

Abstract
The RNA-to-cDNA conversion step in transcriptomics experiments is widely recognised as inefficient and variable, casting doubt on the ability to do quantitative transcriptomics analyses. Multiple studies have focused on ways to optimise this process, resulting in contradictory recommendations. Here we explore the problem of reverse transcription efficiency using digital PCR and the RT method’s impact on subsequent data analysis. Using synthetic RNA standards, an example experiment is presented, outlining a method to (1) determine relevant efficiency and variability values and then to (2) incorporate this information into downstream analyses as a way to improve the accuracy of quantitative transcriptomics experiments.
Abbreviations: qPCR, quantitative PCR; dPCR, digital PCR; RT, reverse transcription; cDNA, complementary DNA
Keywords: Transcriptomics, Quantitative PCR, Digital PCR, Reverse transcription, Efficiency, Complementary DNA, Modelling
1. Introduction
As technology has advanced, transcriptomics at the single cell level has become not only possible but preferable due to greater recognition of sample heterogeneity. Single cell experiments are becoming increasingly common in the form of RNA sequencing, qPCR, and digital PCR (dPCR). It is broadly presumed that the measurements are becoming more accurate with these new methods but one must be preemptively cautious and take note of the variability and uncertainty in transcriptomics data.
Transcriptomics measurements almost invariably include a reverse transcription (RT) step, where RNA transcripts are used as templates to generate cDNA transcripts for quantification. This significantly complicates data interpretation as techniques are not directly measuring RNA transcript number, and results are therefore dependent on the efficiency of the RNA to cDNA conversion. Alternative RT-free methodologies exist and involve direct sequencing of RNA or hybridisation of probes to individual RNA molecules. However, these methods also have limitations as they are currently expensive and still struggling with accuracy and throughput, and poor hybridisation efficiency [[1], [2], [3], [4]].
Problems with the reverse transcription step are many [[5], [6], [7], [8]]. A multitude of research articles have been published that address the effects of modifying individual components or steps of the RT reaction, providing a resource for RT efficiency optimization in experimental design. These modifiable parameters include but are not limited to priming strategy [8,9], choice of RT enzyme [6,7,[9], [10], [11], [12]], choice of PCR priming site [6], target RNA concentration [6,8,9], background RNA concentration [[8], [9], [10]], and RNA quality [6]. Results reported from such studies are often inconsistent; one of the few undisputed findings to come from collating this research is that the effects of changing these parameters appear to be gene-dependent [5,7,8,11,13].
Strategies to improve reverse transcription have been addressed in some detail using population-based RT-qPCR experiments, and many recommendations have been made based on these results. Here, we explore RT methods a step further by examining this problem in the context of single cell analyses using absolute quantification by digital PCR (dPCR). The underlying and consistent experimental and analytical focus is to investigate the efficiency and variability of RT-dPCR in order to determine the consequences of the reverse transcription step in this experimental system.
1.1. The problem of efficiency and variability in reverse transcription
A large proportion of transcriptomics is concerned with relative differences between samples. In such scenarios, simplifying analysis by assuming global 100% efficiency may be justified. The relatively recent release of dPCR with claims of accurate direct quantification point towards the ability to use this system in instances where absolute numbers are important. For example, accurate interpretation of data to attain absolute numbers is both relevant and critical in validating a model especially if low numbers of factors are present and ratios of different factors are important. In this situation, it is imperative to understand the efficiency and variability of the system to properly interpret the data.
Several published articles have addressed this question by attempting to put a value on RNA-to-cDNA conversion efficiency, yet results vary widely with different experimental conditions. Some cited efficiency ranges are 49–114% [14], 50–77% [15], 0–102% [10], and 39–65% [9]. This wide variety effectively illustrates the problem and is likely a combined outcome of the many parameters that are different within and between tests, including the specific transcripts measured.
In addition to variable efficiencies across different transcripts, one must consider the reproducibility of reverse transcription for a single, particular transcript. In a study of RT efficiency variability, Linden et al [7] showed that some genes had much more variability in efficiency than others, and did not correlate with the general transcription efficiency in each reaction. One of these genes was ACTB, a commonly used reference gene. Similar results have been reported elsewhere [8].
