

Abstract
Multiple sclerosis (MS) is an autoimmune disorder manifested via chronic inflammation, demyelination, and neurodegeneration inside the central nervous system. The progressive phase of MS is characterized by neurodegeneration, but unlike classical neurodegenerative diseases, amyloid-like aggregation of self-proteins has not been documented. There is evidence that citrullination protects an immunodominant peptide of human myelin oligodendrocyte glycoprotein (MOG34–56) against destructive processing in Epstein-Barr virus-infected B-lymphocytes (EBV-BLCs) in marmosets and causes exacerbation of ongoing MS-like encephalopathies in mice. Here we collected evidence that citrullination of MOG can also lead to amyloid-like behavior shifting the disease pathogenesis toward neurodegeneration. We observed that an immunodominant MOG peptide, MOG35–55, displays amyloid-like behavior upon site-specific citrullination at positions 41, 46, and/or 52. These amyloid aggregates are shown to be toxic to the EBV-BLCs and to dendritic cells at concentrations favored for antigen presentation, suggesting a role of amyloid-like aggregation in the pathogenesis of progressive MS.
Multiple sclerosis (MS) is a group of autoimmune-driven neuroinflammatory disorders that are pathologically characterized by myelin sheath loss (demyelination) and axonal damage.1 The disease starts in ±85% of the patients with alternating episodes of neurological defects (relapse) and recovery (remission), so-called relapsing–remitting MS (RRMS), which in ±60% of MS patients converts to secondary progressive disease with chronic progression. Approximately 10–15% of MS patients show chronic progression from disease onset, a course called primary progressive multiple sclerosis (PPMS).2 While there are several therapies available for the treatment of RRMS, few therapeutic options yet exist for PPMS. This is due to their fundamentally different pathophysiology.3 Recently, anti-B-cell antibody therapy was approved for the treatment of PPMS,3 which ties in with the hypothesis that—aside from genetic predispositions and well-characterized environmental factors such as smoking and sun exposure (vitamin D)—viral infections play a role in MS pathogenesis, especially in PPMS.4 In particular, Epstein-Barr virus (EBV) infection, which has a selective tropism for B-lymphocytes (BLCs), has been considered to be a prerequisite for MS pathogenesis.5,6 One of the presumed effects of viral infection upon MS emergence is the qualitative alteration of the protein composition of myelin, especially myelin oligodendrocyte glycoprotein (MOG).7,8 Studies in mouse and non-human primate models of experimental autoimmune encephalomyelitis (EAE) revealed that MOG is an essential myelin component for the experimental induction of T- and B-cell autoimmunity in chronic progressive disease.9,10 Moreover, the marmoset EAE model has been reported to share pathological hallmarks of progressive MS when immunized with an immunorelevant CD8+ T-cell epitope of human MOG, MOG34–56, and incomplete Freund’s adjuvant (IFA).11,12
MOG is expressed as a homodimer on the surface of oligodendrocytes, the myelin-forming glial cells of the central nervous system (CNS), and on the outermost lamellae of the myelin sheaths that wrap around axons, forming a protective layer that is essential for fast pulse conduction and trophic support of energy-demanding axons.13 The exact biological function of MOG is not known, but there is evidence that the N-linked glycan at position 31 interacts with the C-type lectin receptor DC-SIGN, which is expressed on microglial cells and on antigen-presenting cells within the brain-draining cervical lymph nodes (CLNs).14 As ligands of DC-SIGN are known to suppress maturation of dendritic cells to a full immunogenic state, we previously posited that MOG may have a role in the avoidance of autoreactive T-cell activation and neuroinflammation.15,16
In addition to the findings described above, there is solid evidence for a link between the chronic inflammation via the production of reactive oxygen/nitrogen species (ROS/RNS)17 and the dysregulation of the ionic balance, especially due to an increase in intracellular Ca2+-ion concentrations.18 The latter event in turn upregulates the activation of peptidyl arginine deiminases (PADs), which then convert arginines (Arg) into citrullines (Cit). Citrullinated self-proteins have been shown to become antigenic, as described by Toes and co-workers for vinculin (VCL) in another autoimmune disease, rheumatoid arthritis (RA).19 Moreover, myelin basic protein (MBP), a major component of the myelin sheath, has been found to be progressively hypercitrullinated20,21 (∼45% citrullinated MBP in MS and ∼100% in the acute neuroinflammatory disorder Marburg’s disease). More recently, Woodroofe and colleagues showed that glial fibrillary acidic protein (GFAP) can be detected as an additional, aberrantly citrullinated protein in MS.22 Finally, the MS relevant murine T-cell epitope of MOG, MOG35–55, has been reported to exacerbate EAE in mice if citrullinated23 at position 41, a contact residue for T-cell receptors (TCRs).24 Moreover, in this study, an exacerbation of disease progression was observed when T-cells specific for citrullinated MOG35–55 were transferred to mice with ongoing, native peptide-induced EAE. Taken together, these data suggested a critical role for citrullination of MOG and other components of the myelin sheath in MS progression.
One essential question that remains unanswered is whether neurodegeneration or peripheral inflammation is the primary cause of PPMS, as neuronal atrophy occurs away from immune cell-containing zones in this disease. Neuronal damage may therefore not be due to direct immune cell–sheath contact. As many other neurodegenerative diseases are characterized by post-translational modification (PTM) of proteins leading to aggregation and accumulation of aggregates within brain tissue [e.g., in transmissible spongiform encephalopathies (TSEs; prion protein, PrP),25 Alzheimer’s disease (amyloid-β and tau protein),26,27 and Parkinson’s disease (α-synuclein)],28 we hypothesized that PPMS may also have an aggregation-induced component to its disease pathology. However, unlike for these classical neurodegenerative diseases, a misfolded autoantigen has not been documented for progressive MS.29,30 Comparing the structures of misfolded PrP models31 with the β-sheet rich crystal structure of native MOG protein (depicted in Figure 1),32 we hypothesized that MOG or a PTM variant of MOG could be the species forming amyloid fibrils in PPMS due to potential structural changes (i.e., conversion from β-sheet to cross-β-sheet) upon citrullination, which are ordered structures of misfolding proteins usually described by seeding nucleation or nucleation-dependent polymerization models.33
Figure 1.

Strategy and proposed mechanism for amyloid aggregation-driven cytotoxicity of MOG. (1) Crystal structure of rMOG (Protein Data Bank entry 1PKO) with critical arginine residues 41 and 46 (highlighted as Arg 41 and 46, respectively). The image was generated with PyMOL. For our strategy, rMOG is degraded into immunorelevant epitope 35–55 with either none or double Arg → Cit mutations at position 41, 46, or 52. (2) MOG35–55 is subjected to misfolding at acidic pH as can be found in microvesicles (MVs) intracellularly and forms amyloid-like aggregates in vitro. (3) In cellulo mechanism of cytotoxicity. Monomeric MOG35–55 is taken up by the cells via MVs and undergoes amyloid-like aggregation in the acidic environment of MVs, and upon release, amyloid aggregates cause cytotoxicity in certain cell types such as phagocytes.
Here we test the hypothesis that PPMS carries an amyloid component by assessing the aggregation characteristics of MOG as well as the key MOG-derived immunodominant peptides containing the relevant T-cell epitopes in vitro. These characteristics were investigated in vitro using a thioflavin T (ThT) assay34 and in vivo using a novel bioorthogonal chemistry-based approach. We found that the recombinant MOG protein was non-immunogenic but that the peptide fragments indeed displayed amyloid behavior (Figure 1). Moreover, they displayed this behavior only upon citrullination at specific positions and showed enhanced aggregation at the pH values found in the B-cell lysosome. The resulting fibrils were shown to be toxic to phagocytes, such as bone marrow-derived dendritic cells (BMDCs) and the BLCs in which they were generated. Furthermore, amyloid-like aggregation altered the antigenicity of citrullinated MOG peptides in a concentration-dependent manner, resulting in high cytotoxicity at physiological concentrations. This study provides the first evidence that amyloid fibril formation may play a role in PPMS pathogenesis and disease progression.
