

Abstract
Background
Pediatric low-grade glioma (pLGG) often initially responds to front-line therapies such as carboplatin, but more than 50% of treated tumors eventually progress and require additional therapy. With the discovery that pLGG often contains mammalian target of rapamycin (mTOR) activation, new treatment modalities and combinations are now possible for patients. The purpose of this study was to determine if carboplatin is synergistic with the mTOR complex 1 inhibitor everolimus in pLGG.
Methods
We treated 4 pLGG cell lines and 1 patient-derived xenograft line representing various pLGG genotypes, including neurofibromatosis type 1 loss, proto-oncogene B-Raf (BRAF)-KIAA1549 fusion, and BRAFV600E mutation, with carboplatin and/or everolimus and performed assays for growth, cell proliferation, and cell death. Immunohistochemistry as well as in vivo and in vitro metabolomics studies were also performed.
Results
Carboplatin synergized with everolimus in all of our 4 pLGG cell lines (combination index <1 at Fa 0.5). Combination therapy was superior at inhibiting tumor growth in vivo. Combination treatment increased levels of apoptosis as well as gamma-H2AX phosphorylation compared with either agent alone. Everolimus treatment suppressed the conversion of glutamine and glutamate into glutathione both in vitro and in vivo. Exogenous glutathione reversed the effects of carboplatin and everolimus.
Conclusions
The combination of carboplatin and everolimus was effective at inducing cell death and slowing tumor growth in pLGG models. Everolimus decreased the amount of available glutathione inside the cell, preventing the detoxification of carboplatin and inducing increased DNA damage and apoptosis.
Keywords: astrocytoma, gamma-H2AX, mTORC1, pediatric brain tumor, RPS6
Importance of the Study
Although most pLGG patients have a favorable prognosis, those with unresectable tumors experience significant morbidity and often require multiple lines of therapy. Mammalian TOR is known to be widely expressed in pLGG, particularly in recurrent/refractory tumors. Clinical trials of the mTORC1 inhibitor everolimus have demonstrated efficacy in some recurrent pLGG patients, but it is unknown if traditional chemotherapies will synergize with everolimus. We here show that the combination of everolimus and carboplatin synergizes in pLGG. We identify a mechanism for this synergy as a decrease in the cell’s ability to detoxify platinum-based chemotherapies. Altogether, this study validates a new therapeutic regimen that can quickly be advanced into phase I/II clinical trials for pediatric patients. Moreover, this study provides a rationale for new treatment combinations for mTORC1-based inhibitors in brain tumors.
Pediatric low-grade glioma (pLGG) is the most common brain tumor in children. Tumors located in surgically favorable sites such as the posterior fossa can often be treated by resection alone.1 However, patients with unresectable tumors or those that progress after surgery will require further treatment. Standard chemotherapy regimens include carboplatin/vincristine or thioguanine/lomustine/procarbazine/vincristine.2,3 While carboplatin-based regimens can be effective for tumors occurring in the context of neurofibromatosis type 1 (NF1) loss, approximately 50% of patients with other molecular drivers will have their tumors recur after chemotherapy.2 The high prevalence of recurrent pLGG tumors indicates that new treatment options should be explored.
Mutations that activate the mitogen-activated protein kinase (MAPK) pathway are a hallmark of pLGG,4,5 and in refractory or recurrent tumors we and others have also identified upregulation of mammalian target of rapamycin (mTOR).6,7 These pathways are highly convergent, as there are multiple inputs of the MAPK pathway into mTOR.8 The mTOR pathway regulates important cell functions, including cell cycle progression, metabolism, and survival.9 Overactivation of this pathway plays a key role in multiple cancers, including pancreatic cancer, high-grade gliomas, and pediatric gliomas.7,10–12
This discovery that relapsed/refractory pLGG activates mTOR has led our group, and others, to examine mTOR inhibitors as a potential therapy.13 We previously identified an mTOR complex 1 (mTORC1) inhibitor as having activity in some pLGG in vitro models.7 Due to the frequent combination of mTORC1 inhibitors with traditional chemotherapies,14 we hypothesized that the mTORC1 inhibitor everolimus would synergize with carboplatin, one of the standard therapies for pLGG. We chose everolimus due to its proven track record as an anticancer agent,15 its demonstrated safety and efficacy against the mTORC1-driven pediatric brain tumor subependymal giant cell astrocytoma (SEGA),16 and its evaluation in phase II trials for patients with pLGG as a single agent (NCT01734512). The combination of everolimus and carboplatin is also known to synergize in breast cancer,17 and in adult phase I and II studies was well tolerated.14,17,18 In this study, we show that carboplatin and everolimus synergize in multiple pLGG models, and provide novel metabolomics evidence for the potential mechanism of synergy.