The issue of reproducibility is of particular concern in single cell studies where there is little scope for replication to help average away technical differences. Reproducibility is also of great relevance to other areas dealing in absolute quantification of RNA, such as the increasing interest in using RT-dPCR for clinical applications (for example the detection of RNA biomarkers) [14,16]. This highlights the importance of characterizing assay variability in order to avoid drawing unreliable conclusions from results [6].
1.2. Sensitivity to variations in reaction conditions
In designing transcriptomics experiments, optimal conditions allow the best efficiency (closest to 100%) with the lowest variability. Considering the range of outcomes reported across different studies, it is clear that the performance of the RT step is greatly influenced by the context of the experiment, dismissing the possibility of a one-size-fits-all approach to RT optimization. Therefore, it is important to note that previous data published in the literature is not always transferrable, and often certain optimization choices are not compatible with the proposed experiment. Examples of such constraints include avoiding gene-specific primers when one needs to store cDNA for later, yet-to-be-decided analysis, or the necessity for random primers when studying non-adenylated transcripts.
In the case of single-cell digital PCR, there are many caveats and constraints. For example, with the direct-lysis single-cell RNA preparation method used for the studies presented here, there is no way to modify or even evaluate some characteristics of the sample, such as target RNA concentration or RNA quality. Another area of significant limitation with single cell analysis is replication. Tichopad et al [17] note that use of sampling replicates can estimate the boundaries of technical noise, however only a single sample is available with single cell studies.
Using dPCR, Sanders et al [15] showed that efficiency problems extend to this method and again, that this effect is gene-dependent. Consequently, while studies have shown the accuracy of digital PCR for DNA quantification [18,19], the technique does not negate the issues identified with reverse transcription. The authors suggest that a calibrant sample with defined value could help account for effects of enzyme efficiency, inhibitors and molecular dropout, while noting that due to the differences between targets, only gene-specific calibrators would be appropriate.
Most efforts in reverse transcription optimization studies up until now have focused on RT-qPCR. Some previously identified improvements could conceivably affect the dPCR output. Previous qPCR findings relevant to single cell studies include that the use of background RNA was shown to exert some of its effect via the qPCR step [10] and that dilution of the RT reaction greatly influenced subsequent qPCR, especially in the presence of low background RNA [10]. The transferability of these effects from qPCR to dPCR is unknown and exploring such space is constrained by the nature of working with single cells, given limited sample and limits of detection at the single-cell scale. It is likely that some variability and reduced efficiency will always remain, regardless of how well a reverse transcription reaction has been optimized. Clearly, rather than relying on previous publications for ‘best-practice’ protocols to allow one to ignore the problem, we should use prior published data as a baseline from which to work to better understand and accept the limitations of our own system and build this into our individual data interpretation.
This study was conducted as an example of how reverse transcription efficiency can be taken into consideration when performing a transcriptomics experiment. First, a non-exhaustive optimization experiment was run using information from the literature to determine optimal RT conditions in our specific system. Subsequently, a range of efficiency and variability values corresponding to a number of genes of interest was determined, which can then be integrated into downstream analyses of data. Finally, this data is used to guide a brief discussion of the implications of the efficiency data in the context of higher-throughput transcriptomics methods.
2. Results and discussion
2.1. Optimization of RT conditions
In this study we explored four genes linked to myeloid haematopoiesis and the common reference gene ACTB. Based on previous literature [9,10], first, a limited optimization experiment was performed, testing a small number of conditions considered most likely to improve the reverse transcription efficiency from our standard protocol. The design included a fully factorial test of three different RT enzymes (SuperScript III VILO Kit, Life Technologies; Superscript II, Life Technologies; Protoscript, New England Biolabs), three concentrations of random hexamer primers (6 uM/as directed; 25 uM; 100 uM), and two concentrations of background yeast RNA (10 pg; 250 ng). Each condition was tested with duplicates of two different transcripts at a known concentration, and measured the efficiency by digital PCR. SSIII VILO LR, LH represents our standard protocol. Results, displayed in Fig. 1, indicated that in our system, SuperScript III VILO was the best performing enzyme, and the addition of extra random hexamers to 25 uM improved upon the efficiency compared with our standard protocol. The addition of high concentration of yeast RNA did not improve efficiency in this case.