Materials and Methods
Fmoc SPPS, Peptide Analysis, and Purification
Peptides were prepared either manually or on automated synthesizer (PTI Tribute UV-IR synthesizer, Gyros Protein Technologies). For manual synthesis, Fmoc deprotection was achieved with 20% piperidine in dimethylformamide (DMF) using two cycles of 3 and 7 min. All amino acids (2.5 equiv) were coupled using HCTU (2.38 equiv) and DIEPA (5 equiv) for 30 mins, unless stated otherwise. For automated peptide synthesis, 5.0 equiv of each amino acid (aa) on a 100–200 μmol scale and 6.0 equiv of each aa on a 50 μmol scale with respect to the resin loading were used. An equimolar quantity of HCTU was used as an activator. Coupling cycles of 1 h were utilized, and unreacted amines were capped after each cycle using a solution of 500 μL of acetic anhydride, 250 μL of DIPEA, and 4.25 mL of DMF for 5 min at room temperature twice. Peptides were globally deprotected and cleaved from dried resin with a mixture of TFA, triisopropylsilane (TIS), and water [92.5:5:2.5 (v/v)] for at least 3 h at room temperature. Precipitation of crude peptides was achieved by addition of cold diethyl ether. Isolated peptides were lyophilized overnight. Peptides were characterized using electrospray ionization mass spectrometry (ESI-MS) on a Thermo Finnigan LCQ Advantage Max LC-MS instrument with a Surveyor PDA plus UV detector or an Agilent 6120 Quadrupole LC-MS instrument with an Agilent 1260 Infinity high-performance liquid chromatography (HPLC) system. On both systems, a Gemini-NX C18 column (Phenomenex, 3 μm, 110 Å, 50 mm × 4.6 mm) was used. Peptides were analyzed using a gradient from 10 to 90% or from 10 to 50% of B over 10–20 min (eluent A, H2O; eluent B, acetonitrile; eluent C, 1% TFA in H2O; eluent D, methanol). Peptide masses were calculated from the experimental mass to charge (m/z) ratios from all of the observed protonation states of the peptides using the Xcalibur Qual Browser or LC-MS_6120B software. Lyophilized peptides were dissolved in a mixture of tert-butanol, acetonitrile (ACN), and water [1:1:1 (v/v)] unless stated otherwise and purified using a preparative HPLC system (Gilson Inc.) with a Gemini-NX C18 semipreparative column (Phenomenex, 5 μm, 110 Å, 250 mm × 10.0 mm) with a linear gradient from 20 to 30% B in 30 min (eluent A, 0.2% TFA in H2O; eluent B, ACN). Fractions containing the peptides were pooled and lyophilized overnight.
Biophysical Characterization
Circular Dichroism (CD) Spectroscopy
Circular dichroism spectroscopy was performed at room temperature using a Jasco J-815 CD spectrometer with a 1 mm path-length cell and a bandwidth of 2.0 nm. The proteins were prepared either in 20 mM Tris buffer (pH 7.5) or in 20 mM NaOAc buffer (pH 5.0). Further analysis was performed via addition of 2–4 mM SDS (β-sheet enhancer) or 50% TFE (α-helix enhancer) with a final peptide concentration of 0.1–0.2 mg/mL. Spectra were recorded from 260 to 190 nm at an interval of 1 nm. Each spectrum was the average of five scans and blank subtraction.
ThT Fluorescence Aggregation Assay
Aggregation assays were modified from those described by Araman et al.67 The experiments were performed in 96-well plates at 37 °C using the Infinite M1000 Pro Tecan plate reader with an excitation wavelength of 444 nm and an emission wavelength of 485 nm with a bandwidth of 10 nm. A mixture of 199 μL of peptide (200 μM to 2 μM) and 1 μL of ThT was measured in each well at a 20000 μm z-position over time with a kinetic interval of 10 min.
Transmission Electron Microscopy (TEM)
TEM imaging was conducted on a JEOL 1010 instrument operated with an accelerating voltage of 70 kV. Samples were diluted to final concentrations of 1–5 μM. A single drop (approximately 1 μL) was applied on the grid (PELCO Center-Marked Grids, 75 mesh, 3.0 mm outside diameter, copper) and drained with the corner of a tissue. The grid was left to dry for 1–3 h prior to measurement.
Dynamic Light Scattering (DLS)
The hydrodynamic diameter (Rhyd) of amyloid fibrils of MOG peptides 5 and 7 was assessed by dynamic light scattering (DLS) spectroscopy on a Zetasizer Nano S (Malvern Instruments, Malvern, U.K.) using PMMA small-volume cuvettes (VWR international, Leuven, Belgium). Hereby, the working concentration was ∼20 μM in 20 mM NaOAc (pH 5).
Cell Biology
B3Z T-Cell Hybridoma Culture
Mouse B3Z T-cell hybridomas were cultured as described previously.68
B-Cell Culture
Human EBV-infected lymphoblastoid cell line BSM (ECACC 88052032) was purchased from Sigma-Aldrich and cultured in polystyrene tissue culture flasks in RPMI 1640 (GIBCO), 2 mM glutamine (GIBCO, catalog no. 35050-038), 10% FCS (TIC Europ, catalog no. 61024-C05), and 100 units/mL penicillin/streptomycin (GIBCO, catalog no. 15140-122) in a 5% CO2 atmosphere at 37 °C and diluted every 3–4 days at a concentration of ∼0.5 × 106 cells/mL.
Bone Marrow Dendritic Cell Culture
Bone marrow (BM) was isolated from 8–12-week-old C57BL/6 mice (strain C57Bl/6NHsd, H-2b haplotype; Envigo Inc., Huntingdon, U.K.) as described previously69 with some modifications; 8–12-week-old mice were kept under specific pathogen-free (SPF) conditions in our own facilities. Femurs and tibiae of female or male mice were removed and cleaned from the surrounding muscle tissue. Then, intact bones were left in 70% ethanol for 2–5 min for disinfection and washed with sterile phosphate-buffered saline (PBS). Next, both ends were cut with scissors, and bone marrow was flushed from femurs and tibiae with prewarmed IMDM (Sigma) using a 20 mL syringe with a 0.45 mm × 23 mm hypodermic needle (G26, Terumo Europe, NN-2623R) through a 70 μm cell strainer (Falcon). Isolated BM cells were centrifuged for 5 min at 350g and resuspended in 10 mL of IMDM supplemented with 8% heat-inactivated fetal calf serum (FCS, Sigma), 2 mM Glutamax (GIBCO), 20 μM 2-mercaptoethanol (Gibco), 50 IU/mL penicillin, and 50 μg/mL streptomycin in the presence of 20 ng/mL rmGM-CSF (Peprotech). The cell suspension was diluted to ∼0.5–1 × 106 cells/mL in complete medium, and cells were incubated in non-adhesive Petri dishes (Sarstedt) at 37 °C and 5% CO2 for 6–7 days.
Cell Viability Assay with 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT)
The cell viability of BMDCs and B3Zs was tested via the MTT assay. For this purpose, the following protocols were used. BMDCs and B3Zs were seeded in a 96-well plate at a density of 2.5 × 104 cells/well and treated with varying amounts of peptides 1, 5, and 7 in IMDM (20, 10, 5, and 3 μM) incubated overnight at 37 °C and 5% CO2. Medium with (control) and medium without 2.5 × 104 cells (blank) were used as controls. After incubation for 24 h, cells were centrifuged (350g for 5 min at 4 °C), the supernatant was discarded, and 100 μL of 0.5 mg/mL MTT [diluted 1:10 (v/v) from a 5 mg/mL MTT stock solution in PBS] was added to each well. Upon incubation for 3–4 h at 37 °C and formation of intracellular formazan crystals, the supernatant was removed and the solubilizing solution (DMSO, 100 μL) was added. The plate was incubated for a further 30 min at 37 °C, and the absorbance was measured at 540 nm (A540) as well as 570 nm (Tecan M1000 plate reader). The following equation was used to assess cell viability:
T-Cell Culture Assay
Mononuclear cells (MNC) isolated from axillary lymph nodes (ALN) were obtained from two adult common marmosets (Callithrix jacchus) that had been enrolled in earlier EAE studies. The cells have been stored deep-frozen directly after being isolated. All marmosets were acquired from the purpose-bred colony at the Biomedical Primate Research Centre and declared in good health by veterinary staff prior to enrollment in the study. Health checks included physical examination and tests for hematological, serological, and microbiological abnormalities. The used EAE model was induced with a synthetic peptide representing residues 34–56 of human myelin oligodendrocyte glycoprotein (MOG34–56; Cambridge Research Biochemicals Ltd., Cleveland, U.K.; sequence of GMEVGWYRPPFSRVVHLYRNGKD) emulsified with IFA (Difco Laboratories, Detroit, MI) as previously described.68 As antigen-presenting cells, EBV-infected marmoset B-lymphoblastoid cells (BLCs) were used. BLC lines were generated by infection of blood MNC with human Epstein-Barr virus propagated in marmoset cell line B95-8.70 Briefly, PBMCs were cultured with supernatants from the EBV-producing B95-8 cell line for 1.5 h at 37 °C. Following incubation, cells were diluted at a 1:1 ratio with RPMI-1640 (GIBCO) containing 1 μg/mL phytohemeagglutinin (PHA) and supplemented with 10% fetal calf serum (FCS), l-alanyl-l-glutamine (GIBCO), and penicillin/streptomycin as antibiotics. Following successful transformation, cells were maintained in culture medium without PHA until further use. For proliferation experiments, marmoset EBV-BLCs were collected, washed in PBS, and irradiated with 70 Gy in RPMI-1640 without FCS. Cells were plated in round-bottom 96-well plates (Greiner) at a concentration of 1 × 103 cells/well together with different concentrations of the peptides (30, 10, 3, and 1 μg/mL; the medium was taken as a negative control) in a humidified atmosphere of 5% CO2 in culture medium with 10% FCS at 37 °C. After 1 h, freshly thawed marmoset ALN cells were added to the BLCs at a density of 1 × 105 cells/well. After 48 h, 25 μL of [methyl-3H]thymidine (0.1 mCi/mL stock; PerkinElmer) was added, and 18 h later, the cells were harvested using a FilterMate Harvester (PerkinElmer) on a microfilter plate. After the plate had dried, 25 μL of microscinttm-E (PerkinElmer) was added, and [3H]thymidine incorporation was measured in a microBeta microplate counter (PerkinElmer).
