0.15 | 0.24 ± 0.22 | Negligibleb |
| UGT2B7 | Negligiblec | N/Ad | 0.45 ± 2.78 |
| UGT1A9 | Negligiblec | N/Ad | 0.22 ± 2.65 |
N/A, not applicable.
The fm values are set to negligible because the adjusted fm result was negative (THC depletion fm: CYP3A = −0.12 ± 0.16, CYP2C19 = −0.53 ± 0.19; 11-OH-THC formation fm: CYP3A = −0.48 ± 0.19).
No significant inhibition of 11-OH-THC depletion observed in the presence of omeprazole and quinidine.
No observed depletion of THC by UGT enzymes.
Not applicable since UGT enzymes did not form 11-OH-THC.
TABLE 2.
Fractional cross-inhibition of P450 enzymes by P450-selective inhibitors in HLMs
Bold values represent the fractional inhibition of enzymes by their corresponding selective inhibitor. Data shown are mean ± S.D. of three independent experiments with each experiment conducted in triplicate.
| Inhibitor | Selective Probe |
|||
|---|---|---|---|---|
| 4-OH-DCL | 6β-OH-TES | 4-OH-MEP | DXO | |
| P450 Enzyme | CYP2C9 | CYP3A | CYP2C19 | CYP2D6 |
| Sulfaphenazole (10 μM) | 0.99 ± 0.00 | 0.28 ± 0.12 | 0.02 ± 0.07 | 0.18 ± 0.17 |
| Itraconazole (2 μM) | 0.44 ± 0.12 | 0.98 ± 0.00 | 0.13 ± 0.07 | 0.27 ± 0.18 |
| Omeprazole (30 μM) | 0.75 ± 0.01 | 0.68 ± 0.07 | 0.82 ± 0.07 | 0.27 ± 0.16 |
| Quinidine (1 μM) | 0.13 ± 0.05 | 0.12 ± 0.19 | −0.33 ± 0.26 | 0.90 ± 0.03 |
DXO, dextrorphan; 4-OH-DCL, 4-OH-diclofenac; 4-OH-MEP, 4-OH-mephenytoin; 6β-OH-TES, 6β-OH-testosterone.
TABLE 1.
Cannabinoid fm valuesa in HLMs determined using selective P450 or UGT inhibitors and quantified by monitoring either THC/11-OH-THC depletion or formation of 11-OH-THC from THC
HLM incubations were conducted with observed concentrations of THC (500 nM) and 11-OH-THC (50 nM) after smoking marijuana. Inhibition of 11-OH-THC depletion by omeprazole and quinidine was negligible (data not shown). Data shown are the mean ± S.D. of three independent experiments with each experiment conducted in duplicate.
| Selective Inhibitor | Enzyme | THC Depletion | 11-OH-THC |
|
|---|---|---|---|---|
| Formation | Depletion | |||
| Sulfaphenazole (10 μM) | CYP2C9 | 0.78 ± 0.03 | 0.90 ± 0.01 | 0.15 ± 0.04 |
| Itraconazole (2 μM) | CYP3A | 0.22 ± 0.05 | −0.11 ± 0.09 | 0.24 ± 0.06 |
| Omeprazole (30 μM) | CYP2C19 | 0.13 ± 0.11 | 0.54 ± 0.07 | Negligible |
| Quinidine (1 μM) | CYP2D6 | 0.15 ± 0.10 | 0.18 ± 0.20 | Negligible |
| Fluconazole (2 mM) | UGT2B7 | Negligible | N/A | 0.36 ± 0.05 |
| Niflumic acid (2.5 μM) | UGT1A9 | Negligible | N/A | 0.41 ± 0.04 |
N/A, not applicable.
Data are not corrected for cross-inhibition of enzyme by the inhibitors.
TABLE 3.
Fractional cross-inhibition of UGT enzymes by UGT-selective inhibitors in HLMs
Bold values represent the fractional inhibition of enzymes by their corresponding selective inhibitor. Data shown are mean ± S.D. of three independent experiments with each experiment conducted in triplicate.
| Inhibitor | Selective Probe |
|
|---|---|---|
| NLX-3-gluc | PPF-gluc | |
| UGT Enzyme | UGT2B7 | UGT1A9 |
| Fluconazole (2 mM) | 0.65 ± 0.19 | 0.27 ± 0.14 |
| Niflumic acid (2.5 μM) | 0.49 ± 0.33 | 0.89 ± 0.02 |
NLX-3-gluc, naloxone-3-glucuronide; PPF-gluc, propofol-glucuronide.
Data Analysis.
