

ABSTRACT
Extracellular vesicles (EVs) are important mediators of cell–cell communication. Intriguingly, EVs can be engineered and thus exploited for the targeted transfer of functional proteins of interest. Thus, engineered EVs may constitute attractive tools for the development of novel therapeutic interventions, like cancer immunotherapies, vaccinations or targeted drug delivery. Here, we describe a novel experimental immunotherapeutic approach for the adjuvant treatment of chronic lymphocytic leukaemia (CLL) based on engineered EVs carrying gp350, the major glycoprotein of Epstein–Barr virus (EBV), CD40L, a central immune accessory molecule and pp65, an immunodominant antigen of the human cytomegalovirus (CMV). We show that these engineered EVs specifically interact with malignant B cells from CLL patients and render these cells immunogenic to allogeneic and autologous EBV- and CMV-specific CD4+ and CD8+ T cells. Collectively, co-opting engineered EVs to re-target the strong herpesviral immunity in CLL patients to malignant cells constitutes an attractive strategy for the adjuvant treatment of a still incurable disease.
Abbreviations: CLL: chronic lymphocytic leukaemia; EBV: Epstein-Barr virus; CMV: cytomegalovirus
KEYWORDS: Extracellular vesicles, immunotherapy, CLL, EBV, CMV
Introduction
Extracellular vesicles (EVs) comprise a mixture of different types of vesicles that are constitutively secreted by most cell types [1]. EVs either fuse to the membrane of recipient cells or are actively engulfed by phagocytic cells such that EV proteins are degraded and the derived peptides are loaded onto MHC class II molecules [2]. EVs can deliver foreign proteins in a very immunogenic manner [1], so that they efficiently reactivate specific CD4+ T-cell clones [3]. But EVs also transfer mRNAs to recipient cells where they can be translated into functional proteins [4]. Upon proteasomal degradation, these proteins are fed into the MHC class I pathway and, consequently, may elicit the activation of specific CD8+ T cells [3,5]. Clearly, specifically engineered EVs are an appealing route for directed transfer of both proteins and nucleic acids [6–9] and may therefore be used as attractive tools for targeted immunotherapeutic interventions.
Chronic lymphocytic leukaemia (CLL) of B-cell origin is the most common adult leukaemia in the Western hemisphere. Despite considerable therapeutic improvements throughout the last years [10–12], the disease remains incurable. CLL is characterized by a number of peculiarities: leukaemic B cells express high levels of surface MHC class I and II molecules but lack expression of important accessory and co-stimulatory molecules [13]. As a consequence, the malignant cells successfully evade the immune system and induce tumour-specific T-cell anergy. In addition, activated T cells from patients show a dramatically reduced expression, or even complete absence, of CD40 ligand (CD154) and therefore fail to activate cells through the CD40 receptor pathway [14], which is crucial for the concomitant induction of additional immune accessory molecules like B7-H1 (CD80) and B7-H2 (CD86), and thus immunogenicity, of normal B cells and malignant CLL cells. Several approaches have been performed to circumvent this T-cell dysfunction by different means in order to specifically stimulate CD40 on CLL, thus rendering the cells “visible” to the immune system [15–19]. In essence, CLL is an attractive prototypic disease for the investigation of CD40L-based experimental therapies aiming at the improvement of the patients’ immune status and clinical outcome.
Infections bear a major influence on the progression of CLL and can also bias the T-cell repertoire of patients. Epstein–Barr virus (EBV) is a widespread human herpes virus that efficiently infects resting human B-lymphocytes where it normally remains asymptomatically. On the other hand, EBV is also associated with different types of cancers of lymphocytic and epithelial origin [20]. Though EBV can also infect CLL cells, and transcription of viral genes has been described [21], the role of EBV in CLL disease progression remains unclear [22,23]. EBV’s B-cell tropism is predominantly mediated by gp350, the major viral envelope glycoprotein that interacts with CD21 (complement receptor 2, CR2) on the cell surface [24]. It has been previously demonstrated, that also EVs released by EBV-transformed B cells express gp350 on their surface and are capable to specifically target B cells through CD21 binding [25].