Fluorescence-Activated Cell Sorting (FACS)-Based Immunotoxicity Assay
For peptide toxicity assessment, EBV-BLCs were prestained with CellTrace (Molecular Probes) for 20 min at room temperature. Labeled cells were plated in round-bottom 96-well plates (Greiner) at a concentration of 1 × 103 cells/well and incubated with different concentrations of the peptides (30, 10, 3, and 1 μg/mL; the medium was taken as a negative control) for 1 h in a humidified atmosphere of 5% CO2 in culture medium with 10% FCS at 37 °C. After 1 h, freshly thawed mononuclear cells isolated from the axillary lymph nodes of an EAE marmoset were added to the BLCs at a density of 1 × 105 cells/well. After incubation for 24 h, the cells were harvested and stained with life/death marker (ebioscience, catalog no. 65-0866-14) to exclude dead cells. Subsequently, cells were stained for apoptosis with AnnexinV-APC (BD, catalog no. 550474). Cells were fixed in 1% PFA/AnnexinV binding buffer (BD Biosciences) prior to measurement of fluorochrome binding. Flow cytometric measurements were performed utilizing the FACS LSRII instrument fitted with FACSDiva 5.0 (BD Biosciences), and data were analyzed with FlowJo software (Treestar, Ashland, OR). On FACS plots, cells were gated as CellTrace+ or CellTrace– cells; both groups were gated for AnnexinV and the LD marker.
Immunofluorescence Microscopy
Cultures of 1.5 × 106 BSM BLCs/well were set up with different concentrations of the peptides (25 and 6.25 μM; the medium was taken as a negative control) in R0 medium. The cells were incubated for 16 h in a humidified atmosphere at 37 °C. Cytospin preparations were made by centrifuging (3 min at 28g) 5 × 104 cells on microscopy glass slides using a Shandon Cytospin 4 Thermo centrifuge. Slides were air-dried overnight at room temperature and stored at −80 °C. Before being stained, slides were thawed and air-dried for 30 min at room temperature. Postfixation was performed with 4% PFA (Affymetrix, catalog no. 19943 1LT) for 15 min, and cells were permeabilized and blocked with block/permeabilization buffer [0.1% fish gelatin (Sigma-Aldrich, catalog no. 67041), 0.1% BSA (Sigma-Aldrich, catalog no. A9647), and 0.5% Triton X-100 (Sigma-Aldrich, catalog no. T8787) in PBS (GIBCO)] for 1 h at room temperature. Cells were stained with LC3 (nanotools, catalog no. 0231-100/LC3-5F10) in a 1:20 ratio in blocking buffer overnight at 4 °C and stained with anti-mouse FITC in a 1:400 ratio in blocking buffer for 1 h at room temperature in the dark. The peptides were detected using a click mixture [0.064 mM CuSO4, 0.038 mM sodium ascorbate, 0.25 mM tris-hydroxypropyltriazolylmethylamine (THPTA), and fluorophore AF-647-azide 1:160 in 100 mM Tris (pH 8.0)] for 1 h at room temperature in the dark, washed, and mounted with prolonged diamond antifade mounting medium with DAPI (life technologies, catalog no. P36971). Pictures were taken using the fluorescence microscope (Leica, catalog no. DFC 365 FX) with Leica application suite X 3.4,2.18368 and analyzed using ImageJ 1.50i (http://imagej.nih.gov/ij).
Results
Citrullination of MOG35–55 Peptides Alters Their Structural Behavior
We first analyzed the aggregation properties of the extracellular portion of recombinant MOG (aa 1–125, hereafter termed rMOG) and its percitrullinated counterpart, cit-rMOG, using a ThT assay. ThT is a fluorogenic dye, which becomes fluorescent upon binding to cross-β-sheet structures, such as those found in amyloid aggregates.34 In brief, proteins were dissolved in 20 mM NaOAc buffer (pH 5.0). Upon addition of ThT and incubation under gentle agitation, the fluorescence emission was recorded at 485 nm over a period of at least 15 h to monitor the propensity of the proteins toward amyloid-like aggregation. rMOG and cit-rMOG were assessed for aggregation under these conditions, but neither protein showed any aggregation (Figure S1). We thus hypothesized that it was not rMOG that was the aggregating species but a fragment produced by proteolytic processing. We focused on the immuno-dominant peptide 35–55 and synthesized eight peptides spanning aa 35–55 of the native MOG sequence carrying TCR contact residues (aa 41–48) and site-specific citrulline residues at the respective (aa 41, 46, and/or 52) positions (Scheme 1 and Table 1, 1–8). Elongated variants (aa 31–55) were also prepared, as were bioorthogonal variants carrying propargylglycine (Pg) as a bioorthogonal handle at position 31 that would allow in-cell visualization (Scheme 1 and Table 1, 9–12). All peptides were synthesized in good yields (5.3–34.9%) and very good purity [>95%; determined by integrating the peak areas on analytical HPLC measurements (Table 1)]. All MOG35–55 peptides were analyzed by liquid chromatography and mass spectrometry (LC–MS) (Figures S2–S9) and circular dichroism (CD) spectroscopy (Figure 2a–c and Figure S10). We observed predominantly random coil structures at two pH values (5.0 and 7.5; chosen for their pathophysiological relevance of being the pH in the endosomal pathway of BLCs and the extracellular pH, respectively), which are in good agreement with the results reported by Albouz-Abo et al.35 for mouse MOG35–55. All our synthetic peptides adopted an α-helical structure upon addition of 50% 2,2,2-trifluoroethanol (TFE), a known stabilizer of α-helical conformations in proteins and peptides36 (Figure 2a–c and Figure S10). In our hands, in the presence of SDS, nonmicellar (β-sheet enhancer)37 peptides 1 and 4 adopted β-sheet rich structures (Figure 2b and Figure S10C). Peptides 2, 3, 6, and 7, however, seemed to adopt a random coil structure under the same conditions (Figure S10A,B,D for 2, 3, and 6 and Figure 2c for 7). Moreover, 5 and 8 adopted α-helical structures. This indicates that the peptides have distinct structures depending on the external solution conditions.
Scheme 1. Schematic Representation of MOG-Derived Peptides MOG35–55 and MOG31–55 with Their Amino Acid Sequences and Positions for Arg → Cit Mutations (colored pink).

Table 1. Synthesized MOG Derivatives.

Figure 2.

(a–c) Biophysical characterization and (d–h) ThT aggregation assay for peptides 1, 5, and 7. (a) CD spectrum of 1 at pH 5.0 and 7.5 and with additives (TFE and SDS) at 50 μM. (b) CD spectrum of 5 at pH 5.0 and 7.5 and with additives (TFE and SDS) at 50 μM. (c) CD spectrum of 7 at pH 5.0 and 7.5 and with additives (TFE and SDS) at 50 μM. All spectra were recorded from 190 to 260 nm and reflect an average of at least five independent measurements: (black circles) sample in 20 mM Tris buffer (pH 7.5), (gray squares) sample in 20 mM sodium acetate (NaOAc) buffer, (red triangles) sample treated with 50% (v/v) TFE, and (blue triangles) sample treated with micellar concentrations of SDS (2–4 mM). (d–f) ThT fluorescence spectra of peptides 1, 5, and 7, respectively. All data were recorded at an excitation wavelength of 444 ± 9 nm and an emission wavelength of 485 ± 9 nm. All samples were used at a pH of 5.0 with varying concentrations: (black circles) 200 μM, (gray squares) 160 μM, (dark blue triangles) 120 μM, (light blue triangles) 80 μM, (purple diamonds) 40 μM, (red circles) 20 μM, (red squares) 10 μM, (pink triangles) 3 μM, and (white triangles) 1 μM. The fluorescence change is normalized to 20 mM NaOAc (pH 5) treated with an equal amount of ThT as in the samples. (g and h) ThT fluorescence spectral data (d–f) of peptides 7 and 5 at time point t = 15 h (representing the end point of aggregation kinetics). All aggregation assays were performed at least three times and with experimental triplicates.