Integration of the chromatographic peaks was performed using Analyst version 1.6 (SCIEX, Framingham, MA). Model fitting of concentration-time curves and all plotting were performed using GraphPad Prism 7 (Graphpad Software Inc., La Jolla, CA). Adjustment of the fraction metabolized was performed using MATLAB R2016b (MathWorks, Inc., Natick, MA).
Results
Reaction Phenotyping Using Recombinant Enzymes.
THC (500 nM) was depleted by recombinant CYP1A1, 1A2, 2C9, 2C19, 2D6, 3A4, 3A5, and 3A7 (Fig. 2A). CYP1A1 and CYP2C9 had the highest THC depletion clearance. Recombinant CYP19, 2A6, 2B6, 2C8, and 2E1 did not significantly deplete THC.
Fig. 2.

Reaction phenotyping using recombinant enzymes. (A) Depletion clearance of THC (500 nM) and (B) formation clearance of 11-OH-THC from THC (500 nM) by recombinant P450 enzymes. (C) Depletion clearance of 11-OH-THC (50 nM) by recombinant P450 enzymes. (D) Enzyme concentration–dependent depletion of 11-OH-THC (50 nM) by recombinant UGT enzymes at 30 minutes. There was no depletion of THC by recombinant UGT enzymes at the highest enzyme concentrations tested (data not shown). Data shown are the mean of duplicate determinations.
11-OH-THC was formed from THC (500 nM) by recombinant CYP1A2, 2C9, 2C19, and 2D6 (Fig. 2B). CYP2C19 and CYP2C9 had the highest 11-OH-THC formation clearance. 11-OH-THC was not formed by recombinant CYP1A1, 3A4, 3A5, and 3A7 even though these enzymes turned over THC. In the chromatographs of THC incubations with recombinant CYP1A1, 2D6, and 3A4/5/7, there was a peak that coeluted with 11-OH-THC in the 331 → 313 mass-to-charge transition at a consistent retention time (data not shown), indicating formation of an additional monohydroxylated THC metabolite. No formation of COOH-THC was detected in the aforementioned experiments.
11-OH-THC (50 nM) was depleted by recombinant CYP1A1, 2C9, 2C19, 2D6, 3A4, and 3A5 (Fig. 2C). Recombinant CYP1A1 and CYP3A4 had the highest 11-OH-THC depletion clearance. Recombinant CYP19, 1A2, 2A6, 2B6, 2C8, 2E1, and 3A7 did not significantly deplete 11-OH-THC. Recombinant CYP2C9 and CYP2C19 were the only enzymes that formed COOH-THC (data not shown).
THC was not significantly depleted by any of the recombinant UGT enzymes tested (data not shown). In contrast, 11-OH-THC (50 nM) was depleted in an enzyme-concentration–dependent manner by recombinant UGT1A9 and UGT2B7 but not by UGT1A10 and UGT1A4 (Fig. 2D).
Reaction Phenotyping Using Pooled HLMs.
Reaction phenotyping at a concentration of THC (500 nM) observed after smoking marijuana (Hunault et al., 2008) was performed in pooled adult HLMs using selective P450 inhibitors for the relevant hepatic enzymes identified from recombinant reaction phenotyping assays (Fig. 3). In control reactions (no inhibitor), CLdep of THC was 3.65 ± 0.36 ml/min per milligram and CLform of 11-OH-THC was 1.74 ± 0.42 ml/min per milligram. Based on the formation clearance of 11-OH-THC in control reactions, approximately 48% of THC was metabolized to 11-OH-THC. THC depletion was inhibited 78% ± 3% by sulfaphenazole (10 μM) and <25% by itraconazole (2 μM), omeprazole (30 μM), and quinidine (1 μM) (Fig. 3A; Table 1). The depletion clearance of THC in the presence of furafylline (10 μM) was larger than in control reactions, and as such furafylline was found not be an inhibitor of THC depletion. The reason for this faster depletion is likely due to initiation of reactions with THC, rather than NADPH, and thus not allowing time for protein and nonspecific binding of THC. 11-OH-THC formation was inhibited 90% ± 0.94% and 54% ± 7.0% by sulfaphenazole (10 μM) and omeprazole (30 μM), respectively, and <25% by itraconazole (2 μM) and quinidine (1 μM) (Fig. 3B; Table 1). There was no COOH-THC detected in any of the aforementioned experiments, likely due to assay detection limitations. There was no THC depletion via UGT enzymes in pooled HLMs (tested using 0.25 mg/ml HLM), confirming previous results observed in recombinant UGT enzymes (data not shown).
Fig. 3.