Fig. 1.

Comparison of efficiency values from RT optimization experiment. SSIII VILO: SuperScriptIII VILO Kit; SSII: SuperScriptII; LR: low yeast RNA (10 pg); HR: high yeast RNA (250 ng); LH: low hexamer (6 uM/as directed); MH: mid hexamer (25 uM); HH: high hexamer (100 uM). Results are presented as mean of duplicate data points with standard deviation. RT efficiency > 100% is possible given random hexamer primers were used.
These results support previous observations that emphasise the gene-dependent nature of reverse transcription efficiency, as there was a doubling in efficiency with one transcript while only minimal improvement with the other using the increased hexamer concentration. This suggests that the determined optimal condition from the tested conditions above may differ for any other transcripts of interest and there will not be a condition optimal across all scenarios. This position underscores the specified goal of identifying best possible conditions given practical and system-based constraints, and in parallel to identify associated efficiency and variability values and incorporate them into the data analysis.
2.2. Identifying variability and efficiency values
Once the ‘optimal’ RT conditions were defined, variability and efficiency tests with IVRS (in vitro RNA synthesis)-produced transcripts were run for our five genes of interest. ACTB was included as a comparison given its wide use as a reference gene despite having been shown to exhibit high RT variability [7].
First, 10 RT replicates per transcript were measured using a single dilution at three concentrations (10 fg, 1 fg and 100 ag/reaction) to determine inherent variability of the RT step for each transcript. The coefficient of variation (CV) was less than 12% for all transcripts at the 10 fg and 1 fg level (see Fig. 2A and Table 1). Despite the fact that these values incorporate both RT and PCR variability, they compare favourably with PCR component-only CV values reported in the literature [14,16] and are within the guidelines for dPCR equipment specifications for repeated readings (Bio-Rad specifies QX200 precision as ±10%). However, at the 100 ag level the CV ranged from 12 to 35% depending on the transcript, indicating an increased variability at this concentration. It is uncertain whether this increase is primarily driven by an inherent increase in RT variability at the lower concentration or stochasticity associated with small molecule numbers, for example when pipetting from the master mix, and is likely to be some combination of the two factors.
Fig. 2.

Variability and efficiency tests for transcripts of interest using EvaGreen dPCR. A) Variability. Ten replicates were performed per concentration for each transcript (nine for ACTB 100 ag), and coefficient of variation calculated for each. B) Efficiency. Five dilution replicates were performed per concentration for each transcript (four for ACTB 10 fg, ACTB 1 fg, ACTB 100 ag, CEBPA 1 fg, PU.1 1 fg), and mean efficiency values calculated for each. More information can be found in Table 1 and values used for calculations are included in the Supplementary Information.
Table 1.
Variability and efficiency values for transcripts of interest using EvaGreen dPCR. Data used for graphs in Fig. 2 are shaded.
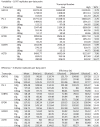 |
Based on these results, RT variability was considered to be minimal for the majority of the reactions, and RT replicates were not performed for the following steps. It is important to note for data analysis that measurements at the 100 ag level (54–130 transcripts/reaction for our particular genes) carry significant variability and modest differences between cells should be approached with caution.
Subsequently, to determine efficiency values while keeping in mind the possibility of pipetting inconsistencies during dilution, five dilution replicates of each transcript were measured over a range of three concentrations, corresponding to a theoretical medium, high and very high transcript expression level in a single cell [20]. The medium level of expression was considered to be towards the lower limit of what was detectable using EvaGreen dPCR with a reasonable signal-to-noise ratio (unpublished results).
Replicate average efficiency values were similar across concentrations for each transcript (Fig. 2B), although the error is much more pronounced at low concentration (100 ag/transcript). Similar to results reported above, CV of all transcripts at 10 fg and 1 fg level is less than 15%, indicating minimal impact from dilution uncertainty when compared with RT replicates and giving confidence in calculated efficiency values. However, at the 100 ag level the coefficient of variation for some transcripts was above 60%, showing additional stochasticity introduced with dilution in addition to RT, and casting doubt on the utility of using standards for measuring transcript levels at this low concentration.