Citrullinated MOG Peptides Show Amyloid-like Aggregation
Next, we asked whether the peptides were prone to amyloid-like aggregation. To investigate amyloid-like aggregation using the aforementioned ThT fluorescence assay, peptides (200 μM, Figure 2 and Figure S11) were dissolved in buffers with a pH range of 3.0–7.0. Noncitrullinated peptide 1 did not show any aggregation at any pH or at any of the concentrations tested (Figure S11 and Figure 2d). Citrullinated peptides 2–7, on the other hand, showed pH-dependent aggregation characteristics with the strongest aggregation observed at pH 4.0 or 5.0, which is the pH range found within the B-cell lysosomal pathway (Figure S11). At all other pH values tested, either no increase or only a slight (<2-fold) increase in fluorescence intensity was observed, suggesting that the lysosome-like pH value of 4.0–5.0 was essential for amyloid-like behavior of the citrullinated MOG peptides. Note that peptide 8 showed aggregation behavior different from those of all of the other peptides tested aggregating at pH 6 (∼3-fold compared to pH 5) and pH 7 (∼4-fold compared to pH 5) the fastest. On the basis of the results presented above, we subsequently analyzed the concentrations at which amyloid-like aggregation was observed for the citrullinated MOG35–55 variants 2–8. Each of the citrullinated peptides displayed distinct aggregation behavior, underlining the hypothesis that specific arginines are responsible for characteristics of amyloid aggregation for MOG35–55. Peptide 2 aggregated only at concentrations of >4 μM (Figure S12A). Peptides 3, 5, and 8 on the other hand did aggregate at concentrations of >4 μM but not at a final concentration of 1 μM (Figure S12B,E and Figure 2). Peptides 6 and 7 showed aggregation at all concentrations tested (Figure S12D and Figure 2f). Peptide aggregation at lower concentrations is regarded a hallmark of nucleation-dependent aggregation and is thus a strong indication to prove our hypothesis. The most prominent aggregation behavior at low concentrations (<10 μM) was observed with peptides 5 and 7 (plotted data, Figure 2g,h and Figure S25). Hence, these peptides were used for further analysis.
Citrullinated Peptides Induce Seeded Nucleation and Show a Heterogeneous Fibril Morphology
A further characteristic of amyloid behavior is the observation of seeded nucleation,33 in which amyloid precipitation can be accelerated by the addition of amyloid seeds. In the lag phase, only negligible amounts of self-propagated amyloid fibrils (also called seeds or nuclei) are formed, whereas upon their formation, an exponential increase in the level of fibril growth can be explored, which ends up reaching a plateau phase.
Peptides 1 (Arg41, Arg46, and Arg52), 5 (Cit41, Cit46, and Arg52), and 7 (Arg41, Cit46, and Cit52) were therefore dissolved in 20 mM NaOAc (pH 5.0) at a concentration of 0.1 mg/mL (40 μM) and incubated for at least 24 h at 37 °C under constant agitation to produce seeds for the experiment. We then separated the supernatant and pellet via centrifugation (20000g for 30 min) and analyzed by CD spectroscopy (Figure S13). The seeds were then homogenized in 20 mM NaOAc buffer (the same amount of buffer that was used for the incubation reaction) to an estimated final concentration of 0.1 mg/mL for further use in seeded nucleation assays.
The seed homogenate of peptide 5 formed a β-sheet-like structure, while the supernatant contained only random coil structures (Figure S13A). However, the minimum of the spectrum was observed at a wavelength (222 nm) higher than that normally observed for β-sheets (218 nm), which has also been observed for certain Aβ peptides38 and scrapie PrP (PrPSc).39 The seed homogenate of peptide 7 seems to reach a transition state-like conformation with characteristics of both α-helices and β-sheets (Figure S13B), and similar to that of 5, the peptide in the supernatant fraction of 7 was mainly random coil. As expected, no seeds for peptide 1 could be collected.
The collected seeds (3–300 ng per experiment) were then incubated with solutions of peptides 1, 5, and 7 (40 μM, 200 μL) to determine whether an acceleration of aggregation kinetics was observed (Figure 3 and Figure S14). This would confirm the amyloid characteristic of small amounts (submicrograms) of already aggregated species being able to act as seeds and drive faster amyloid aggregation by interacting with template peptides in solution. Indeed, faster aggregation kinetics for peptides 5 and 7 but not for 1 were observed. Sigmoidal curves were interpolated (eq E1 and Tables S1 and S2) and used to determine the time shift of aggregation at a relative intensity of 0.9-fold with all peptides starting to aggregate at this value. For seeded aggregation of peptide 1, under all conditions, no aggregation was observed (Figure S14A). For peptide 5, however, a positive time shift (thus faster aggregation) was observed. The most pronounced time shift was observed upon seeding with 0.3 μg of peptide 7 [Δt1/2 = 2.9 h (Figure 3b)]. In addition, seeding with 0.03 μg of 5 resulted in significantly faster aggregation [Δt1/2 = 1.1 h (Figure 3a)], indicating a synergistic effect of both seeds with peptide 5.
Figure 3.

Seeded aggregation of peptides 5 and 7. Peptides 1, 5, and 7 were subjected to aggregation in the presence of seeds from either peptide 5 (depicted as “seed 5”) or 7 (depicted as “seed 7”) with different amounts (3, 30, and 300 ng in 200 μL, depicted in corresponding diagrams in brackets). For aggregation-prone peptides 5 (a and b) and 7 (c and d), aggregation curves were fitted according to eq E1 in the Supporting Information (depicted as “fit”) to determine the change in t1/2 as a measure of aggregation kinetics. All data were recorded at an excitation wavelength of 444 ± 9 nm and an emission wavelength of 485 ± 9 nm. All samples were used at a pH of 5.0 and a concentration of 40 μM.
Likewise, peptide 7 showed a prominent shift in time upon seeding with 0.003 μg of 5 (Figure 3c) (Δt1/2 = 3.5 h). Additionally, seeding with 0.03 μg of peptide 7 resulted in significantly faster aggregation [Δt1/2 = 1.2 h (Figure 3d)]. This indicates a synergistic effect of both seeds with 7. It is therefore likely that peptides 5 and 7 act as a substrate for either seed leading to faster aggregation kinetics. This kind of seeded aggregation is also observed for other amyloids, as has been shown for Aβ peptides, PrP, and α-syn.40−42 TEM was next used to obtain information about the morphology of the fibrils (Figure 4a,b). Both fibrils showed different morphologies. Peptide 7 formed needle-like, long (>100 nm) assemblies of fibrils with partially amorphous structures (Figure 4b), while fibrils of peptide 5 showed shorter (<100 nm) assemblies with tight packing corresponding to the higher susceptibility to aggregation over peptide 7in vitro (Figure 4a). Additionally, DLS measurements were performed to determine the size of the fibrils (Figure 4c,d). The fibrils have hydrodynamic radii (Rhyd) of 78 ± 3.1 and 175 ± 2.5 nm for peptide 5 and 7 seeds, respectively. The Rhyd values are larger than expected for amyloid-like structures, which typically have a diameter of ∼10 nm in an oligomeric state.43 Therefore, we concluded that the seeds of peptides 5 and 7 can be considered as mixed fibrils with amyloid-like structural characteristics as reported previously.44,45
Figure 4.
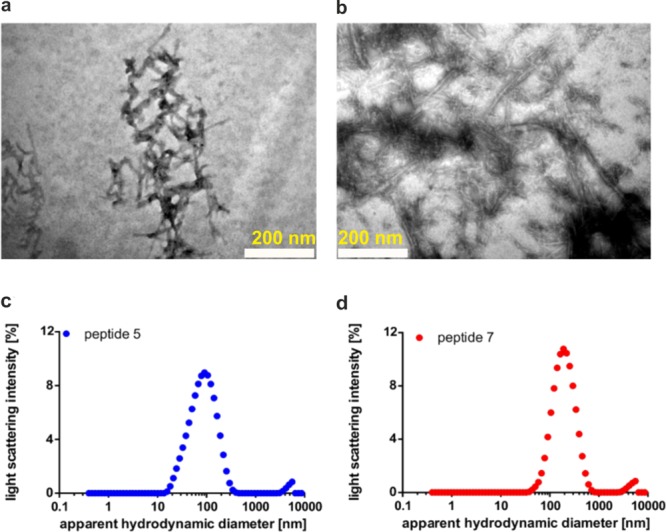
Morphology of amyloid fibrils of 5 and 7. (a) Image of seeds generated from peptide 5 (scale bar, 200 nm). (b) TEM image of seeds generated from peptide 7 (scale bar, 200 nm). (c) Determination of the apparent hydrodynamic diameter (Rhyd) of seeds generated from peptide 5 with dynamic light scattering (DLS). (d) Determination of Rhyd values of seeds generated from peptide 7 with DLS.