THC reaction phenotyping using pooled adult HLMs. Representative depletion of (A) 500 nM THC, a concentration observed after smoking marijuana, and formation of (B) 11-OH-THC from THC (500 nM) was monitored in the presence and absence of selective P450 inhibitors. Sulfaphenazole (10 μM) inhibited THC depletion and 11-OH-THC formation by the greatest extent, indicating CYP2C9 is the major enzyme responsible for THC turnover to 11-OH-THC. Panels show data from one representative experiment with duplicate determinations and fit with a model (eqs. 1 and 2 for kdep and kform, respectively) that includes the observed time lag. The fm mean ± S.D. values calculated using kdep and kform (eqs. 3 and 4) from three independent experiments are given in Table 1.
Reaction phenotyping at a concentration of 11-OH-THC (50 nM) observed after smoking marijuana (Hunault et al., 2008) was performed in pooled adult HLMs using selective P450 and UGT inhibitors (Fig. 4). In control reactions, 11-OH-THC was depleted by UGT and P450 enzymes with CLdep of 0.458 ± 0.025 and 0.299 ± 0.096 ml/min per milligram, respectively. As such, UGT and P450 enzymes accounted for 60% ± 4.5% and 40% ± 4.5% of 11-OH-THC depletion in HLMs, respectively (Fig. 4A). In P450-mediated reactions, 11-OH-THC depletion was inhibited 37% ± 8.3% and 61% ± 12% by sulfaphenazole (10 μM) and itraconazole (2 μM), respectively (Fig. 4B). Omeprazole (30 μM) and quinidine (1 μM) did not significantly inhibit depletion of 11-OH-THC (data not shown). COOH-THC was not detected in any of the aforementioned experiments. In UGT-mediated reactions, 11-OH-THC depletion was inhibited 60% ± 5.5% and 67% ± 3.6% by fluconazole (2 mM) and niflumic acid (2.5 μM), respectively (Fig. 4C).
Fig. 4.

11-OH-THC reaction phenotyping using pooled adult HLMs. (A) Representative depletion of 50 nM 11-OH-THC, a concentration observed after smoking marijuana, is greater by UGT than P450 enzymes. (B) Itraconazole (2 μM) and sulfaphenazole (10 μM) inhibited the depletion of 11-OH-THC, indicating CYP3A4 and CYP2C9 are both important in the turnover of 11-OH-THC. Of note, no COOH-THC formation was observed in any of the HLM assays. (C) Fluconazole (2 mM) and niflumic acid (2.5 μM) inhibited UGT depletion of 11-OH-THC to a similar degree, indicating that UGT2B7 and UGT1A9 are equally important in the depletion of 11-OH-THC. High and low percentages of organic solvent controls were conducted because of the differential organic solvent content in the presence of the UGT inhibitors. Panels show data from one representative experiment with duplicate determinations and fit with a monoexponential decay curve (eq. 1 for kdep). The fm mean ± S.D. values calculated using kdep (eq. 3) from three independent experiments are given in Table 1.
Cross-inhibition of Selective Enzyme Inhibitors.
To accurately determine the fractional contribution of enzymes involved in cannabinoid disposition, the cross-inhibition of the selective P450 and UGT inhibitors was determined. While the inhibitors used efficiently inhibited their respective probe (82%–99% inhibition; see the bold values in Table 2), there was also significant cross-inhibition, ranging from 2% to 75% (Table 2). For example, sulfaphenazole (10 μM) inhibited CYP3A4-mediated 6β-OH-testosterone formation by 28% ± 12% while itraconazole (2 μM) inhibited CYP2C9-mediated 4-OH-diclofenac formation by 44% ± 12%. Omeprazole (30 μM) showed the highest degree of cross-inhibition (range 27%–75%) while quinidine (1 μM) showed the least degree of cross-inhibition (range −33% to 13%).
There was severe cross-inhibition by the UGT selective inhibitors (Table 3). Fluconazole (2 mM) inhibited UGT1A9-mediated formation of propofol-glucuronide by 27% ± 14% while niflumic acid (2.5 μM) inhibited UGT2B7-mediated formation of naloxone-3-glucuronide by 49% ± 33%.
Using a previously established methodology described by Njuguna et al. (2016), the fm values determined using selective inhibitors (unadjusted) in Table 1 were adjusted for their P450 and UGT inhibitor cross-inhibition given in Tables 2 and 3, respectively, using eq. 6. Table 4 gives the adjusted fm values and S.D. using error propagation via stochastic simulation. CYP2C9 and CYP2D6 were the major and minor (fm = 0.82 ± 0.08 and 0.17 ± 0.15, respectively) DMEs responsible for THC depletion. CYP2C9 was the major DME that formed 11-OH-THC (fm = 0.99 ± 0.10); however, there was a minor contribution from CYP2C19 and CYP2D6 (fm = 0.07 ± 0.18 and 0.24 ± 0.22, respectively). Of the total UGT depletion of 11-OH-THC, the UGT2B7 and UGT1A9 fm values were 0.67 ± 4.14 and 0.33 ± 3.98, respectively. Of the total P450 depletion of 11-OH-THC, the CYP3A and CYP2C9 fm values were 0.69 ± 0.28 and 0.31 ± 0.18, respectively. When combining UGT- and P450-mediated 11-OH-THC depletion, UGT2B7 was the major DME (fm = 0.45 ± 2.78) followed by CYP3A (fm = 0.20 ± 0.08). The sum of the adjusted fm values approximates 1 for THC and 11-OH-THC depletion, but is larger than 1 for the 11-OH-THC formation (Fig. 5).