Cytomegalovirus (CMV) is yet another member of the family of herpes viruses. The virus is uniquely immunodominant in infected individuals leading to the development of oligoclonal CMV-specific T cells constituting up to 25% of the total CD8+ T-cell population [26]. Expansion of CMV-specific T cells is even more pronounced in CLL patients where increased numbers of both CMV-specific CD4+ and CD8+ T cells have been observed [27,28]. Interestingly enough, whereas the overall T-cell population in CLL patients reveal signs of exhaustion, the cytotoxic function of CMV-specific CD8+ T cells remains intact even at advanced stages of the disease [29].
We present here a novel approach for the specific transfer of functional proteins to primary CLL cells via engineered EVs. We show that EVs carrying CD40L and the immunodominant herpesviral proteins gp350 and pp65 target CLL cells, reactivate their antigen-presenting capacity and induce the reactivation of autologous cytolytic gp350- and pp65-specific T cells.
Materials and methods
We have submitted all relevant data of our experiments to the EV-TRACK knowledgebase (EV-TRACK ID: EV180011 [30]).
Blood samples and cell culture
HEK293 cells [31] and mini-EBV-immortalized B-cell lines (mini-LCLs) [32] were grown in RPMI 1640 medium supplemented with 8% heat-inactivated foetal calf serum (FCS, Bio & Sell, Feucht, Germany), penicillin (100 U/ml) and streptomycin (100 mg/ml). The cell cultures were tested on a regular basis to exclude mycoplasma contamination using the MycoAlert mycoplasma detection kit (Lonza, Basel, Switzerland). Peripheral blood samples were obtained from patients with confirmed CLL after written informed consent approved by the Institutional Ethics Committee. Mononuclear cells were isolated by Ficoll Hypaque (PAN Biotech, Aidenbach, Germany) gradient centrifugation, maintained in supplemented RPMI medium and the percentage of CD5+/CD19+ CLL cells was analysed by flow cytometry. Gp350- (HLA-DRB1*1301-restricted CD4+, epitope FGQLTPHTKAVYQPR) and pp65-specific (HLA-DQB1*0501-restricted CD4+, epitope KYQEFFWDAND; HLA-A*0201-restricted CD8+, epitope NLVPMVATV) T-cell clones used in this study were established from PBMCs isolated from EBV- or CMV-positive voluntary blood donors and kindly provided by A. Moosmann and J. Mautner, Helmholtz Centre Munich, Germany. All cells were cultivated at 37°C and 5% CO2 in a humified atmosphere. Cell viability was checked by trypan blue exclusion and cultures with viabilities above 95% were used for experiments.
Isolation of EVs
HEK293 cells were transfected by lipofection with expression plasmids for gp350 (BLLF1), CD40L and/or pp65. For the generation of EVs, RPMI supplemented with EV-depleted FCS was used, as described elsewhere [33]. Briefly, medium with 20% FCS was prepared and centrifuged for 16 h at 100,000 × g, 4°C, in a Beckman LE-80K ultracentrifuge in a SW28 or SW32 swing-out rotor (Beckman Coulter, Brea, CA) to deplete contaminating serum-derived EVs. Afterwards the supernatant was filter-sterilized (pore size 0.22 µm) and diluted with serum-free medium to reach the final FCS concentration. Cells were cultivated for three days in EV-depleted medium and EVs were isolated by serial centrifugation and density gradient centrifugation. 180 ml of conditioned supernatant were collected, subjected to repeated centrifugations at increasing centrifugal force (10 min at 300 × g, 4°C, and 20 min at 5000 × g, 4°C), filtrated (pore size 0.45 µm) and finally precipitated for two hours at 100,000 × g, 4°C, in a SW28 or SW32 rotor. Pelleted vesicles were resuspended in 500 µl volume of PBS and further purified by flotation into a two-layer bottom-up iodixanol gradient (OptiPrep™, Sigma Aldrich, Taufkirchen, Germany). The EVs were mixed with iodixanol to reach a final concentration of 44%, and carefully overlayed in 4-ml tubes (Ultra-Clear Thinwall, Beckman Coulter) with 2.3 ml 30% iodixanol and 600 µl PBS. The gradient was centrifuged for 16 h at 160,000 × g, 4°C, in a SW60Ti swing-out rotor and eight fractions of 500 µl each were collected from the top of each tube. The refractive density of each fraction was measured with a refractometer (Reichert Technologies, Unterschleißheim, Germany) and the EV concentration was enumerated by nanoparticle tracking analysis (NTA), as described later. EV-containing fractions were pooled, filled up with PBS to 30 ml and washed by ultracentrifugation in a SW28 or SW32 rotor (100,000 × g, 4°C, 2 h). The final EV pellet was resuspended in 500 µl PBS supplemented with protease inhibitors (cOmplete™ Mini, EDTA-free, Roche, Penzberg, Germany). EVs were used immediately or stored at 4°C up to 4 weeks.