Results
Everolimus Decreases mTORC1 Signaling and Suppresses Growth in Pediatric Low-Grade Astrocytoma Lines
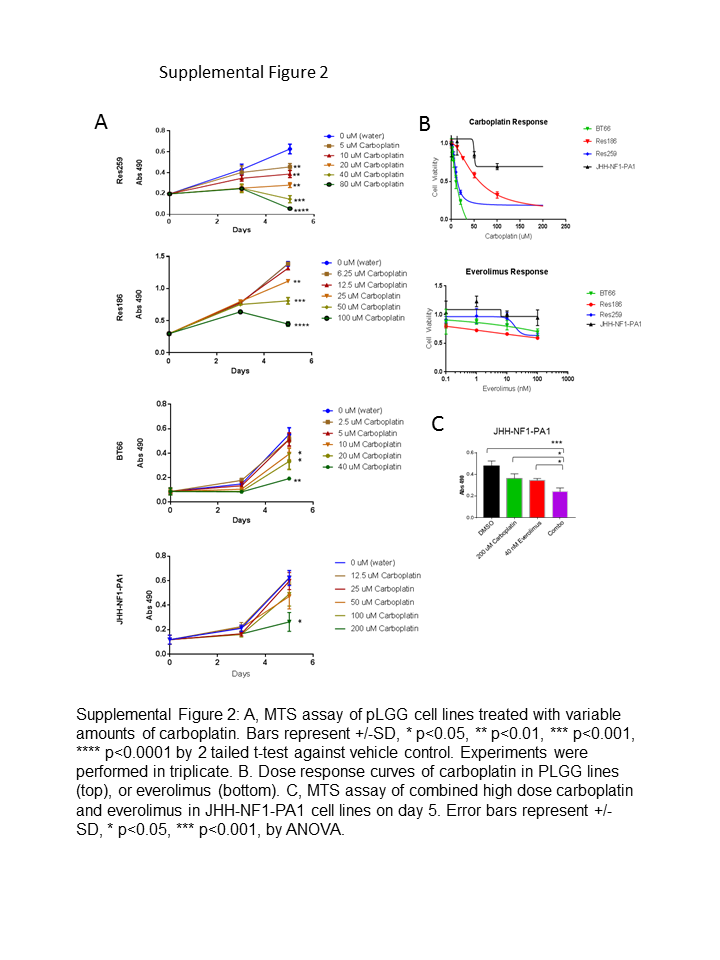
To evaluate everolimus in pLGG, we treated cell lines derived from patients and characterized by a variety of mutations commonly found in pLGG (Supplementary Table 1), as well as expression of both MAPK and mTORC1 pathway activation (Supplementary Fig. 1A). The lines include: Res186, a grade I pilocytic astrocytoma cell line; Res259, a pediatric grade II diffuse-astrocytoma cell line19; BT66, a grade I pilocytic-astrocytoma cell line harboring the proto-oncogene B-Raf (BRAF)-KIAA1549 fusion and immortalized with an inducible large T-antigen20 (Supplementary Fig. 1B); and JHH-NF1-PA1, a grade I pilocytic-astrocytoma line that lacks NF1 protein expression (Supplementary Fig. 2C), derived from a patient with type 1 neurofibromatosis. The JHH-NF1-PA1 line was developed from a biopsy performed on an optic pathway tumor that progressed after treatment with first carboplatin and then everolimus as single agents.
Six hours post everolimus treatment, Res186 and JHH-NF1-PA1 exhibited a dose-dependent decrease in mTOR activation, with significant phospho-S6 reductions (>50%) seen at 0.1 nM. Res259 and BT66 exhibited a phospho-S6 decrease at 1 nM and 10 nM, respectively (Fig. 1A). Growth was inhibited by everolimus for Res186 in picomolar range (P < 0.0001 by t-test) and in the nanomolar range for BT66 (P < 0.05 by t-test). Res259 and JHH-NF1-PA1 did not show any significant growth decrease (Fig. 1B). Bromodeoxyuridine (BrdU) incorporation assay also found that everolimus significantly decreased the proliferation of Res186 at 1 nM (P = 0.0006) and BT66 at 10 nM (P = 0.015 by t-test against vehicle control). Res259 and JHH-NF1-PA1 showed no decrease in proliferation at any of the doses used (Fig. 1C). These results were concordant with our previous paper in which Res186 showed sensitivity to the mTORC1 inhibitor ridaforolimus, while Res259 was resistant.7 Upon everolimus treatment, both Res186 and Res259 exhibited mTORC2 pathway activation, even while mTORC1 was inhibited (Supplementary Fig. 1D).
Fig. 1 .

Everolimus inhibits the growth of pLGG lines. (A) Western blot of phospho-S6, total S6, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in Res186, Res259, BT66, and JHH-NF1-PA1. Cells were incubated with 0, 0.1, 1, or 10 nM of everolimus for 6 hours. (B) MTS assay of cellular proliferation after cells were treated with 0.1–10 nM everolimus. Each condition was performed in triplicate. (C) Quantification of BrdU. Bars represent ±SD. Experiment was performed in 3 distinct biological replicates. Bars represent ±SD; *P < 0.05, ***P < 0.001, ****P < 0.001 against 0 nM by 2-tailed t-test.