Fig. 5.

Final hepatic enzyme contributions to THC and 11-OH-THC disposition. The sum of the fm values for THC and 11-OH-THC depletion after cross-inhibition adjustment is close to 1 but the sum for 11-OH-THC formation is greater than 1 (likely due to error propagation caused by the cross-inhibition adjustment). Data shown are mean fm values after cross-inhibition adjustment (eq. 6; Table 4). Of note, while the UGT2B7 and UGT1A9 fm values were estimated with poor confidence (see Table 4) due to severe cross-inhibition of the selective inhibitors, the total UGT contribution (fm = 0.60 ± 0.05) was estimated with confidence.
Discussion
The aims of this work were to 1) investigate the recombinant enzymes that can metabolize THC and 11-OH-THC and 2) use this information as a guide to quantify the fm value of the relevant maternal hepatic enzymes at concentrations of THC and 11-OH-THC observed after smoking marijuana. The mechanistic data generated represent the first set of drug-dependent parameters necessary to build a linked THC/11-OH-THC PBPK model to prospectively predict maternal-fetal cannabinoid exposure during pregnancy.
THC is metabolized to more than 40 metabolites (Agurell et al., 1986). Standards for the majority of these metabolites are not readily available; therefore, to elucidate the enzymes important in the disposition of THC and 11-OH-THC, one needs to conduct substrate depletion studies as well as monitor the formation of 11-OH-THC from THC. Furthermore, to capture enzyme contributions that are clinically relevant, the concentration of substrate chosen in depletion experiments needs to be representative of those observed in vivo. After smoking a marijuana cigarette containing 9.8%, 16.4%, and 23.1% w/w THC, the maximum THC plasma concentration (Cmax) values were 430, 645, and 735 nM while the 11-OH-THC Cmax values were 28, 50, and 48 nM, respectively (Hunault et al., 2008). The average THC potency was 12% in 2014 (ElSohly et al., 2016). Therefore, the chosen 500 nM THC and 50 nM 11-OH-THC reflect plasma concentrations after smoking marijuana of contemporary potency. Furthermore, the chosen THC concentration was lower than the estimated Km for THC depletion of 3.76 ± 1.28 μM (Benito-Gallo et al., 2016) and the estimated Km for 11-OH-THC formation of 0.80 ± 0.12 μM (Bland et al., 2005).
Our reaction phenotyping studies with recombinant enzymes generated novel data indicating that CYP1A1, 1A2, 3A5, and 3A7 depleted THC; however, of these enzymes only CYP1A2 formed 11-OH-THC. Our data corroborate previous reports that found CYP2C9, 2C19, 2D6, and 3A4 are important in the metabolism of THC (Watanabe et al., 2007). Interestingly, the THC depletion clearance of CYP1A1 was higher than CYP2C9, the major DME of THC (Fig. 2A). While the contribution of CYP1A1 is negligible in the liver, CYP1A1, the major DME in the lung (Zhang et al., 2006), may be important for cannabinoid lung disposition. There is no current direct evidence of lung cannabinoid metabolism in human; however, biotransformation of THC to 11-OH-THC is observed in rat lung homogenate and dog and guinea pig perfused lung (Nakazawa and Costa, 1971; Widman et al., 1975; Halldin et al., 1984). Since metabolism of THC by the human lung may be an important drug parameter in the cannabinoid PBPK, further studies need to be conducted to establish the contribution of lung metabolism to THC disposition, especially if CYP1A1 is induced by smoking marijuana.
Maternal THC exposure drives fetal THC exposure. Therefore, we used the recombinant enzyme data to guide reaction phenotyping studies of hepatic enzymes that are responsible for the maternal hepatic clearance of THC. Our data corroborate previous studies that demonstrated CYP2C9 is the major DME responsible for THC metabolism in HLMs (Bornheim et al., 1992; Bland et al., 2005; Watanabe et al., 2007). We adjusted the apparent fm values determined via selective inhibitors (Table 1) with the inhibitor cross-inhibition (Tables 2 and 3). This was to account for potential overestimation of the fm values when the inhibitor selectivity is not optimum. After adjusting for inhibitor cross-inhibition, CYP2C9 was the main DME responsible for THC depletion (fm = 0.82 ± 0.08) with the remaining contribution from CYP2D6 (Table 4). While there was turnover of THC by CYP3A4 and CYP2C19 in recombinant enzymes, as well as significant inhibition by itraconazole and omeprazole (22% ± 5% and 13% ± 11%, respectively), this inhibition was primarily due to cross-inhibition of CYP2C9 (44% ± 12% and 75% ± 1%, respectively) (Table 2). It should be noted that due to error propagation, there is less confidence when estimating small fm values, such as those for CYP2D6, 3A4, and 2C19.