Western blot analysis
To detect EV-associated proteins in density gradient fractions, 15 µl of each OptiPrep™ fraction were mixed with 5x Laemmli protein sample buffer and boiled for 10 min at 95°C. Corresponding cell lysates were prepared by adding ice-cold RIPA lysis buffer (0.1% SDS, 50 mM Tris-HCl pH 8.0, 0.5% DOC, 1% NP-40, 150 mM NaCl) with protease inhibitor to pelleted cells, followed by incubation on ice for 15 min. Protein concentration was measured by Bradford protein assay (Bio-Rad Laboratories, Munich, Germany) and 10 µg of each cell lysate were used for SDS-PAGE. Protein samples were separated on 6–10% bis-tris/acrylamide gels, electroblotted onto a 0.45-µm pore size nitrocellulose membrane (GE Healthcare, Little Chalfont, UK), followed by blocking for 1 h in 5% non-fat milk and an incubation overnight with primary antibodies at 4°C under gentile shaking. The membrane was washed three times in tris-buffered saline with 0.05% Tween-20, incubated with HRP-coupled secondary antibodies at room temperature for 1 h and developed with the ECL system (GE Healthcare). The following antibodies were used for immunostaining: mouse anti-TSG101 (1:1,000; 4A10; GeneTex, Irvine, CA), mouse anti-Alix (1:250; 3A9; BioLegend, London, UK), mouse anti-Calnexin (1:1000; 610523; BD Biosciences, Heidelberg, Germany), mouse anti-pp65 (1:1000; ab49214; Abcam, Cambridge, UK), rabbit anti-CD40L (1:1000; C-20, Santa Cruz, Heidelberg, Germany), rabbit anti-ERK1/2 (1:2000; 9102; Cell Signaling, Frankfurt, Germany), rabbit anti-phospho-ERK1/2 (1:2000; 9101; Cell Signaling), rabbit anti-JNK1/3 (1:500; C-17; Santa Cruz) and rabbit anti-phospho-JNK1/3 (1:1000; ab124956; Abcam). The rat anti-CD63 antibody 24F9 was developed in our own group (Helmholtz Centre Munich, Germany), the mouse anti-gp350 OT6 antibody was a gift from Jaap Middledorp, VU University Medical Centre, The Netherlands. Please note: non-reducing sample buffer without β-mercaptoethanol was used for the detection of CD63.
Nanoparticle tracking analysis
EVs were enumerated by NTA using the ZetaView PMX110 instrument (Particle Metrix, Inning, Germany) and its corresponding software (ZetaView 8.02.31). Purified EVs were diluted in PBS to a concentration of 100–200 particles per video frame. The instrument was calibrated with polystyrene beads of known size and concentration (100 nm NanoStandards, Applied Microspheres, Leusden, The Netherlands). Each EV sample was measured at 11 positions with three reading cycles at each position. Temperature was kept constant throughout the measurements at 22°C. The pre-acquisition parameters were set to a sensitivity of 70, a shutter of 50, a frame rate of 30 frames per second and a trace length of 15. The post-acquisition parameters were set to a minimum brightness of 20, a minimum size of 5 pixels and a maximum size of 1000 pixels.