Citrullinated Peptides Aggregate inside BLCs
We next asked whether this aggregation of citrullinated peptides also occurred within EBV-infected BLCs. To date, visualization of protein and peptide aggregation inside cells is a challenging task as fluorescence microscopy with thioflavin S (ThS), a derivative of ThT, is difficult due to aspecific binding to all amyloid proteins.46 Super-resolution fluorescence microscopy47 and cryo-electron microscopy48 have recently been used but are yet too complex for routine analysis. Having recently shown that bioorthogonal modifications affect peptide behavior only minimally inside antigen-presenting cells,49,50 we opted for a bioorthogonal approach to visualize MOG–peptide aggregation in the EBV-infected BLCs. Bioorthogonal reactions are chemoselective reactions in living systems, which do not interfere with other biochemical processes.51,52 Four new peptides that are N-terminally extended (aa 31–55) harboring an N → Pg mutation at position 31 were generated (Scheme 1 and Table 1, peptides 10–12). Position 31 was chosen to avoid any interference with the immunorelevant epitope 35–55 by using three additional amino acids of the native MOG sequence as linkers (aa 32–34). Aside from the traceable Pg, the peptides were synthesized without Cit (10) or double citrulline substitutions at critical positions (positions 41 and 46 and positions 46 and 52 for 11 and 12, respectively), and a nonbioorthogonal variant of noncitrullinated 10, peptide 9, was additionally generated. Neither 9 nor 10 aggregated (Figures S9, S17, and S20), and the aggregation behavior of 11 and 12 was not affected by the four-amino acid extension or by the presence of Pg compared to 5 and 7 (Figures S15–S20), confirming their suitability for tracing amyloid aggregation in cells. EBV-infected BLCs were therefore incubated under serum-free conditions with 10, 11, and 12 (0, 6.2, and 25 μM depicted in Figure 5; 3.1 and 12.5 μM depicted in Figures S21 and 22) for 48 h. After incubation, cells were centrifuged onto glass slides and fixed with PFA. The peptides were visualized by the copper-catalyzed azide alkyne cycloaddition (CuAAC)53,54 reaction with Alexa647-azide (Thermo Fisher). Cells were co-stained with DAPI (blue channel) and LC3 (green channel), as this protein—which becomes post-translationally lipidated during autophagy as a marker of autophagic activity—is recruited to phagosomes containing aggregates (Figure 5). Peptides 11 and 12 in the absence of cells showed large aggregates, while peptide 10 had formed no aggregates. When peptides 11 and 12 were incubated with EBV-infected BLCs, spherical structures of different sizes (small spheres for 11 and large agglomerates for 12) were found, contained within LC3 positive vesicles, indicating intracellularly aggregated peptide. To our surprise, noncitrullinated peptide 10 also aggregated, once it was taken up by BLCs. This may be due to citrullination of peptide 10 by elevated levels of PAD enzymes present in EBV-infected B-cells.18 Earlier work from our group showed that EBV infection of B-cells increases the level of degradation of MOG35–55 in those cells. Thus, the aggregation behavior might not be restricted to B-cells but is likely a consequence of elevated PAD2 or PAD4 levels.71
Figure 5.

Monitoring the uptake and aggregation of bioorthogonal, site-specific citrullinated MOG peptides via confocal microscopy. Human EBV-infected BLCs were incubated for 48 h with either no peptide (0 μM) or 6.2 or 25 μM peptide 10, peptide 11, or peptide 12 (highlighted in yellow). Cells were fixed with 4% PFA and processed for immunofluorescence with the following primary antibodies. The nucleus was stained with DAPI (blue), and LC3 was used as an autophagosome marker (green). The bioorthogonal peptides were stained using CuAAC chemistry with azide Alexa-647 (Thermo Fisher). The scale bar is 25 μm (white bar).
Citrullination Converts Antigenic MOG Peptides into Cytotoxic Aggregates in Distinct Cell Types
We next tested whether amyloid aggregation of the peptides resulted in them becoming toxic to cells. We aimed to assess the toxicity of the peptides toward the core autoimmune process, which is the antigen-mediated cross-talk of EBV-infected BLCs with autoaggressive T-cells. In a first experiment, lethally irradiated marmoset EBV-BLCs (104 per milliliter) were incubated for 1 h with titrating doses of peptides 10–12 or MOG14–36 as an irrelevant peptide (Figure 6a). Subsequently, a mixed lymphocyte population harvested from ALN of a marmoset immunized with MOG34–56 was added and cultured for 72 h. Proliferation of the lymphocytes was assayed via the incorporation of [3H]thymidine during the final 18 h. Figure 6a shows a dose-dependent stimulation of proliferation by the nonmodified peptide 10, while both citrullinated peptides (11 and 12) induced dose-dependent inhibition of proliferation even beneath the background response against antigens expressed by the EBV-BLCs. Peptide 10 showed stimulation of T-cell proliferation at all concentrations tested. To assess the targets of the citrullinated peptides, we stained the EBV-BLCs with the CellTrace dye, prior to the incubation with peptides and lymphocytes. After 24 h, cells were harvested, stained with Annexin V, a cellular probe binding phosphatidylserine, a marker of apoptotic and dead cells, and analyzed via fluorescence-activated cell sorting (FACS). Figure 6b shows that Annexin V staining, marking cytotoxicity, stains the CellTrace+ve and CellTrace–ve fraction of cells incubated with the aggregation-prone peptides 11 and 12. As expected, for 11 and 12, an increased level of apoptosis was observed at concentrations of >1 μg/mL (Figure 6b, white circles), whereas peptide 10 did not show any cytotoxic effects in EBV-BLCs or in a co-culture with the lymphocytes. Furthermore, the toxicity of MOG35–55 peptides 5 and 7 was assessed toward murine bone marrow-derived dendritic cells (mBMDCs), which have the capacity to phagocytose aggregates, and to the T-cell hybridoma cell line B3Z55 as a nonphagocytosing cell line (Figure S24). Four different concentrations of peptides were tested (40, 20, 10, and 3 μM). Peptides 5 and 7 were toxic to mBMDCs [cell viability down to 70% for 5 and 62% for 7 (Figure 6c)] when compared to a nontreated control (Figure 6c, control). Both 5 and 7 showed cytotoxicity at concentrations of ≤10 μM. Additionally, 5 showed significant cytotoxicity at 40 μM. As expected, no decrease in cell viability was measured when B3Zs were incubated with increasing concentrations of peptides 5 and 7.
Figure 6.

Cytotoxicity of MOG derived peptides (a and b) 10–12 and (c) 5 and 7 in EBV-BLC co-cultures with lymph node cells from EAE marmosets and in mBMDCs. (a) Marmoset EBV-induced BLCs were lethally irradiated and incubated for 1 h with titrating concentrations of peptide 10, 11, or 12 or an irrelevant peptide. Subsequently, lymph node or spleen cells from marmosets immunized with MOG34–56 were added. The responses of T-cells to the peptides were assayed by proliferation and are expressed as the stimulation index per culture condition. The experiment was conducted six times (marmosets) and with three biological replicates. Data are presented as means ± the standard error of the mean. (b) To test which cell type is targeted by the peptides, EBV-BLCs (from two marmosets, M1 and M2) were incubated with CellTrace dye before incubation with peptide (white circles) and a mixture with the spleen/lymph node cells (red triangles). Lymphocytes that are not subjected to co-culturing were used as controls (black circles). Cultured cells were harvested and stained for Annexin V as a marker of late apoptotic/dead cells. The final analysis was done utilizing FACS. (c) Cytotoxicity assays with citrullinated MOG peptides in BMDCs. *p < 0.05, **p < 0.005, ***p < 0.0005, and ****p < 0.00005. n.s., not significant. The experiment was conducted twice and with three biological replicates. Group mean values were analyzed by one-way analysis of variance with the Bonferroni post hoc significant difference test using GraphPad Prism 6.0. Data are represented as means ± the standard deviation.
Discussion
MOG has been accepted as a key autoantigen in MS. Nevertheless, the pathophysiological role of MOG remains complex. The response of T-cells to MOG epitopes depends on the genetic background of the species (human, marmoset, and mouse) as well as on environmental factors (especially EBV infection) and post-translational modifications (e.g., N-glycosylation and glucosylation).14,56,57 In the marmoset model, two key epitopes were identified, an MHC class II restricted epitope of inflammatory T-cells (MOG24–36) and an MHC class Ib restricted epitope of cytotoxic T-cells (MOG40–48).58 The latter pathway is particularly interesting for MS, as it leads to lesions in brain gray matter,12 which have the strongest impact on the disease.59
A key finding underlining this complexity has been that the antigenicity of the MOG40–48 epitope is dependent on pathogenic post-translational citrullination. We have shown that citrullination of a peptide containing the MOG40–48 epitope at position 46 alters its proteolytic sensitivity to the endolysosomal serine protease cathepsin G in BLCs.60 Interestingly, it was shown in EBV-infected BLCs that expression levels of PAD2 and PAD4 are increased and that the autophagic flux is activated in infected cells, providing a protection mechanism for the peptide against rapid destruction. This mechanism putatively explains the requisite involvement of EBV-infected BLCs in the activation of autoreactive CD8+CD56+ T-cells, which drive disease progression in the marmoset EAE model.61 On the other hand, earlier in vivo experiments in mice showed that the two arginine residues in the MOG40–48 epitope are TCR contact residues of the pro-inflammatory CD4+ T-cells.23 The study presented here shows that this may also be the case in the marmoset EAE model, as T-cells from marmosets immunized with MOG34–56 are not stimulated (proliferation) by EBV-infected BLCs presenting citrullinated MOG34–56. In this study, we synthesized and characterized site-specifically citrullinated peptides as well as three bioorthogonal variants thereof to analyze processing and presentation of MOG-derived epitopes and their contribution to neurodegeneration. We showed that site-specific citrullination of MOG35–55 peptides resulted in amyloid-like aggregation behavior with formation of heterogeneous fibrils. This aggregation behavior was strongest at pH 5.0 (Figure S11), highlighting the potential involvement of intracellular vesicular proteolysis, like that found in the autophagy pathway.62 Moreover, above peptides aggregated at low concentrations (≤10 μM), the best and the fastest renders this concentration the critical aggregation concentration. One possible explanation for high concentrations (≥160 μM) not yielding to higher/faster aggregation susceptibility is that those are not within the critical aggregation concentration range and therefore might slow aggregation as shown for other proteins.63 Furthermore, Hevehan et al. reported refolding of aggregated lysozyme at high concentrations. These findings might explain the altered aggregation behavior of aforementioned MOG35–55 variants at high concentrations.64 Two doubly citrullinated peptides, 5 and 7, also showed characteristics of seeded nucleation,33 thus supporting the hypothesis that these peptides display amyloid behavior. Collectively, these findings may shed new light on the role of EBV-infected BLCs and the immunodominant MOG34–56 peptide in the pathogenesis of MS and the role of EBV-infected BLCs and the immunodominant MOG34–56 peptide in the pathogenesis of MS.