Significantly, there is a wide range of efficiency values across the different transcripts, ranging from a combined average of 120% for EPOR to 55% for GCSFR. This illustrates how important knowledge of this value is for interpreting data in a quantitative setting. It is clear that data obtained without adjustment of this discrepancy is inaccurate. This is especially problematic in a quantitative experiment where the aim is to enumerate absolute numbers of transcripts rather than relative values.
2.3. Incorporation into downstream analyses
The specific calculated efficiency values for synthetic transcripts can be incorporated as a normalisation factor into dPCR experiments performed in an identical manner. To arrive at a corrected absolute transcript number, measured transcript numbers from the dPCR experiment can be divided by the efficiency value for each transcript. For single cell experiments, the measured transcript numbers are likely to be at the low end of the concentrations tested where the efficiency values are highly variable. Therefore, we would recommend using the mean efficiency value for all concentrations tested. Of course, this also indicates large variability in the single cell reactions, and all data should be approached with this presumption. With an estimate of the variability present in the reaction at a defined expression level, the information can be incorporated into significance calculations and interpretation of results by both using the variance as bounds on model input variables and by capturing the effect this may have on the model output. In measuring higher transcript numbers (i.e. a very highly expressed transcript at single cell level or an experiment involving more than one cell per reaction) it might be preferable to use the efficiency value determined for the concentration closest to that being measured, assuming low variability for that particular concentration.
2.4. Comparison across assays
Digital PCR experiments may be run using two different detection chemistries, EvaGreen and probes. It is easy to switch between the two methods as they are performed in a highly similar manner with the same hardware and procedure. These parallel methods provide a means to explore the impact of detection chemistry on efficiency and variability values. To test the applicability of efficiency values as a useful tool across PCR detection methods, probe-based dPCR assays were run for three targets (GATA1, PU.1 and CEBPA) using the same dilution series as the EvaGreen experiment, with the addition of a 10 ag concentration to leverage the improved signal-to-noise ratio of the probe assay. Substantial differences were observed between the efficiency values obtained from the two experiments, as seen in Fig. 3; data for the probe experiment is shown in Supplementary Figure S1 and Supplementary Table S1.
Fig. 3.

Comparison of efficiency values obtained with EvaGreen and probe assays. Results are presented as mean of replicate data points with standard deviation. There is a discrepancy in GATA1/PU.1 ratio in the higher concentration probe results not evident in the EvaGreen results.
While the values for CEBPA are consistent, GATA1 and PU.1 show a divergence in efficiency values between the two assays. While the efficiencies of reverse transcription of the two transcripts are reasonably similar using the EvaGreen assay, they are markedly different using the probe-based assay, such that the ratio of GATA1 to PU.1 is highly affected depending on the approach used. Based on these values, adoption of the EvaGreen efficiency value for probe assay data would overestimate GATA1 concentration by 60% relative to PU.1 concentration (ratio of GATA1/PU.1 normalisation factors = 0.86 for EvaGreen assay, 0.53 for probe assay). This would substantially bias the outcome of any model where quantitative data is expected. This calculation illustrates the necessity of determining empirically the performance of the assay in each specific situation and negates the possibility of broadly applying efficiency values between platforms. This is not unexpected given the evidence outlined above regarding the sensitivity of RT reactions to modifications in protocol.
2.5. Implications for high-throughput technologies
As demonstrated here, measurement of reverse transcription efficiency should be incorporated into any experiment involving quantitative RNA assessment. While adopting this recommendation is relatively easy for smaller scale experiments, it becomes unworkable for more high-throughput approaches such as single cell sequencing. As single-cell sequencing experiments are becoming more common, there is increasing discussion about the appropriate method of analysing such data. Using a well-studied, homogeneous cell line, Marinov et al. [20] demonstrated a 6-fold range in RNA content between single cells, leading to the assertion that in such a single-cell setting the often-used expression unit ‘fragments per kilobase per million mapped reads’ (FKPM) is misleading and absolute transcripts should be counted. Consequently, this and other studies [21,22] have attempted to calculate absolute transcript numbers from sequencing data using spike-in standards.