Electron microscopy
For negative staining, a drop (20 µm) of EV samples was placed onto a carbon coated copper grid (400 mesh), freshly lyophilized by glow discharge. After incubation for 1 min, the drop was quickly removed with a Pasteur pipette and air dried, blocked with 0.1% BSA in PBS for 5 min, followed by incubation for 1 h at RT with a primary antibody: rabbit anti-CD40L (H-215; Santa Cruz), rat anti-CD63 24F9 or rat anti-gp350 6G4 (produced and provided by Helmholtz Centre Munich, Germany), with a concentration of 5 µg/ml in 0.1% BSA in PBS. Grids were washed 5 × 3 min with PSB and incubated with a 12 nm gold particle-conjugated secondary antibodies (Dianova, Hamburg, Germany), 1:20 dilution in PBS for 1 h. Grids were washed 3 × 3 min with PSB, 2 × 3 min with ddH2O, air-dried and finally stained with 2% phosphotungstic acid and 0.05% glucose for 5 min. Micrographs were taken with an EM 912 electron microscope (Zeiss, Oberkochen, Germany) equipped with an integrated Omega energy filter operated in the zero-loss mode.
Labelling of EVs with PKH26 membrane dye
For visualization of EV-cell interactions, EVs were stained with the PKH26 membrane dye by using the PKH26 Red Fluorescent Cell Linker Kit for General Cell Membrane Labelling (Sigma Aldrich). After ultracentrifugation at 100,000 × g, 100 µl of pelleted EVs were mixed with 100 µl Diluent C (provided by the kit). As control, 100 µl EV-free PBS were mixed with Diluent C. 0.5 µl of PKH26 dye were added and incubated for 5 min with periodic mixing. The staining reaction was stopped by adding 200 µl of 1% BSA for another minute. Finally, the PKH26-labeled vesicles were loaded on an OptiPrep™ density gradient to purify the EVs and to remove residual excess dye.
Cell analysis by flow cytometry
500,000 B-CLL cells were cultivated with 500 ng of EVs in a final volume of 2 ml in a 6-well plate for 48 h. Induction of surface accessory molecules was measured by flow cytometry using a FACSCanto flow cytometer (Becton Dickinson, Franklin Lanes, NJ,) and analysed with FlowJo (Tree Star Inc., Ashland, ORE). The cells were washed in PBS and incubated for 20 min at 4°C with the following antibodies diluted 1:50 in PBS/3% FCS: mouse anti-CD19–APC (HIB19; BioLegend), mouse anti-CD80-PE (2D10; BioLegend), mouse anti-CD86-PE (37301; R&D Systems, Wiesbaden, Germany), mouse anti-CD54-PE (HCD54; BioLegend), mouse anti-CD95-PE (DX2; BD Pharmingen). The gp350 antibody 72A1 was developed in our own group. To study the activation of bystander CLL cells, cells were stained with CellTrackerTM Green CFMDA dye (Thermo Fisher Scientific, Waltham, MA), according to the manufacturer’s protocol. T cells were detected by staining with mouse anti-CD4-PE (RPA-T4; BioLegend), mouse anti-CD8-APC (RPA-T8; BioLegend), unlabelled Pro5 HLA-A*02:01/NLV pentamer (1:30; pp65 495–504; NLVPMVATV; ProImmune, Oxford, UK) and PE-labelled Pro5 Fluorotag (1:10; ProImmune). Cell surface expression of the human leucocyte antigens on primary CLL cells was analysed by using the antibodies mouse anti-HLA-A2-PE (BB7.2; BioLegend) and FITC-coupled mouse anti-HLA-DR, DP, DQ (Tu39; BD Pharmingen).