This finding was further supported when using bioorthogonal variants of MOG31–55 in EBV-infected BLCs. Peptides were shown to aggregate within 48 h in these cells in a concentration-dependent manner (the higher the concentration, the larger the number of aggregates). Additionally, we could show that the autophagy pathway was stimulated, when using concentrations of ≤6.2 μM (Figure 5). We hypothesized that above this concentration, the aggregates are formed already before uptake in the cells and hence only a small portion of the peptides aggregate intracellularly and enter the autophagic pathway; instead, the aggregates may be taken up via alternative routes such as phagocytosis, endocytosis, or micropinocytosis as observed for amyloid β (Aβ)-40 and -42.65,66
To provide further evidence for a link between the amyloid behavior of these citrullinated MOG fragments and PPMS, the aggregates showed enhanced cytotoxicity toward phagocytes (EBV-BLCs and mBMDCs) in a concentration-dependent manner (≥10 μM for BLCs and ≤10 μM for mBMDCs).
In summary, we were able to show for the first time that citrullination is a protection mechanism against fast, physiological degradation of MOG peptides harboring T-cell epitopes and enhances antigenicity but promotes amyloid aggregation as a potential neurotoxic mechanism. These results provide evidence for the theory that a pathogenic mechanism initiated by EBV infection is one of the driving forces behind autoimmunity in MS. Our future work is directed toward live cell imaging amyloid-like aggregation, antigen processing, and presentation events in APCs incubated with MOG-derived peptides in marmoset EAE.
Acknowledgments
The authors thank Sanne Vonk for the assistance with MTT assays and G. E. M. Lemmers for the technical support with TEM. Additionally, the authors acknowledge A. Kross for granting us the access to the JASCO J-815 CD spectrometer, the Infinite M1000 Pro Tecan plate reader, and the dynamic light scattering (DLS) spectrometer Zetasizer Nano S.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.8b00852.
Tables S1 and S2, eq E1, Figures S1–S25, and supporting materials (PDF)
C.A. was supported by The Netherlands Organization for Scientific Research (NWO)-CW-ECHO Grant program 2016. M.H.S.M. was supported by a Lundbeck Postdoc Abroad grant. S.I.v.K. acknowledges the European Research Council (ERC-2014-StG-639005) for financial support.
The authors declare no competing financial interest.
Supplementary Material
References
- Compston A.; Coles A. (2008) Multiple sclerosis. Lancet 372 (9648), 1502–17. 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- Lublin F. D.; Reingold S. C.; Cohen J. A.; Cutter G. R.; Sørensen P. S.; Thompson A. J.; Wolinsky J. S.; Balcer L. J.; Banwell B.; Barkhof F.; Bebo B.; Calabresi P. A.; Clanet M.; Comi G.; Fox R. J.; Freedman M. S.; Goodman A. D.; Inglese M.; Kappos L.; Kieseier B. C.; Lincoln J. A.; Lubetzki C.; Miller A. E.; Montalban X.; O’Connor P. W.; Petkau J.; Pozzilli C.; Rudick R. A.; Sormani M. P.; Stüve O.; Waubant E.; Polman C. H. (2014) Defining the clinical course of multiple sclerosis. Neurology 83 (3), 278–286. 10.1212/WNL.0000000000000560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciotti J. R.; Cross A. H. (2018) Disease-Modifying Treatment in Progressive Multiple Sclerosis. Curr. Treat Option. Ne. 20 (5), 12. 10.1007/s11940-018-0496-3. [DOI] [PubMed] [Google Scholar]
- Olsson T.; Barcellos L. F.; Alfredsson L. (2017) Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat. Rev. Neurol. 13, 25. 10.1038/nrneurol.2016.187. [DOI] [PubMed] [Google Scholar]
- Sumaya C. V.; Myers L.; Ellison G. W. (1976) Epstein-Barr virus antibodies in multiple sclerosis. Trans. Am. Neurol. Assoc. 101, 300. [PubMed] [Google Scholar]
- Ascherio A.; Munger K. L. (2010) Epstein–Barr Virus Infection and Multiple Sclerosis: A Review. J. Neuroimmune Pharmacol. 5 (3), 271–277. 10.1007/s11481-010-9201-3. [DOI] [PubMed] [Google Scholar]
- Linnington C.; Webb M.; Woodhams P. L. (1984) A novel myelin-associated glycoprotein defined by a mouse monoclonal antibody. J. Neuroimmunol. 6 (6), 387–396. 10.1016/0165-5728(84)90064-X. [DOI] [PubMed] [Google Scholar]
- Lee D.-H.; Linker R. A. (2012) The role of myelin oligodendrocyte glycoprotein in autoimmune demyelination: a target for multiple sclerosis therapy?. Expert Opin. Ther. Targets 16 (5), 451–462. 10.1517/14728222.2012.677438. [DOI] [PubMed] [Google Scholar]
- Jagessar S. A.; Smith P. A.; Blezer E.; Delarasse C.; Pham-Dinh D.; Laman J. D.; Bauer J.; Amor S.; ’t Hart B. (2008) Autoimmunity against myelin oligodendrocyte glycoprotein is dispensable for the initiation although essential for the progression of chronic encephalomyelitis in common marmosets. J. Neuropathol. Exp. Neurol. 67 (4), 326–40. 10.1097/NEN.0b013e31816a6851. [DOI] [PubMed] [Google Scholar]
- Smith P. A.; Heijmans N.; Ouwerling B.; Breij E. C.; Evans N.; van Noort J. M.; Plomp A. C.; Delarasse C.; ’t Hart B.; Pham-Dinh D.; Amor S. (2005) Native myelin oligodendrocyte glycoprotein promotes severe chronic neurological disease and demyelination in Biozzi ABH mice. Eur. J. Immunol. 35 (4), 1311–9. 10.1002/eji.200425842. [DOI] [PubMed] [Google Scholar]
- Jagessar S. A.; Kap Y. S.; Heijmans N.; van Driel N.; van Straalen L.; Bajramovic J. J.; Brok H. P. M.; Blezer E. L. A.; Bauer J.; Laman J. D.; ’t Hart B. A. (2010) Induction of Progressive Demyelinating Autoimmune Encephalomyelitis in Common Marmoset Monkeys Using MOG34–56 Peptide in Incomplete Freund Adjuvant. J. Neuropathol. Exp. Neurol. 69 (4), 372–385. 10.1097/NEN.0b013e3181d5d053. [DOI] [PubMed] [Google Scholar]
- ’t Hart B. A.; Dunham J.; Faber B. W.; Laman J. D.; van Horssen J.; Bauer J.; Kap Y. S. (2017) A B Cell-Driven Autoimmune Pathway Leading to Pathological Hallmarks of Progressive Multiple Sclerosis in the Marmoset Experimental Autoimmune Encephalomyelitis Model. Front. Immunol. 8, 804. 10.3389/fimmu.2017.00804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardinier M. V.; Amiguet P.; Linington C.; Matthieu J. M. (1992) Myelin/oligodendrocyte glycoprotein is a unique member of the immunoglobulin superfamily. J. Neurosci. Res. 33 (1), 177–87. 10.1002/jnr.490330123. [DOI] [PubMed] [Google Scholar]
- García-Vallejo J. J.; Ilarregui J. M.; Kalay H.; Chamorro S.; Koning N.; Unger W. W.; Ambrosini M.; Montserrat V.; Fernandes R. J.; Bruijns S. C. M.; van Weering J. R. T.; Paauw N. J.; O’Toole T.; van Horssen J.; van der Valk P.; Nazmi K.; Bolscher J. G. M.; Bajramovic J.; Dijkstra C. D.; ’t Hart B. A.; van Kooyk Y. (2014) CNS myelin induces regulatory functions of DC-SIGN–expressing, antigen-presenting cells via cognate interaction with MOG. J. Exp. Med. 211 (7), 1465–1483. 10.1084/jem.20122192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geijtenbeek T. B.; Van Vliet S. J.; Engering A.; ’t Hart B. A.; Van Kooyk Y. (2004) Self- and Nonself-Recognition by C-Type Lectins on Dendritic Cells. Annu. Rev. Immunol. 22, 33–54. 10.1146/annurev.immunol.22.012703.104558. [DOI] [PubMed] [Google Scholar]
- ’t Hart B. A.; Weissert R. (2016) Myelin oligodendrocyte glycoprotein has a dual role in T cell autoimmunity against central nervous system myelin. Mult. Scler. J. 2, 205521731663099. 10.1177/2055217316630999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider L.; Fischer M. T.; Frischer J. M.; Bauer J.; Höftberger R.; Botond G.; Esterbauer H.; Binder C. J.; Witztum J. L.; Lassmann H. (2011) Oxidative damage in multiple sclerosis lesions. Brain 134 (7), 1914–1924. 10.1093/brain/awr128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dendrou C. A.; Fugger L.; Friese M. A. (2015) Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 15, 545. 10.1038/nri3871. [DOI] [PubMed] [Google Scholar]