The formation fm values of enzymes responsible for the formation of 11-OH-THC, the main and active metabolite of THC, were reflective of THC enzyme contributions. After adjusting for inhibitor cross-inhibition CYP2C9 was the main DME responsible for 11-OH-THC formation (fm = 0.99 ± 0.10) with minor contributions from CYP2D6 and CYP2C19. The sum of the 11-OH-THC formation fm values was greater than 1 (Fig. 5). This may be due to the error propagation during fm adjustment, particularly for the smaller fm values for CYP2D6 and CYP2C19.
Reaction phenotyping studies were extended to include the depletion of 11-OH-THC. All of the P450 enzymes that metabolized THC also turned over 11-OH-THC except for CYP1A2 and CYP3A7. Like THC, recombinant CYP1A1 had the largest 11-OH-THC depletion clearance (Fig. 1C). A previous extensive screen using recombinant UGT enzymes found that only UGT1A9 and UGT1A10 are relevant to the formation of 11-OH-THC-glucuronide (Mazur et al., 2009). As such, we verified the 11-OH-THC depletion by UGT1A9 and UGT1A10, and we additionally included UGT1A4, an enzyme that is inducible during pregnancy (Anderson, 2005; Ke et al., 2014b), and UGT2B7, the major UGT expressed in the liver (Achour et al., 2014). We tested 11-OH-THC depletion at protein concentrations five times higher for UGT1A10 compared with UGT1A9 because in a previous study by Mazur et al. (2009) the clearance of 11-OH-THC (Vmax/Km) by UGT1A9 was approximately 5-fold higher than UGT1A10. Even so, we did not observe significant depletion of 11-OH-THC by UGT1A10. Unlike the previous report, we observed depletion of 11-OH-THC by UGT2B7. In HLMs, UGT enzymes depleted 11-OH-THC at a greater extent than P450 enzymes (fm = 0.60 ± 0.05 vs. 0.40 ± 0.05). In contrast to THC, CYP3A has a larger contribution to 11-OH-THC depletion than CYP2C9 (fm = 0.20 ± 0.08 vs. 0.09 ± 0.05) (Table 4). Furthermore, the reaction phenotyping studies in HLMs identified UGT2B7 as the major DME (fm = 0.45 ± 2.78). It should be noted that there was significant cross-inhibition of the UGT selective inhibitors (Table 3). Because of this, there was a large uncertainty associated with the adjusted UGT2B7 and UGT1A9 fm values (Table 4). Selective inhibitors of UGTs are needed to better define these fm values. As such, the total UGT contribution (fm = 0.60 ± 0.05) rather than the individual UGT fm values should be used for PBPK modeling and simulation.
Clinical data further confirm that CYP2C9 and CYP3A4 are major contributors in the disposition of THC and 11-OH-THC. After oral administration of 15 mg THC in CYP2C9*3/*3 homozygotes, which confers decreased CYP2C9 function, there is a 3-fold increase in THC area under the curve (AUC)(0-inf) and no change in 11-OH-THC AUC(0-inf) when compared with wild type (Sachse-Seeboth et al., 2009). CYP2C9 inhibition leads to an increase in the combined THC/11-OH-THC exposure because CYP2C9 forms and depletes 11-OH-THC. When an oromucosal spray containing 10.8 mg THC/10 mg cannabidiol is coadministered with ketoconazole (CYP3A inhibitor), the THC and 11-OH-THC AUC(0-inf) is increased 1.84- and 3.62-fold, respectively (Stott et al., 2013). CYP3A THC fm in the liver is insignificant but may be significant in the gut where expression of CYP3A is much greater than CYP2C9. As such, the THC drug-drug interaction with ketoconazole may be due to presystemic interactions. Unlike inhibition of CYP2C9, inhibition of CYP3A4 significantly increases the 11-OH-THC AUC because CYP3A4 depletes but does not form 11-OH-THC. In both examples, the inhibition of CYP2C9 and CYP3A4 led to changes in the pharmacodynamic effect (increased sedation and euphoric mood). Because 11-OH-THC is equally or even more potent than THC (Perez-Reyes et al., 1972), the combined exposure of THC/11-OH-THC needs to be considered when establishing cannabinoid dose-risk relationship during pregnancy.