While spike-in RNA standards (such as ERCC spike-ins [23], Sequins [24], and SIRVs [25]) allow calculation of a global efficiency value based on a pre-determined subset of synthetic genes for assay performance and quality control purposes, the data presented here indicate this can not be extrapolated to estimate efficiency values for specific genes of interest in the data. Indeed, it has previously been reported using bulk RNA sequencing that reproducible transcript-dependent discrepancies show absolute measurements using RNA-seq are inaccurate [26].
Therefore, while the addition of spike-ins to calculate global efficiency may allow an improvement on current methods for analysing single cell gene expression, it still only gives a relative value for comparing between samples that cannot be compared across transcripts, and is not able to give absolute quantitative information about RNA molecules in a cell. Instead, synthetic standards should be incorporated to gain efficiency values for subsequent validation tests for significant findings.
3. Conclusions
The data presented in this publication and other works cited here reiterate gene-to-gene variability in reverse transcription efficiency and highlight the necessity of considering RT-efficiency when working with quantitative data. The wide range of values calculated for the six synthetic RNAs tested may have a significant impact on the quantitative analysis of the downstream transcriptomics data. Despite the between-gene variability, efficiency values are sufficiently reproducible at all but the lowest concentration within the confines of a particular protocol such that this variability can be mitigated through incorporation of synthetic standards as controls. Standards specific to each experiment are necessary as the efficiency values have been shown to be highly sensitive to alterations of even a single component of the workflow.
Certain assays are limited in throughput (for example dPCR which allows for low-level multiplexing) and it is reasonable to include controls for all the genes investigated in an assay. We suggest including a synthetic transcript for each transcript measured across at least 3 biologically relevant concentrations in technical replication to gain a metric for normalisation of data, and find the lower limit for reproducibility and detection of signal above noise. While this is not possible for higher-throughput technologies such as single-cell sequencing, standards should be included in targeted follow-up validation assays if quantitative claims are made.
Measurement of lowly-expressed transcripts using dPCR at the single cell level is likely to be highly variable, and analysis of technical variability should be factored in to final conclusions.
4. Materials and methods
4.1. Synthetic RNA standards
4.1.1. Construction of synthetic standards
Synthetic RNA standards were made for the following transcripts: GATA1, PU.1, CEBPA, GCSFR, EPOR, ACTB. Lengths of RNA transcripts ranged from 1428 to 3414 nucleotides.
Full-length mRNA has been shown to be the most accurate method of setting up RNA standards [9]. Therefore, transcripts were amplified using Phusion High-Fidelity DNA Polymerase (New England Biolabs) from cDNA isolated from haematopoietic cells with primers designed at the ends of the mRNA transcript with leeway to ensure acceptable primer design. Primers used for amplification are outlined in Supplementary Info.
cDNA was cloned into pGem-T Easy plasmid (Promega) and IVRS performed using linearized plasmid and HiScribe T7 High-Yield RNA Synthesis Kit (New England Biolabs). In vitro-synthesised RNA was isolated using Nucleospin RNA II kit (Macherey-Nagel), treated with TURBO DNase (Ambion), and cleaned up using RNA Clean & Concentrator Kit (Zymo). Sample quantity and quality were assessed using Qubit RNA Assay (ThermoFisher Scientific) and RNA Pico Bioanalyzer chip (Agilent) respectively, and stored in Lo-Bind tubes (Eppendorf) at -80°C. Stock concentrations were between 100 ng/uL and 700 ng/uL. All experiments were performed within six months of RNA generation.
4.1.2. Dilution of synthetic standards
Evagreen and probe dPCR efficiency test: A single dilution series was performed for each transcript for the 10 fg and 1 fg variability test. Samples were diluted to ˜10 ng/uL in water and measured again with Qubit RNA Assay (ThermoFisher Scientific). From this working stock, aliquots of 1 ng/uL were prepared for all transcripts. A series of 1 in 10 dilutions in a final volume of 20 u L was performed until desired concentrations were reached. All dilutions were performed in Lo-Bind tubes (Eppendorf).