- van Heemst J.; Jansen D. T. S. L.; Polydorides S.; Moustakas A. K.; Bax M.; Feitsma A. L.; Bontrop-Elferink D. G.; Baarse M.; van der Woude D.; Wolbink G.-J.; Rispens T.; Koning F.; de Vries R. R. P.; Papadopoulos G. K.; Archontis G.; Huizinga T. W.; Toes R. E. (2015) Crossreactivity to vinculin and microbes provides a molecular basis for HLA-based protection against rheumatoid arthritis. Nat. Commun. 6, 6681. 10.1038/ncomms7681. [DOI] [PubMed] [Google Scholar]
- Wood D. D.; Moscarello M. A.; Bilbao J. M.; O’Connors P. (1996) Acute multiple sclerosis (marburg type) is associated with developmentally immature myelin basic protein. Ann. Neurol. 40 (1), 18–24. 10.1002/ana.410400106. [DOI] [PubMed] [Google Scholar]
- Wood D. D.; Moscarello M. A. (1989) The isolation, characterization, and lipid-aggregating properties of a citrulline containing myelin basic protein. J. Biol. Chem. 264 (9), 5121–7. [PubMed] [Google Scholar]
- Bradford C. M.; Ramos I.; Cross A. K.; Haddock G.; McQuaid S.; Nicholas A. P.; Woodroofe M. N. (2014) Localisation of citrullinated proteins in normal appearing white matter and lesions in the central nervous system in multiple sclerosis. J. Neuroimmunol. 273 (1), 85–95. 10.1016/j.jneuroim.2014.05.007. [DOI] [PubMed] [Google Scholar]
- Carrillo-Vico A.; Leech M. D.; Anderton S. M. (2010) Contribution of Myelin Autoantigen Citrullination to T Cell Autoaggression in the Central Nervous System. J. Immunol. 184 (6), 2839–2846. 10.4049/jimmunol.0903639. [DOI] [PubMed] [Google Scholar]
- Petersen T. R.; Bettelli E.; Sidney J.; Sette A.; Kuchroo V.; Backstrom B. T. (2004) Characterization of MHC- and TCR-binding residues of the myelin oligodendrocyte glycoprotein 38–51 peptide. Eur. J. Immunol. 34 (1), 165–73. 10.1002/eji.200324669. [DOI] [PubMed] [Google Scholar]
- Collinge J. (2016) Mammalian prions and their wider relevance in neurodegenerative diseases. Nature 539, 217. 10.1038/nature20415. [DOI] [PubMed] [Google Scholar]
- Skovronsky D. M.; Lee V. M. Y.; Trojanowski J. Q. (2006) NEURODEGENERATIVE DISEASES: New Concepts of Pathogenesis and Their Therapeutic Implications. Annu. Rev. Pathol.: Mech. Dis. 1 (1), 151–170. 10.1146/annurev.pathol.1.110304.100113. [DOI] [PubMed] [Google Scholar]
- Iqbal K.; Liu F.; Gong C.-X. (2016) Tau and neurodegenerative disease: the story so far. Nat. Rev. Neurol. 12, 15. 10.1038/nrneurol.2015.225. [DOI] [PubMed] [Google Scholar]
- Wong Y. C.; Krainc D. (2017) α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat. Med. 23, 1. 10.1038/nm.4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X.; Jickling G. C.; Ander B. P.; Stamova B.; Liu D.; Kao P. F.; Zelin M. A.; Jin L.-W.; DeCarli C.; Sharp F. R. (2015) Myelin Basic Protein Associates with AβPP, Aβ(1–42), and Amyloid Plaques in Cortex of Alzheimer’s Disease Brain. J. Alzheimer's Dis. 44 (4), 1213–1229. 10.3233/JAD-142013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matías-Guiu J. A.; Oreja-Guevara C.; Cabrera-Martín M. N.; Moreno-Ramos T.; Carreras J. L.; Matías-Guiu J. (2016) Amyloid Proteins and Their Role in Multiple Sclerosis. Considerations in the Use of Amyloid-PET Imaging. Front. Neurol. 7, 53. 10.3389/fneur.2016.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vázquez-Fernández E.; Young H. S.; Requena J. R.; Wille H. (2017) Chapter Seven - The Structure of Mammalian Prions and Their Aggregates. Int. Rev. Cell Mol. Biol. 329, 277–301. 10.1016/bs.ircmb.2016.08.013. [DOI] [PubMed] [Google Scholar]
- Clements C. S.; Reid H. H.; Beddoe T.; Tynan F. E.; Perugini M. A.; Johns T. G.; Bernard C. C. A.; Rossjohn J. (2003) The crystal structure of myelin oligodendrocyte glycoprotein, a key autoantigen in multiple sclerosis. Proc. Natl. Acad. Sci. U. S. A. 100 (19), 11059–11064. 10.1073/pnas.1833158100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles T. P. J.; Vendruscolo M.; Dobson C. M. (2014) The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384. 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- Vassar P. S.; Culling C. F. (1959) Fluorescent stains, with special reference to amyloid and connective tissues. Arch. Pathol. 68, 487–98. [PubMed] [Google Scholar]
- Albouz-Abo S.; Wilson J. C.; Bernard C. C.; von Itzstein M. (1997) A conformational study of the human and rat encephalitogenic myelin oligodendrocyte glycoprotein peptides 35–55. Eur. J. Biochem. 246 (1), 59–70. 10.1111/j.1432-1033.1997.t01-2-00059.x. [DOI] [PubMed] [Google Scholar]
- Nelson J. W.; Kallenbach N. R. (1986) Stabilization of the ribonuclease S-peptide alpha-helix by trifluoroethanol. Proteins: Struct., Funct., Genet. 1 (3), 211–7. 10.1002/prot.340010303. [DOI] [PubMed] [Google Scholar]
- Wu C.-S. C.; Yang J. T. (1981) Sequence-dependent conformations of short polypeptides in a hydrophobic environment. Mol. Cell. Biochem. 40 (2), 109–122. 10.1007/BF00224754. [DOI] [PubMed] [Google Scholar]
- Barrow C. J.; Zagorski M. G. (1991) Solution structures of beta peptide and its constituent fragments: relation to amyloid deposition. Science 253 (5016), 179–82. 10.1126/science.1853202. [DOI] [PubMed] [Google Scholar]
- Safar J.; Roller P. P.; Gajdusek D. C.; Gibbs C. J. Jr. (1993) Conformational transitions, dissociation, and unfolding of scrapie amyloid (prion) protein. J. Biol. Chem. 268 (27), 20276–84. [PubMed] [Google Scholar]
- Harper J. D.; Lansbury P. T. Jr. (1997) Models of amyloid seeding in Alzheimer’s disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 66, 385–407. 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- Jarrett J. T.; Lansbury P. T. Jr. (1993) Seeding ″one-dimensional crystallization″ of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie?. Cell 73 (6), 1055–8. 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- Wood S. J.; Wypych J.; Steavenson S.; Louis J. C.; Citron M.; Biere A. L. (1999) alpha-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson’s disease. J. Biol. Chem. 274 (28), 19509–12. 10.1074/jbc.274.28.19509. [DOI] [PubMed] [Google Scholar]
- Yong W.; Lomakin A.; Kirkitadze M. D.; Teplow D. B.; Chen S.-H.; Benedek G. B. (2002) Structure determination of micelle-like intermediates in amyloid β-protein fibril assembly by using small angle neutron scattering. Proc. Natl. Acad. Sci. U. S. A. 99 (1), 150–154. 10.1073/pnas.012584899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitan G.; Kirkitadze M. D.; Lomakin A.; Vollers S. S.; Benedek G. B.; Teplow D. B. (2003) Amyloid beta-protein (A beta) assembly: A beta 40 and A beta 42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. U. S. A. 100 (1), 330–335. 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzen N.; Nielsen S. B.; Buell A. K.; Kaspersen J. D.; Arosio P.; Vad B. S.; Paslawski W.; Christiansen G.; Valnickova-Hansen Z.; Andreasen M.; Enghild J. J.; Pedersen J. S.; Dobson C. M.; Knowles T. P. J.; Otzen D. E. (2014) The Role of Stable α-Synuclein Oligomers in the Molecular Events Underlying Amyloid Formation. J. Am. Chem. Soc. 136 (10), 3859–3868. 10.1021/ja411577t. [DOI] [PubMed] [Google Scholar]