The reported cannabinoid metabolic profiling can be extrapolated to pregnant women. During pregnancy, CYP3A4- and CYP2C9-mediated clearance is increased 2- and 1.6-fold, respectively, UGT2B7 activity does not change, and UGT1A9 changes are unknown (Anderson, 2005; Ke et al., 2014b). The changes to DME expression and activity during pregnancy are likely to increase THC clearance and 11-OH-THC formation/clearance. The fetal-to-maternal concentration ratio of THC in chronic marijuana users ranged from 0.16 to 0.38 (Blackard and Tennes, 1984). Because the fetal-to-maternal ratio is less than 1, there may be extraction of cannabinoids by the placenta or the fetal liver or both. As such, placental CYP1A1, UGT2B7, and fetal CYP3A7 may limit fetal exposure of THC and 11-OH-THC. Of particular interest is the contribution of placental CYP1A1 to cannabinoid fetal exposure since marijuana tar induces CYP1A1 mRNA (Roth et al., 2001). Studies need to be conducted with placental microsomes/S9 fractions to elucidate the contribution of placental metabolism of THC/11-OH-THC in fetal exposure to these drugs.
The relevant hepatic enzymes and their fractions metabolized have been established for THC and 11-OH-THC. CYP2C9 is the most important DME for THC depletion and formation of 11-OH-THC, while UGT enzymes represent the major pathway of 11-OH-THC depletion. The mechanistic information presented here is necessary to build a THC/11-OH-THC PBPK model that can be used to predict cannabinoid exposure during pregnancy. Studies need to be conducted to determine the contribution of other enzymes in lungs or the placenta in the clearance of THC/11-OH-THC. In addition, to build a PBPK model of these cannabinoids after oral administration of THC, studies to determine the contribution of intestinal enzymes in the first pass metabolism of THC/11-OH-THC will need to be conducted.
Acknowledgments
We thank Bhagwat Prasad and Haeyoung Zhang for providing the UGT cocktail, and Neha Maharao for the P450 cocktail.
Abbreviations
- AUC
area under the curve
- BSA
bovine serum albumin
- COOH-THC
11-nor-9-carboxy-∆9-tetrahydrocannabinol
- DME
drug metabolizing enzyme
- fm
fraction metabolized
- HLM
human liver microsome
- LB
low-binding
- LC
liquid chromatography
- MS
mass spectrometry
- 11-OH-THC
11-hydroxy-∆9-tetrahydrocannabinol
- P450
cytochrome P450
- PBPK
physiologically based pharmacokinetic
- rCYP
recombinant cytochrome P450
- THC
(−)-∆9-tetrahydrocannabinol
- UGT
UDP-glucuronosyltransferase
Authorship Contributions
Participated in research design: Patilea-Vrana, Anoshchenko, Unadkat.
Conducted experiments: Patilea-Vrana, Anoshchenko.
Contributed new reagents or analytic tools: Patilea-Vrana.
Performed data analysis: Patilea-Vrana, Anoshchenko.
Wrote or contributed to the writing of the manuscript: Patilea-Vrana, Anoshchenko, Unadkat.
Footnotes
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
This work was supported in part by National Institutes of Health National Institute on Drug Abuse [Grant P01DA032507 to G.P.V., O.A., J.D.U.]; National Institutes of Health National Center for Advancing Translational Sciences [Grant TL1 TR000422 to G.P.V.]; Elmer M. Plein Endowed Research Fund (G.P.V.), and Rene Levy Fellowship (G.P.V.).