5 dilution replicates: A separate dilution series comprising five replicates per transcript, beginning with five aliquots at 1 ng/uL, was performed for the EvaGreen and probe efficiency tests as outlined above. Dilution 1 for each of these transcripts was also used for the 100 ag variability test. Dilutions were prepared on the same day as each of the studies were conducted.
4.2. cDNA synthesis
4.2.1. cDNA synthesis for RT optimisation
Reverse transcription reactions were set up and run in 96-well Twin-tec semi-skirted LoBind plates (Eppendorf). Reactions were designed to measure 100 ag of PU.1 and GATA1 RNA in 5 uL reactions containing 5 mg/mL UltraPure BSA (Ambion), 5U SUPERaseIn RNase Inhibitor (ThermoFisher Scientific), 1X RT Reaction Mix/Buffer, 0.5X RT enzyme, variable concentrations of yeast RNA (10 pg and 250 ng; Ambion), and variable concentrations of random hexamers (6 uM/as directed, 25 uM and 100 uM; Integrated DNA Technologies). Primer concentration in the 1× SSIII VILO Reaction Buffer was not specified and for the purposes of this experiment was assumed to be 6 uM. SuperscriptII and Protoscript reactions also required the addition of 500uM dNTP Mix (New England Biolabs) and 10 mM DTT (ThermoFisher Scientific). A denaturation step at 65 °C for 5 min was performed before the addition of RT enzyme. The temperature profile of the SSIII VILO reaction was 25°C for 10 min, 50°C for 30 min, 55°C for 25 min, 60°C for 5 min, and 70°C 15 min. Temperature profile of SSII and Protoscript reactions were 25°C for 10 min, 42 °C for 30 min, 48°C for 25 min, 50°C for 5 min and 70°C for 15 min. Upon completion of each reverse transcription (RT) reaction, 96-well plates were spun to recover individual reaction volumes and cDNA was stored at −80 °C for up to two weeks. After thawing on ice, all RT reactions were transferred to a single new plate for dPCR.
4.2.2. cDNA Synthesis for RT efficiency/variability test
Reverse transcription reactions were set up and run in 96-well Twin-tec semi-skirted LoBind plates (Eppendorf). Reactions were designed to measure 10 technical replicates of three concentrations (100 ag, 1 fg, 10 fg) of six target transcripts (PU.1, GATA1, CEBPA, GCSFR, EPOR, ACTB) in 5 uL reactions containing 5 mg/mL UltraPure BSA (Ambion), 5U SUPERaseIn RNase Inhibitor (ThermoFisher Scientific), 1X VILO Reaction Mix, 0.5× SSIII enzyme, 10 pg yeast RNA (Ambion), and 19 uM additional random hexamers (Integrated DNA Technologies). A denaturation step at 65 °C for 5 min was performed before the addition of RT enzyme. The temperature profile was 25°C for 10 min, 50°C for 30 min, 55°C for 25 min, 60°C for 5 min, and 70°C 15 min. Upon completion of each cDNA synthesis reaction, 96-well plates were spun to collect volume and cDNA was stored at 4 °C for up to four days.
4.3. Digital PCR
4.3.1. EvaGreen dPCR
Samples were prepared for dPCR in 22 uL reactions containing cDNA, 1× ddPCR™ EvaGreen Supermix (Bio-Rad), and primers. For targets PU.1, GATA1, CEBPA, and ACTB primers were designed and validated in-house and used at 200 nM for each oligo (Integrated DNA Technologies). For targets GCSFR and EPOR, PrimePCR™ EvaGreen Assay (Bio-Rad) were used at 1× primer mix. Primer sequences (or context sequences for commercial assays) are provided in the Supplementary Information. No template RT controls contained only yeast RNA and were included for each primer set. No-RT controls were performed previously for each transcript and confirmed absence of DNA.
Droplets were created using an Automated Droplet Generator (BioRad) followed by the recommended PCR thermocycling protocol using a C1000 Thermal Cycler (Bio-Rad): 95 °C for 10 min, followed by 40 cycles of 95 °C for 30 s and 58 °C for 60 s, and a final signal stabilization cycle of 4 °C for 5 min and 90 °C for 5 min. A QX200™ Droplet Reader (BioRad) was used for signal detection.