Everolimus Is Synergistic with Carboplatin in pLGG Cell Lines
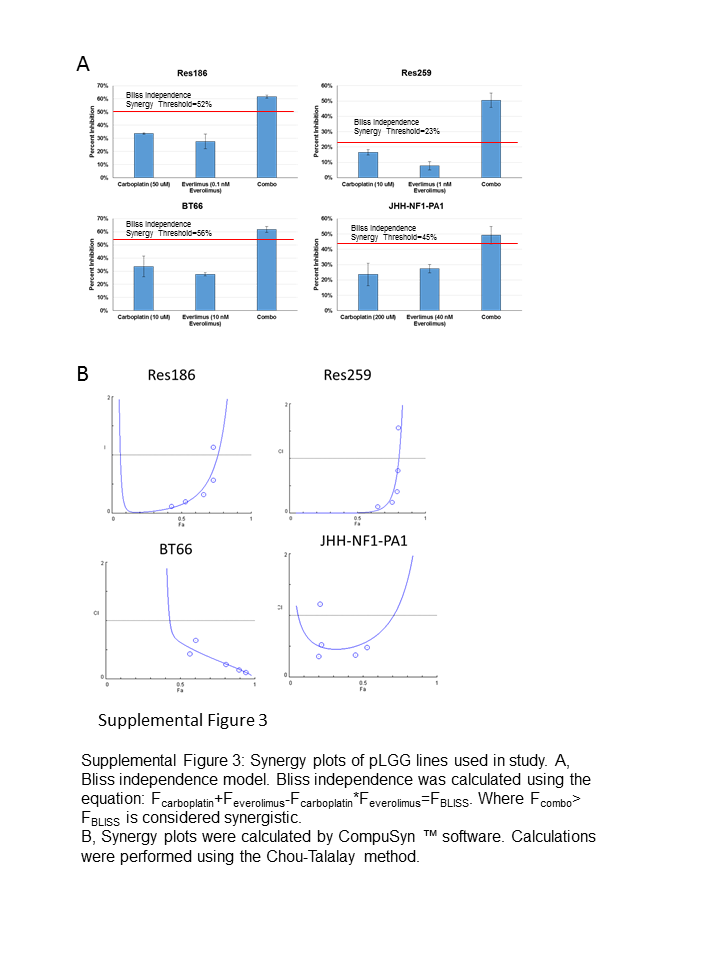
To determine if carboplatin synergized with everolimus, we first identified the inhibitory concentration of 25% (IC25) and the half-maximal inhibitory concentration (IC50) of each cell line for carboplatin using 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay. Res186 was relatively resistant to carboplatin, while Res259 and BT66 were more sensitive. JHH-NF1-PA1 was the most resistant, with an IC50 >100 μM (Supplementary Fig. 2A, B), consistent with these cells being derived from a tumor that progressed after carboplatin treatment. While Res259 showed resistance to everolimus, 1 nM everolimus in combination with 10 μM carboplatin resulted in dramatic growth inhibition (69% reduction, P < 0.0001 via ANOVA against vehicle control). Both Res186 and BT66 also showed growth inhibition when low concentrations of everolimus were combined with carboplatin (Fig. 2A, Res186: 52% reduction, P = 0.003 and BT66: 68% reduction, P < 0.0001 via ANOVA against vehicle control). The doses used were found to be synergistic according to the Bliss independence model (Supplementary Fig. 3A).21 Everolimus concentrations for Chou–Talalay synergy studies in Res186, Res259, and BT66 were determined based on the approximate IC50 of p-S6. Res186, Res259, and BT66 demonstrated formal carboplatin and everolimus synergy with a combination index >1 for most concentrations (Supplementary Fig. 3B, Supplementary Table 2).22 The JHH-NF1-PA1 cell line did not show synergy at low concentrations of carboplatin and everolimus (Fig. 2A); however, it did show synergy at higher concentrations (Supplementary Fig. 2C). Experiments using BrdU incorporation were concordant with the assay by MTS and demonstrated decreased proliferation for Res186, Res259, and BT66 cell lines after 96 hours (P < 0.001 for all cell lines compared with single treated cells and control cells via ANOVA) (Fig. 2B–D).
Fig. 2.

Everolimus synergizes with carboplatin in pLGG lines. (A) MTS assay of carboplatin and everolimus in Res186, Res259, BT66, and JHH-NF1-PA1 cell lines on day 5. Carboplatin concentrations were determined based on IC25 from previous single dose experiments. Error bars represent ±SD; **P < 0.01, ***P < 0.001, ****P < 0.0001 by ANOVA. (B) Left: 200× fluorescent images of Res186 treated with DMSO, 0.1 nM everolimus, 50 μM carboplatin, or combination for 96 hours. Right: quantification of BrdU positive cells. (C) Left: 200× fluorescent images of Res259 treated with DMSO, 1 nM everolimus, 10 μM carboplatin, or combination for 96 hours. (D) Left: 200× fluorescent images of BT66 treated with DMSO, 10 nM everolimus, 10 uM carboplatin, or combination for 96 hours. Stain of 4′,6′-diamidino-2-phenylindole (blue) and BrdU (red). Right: quantification of BrdU. Bars represent ±SD; *P < 0.05, **P < 0.01, ****P < 0.0001 by ANOVA. Experiment was performed in 3 distinct biological replicates. Scale bar is 50 microns.
Everolimus Is Synergistic with Carboplatin in a pLGG In Vivo Model
To further investigate whether carboplatin is better at reducing tumor growth in combination with everolimus, we treated mice bearing BT40 gliomas. The BT40 patient-derived xenograft line is a previously characterized pediatric pilocytic astrocytoma tumor model containing a BRAFV600E mutation23–25 (Supplementary Fig. 1A). BT40 grows best as a flank tumor, and all experiments were performed using this modality.25 Mice received 50 mg/kg carboplatin weekly26 and/or 5 mg/kg everolimus 3 times a week27,28 or vehicle. The combination of carboplatin and everolimus reduced tumor growth better than either agent alone (P < 0.001 by ANOVA). The final tumor volume of the combination-treated mice was also reduced compared with either agent alone (P < 0.005, and P < 0.05 against either agent alone by ANOVA; Fig. 3B). Mice did not show any overt signs of toxicity from carboplatin, everolimus, or the combination at the doses used.
Fig. 3 .

Carboplatin plus everolimus suppresses the growth of the BRAFV600E-driven glioma model BT40. (A) Fold change of tumor growth with 50 mg/kg carboplatin 1/week i.p. (n = 17), 5 mg/kg 3×/week p.o. (n = 16), vehicle (n = 9), or combination (n = 20). Treatment was initiated when tumor volume was ~200 mm3. Mice were sacrificed on day 18 and tumors flash frozen for protein. (B) Chart of tumor volume at time of sacrifice. Each data point represents an individual tumor. Bars represent ±SD; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by ANOVA. (C) Western blot of p-S6, total-S6, p-Rb (Serine780), total Rb, gamma-H2AX, and GAPDH from tumors. Values were normalized to GAPDH expression and vehicle control.
We next assessed the downstream targets of everolimus and carboplatin in BT40 tumors in vivo. Western blotting and immunohistochemistry (IHC) showed decreased phospho-S6 in everolimus-treated tumors compared with vehicle or carboplatin-treated ones (Fig. 3C, Supplementary Fig. 4). To measure cell cycle progression, we probed for phospho-retinoblastoma protein (p-Rb; ser780) and found that everolimus or combination therapy decreased p-Rb greater than carboplatin or control (Fig. 3C). We also measured gamma-H2AX, a marker for DNA double stranded breaks that is often used as a damage indicator of platinum-based chemotherapy.29,30 We detected increased gamma-H2AX via western blot in mice treated with carboplatin plus everolimus compared with either agent alone (Fig. 3C).