References
- Achour B, Rostami-Hodjegan A, Barber J. (2014) Protein expression of various hepatic uridine 5′-diphosphate glucuronosyltransferase (UGT) enzymes and their inter-correlations: a meta-analysis. Biopharm Drug Dispos 35:353–361. [DOI] [PubMed] [Google Scholar]
- Agurell S, Halldin M, Lindgren JE, Ohlsson A, Widman M, Gillespie H, Hollister L. (1986) Pharmacokinetics and metabolism of delta 1-tetrahydrocannabinol and other cannabinoids with emphasis on man. Pharmacol Rev 38:21–43. [PubMed] [Google Scholar]
- Anderson GD. (2005) Pregnancy-induced changes in pharmacokinetics: a mechanistic-based approach. Clin Pharmacokinet 44:989–1008. [DOI] [PubMed] [Google Scholar]
- Benito-Gallo P, Marlow M, Zann V, Scholes P, Gershkovich P. (2016) Linking in vitro lipolysis and microsomal metabolism for the quantitative prediction of oral bioavailability of BCS II drugs administered in lipidic formulations. Mol Pharm 13:3526–3540. [DOI] [PubMed] [Google Scholar]
- Bhatt DK, Mehrotra A, Gaedigk A, Chapa R, Basit A, Zhang H, Choudhari P, Boberg M, Pearce RE, Gaedigk R, et al. (2018) Age- and genotype-dependent variability in the protein abundance and activity of six major uridine diphosphate-glucuronosyltransferases in human liver. Clin Pharmacol Ther DOI: 10.1002/cpt.1109 [published ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt DK, Mehrotra A, Gaedigk A, Chapa R, Basit A, Zhang H, Choudhari P, Boberg M, Pearce RE, Gaedigk R, Broeckel U, Leeder JS, Prasad B. (2019) Age- and Genotype-Dependent Variability in the Protein Abundance and Activity of Six Major Uridine Diphosphate-Glucuronosyltransferases in Human Liver. Clin Pharmacol Ther 105:131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackard C, Tennes K. (1984) Human placental transfer of cannabinoids. N Engl J Med 311:797. [DOI] [PubMed] [Google Scholar]
- Bland TM, Haining RL, Tracy TS, Callery PS. (2005) CYP2C-catalyzed delta9-tetrahydrocannabinol metabolism: kinetics, pharmacogenetics and interaction with phenytoin. Biochem Pharmacol 70:1096–1103. [DOI] [PubMed] [Google Scholar]
- Bornheim LM, Lasker JM, Raucy JL. (1992) Human hepatic microsomal metabolism of delta 1-tetrahydrocannabinol. Drug Metab Dispos 20:241–246. [PubMed] [Google Scholar]
- Brown QL, Sarvet AL, Shmulewitz D, Martins SS, Wall MM, Hasin DS. (2017) Trends in marijuana use among pregnant and nonpregnant reproductive-aged women, 2002–2014. JAMA 317:207–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZH, Zhang SX, Long N, Lin LS, Chen T, Zhang FP, Lv XQ, Ye PZ, Li N, Zhang KZ. (2016) An improved substrate cocktail for assessing direct inhibition and time-dependent inhibition of multiple cytochrome P450s. Acta Pharmacol Sin 37:708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook CE, Hawos ML, Amerson EW, Pitt CG, Williams D. (1976) Radiommunoassay of delta9-tetrahydrocannabinol. NIDA Res Monogr 7:15–27. [PubMed] [Google Scholar]
- ElSohly MA, Mehmedic Z, Foster S, Gon C, Chandra S, Church JC. (2016) Changes in cannabis potency over the last 2 decades (1995–2014): analysis of current data in the United States. Biol Psychiatry 79:613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett ER, Hunt CA. (1974) Physiochemical properties, solubility, and protein binding of delta9-tetrahydrocannabinol. J Pharm Sci 63:1056–1064. [DOI] [PubMed] [Google Scholar]
- Halldin MM, Isaac H, Widman M, Nilsson E, Ryrfeldt A. (1984) A comparison between the metabolism of Δ1-tetrahydrocannabinol by perfused lung and liver of rat and guinea-pig. Xenobiotica 14:277–282. [DOI] [PubMed] [Google Scholar]
- Hudson J, Hutchings J, Wagner R, Harper C, Friel P. (2013) Cannabinoid Quantification using an Agilent 6430 LC-MS/MS, Agilent Technologies, http://www.chem.agilent.com/Library/applications/5991-2521EN.pdf. [Google Scholar]
- Hunault CC, Mensinga TT, de Vries I, Kelholt-Dijkman HH, Hoek J, Kruidenier M, Leenders ME, Meulenbelt J. (2008) Delta-9-tetrahydrocannabinol (THC) serum concentrations and pharmacological effects in males after smoking a combination of tobacco and cannabis containing up to 69 mg THC. Psychopharmacology (Berl) 201:171–181. [DOI] [PubMed] [Google Scholar]
- Jones HM, Houston JB. (2004) Substrate depletion approach for determining in vitro metabolic clearance: time dependencies in hepatocyte and microsomal incubations. Drug Metab Dispos 32:973–982. [DOI] [PubMed] [Google Scholar]