4.3.2. Probe dPCR
Samples were prepared for dPCR in 22 uL reactions containing cDNA, 1X ddPCR™ Supermix for Probes (No dUTP; Bio-Rad) and 1X PrimePCR™ ddPCR™ Expression Probe Assay primers/probe mix (Bio-Rad): FAM-PU.1, HEX-GATA1 and HEX-CEBPA. PU.1 and GATA1 were run as duplex samples, CEBPA as singleplex. Context sequences are provided in the Supplementary Information. No template RT controls contained only yeast RNA and were included for each primer set.
Droplets were created using an Automated Droplet Generator (BioRad) followed by the recommended PCR thermocycling protocol using a C1000 Thermal Cycler (Bio-Rad): 95 °C for 10 min, followed by 40 cycles of 94 °C for 30 s and 55 °C for 60 s, and a final incubation at 98 °C for 10 min. A QX200™ Droplet Reader (BioRad) was used for signal detection.
4.3.3. dPCR data analysis
QuantaSoft™ Analysis Pro analysis software (Bio-Rad) was used to determine absolute transcript numbers. A threshold for defining positive droplets was set manually by comparison with control samples. The number of positive droplets was used by the software to perform a Poisson correction to give an absolute number of transcripts per microliter. These results were multiplied by the total sample reaction volume of 22 uL for a final absolute quantification of a given target.
4.4. Calculations
4.4.1. Calculation of efficiency values
The exact sequence of each transcript were determined by taking the plasmid sequence starting at the final G nucleotide of the T7 promoter and continuing through to the final base before cleavage at the linearization site. Molecular weights of each synthetic RNA standard was calculated according to transcript sequence using the following formula:
| M.W. of ssRNA transcript (g/mol) = (An * 329.2) + (Un* 306.2) + (Cn *305.2) + (Gn * 345.2) + 159, |
where An, Un, Cn, and Gn are the numbers of A, U, C, and G bases, respectively, and the additional 159 corresponds to the weight of the 5′ triphosphate group.
This value was used to determine the number of transcripts present in each tested concentration. The numbers calculated for each tested concentration can be found in the Supplementary Information. Numbers of transcripts detected by dPCR for each sample were divided by the theoretical number in the reaction to arrive at an efficiency value.
4.4.2. Calculation of variability values
The coefficient of variation for each transcript was calculated by dividing the standard deviation of the number of transcripts per reaction for 10 replicates by the mean, expressed as a percentage.
Funding source
This work was supported by Stem Cells Australia.
Handled by Dr S. Bustin
Footnotes
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.bdq.2018.12.002.
Appendix A. Supplementary data
The following are Supplementary data to this article:
References
- 1.Garalde D.R. Highly parallel direct RNA sequencing on an array of nanopores. bioRxiv. 2016 doi: 10.1038/nmeth.4577. [DOI] [PubMed] [Google Scholar]
- 2.Geiss G.K. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat. Biotechnol. 2008;26(3):317–325. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 3.Materna S.C., Nam J., Davidson E.H. High accuracy, high-resolution prevalence measurement for the majority of locally expressed regulatory genes in early sea urchin development. Gene Expr. Patterns. 2010;10(4-5):177–184. doi: 10.1016/j.gep.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kulkarni M.M. Digital multiplexed gene expression analysis using the NanoString nCounter system. Curr. Protoc. Mol. Biol. 2011:10. doi: 10.1002/0471142727.mb25b10s94. Chapter 25: p. Unit25B. [DOI] [PubMed] [Google Scholar]
- 5.Bustin S.A., Nolan T. Pitfalls of quantitative real-time reverse-transcription polymerase chain reaction. J. Biomol. Tech. 2004;15(3):155–166. [PMC free article] [PubMed] [Google Scholar]
- 6.Bustin S. Variability of the reverse transcription step: practical implications. Clin. Chem. 2015;61(1):202–212. doi: 10.1373/clinchem.2014.230615. [DOI] [PubMed] [Google Scholar]