Combination of Carboplatin and Everolimus Induces Apoptosis and DNA Damage
To determine the potential mechanism of synergy, we examined different apoptotic and DNA damage markers post treatment. We found that for Res186 and Res259, the combination of carboplatin and everolimus resulted in increased cleaved caspase-3 (CC3) staining, which is a marker for apoptosis (Res186: 921% increase, P < 0.0001 by ANOVA; Res259: 959% increase, P < 0.0001 by ANOVA compared with vehicle control) (Fig. 4A, B). Everolimus alone did not increase apoptosis.
Fig. 4 .

Carboplatin and everolimus induce apoptosis and increased DNA damage. (A) Left: 200× fluorescent images of Res186 treated with DMSO, 0.1 nM everolimus, 50 μM carboplatin, or combination for 96 hours. Stains of 4′,6′-diamidino-2-phenylindole (DAPI) nuclei (blue) and CC3 (red) indicate apoptotic cells. Right: quantification of cells that were CC3 positive. (B) Left: 200× fluorescent images of Res259 treated with DMSO, 1 nM everolimus, 10 μM carboplatin, or combination for 96 hours. DAPI stains nuclei (blue) and CC3 (red) indicate apoptotic cells. Right: quantification of cells that were CC3 positive. Bars represent SD; **P < 0.01, ***P < 0.001, ****P < 0.0001 by ANOVA. (C) Western blot of gamma-H2AX, cleaved PARP, phospho-S6, and (GAPDH) for Res186, Res259, and BT66 cell lines treated for 96 hours. Normalized quantification of fold induction is indicated under blots. Scale bar is 50 microns.
We then probed Res186, Res259, and BT66 by western blot for cleaved poly(ADP-ribose) polymerase (PARP), an additional apoptotic marker, and gamma-H2AX. In all cell lines, carboplatin induced elevated levels of cleaved PARP, while everolimus did not. We found that combination therapy increased cleaved PARP expression over that of carboplatin alone (Fig. 4C, Res186: 220% increase, Res259: 230% increase, BT66: 930% increase, compared with carboplatin alone). We also found that the combination of carboplatin and everolimus increased levels of gamma-H2AX in all cell lines, more so than carboplatin alone (Fig. 4C, Res186: 1800% increase, Res259: 154% increase, BT66: 280% increase, compared with carboplatin treated condition). Due to the increased presence of gamma-H2AX post combination therapy and the lack of CC3 and cleaved PARP increase from everolimus alone, we hypothesized that everolimus increased the effectiveness of carboplatin.
Everolimus Decreases Intracellular Glutathione Levels
A key mechanism for detoxification of platinum-containing drugs is glutathione-mediated reduction of the reactive platinum moiety.31 Because mTORC1 is known to promote glutathione production,32,33 we hypothesized that everolimus would decrease intracellular glutathione to potentiate carboplatin-mediated DNA damage. To explore the mechanisms for carboplatin’s synergy with everolimus, we measured glutathione levels after treatment.
To test if glutathione production was diminished in pLGG cell lines after everolimus treatment, we performed flux metabolomic analysis of cells incubated with stable isotopically labeled glutamine (13C5, 15N2). We used isotopically labeled glutamine because it is the primary source of glutamate inside most cells, which is a precursor to glutathione. If glutathione is to be directly synthesized from the glutamine (M+7) label, it would be converted into a glutamate with 5 isotopically labeled carbons and 1 isotopically labeled nitrogen (M+6) (Fig. 5A). This isotope distribution should remain in glutathione (M+6), because none of glutamate’s labeled carbon or nitrogen is removed in subsequent metabolic reactions. We found a significant decrease of the glutamine (M+7) label going into glutamate (M+6), indicating glutaminase inhibition from everolimus treatment. Glutathione (M+6) itself was approximately 50% reduced in both Res186 and Res259 cell lines (P < 0.0001) (Fig. 5B).
Fig. 5.

Everolimus depletes glutathione in pLGG cells. (A) Diagram showing the pathway for isotopically labeled carbon (13C) and nitrogen (15N) derived from fully labeled glutamine. (B) Evaluation of isotopically labeled glutamine (M+7), glutamate (M+6), and glutathione (M+6) relative intensities 24 hours post everolimus treatment (Res259 1 nM everolimus, Res186 0.1 nM everolimus). (C) Comparison of un-labeled glutamine, glutamate, and glutathione in BT40 tumors. Tumors were treated with 5 mg/kg everolimus. Bars represent ±SD, **P < 0.01, ***P < 0.001, ****P < 0.0001 against vehicle control by 2-tailed t-test. Each data point represents a cell culture replicate (B) or xenograft tumor (C).
We also evaluated the metabolic alterations that occurred in BT40 tumors treated with everolimus in vivo. The results were concordant with our in vitro stable isotope results. Glutamine remained unchanged; however, glutamate pools were reduced by 35% (P < 0.0001) and glutathione pools were reduced by 42% (P < 0.0001) (Fig. 5C).
Glutathione Rescues Cells from Carboplatin and Everolimus Combination Treatment
We next wanted to test if the gamma-glutamyl-cysteine synthetase inhibitor buthionine sulfoximine (BSO), which depletes glutathione,34 would mimic the effect of everolimus treatment. When carboplatin was combined with BSO in all pLGG cell lines, there was a greater phosphorylation of gamma-H2AX in all cell lines except for JHH-NF1-PA1, which never showed an increase in gamma-H2AX (Fig. 6A, Res186: 150% increase, Res259: 760% increase, BT66: 290% increase compared with carboplatin treatment). Res186 and Res259 exhibited significant reduced growth after combined treatment compared with either agent alone (P < 0.05 by ANOVA compared with carboplatin alone for both cell lines). In JHH-NF1-PA1, however, combination therapy did not reduce growth compared with BSO or carboplatin (Supplementary Fig. 5).