- LeVine H. (1999) [18] Quantification of β-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 309, 274–284. 10.1016/S0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- Kaminski Schierle G. S.; van de Linde S.; Erdelyi M.; Esbjörner E. K.; Klein T.; Rees E.; Bertoncini C. W.; Dobson C. M.; Sauer M.; Kaminski C. F. (2011) In Situ Measurements of the Formation and Morphology of Intracellular β-Amyloid Fibrils by Super-Resolution Fluorescence Imaging. J. Am. Chem. Soc. 133 (33), 12902–12905. 10.1021/ja201651w. [DOI] [PubMed] [Google Scholar]
- Gremer L.; Schölzel D.; Schenk C.; Reinartz E.; Labahn J.; Ravelli R. B. G.; Tusche M.; Lopez-Iglesias C.; Hoyer W.; Heise H.; Willbold D.; Schröder G. F. (2017) Fibril structure of amyloid-β(1–42) by cryo–electron microscopy. Science 358 (6359), 116–119. 10.1126/science.aao2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlak J. B.; Hos B. J.; van de Graaff M. J.; Megantari O. A.; Meeuwenoord N.; Overkleeft H. S.; Filippov D. V.; Ossendorp F.; van Kasteren S. I. (2016) The Optimization of Bioorthogonal Epitope Ligation within MHC-I Complexes. ACS Chem. Biol. 11 (11), 3172–3178. 10.1021/acschembio.6b00498. [DOI] [PubMed] [Google Scholar]
- van der Gracht A. M. F.; de Geus M. A. R.; Camps M. G. M.; Ruckwardt T. J.; Sarris A. J. C.; Bremmers J.; Maurits E.; Pawlak J. B.; Posthoorn M. M.; Bonger K. M.; Filippov D. V.; Overkleeft H. S.; Robillard M. S.; Ossendorp F.; van Kasteren S. I. (2018) Chemical Control over T-Cell Activation in Vivo Using Deprotection of trans-Cyclooctene-Modified Epitopes. ACS Chem. Biol. 13 (6), 1569–1576. 10.1021/acschembio.8b00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxon E.; Bertozzi C. R. (2000) Cell Surface Engineering by a Modified Staudinger Reaction. Science 287 (5460), 2007–2010. 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. (2009) Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem., Int. Ed. 48 (38), 6974–6998. 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. (2002) A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective ″ligation″ of azides and terminal alkynes. Angew. Chem., Int. Ed. 41 (14), 2596–9. . [DOI] [PubMed] [Google Scholar]
- Tornøe C. W.; Christensen C.; Meldal M. (2002) Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 67 (9), 3057–3064. 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- Karttunen J.; Sanderson S.; Shastri N. (1992) Detection of rare antigen-presenting cells by the lacZ T-cell activation assay suggests an expression cloning strategy for T-cell antigens. Proc. Natl. Acad. Sci. U. S. A. 89 (13), 6020–6024. 10.1073/pnas.89.13.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolli F.; Mulinacci B.; Carotenuto A.; Bonetti B.; Sabatino G.; Mazzanti B.; D’Ursi A. M.; Novellino E.; Pazzagli M.; Lovato L.; Alcaro M. C.; Peroni E.; Pozo-Carrero M. C.; Nuti F.; Battistini L.; Borsellino G.; Chelli M.; Rovero P.; Papini A. M. (2005) An N-glucosylated peptide detecting disease-specific autoantibodies, biomarkers of multiple sclerosis. Proc. Natl. Acad. Sci. U. S. A. 102 (29), 10273–10278. 10.1073/pnas.0503178102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzucco S.; Matà S.; Vergelli M.; Fioresi R.; Nardi E.; Mazzanti B.; Chelli M.; Lolli F.; Ginanneschi M.; Pinto F.; Massacesi L.; Papini A. M. (1999) A synthetic glycopeptide of human myelin oligodendrocyte glycoprotein to detect antibody responses in multiple sclerosis and other neurological diseases. Bioorg. Med. Chem. Lett. 9 (2), 167–172. 10.1016/S0960-894X(98)00698-2. [DOI] [PubMed] [Google Scholar]
- ’t Hart B. A.; Gran B.; Weissert R. (2011) EAE: imperfect but useful models of multiple sclerosis. Trends Mol. Med. 17 (3), 119–125. 10.1016/j.molmed.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Eshaghi A.; Marinescu R. V.; Young A. L.; Firth N. C.; Prados F.; Jorge Cardoso M.; Tur C.; De Angelis F.; Cawley N.; Brownlee W. J.; De Stefano N.; Laura Stromillo M.; Battaglini M.; Ruggieri S.; Gasperini C.; Filippi M.; Rocca M. A.; Rovira A.; Sastre-Garriga J.; Geurts J. J. G.; Vrenken H.; Wottschel V.; Leurs C. E.; Uitdehaag B.; Pirpamer L.; Enzinger C.; Ourselin S.; Gandini Wheeler-Kingshott C. A.; Chard D.; Thompson A. J.; Barkhof F.; Alexander D. C.; Ciccarelli O. (2018) Progression of regional grey matter atrophy in multiple sclerosis. Brain 141 (6), 1665–1677. 10.1093/brain/awy088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagessar S. A.; Holtman I. R.; Hofman S.; Morandi E.; Heijmans N.; Laman J. D.; Gran B.; Faber B. W.; van Kasteren S. I.; Eggen B. J. L.; t Hart B. A. (2016) Lymphocryptovirus Infection of Nonhuman Primate B Cells Converts Destructive into Productive Processing of the Pathogenic CD8 T Cell Epitope in Myelin Oligodendrocyte Glycoprotein. J. Immunol. 197, 1074–1088. 10.4049/jimmunol.1600124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- t Hart B. A.; Kap Y. S.; Morandi E.; Laman J. D.; Gran B. (2016) EBV Infection and Multiple Sclerosis: Lessons from a Marmoset Model. Trends Mol. Med. 22 (12), 1012–1024. 10.1016/j.molmed.2016.10.007. [DOI] [PubMed] [Google Scholar]
- Kaminskyy V.; Zhivotovsky B. (2012) Proteases in autophagy. Biochim. Biophys. Acta, Proteins Proteomics 1824 (1), 44–50. 10.1016/j.bbapap.2011.05.013. [DOI] [PubMed] [Google Scholar]
- Hevehan D. L.; De Bernardez Clark E. (1997) Oxidative renaturation of lysozyme at high concentrations. Biotechnol. Bioeng. 54 (3), 221–30. . [DOI] [PubMed] [Google Scholar]
- Roberts C. J. (2007) Non-native protein aggregation kinetics. Biotechnol. Bioeng. 98 (5), 927–38. 10.1002/bit.21627. [DOI] [PubMed] [Google Scholar]
- Jin S.; Kedia N.; Illes-Toth E.; Haralampiev I.; Prisner S.; Herrmann A.; Wanker E. E.; Bieschke J. (2016) Amyloid-β(1–42) Aggregation Initiates Its Cellular Uptake and Cytotoxicity. J. Biol. Chem. 291 (37), 19590–19606. 10.1074/jbc.M115.691840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesén E.; Jeffries G. D. M.; Matson Dzebo M.; Esbjörner E. K. (2017) Endocytic uptake of monomeric amyloid-β peptides is clathrin- and dynamin-independent and results in selective accumulation of Aβ(1–42) compared to Aβ(1–40). Sci. Rep. 7 (1), 2021. 10.1038/s41598-017-02227-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araman C.; Thompson R. E.; Wang S.; Hackl S.; Payne R. J.; Becker C. F. W. (2017) Semisynthetic prion protein (PrP) variants carrying glycan mimics at position 181 and 197 do not form fibrils. Chem. Sci. 8 (9), 6626–6632. 10.1039/C7SC02719B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlak J. P.; Gential G. P. P.; Ruckwardt T. J.; Bremmers J. S.; Meeuwenoord J. N.; Ossendorp F. A.; Overkleeft S. H.; Filippov D. V.; van Kasteren S. I. (2015) Bioorthogonal Deprotection on the Dendritic Cell Surface for Chemical Control of Antigen Cross-Presentation. Angew. Chem., Int. Ed. 54 (19), 5628–5631. 10.1002/anie.201500301. [DOI] [PubMed] [Google Scholar]
- Lutz M. B.; Kukutsch N.; Ogilvie A. L. J.; Rößner S.; Koch F.; Romani N.; Schuler G. (1999) An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods 223 (1), 77–92. 10.1016/S0022-1759(98)00204-X. [DOI] [PubMed] [Google Scholar]
- Jagessar S. A.; Vierboom M.; Blezer E. L. A.; Bauer J.; Hart B. A. t.; Kap Y. S. (2013) An Overview of Models, Methods, and Reagents Developed for Translational Autoimmunity Research in the Common Marmoset (Callithrix jacchus). Exp. Anim. 62 (3), 159–171. 10.1538/expanim.62.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morandi E.; Jagessar S. A.; ’t Hart B. A.; Gran B. (2017) EBV Infection Empowers Human B Cells for Autoimmunity: Role of Autophagy and Relevance to Multiple SclerosisJ. J. Immunol. 199, 435–448. 10.4049/jimmunol.1700178. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.