- Ke AB, Nallani SC, Zhao P, Rostami-Hodjegan A, Unadkat JD. (2012) A PBPK model to predict disposition of CYP3A-metabolized drugs in pregnant women: verification and discerning the site of CYP3A induction. CPT Pharmacometrics Syst Pharmacol 1:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke AB, Nallani SC, Zhao P, Rostami-Hodjegan A, Unadkat JD. (2014a) Expansion of a PBPK model to predict disposition in pregnant women of drugs cleared via multiple CYP enzymes, including CYP2B6, CYP2C9 and CYP2C19. Br J Clin Pharmacol 77:554–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke AB, Rostami-Hodjegan A, Zhao P, Unadkat JD. (2014b) Pharmacometrics in pregnancy: an unmet need. Annu Rev Pharmacol Toxicol 54:53–69. [DOI] [PubMed] [Google Scholar]
- Mazur A, Lichti CF, Prather PL, Zielinska AK, Bratton SM, Gallus-Zawada A, Finel M, Miller GP, Radomińska-Pandya A, Moran JH. (2009) Characterization of human hepatic and extrahepatic UDP-glucuronosyltransferase enzymes involved in the metabolism of classic cannabinoids. Drug Metab Dispos 37:1496–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa K, Costa E. (1971) Metabolism of delta 9 -tetrahydrocannabinol by lung and liver homogenates of rats treated with methylcholanthrene. Nature 234:48–49. [DOI] [PubMed] [Google Scholar]
- Nath A, Atkins WM. (2006) A theoretical validation of the substrate depletion approach to determining kinetic parameters. Drug Metab Dispos 34:1433–1435. [DOI] [PubMed] [Google Scholar]
- Njuguna NM, Umehara KI, Huth F, Schiller H, Chibale K, Camenisch G. (2016) Improvement of the chemical inhibition phenotyping assay by cross-reactivity correction. Drug Metab Pers Ther 31:221–228. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes M, Timmons MC, Lipton MA, Davis KH, Wall ME. (1972) Intravenous injection in man of 9-tetrahydrocannabinol and 11-OH-9-tetrahydrocannabinol. Science 177:633–635. [DOI] [PubMed] [Google Scholar]
- Prasad B, Singh S. (2009) In vitro and in vivo investigation of metabolic fate of rifampicin using an optimized sample preparation approach and modern tools of liquid chromatography-mass spectrometry. J Pharm Biomed Anal 50:475–490. [DOI] [PubMed] [Google Scholar]
- Roth MD, Marques-Magallanes JA, Yuan M, Sun W, Tashkin DP, Hankinson O. (2001) Induction and regulation of the carcinogen-metabolizing enzyme CYP1A1 by marijuana smoke and Δ9-tetrahydrocannabinol. Am J Respir Cell Mol Biol 24:339–344. [DOI] [PubMed] [Google Scholar]
- Sachse-Seeboth C, Pfeil J, Sehrt D, Meineke I, Tzvetkov M, Bruns E, Poser W, Vormfelde SV, Brockmöller J. (2009) Interindividual variation in the pharmacokinetics of delta9-tetrahydrocannabinol as related to genetic polymorphisms in CYP2C9. Clin Pharmacol Ther 85:273–276. [DOI] [PubMed] [Google Scholar]
- Stott C, White L, Wright S, Wilbraham D, Guy G. (2013) A phase I, open-label, randomized, crossover study in three parallel groups to evaluate the effect of rifampicin, ketoconazole, and omeprazole on the pharmacokinetics of THC/CBD oromucosal spray in healthy volunteers. Springerplus 2:236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout SM, Cimino NM. (2014) Exogenous cannabinoids as substrates, inhibitors, and inducers of human drug metabolizing enzymes: a systematic review. Drug Metab Rev 46:86–95. [DOI] [PubMed] [Google Scholar]
- Substance Abuse and Mental Health Services Administration (2017) Key Substance Use and Mental Health Indicators in the United States: Results from the 2016 National Survey on Drug Use and Health, Center for Behavioral Health Statistics and Quality, Substance Abuse and Mental Health Services Administration, Rockville, MD. [Google Scholar]
- Watanabe K, Yamaori S, Funahashi T, Kimura T, Yamamoto I. (2007) Cytochrome P450 enzymes involved in the metabolism of tetrahydrocannabinols and cannabinol by human hepatic microsomes. Life Sci 80:1415–1419. [DOI] [PubMed] [Google Scholar]
- Widman M, Nordqvist M, Dollery CT, Briant RH. (1975) Metabolism of Δ1-tetrahydrocannabinol by the isolated perfused dog lung. Comparison with in vitro liver metabolism. J Pharm Pharmacol 27:842–848. [DOI] [PubMed] [Google Scholar]
- Zhang JY, Wang Y, Prakash C. (2006) Xenobiotic-metabolizing enzymes in human lung. Curr Drug Metab 7:939–948. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Imperial MZ, Patilea-Vrana GI, Wedagedera J, Gaohua L, Unadkat JD. (2017) Development of a novel maternal-fetal physiologically based pharmacokinetic model I: insights into factors that determine fetal drug exposure through simulations and sensitivity analyses. Drug Metab Dispos 45:920–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Unadkat JD. (2017) Development of a novel maternal-fetal physiologically based pharmacokinetic model II: verification of the model for passive placental permeability drugs. Drug Metab Dispos 45:939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]