Fig. 6 .

Glutathione rescues cells treated with carboplatin and everolimus. (A) Western blot of gamma-H2AX of pLGG cell lines treated with carboplatin or BSO for 96 hours. Numbers represent quantification of western blot expression normalized to GAPDH and vehicle. (B) Phase contrast 200× photomicrographs of cell treated with DMSO, carboplatin plus everolimus, or carboplatin, everolimus, and 4 mM glutathione for 96 hours in Res259, showing induction of cell death in combination therapy, which is prevented by glutathione supplementation. (C) Quantification of Res186 and Res259 cells that were BrdU positive by immunofluorescence 96 hours post carboplatin, everolimus, and/or glutathione treatment. (D) Quantification of Res186 and Res259 cells that were CC3 positive by immunofluorescence 96 hours post treatment. Bars represent range of samples with interquartile distribution; **P < 0.01, ****P < 0.0001 by ANOVA. (E) Cartoon of proposed mechanism in which everolimus decreases the available glutathione pool, which would normally prevent carboplatin from inducing DNA damage. Suppression of glutathione production leads to increased carboplatin-induced DNA damage and increased cell death. Scale bar is 50 microns.
Finally, we examined whether exogenous glutathione could rescue cells from carboplatin and everolimus. Exogenous glutathione visibly rescued cells from the effect of carboplatin and everolimus combination in Res259 cells (Fig. 6B). Combining glutathione with carboplatin and everolimus rescued Res186 and Res259 in our BrdU assay as well (P < 0.01 by ANOVA compared with carboplatin/everolimus treated cells). Glutathione alone had no effect on overall BrdU incorporation (Fig. 6C, Res186: P = 0.99, Res259: P = 0.90 by ANOVA against vehicle control). Glutathione also rescued cells from apoptosis, with the combination of carboplatin and everolimus showing significantly higher CC3 expression than any group treated with glutathione (P < 0.0001 by ANOVA compared with carboplatin/everolimus treated cells). Overall, our data suggest that everolimus diminishes the available glutathione pool, increasing the efficacy of carboplatin inside the cell (summarized in cartoon in Fig. 6E).
Discussion
While pLGG has a favorable overall prognosis, many patients suffer lifelong disability due to the proximity of the tumor to the optic pathway or midline structures.35 With the discovery that many relapsed/refractory pLGGs have activation of mTOR,7,36 more treatment options may be possible for patients, including everolimus, a brain-penetrant drug already FDA approved for treatment of SEGA in children.16 Due to the success of mTORC1 inhibitors being used in combination with traditional chemotherapy in other tumor types, we hypothesized that everolimus would synergize with carboplatin, one of the first-line therapies for pLGG.
To this end, we examined the effects of everolimus alone in pLGG cell lines, including those representing BRAFV600E-activated tumors (BT40), NF1 deleted tumors (JHH-NF1), and BRAF-KIAA1549 fusion (BT66). The lines showed variable response to everolimus, with Res186 the most sensitive, and JHH-NF1-PA1 and Res259 being the most resistant. The source of Res186’s sensitivity could be due to its deletion of phosphatase and tensin homolog (PTEN), which is a known factor for sensitivity to mTORC1 inhibition in certain cell lines.37 However, it is also possible that Res259 is more dependent upon mTORC2 for proliferation, rather than mTORC1, as evidenced by the robustly increased phospho-AKT expression we observe upon everolimus treatment (Supplementary Fig. 1D). However, while all cell lines had variable responses to everolimus, the combination of everolimus and carboplatin was effective at low concentrations in all but the JHH-NF1-PA1 cell line. For the Res259, Res186, and BT66 cell lines, the combination index at Fa 0.5 was <1, showing that the drugs work synergistically. This combination therapy also worked in our BT40 model, reducing tumor growth and tumor volume compared with monotherapy. And while the JHH-NF1-PA1 did show synergy at higher concentrations, these concentrations exceed what can safely be achieved in patients. However, it is unsurprising that JHH-NF1-PA1 was resistant to the combination considering that the patient from whom this line was derived received both carboplatin and everolimus as single agents. Thus, JHH-NF1-PA1 likely became resistant to everolimus treatment and is not dependent upon mTORC1 for growth. In addition, the media for JHH-NF1-PA1 contain the Rho kinase (ROCK) inhibitor Y-27632, which inhibits apoptosis through suppression of caspase-3 activity.38 Both of these reasons could explain why carboplatin and everolimus did not show synergy until high concentrations were used in this line.
Platinum-based drugs work by inducing DNA adducts, causing the cell to try and repair the damage, senesce, or undergo apoptosis.39 A hallmark of this injury is increased phosphorylation of a double stranded DNA damage marker, gamma-H2AX.29 We found that the combination of everolimus with carboplatin induced higher phosphorylation of gamma-H2AX than carboplatin alone. This was not an additive effect, because everolimus alone did not induce increased gamma-H2AX phosphorylation. We found elevated levels of gamma-H2AX in our BT40 mouse derived xenografts as well. Thus, we hypothesized that everolimus increases the ability of carboplatin to induce DNA damage.
We then established that everolimus reduced the amount of available glutathione using analysis of isotopically labeled glutamine incorporation into glutathione in vitro, and unlabeled glutathione levels in vivo. The isotope tracing suggests that everolimus decreased the catabolism of glutamine, thus reducing the available glutamate pool that can be converted into glutathione. In further support of this mechanism, we found that the glutathione depleting drug BSO phenocopied everolimus in combination with carboplatin. Furthermore, exogenous glutathione completely mitigated the effects of carboplatin and everolimus in pLGG lines. This suggests that carboplatin and everolimus synergy is at least partially mediated through glutathione metabolism.