- 7.Linden J., Ranta J., Pohjanvirta R. Bayesian modeling of reproducibility and robustness of RNA reverse transcription and quantitative real-time polymerase chain reaction. Anal. Biochem. 2012;428(1):81–91. doi: 10.1016/j.ab.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 8.Stahlberg A. Properties of the reverse transcription reaction in mRNA quantification. Clin. Chem. 2004;50(3):509–515. doi: 10.1373/clinchem.2003.026161. [DOI] [PubMed] [Google Scholar]
- 9.Miranda J.A., Steward G.F. Variables influencing the efficiency and interpretation of reverse transcription quantitative PCR (RT-qPCR): an empirical study using Bacteriophage MS2. J. Virol. Methods. 2017;241:1–10. doi: 10.1016/j.jviromet.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 10.Levesque-Sergerie J.P. Detection limits of several commercial reverse transcriptase enzymes: impact on the low- and high-abundance transcript levels assessed by quantitative RT-PCR. BMC Mol. Biol. 2007;8:93. doi: 10.1186/1471-2199-8-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stahlberg A., Kubista M., Pfaffl M. Comparison of reverse transcriptases in gene expression analysis. Clin. Chem. 2004;50(9):1678–1680. doi: 10.1373/clinchem.2004.035469. [DOI] [PubMed] [Google Scholar]
- 12.Sieber M.W. Substantial performance discrepancies among commercially available kits for reverse transcription quantitative polymerase chain reaction: a systematic comparative investigator-driven approach. Anal. Biochem. 2010;401(2):303–311. doi: 10.1016/j.ab.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 13.Bengtsson M. Quantification of mRNA in single cells and modelling of RT-qPCR induced noise. BMC Mol. Biol. 2008;9:63. doi: 10.1186/1471-2199-9-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hindson C.M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods. 2013;10(10):1003–1005. doi: 10.1038/nmeth.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanders R. Evaluation of digital PCR for absolute RNA quantification. PLoS One. 2013;8(9):e75296. doi: 10.1371/journal.pone.0075296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campomenosi P. A comparison between quantitative PCR and droplet digital PCR technologies for circulating microRNA quantification in human lung cancer. BMC Biotechnol. 2016;16(1):60. doi: 10.1186/s12896-016-0292-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tichopad A. Design and optimization of reverse-transcription quantitative PCR experiments. Clin. Chem. 2009;55(10):1816–1823. doi: 10.1373/clinchem.2009.126201. [DOI] [PubMed] [Google Scholar]
- 18.Dong L. Comparison of four digital PCR platforms for accurate quantification of DNA copy number of a certified plasmid DNA reference material. Sci. Rep. 2015;5:13174. doi: 10.1038/srep13174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanders R. Evaluation of digital PCR for absolute DNA quantification. Anal. Chem. 2011;83(17):6474–6484. doi: 10.1021/ac103230c. [DOI] [PubMed] [Google Scholar]
- 20.Marinov G.K. From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing. Genome Res. 2014;24(3):496–510. doi: 10.1101/gr.161034.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Owens N.D. Measuring absolute RNA copy numbers at high temporal resolution reveals transcriptome kinetics in development. Cell Rep. 2016;14(3):632–647. doi: 10.1016/j.celrep.2015.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fan X.Y. Single-cell RNA-seq transcriptome analysis of linear and circular RNAs in mouse preimplantation embryos. Genome Biol. 2015;16 doi: 10.1186/s13059-015-0706-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang L. Synthetic spike-in standards for RNA-seq experiments. Genome Res. 2011;21(9):1543–1551. doi: 10.1101/gr.121095.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hardwick S.A. Spliced synthetic genes as internal controls in RNA sequencing experiments. Nat. Methods. 2016;13(9):792–798. doi: 10.1038/nmeth.3958. [DOI] [PubMed] [Google Scholar]
- 25.Paul L. SIRVs: spike-in RNA variants as external isoform controls in RNA-Sequencing. bioRxiv. 2016 [Google Scholar]
- 26.Consortium S.M.-I. A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the sequencing quality control consortium. Nat. Biotechnol. 2014;32(9):903–914. doi: 10.1038/nbt.2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.