It is important to recognize that the cell lines used in this manuscript, while they carry driver mutations found in pLGG, such as BRAF mutations and deletions of cyclin-dependent kinase inhibitor 2A,40 also have growth characteristics not normally found in pLGG, such as a fast doubling time and, in the case of BT40, aggressive tumor formation in mice. Also, as evidenced by our development of the NF1-derived low-grade glioma cell line used in this manuscript, we are actively attempting to develop new, high-fidelity models for drug testing. Acknowledging the limitations of the available models, the experiments presented here indicate the potential therapeutic benefit of combining carboplatin with everolimus in pLGG patients. Mechanistically, everolimus decreases the synthesis of glutathione inside the cell, explaining at least partially why carboplatin synergized with the mTORC1 inhibitor everolimus. Evidence that everolimus works with carboplatin in pLGG could help shape future treatment options for children with low-grade glioma.
Materials and Methods
Cell Culture
Res259 and Res186 cell lines were provided by Dr Chris Jones (Institute of Cancer Research, Sutton, UK).19 Lines were maintained as monolayers in Dulbecco’s modified Eagle’s medium (DMEM)/F12 Ham’s medium containing 10% fetal bovine serum and 1% penicillin-streptomycin. Medium containing 2% fetal bovine serum was used in all drug treatment assays. BT66 was provided by Drs Till Milde and Olaf Witte (DKFZ, Heidelberg, Germany) and grown in ABM basal medium (Lonza; CC-3187, US) supplemented with AGM SingleQuot Kit Supplements & Growth Factors (Lonza; CC-4123, US) with 1 μg/mL doxycycline. Confirmation of BRAF-KIAA1549 fusion and BRAFV600E mutation was determined through PCR, as previously described20,41 (Supplementary Fig. 1B, C). JHH-NF1-PA1 was generated by our group and will be described in detail elsewhere (M. Yuan, unpublished data). Briefly, the line was derived from a 16-year-old male with germline NF1 with a refractory hypothalamic/optic pathway pilocytic astrocytoma. The patient was treated first with carboplatin, then with bevacizumab, and lastly with everolimus, with progression prior to biopsy and placement of cells into culture. This line was created from a patient biopsy under an institutional review board–approved consent protocol. Immunoblot confirmed 100% loss of NF1 protein in cultured cells. JHH-NF1-PA1 cells were grown in 50% DMEM/F12 and 50% DMEM/F12 NIH 3T3-conditioned media with the addition of ROCK inhibitor (Selleckchem # S1049).42 All cell lines were verified to be Mycoplasma free by periodic PCR testing. Cell line identity testing is reported in Supplementary Fig. 6. Cells were maintained in a humidified 37°C incubator with 5% CO2. Carboplatin (# S1215) and everolimus (# S1120) were purchased through Selleckchem.
Synergy Calculations
Synergy calculations were determined using the Chou‒Talalay method22 or Bliss independence model.21 Data analysis was conducted using CompuSyn software (http://www.combosyn.com).
Metabolomic and Stable Isotope Analysis
Cells were treated 24 hours with everolimus then incubated 2 hours with 4 mM glutamine (13C5, 15N2, 99% purity) label from Cambridge Isotope (# CNLM-1275-H-0.5). Samples were then washed with phosphate buffered saline and metabolites extracted with pre-chilled 80% high performance liquid chromatography (HPLC) grade methanol (MeOH).
Tumors were treated with 5 mg/kg everolimus or vehicle 6 hours before being sacrificed. Tumors were flash frozen in liquid nitrogen and ground with mortar and pestle. Fifty to 100 milligrams of tumor powder was added to 5 bed volumes of pre-chilled 80% HPLC grade MeOH.
Samples were centrifuged at 14000 × rpm for 10 minutes at 4°C, and the supernatants were transferred to glass insert liquid chromatography vials. Analyses occurred on an Agilent 1290 liquid chromatography system coupled to an Agilent 6520 quadrupole time of flight mass spectrometer. Samples (5 µL) were injected and separated on a Waters Acquity UPLC BEH (bridged ethyl hybrid) Amide 1.7 µm 2.1 × 100 mm HILIC (hydrophilic interaction liquid chromatography) column with a flow rate of 0.3 mL/minute. Mobile phases consisted of A (water +0.1% formic acid) and B (acetonitrile+0.1% formic acid). The column was equilibrated at 2.5/97.5 (A/B) and maintained for 1 minute post injection. Mobile-phase A increased in a linear gradient from 2.5% to 65% from 1 to 9 minutes post injection then stepped to 97.5% A from 9 to 11 minutes to wash the column. Column was equilibrated in starting condition for 3 minutes before the next injection. The mass spectrometer, equipped with a dual electrospray ionization source, was run in negative ion and then positive ion mode. The scan range was 50–1600 m/z. The source settings consisted of: drying gas flow rate: 11 L/min; nebulizer: 40 pounds per square inch gauge; gas temp: 350ºC; capillary voltage: 3000 V (neg), 2500 V (pos).
Liquid chromatography–mass spectrometry data were analyzed using Agilent Qualitative Analysis B.07.00 and Metabolomic Analysis and Visualization ENgine (MAVEN).43 Metabolite identification was determined using standards and fragmentation. Cell based experiments were normalized based on total ion chromatogram, and in vivo based experiments were normalized based on weight of tumor sample extracted.
In Vitro Growth, Proliferation, and Apoptosis Assays
MTS growth assay (Promega) was performed as previously described.44 Cleaved caspase-3 and BrdU incorporation fluorescence assays were performed as previously described.45,46
Immunoblotting
Western blot and protein extraction procedures were performed as previously described.44 Antibodies used and dilutions are listed in Supplementary Table 3.
In Vivo Studies
Four- to 6-week-old female athymic nu/nu mice from Charles River were injected with 1 × 106 BT40 cells. When tumors reached ~200 mm3, mice received 50 mg/kg i.p. carboplatin weekly,26 and/or 5 mg/kg everolimus p.o. 3 times/week,27,28 or vehicle (0.5% dimethyl sulfoxide [DMSO] in water, p.o.). Dosing represents the standard preclinical doses of these medications and translates to approximately 150 mg/m2 of carboplatin and 15 mg/m2 of everolimus.47 Higher dosing in mice compared with humans (pediatric maximum tolerated dose is 5 mg/m2/d) is likely related to the much shorter half-life of everolimus in mice (~5 hours in mice compared with 18 hours in pediatric brain tumor patients) and differences in dosing scheme (3× weekly in mice versus daily in humans).27,48 Mice were sacrificed 18 days post treatment and tumors flash frozen in liquid nitrogen for protein and metabolomics studies. Tumor volumes were measured using the following formula: V = (largest tumor dimension) × (smallest tumor dimension)2 × 0.52. The “Principles of Laboratory Animal Care” (NIH publication no. 8623, revised 1985) was followed, with protocol approved by the Johns Hopkins Animal Care and Use committee. All studies performed were in compliance with the United States Animal Welfare Act regulations and Public Health Service Policy. Tumors were fixed with 4% paraformaldehyde for IHC post treatment. IHC was performed as previously described.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism or Microsoft Excel. Single group comparisons were done using 2-sided Student’s t-test. Multiple group comparisons were done using one-way ANOVA with Dunnett multiple comparisons post-test. P-values < 0.05 were considered significant.
Supplementary Material
Supplementary data are available at Neuro-Oncology online.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Funding
This work was supported by Johns Hopkins Institute of Clinical and Translational Sciences (NCATS 5UL1TR001079) (to B.S.S); NCI Core Grant to the Sidney Kimmel Comprehensive Cancer Center (3P30CA006973-52S3) (E.H.R); Giant Food Pediatric Cancer Research Fund (E.H.R); Imagine an Answer to Kid’s Brain Cancer Foundation (E.H.R); and A Kid’s Brain Tumor Cure Foundation (E.H.R).
Conflict of interest statement. No conflicts.
References
- 1. Terashima K, Chow K, Jones J, et al. Long-term outcome of centrally located low-grade glioma in children. Cancer. 2013;119(14):2630–2638. [DOI] [PubMed] [Google Scholar]
- 2. Ater JL, Zhou T, Holmes E, et al. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(21):2641–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lassaletta A, Scheinemann K, Zelcer SM, et al. Phase II weekly vinblastine for chemotherapy-naïve children with progressive low-grade glioma: a Canadian Pediatric Brain Tumor Consortium Study. J Clin Oncol. 2016;34(29):3537–3543. [DOI] [PubMed] [Google Scholar]
- 4. Bar EE, Lin A, Tihan T, Burger PC, Eberhart CG. Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J Neuropathol Exp Neurol. 2008;67(9):878–887. [DOI] [PubMed] [Google Scholar]
- 5. Raabe E, Kieran MW, Cohen KJ. New strategies in pediatric gliomas: molecular advances in pediatric low-grade gliomas as a model. Clin Cancer Res. 2013;19(17):4553–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mueller S, Phillips J, Onar-Thomas A, et al. PTEN promoter methylation and activation of the PI3K/Akt/mTOR pathway in pediatric gliomas and influence on clinical outcome. Neuro Oncol. 2012;14(9):1146–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hütt-Cabezas M, Karajannis MA, Zagzag D, et al. Activation of mTORC1/mTORC2 signaling in pediatric low-grade glioma and pilocytic astrocytoma reveals mTOR as a therapeutic target. Neuro Oncol. 2013;15(12):1604–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36(6):320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yao JC, Shah MH, Ito T, et al. ; RAD001 in Advanced Neuroendocrine Tumors, Third Trial (RADIANT-3) Study Group Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Akhavan D, Cloughesy TF, Mischel PS. mTOR signaling in glioblastoma: lessons learned from bench to bedside. Neuro Oncol. 2010;12(8):882–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Matsubara S, Ding Q, Miyazaki Y, Kuwahata T, Tsukasa K, Takao S. mTOR plays critical roles in pancreatic cancer stem cells through specific and stemness-related functions. Sci Rep. 2013;3:3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kaul A, Toonen JA, Cimino PJ, Gianino SM, Gutmann DH. Akt- or MEK-mediated mTOR inhibition suppresses Nf1 optic glioma growth. Neuro Oncol. 2015;17(6):843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hauke RJ, Infante JR, Rubin MS, Shih KC, Arrowsmith ER, Hainsworth JD. Everolimus in combination with paclitaxel and carboplatin in patients with metastatic melanoma: a phase II trial of the Sarah Cannon Research Institute Oncology Research Consortium. Melanoma Res. 2013;23(6):468–473. [DOI] [PubMed] [Google Scholar]
- 15. Oudard S, Medioni J, Aylllon J, et al. Everolimus (RAD001): an mTOR inhibitor for the treatment of metastatic renal cell carcinoma. Expert Rev Anticancer Ther. 2009;9(6):705–717. [DOI] [PubMed] [Google Scholar]
- 16. Franz DN, Agricola K, Mays M, et al. Everolimus for subependymal giant cell astrocytoma: 5-year final analysis. Ann Neurol. 2015;78(6):929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Singh J, Novik Y, Stein S, et al. Phase 2 trial of everolimus and carboplatin combination in patients with triple negative metastatic breast cancer. Breast Cancer Res. 2014;16(2):R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schwarzlose-Schwarck S, Scholz CW, Regierer AC, et al. The mTOR inhibitor everolimus in combination with carboplatin in metastatic breast cancer—a phase I trial. Anticancer Res. 2012;32(8):3435–3441. [PubMed] [Google Scholar]
- 19. Bax DA, Little SE, Gaspar N, et al. Molecular and phenotypic characterisation of paediatric glioma cell lines as models for preclinical drug development. PLoS One. 2009;4(4):e5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Selt F, Hohloch J, Hielscher T, et al. Establishment and application of a novel patient-derived KIAA1549:BRAF-driven pediatric pilocytic astrocytoma model for preclinical drug testing. Oncotarget. 2017;8(7):11460–11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Foucquier J, Guedj M. Analysis of drug combinations: current methodological landscape. Pharmacol Res Perspect. 2015;3(3):e00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2):440–446. [DOI] [PubMed] [Google Scholar]
- 23. Kolb EA, Gorlick R, Houghton PJ, et al. Initial testing (stage 1) of AZD6244 (ARRY-142886) by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. 2010;55(4):668–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schiffman JD, Hodgson JG, VandenBerg SR, et al. Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Cancer Res. 2010;70(2):512–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bid HK, Kibler A, Phelps DA, et al. Development, characterization, and reversal of acquired resistance to the MEK1 inhibitor selumetinib (AZD6244) in an in vivo model of childhood astrocytoma. Clin Cancer Res. 2013;19(24):6716–6729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kulkarni-Datar K, Orsulic S, Foster R, Rueda BR. Ovarian tumor initiating cell populations persist following paclitaxel and carboplatin chemotherapy treatment in vivo. Cancer Lett. 2013;339(2):237–246. [DOI] [PubMed] [Google Scholar]
- 27. O’Reilly T, McSheehy PM, Kawai R, et al. Comparative pharmacokinetics of RAD001 (everolimus) in normal and tumor-bearing rodents. Cancer Chemother Pharmacol. 2010;65(4):625–639. [DOI] [PubMed] [Google Scholar]
- 28. Crazzolara R, Cisterne A, Thien M, et al. Potentiating effects of RAD001 (everolimus) on vincristine therapy in childhood acute lymphoblastic leukemia. Blood. 2009;113(14):3297–3306. [DOI] [PubMed] [Google Scholar]
- 29. Olive PL, Banáth JP. Kinetics of H2AX phosphorylation after exposure to cisplatin. Cytometry B Clin Cytom. 2009;76(2):79–90. [DOI] [PubMed] [Google Scholar]
- 30. Stefanou DT, Bamias A, Episkopou H, et al. Aberrant DNA damage response pathways may predict the outcome of platinum chemotherapy in ovarian cancer. PLoS One. 2015;10(2):e0117654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7(8):573–584. [DOI] [PubMed] [Google Scholar]
- 32. Karmaus PWF, Herrada AA, Guy C, et al. Critical roles of mTORC1 signaling and metabolic reprogramming for M-CSF-mediated myelopoiesis. J Exp Med. 2017;214(9):2629–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li J, Shin S, Sun Y, et al. mTORC1-driven tumor cells are highly sensitive to therapeutic targeting by antagonists of oxidative stress. Cancer Res. 2016;76(16):4816–4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Drew R, Miners JO. The effects of buthionine sulphoximine (BSO) on glutathione depletion and xenobiotic biotransformation. Biochem Pharmacol. 1984;33(19):2989–2994. [DOI] [PubMed] [Google Scholar]
- 35. Sievert AJ, Fisher MJ. Pediatric low-grade gliomas. J Child Neurol. 2009;24(11):1397–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rodriguez FJ, Raabe EH. mTOR: a new therapeutic target for pediatric low-grade glioma?CNS Oncol. 2014;3(2):89–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Neshat MS, Mellinghoff IK, Tran C, et al. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci U S A. 2001;98(18):10314–10319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu Y, Shu J, He C, et al. ROCK inhibitor Y27632 promotes proliferation and diminishes apoptosis of marmoset induced pluripotent stem cells by suppressing expression and activity of caspase 3. Theriogenology. 2016;85(2):302–314. [DOI] [PubMed] [Google Scholar]
- 39. Jung Y, Lippard SJ. Direct cellular responses to platinum-induced DNA damage. Chem Rev. 2007;107(5):1387–1407. [DOI] [PubMed] [Google Scholar]
- 40. Raabe EH, Lim KS, Kim JM, et al. BRAF activation induces transformation and then senescence in human neural stem cells: a pilocytic astrocytoma model. Clin Cancer Res. 2011;17(11):3590–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huang T, Zhuge J, Zhang WW. Sensitive detection of BRAF V600E mutation by Amplification Refractory Mutation System (ARMS)-PCR. Biomark Res. 2013;1(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu X, Ory V, Chapman S, et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol. 2012;180(2):599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Melamud E, Vastag L, Rabinowitz JD. Metabolomic analysis and visualization engine for LC-MS data. Anal Chem. 2010;82(23):9818–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kaur H, Hütt-Cabezas M, Weingart MF, et al. The chromatin-modifying protein HMGA2 promotes atypical teratoid/rhabdoid cell tumorigenicity. J Neuropathol Exp Neurol. 2015;74(2):177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Weingart MF, Roth JJ, Hutt-Cabezas M, et al. Disrupting LIN28 in atypical teratoid rhabdoid tumors reveals the importance of the mitogen activated protein kinase pathway as a therapeutic target. Oncotarget. 2015;6(5):3165–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hanaford AR, Archer TC, Price A, et al. DiSCoVERing innovative therapies for rare tumors: combining genetically accurate disease models with in silico analysis to identify novel therapeutic targets. Clin Cancer Res. 2016;22(15):3903–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7(2):27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fouladi M, Laningham F, Wu J, et al. Phase I study of everolimus in pediatric patients with refractory solid tumors. J Clin Oncol. 2007;25(30):4806–4812. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.