T-cell reactivation assays
To determine the immunogenicity of recombinant EVs, mini-LCLs were incubated with EVs overnight in 96-well round bottom plates (10,000 cells/well). The cells were then mixed with T-cell clones at a 1:1 ratio in 96-well V-bottom plates. The supernatants were collected 24 h later and IFN-γ and Granzyme B ELISA assays were performed according to the manufacturer’s protocol (Mabtech, Nacka Strand, Sweden). To reactivate autologous pp65-specific T cells, 500,000 PBMCs from CLL patients were seeded on ELISpot filter microwells (0.45 µm pore size; Merck Millipore, Darmstadt, Germany) coated with a IFN-γ capture antibody and challenged with TPA (50 ng/ml) and ionomycin (0.5 µM), NLV peptide (2 µg/ml; JPT Peptide Technologies, Berlin, Germany) or 500 ng of recombinant EVs. IFN-γ producing cells were detected after 24-h incubation by human ELISpot assay with alkaline phosphatase (Mabtech). In order to reactivate CMV-specific T-cells in B-CLL blood samples, 1 × 107 PBMCs were cultivated with 500 ng of recombinant CD40L+/gp350+/pp65+ EVs in the presence of IL-2 (1000 U/ml) in a final volume of 2 ml. As negative control, cells were left untreated or incubated with HEK293 control EVs. Re-stimulations were performed after seven and 14 days. After 25 days, the cells were harvested and analysed by flow cytometry with the indicated antibodies or mixed with 10,000 freshly thawed autologous PBMCs loaded with EVs, a pp65 peptide pool (0.5 µg/µl final concentration of each peptide; JPT) or an Adenovirus-derived control peptide pool (AdV-3; JPT). The cells were seeded on ELISpot filter plates to analyse IFN-γ and Granzyme B secretion after overnight incubation. The MHC class II blocking antibody L243 and the MHC class I blocking antibody W6/32 were provided by Regina Feederle (Helmholtz Centre Munich, Germany).
RNA analysis
RNA from purified EVs and corresponding HEK293 producer cells were isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. To eliminate free RNA, fresh EV preparations were incubated with RNase A (10µg/ml; Promega) at 37°C for 30 min prior to RNA isolation. The reaction was stopped by adding RNasin (1 U/µl; Promega, Madison, WI). Purified RNA was analysed with the Agilent RNA 6000 Pico kit in the Agilent 2100 bioanalyzer instrument (Agilent Technologies, Santa Clara, CA). One microlitre of each EV RNA sample was used; cellular RNA was diluted to a concentration of 2 ng/µl. The QuantiTect Reverse Transcription Kit (Qiagen) was used for the synthesis of cDNA. For each RNA sample, a reaction mix without reverse transcription was included as negative control. GAPDH and pp65 sequences were afterwards detected by standard PCR using gene-specific primers (GAPDH: 5ʹ-TTCTTTTGCGTCGCCAGC-3ʹ and 5ʹ-GTGACCAGGCGCCCAATACGA-3ʹ; pp65: 5ʹ-CTTGGTATCGCAGTACACGC-3ʹ and 5ʹ-GCGCGTACACATAGATCGAC-3ʹ).
Results
CD40 ligand and EBV gp350 are functionally packaged into engineered EVs
EV engineering allows for the controlled integration of functional proteins and immunogenic antigens into EVs. To obtain EVs carrying CD40L and EBV gp350, we transfected HEK293 cells with the corresponding expression plasmids. After 3 days of incubation in EV-depleted medium, we isolated EVs from conditioned cell culture supernatants of the transfected cells by serial centrifugation steps. The EVs contained in the 100,000 × g pellet were further purified by floating into a bottom-up iodixanol gradient. Eight fractions were collected, and Western blot analysis showed that gp350 and CD40L co-localize with the EV markers CD63, TSG101 and Alix mainly in fractions 2 and 3, corresponding to a buoyant density of 1.06–1.13 g/ml (Figure 1(a)). Calnexin, a protein of the endoplasmic reticulum, was absent in all fractions of the gradient, indicative for the purity of the EV preparations. Additional characterization of all fractions of the gradient by NTA and Bradford protein assay confirmed the complete separation of EVs from contaminating free proteins (Suppl. Figure 1). EV-containing fractions were pooled, washed in PBS and pelleted by centrifugation at 100,000 × g. The presence of CD40L and gp350 in final EV preparations (positive EVs and control EVs) was confirmed by Western Blotting (Supplemental Figure 2). Analysis of EV preparations by electron microscopy demonstrated that the isolated material contained round, cup-shaped membrane vesicles of a mean diameter of 100–150 nm (Figure 1(b)). Beyond that, immunogold labelling of CD40L+/gp350+ EVs with antibodies against CD63, gp350 and CD40L confirmed the incorporation of these molecules into the EV membrane.
Figure 1.
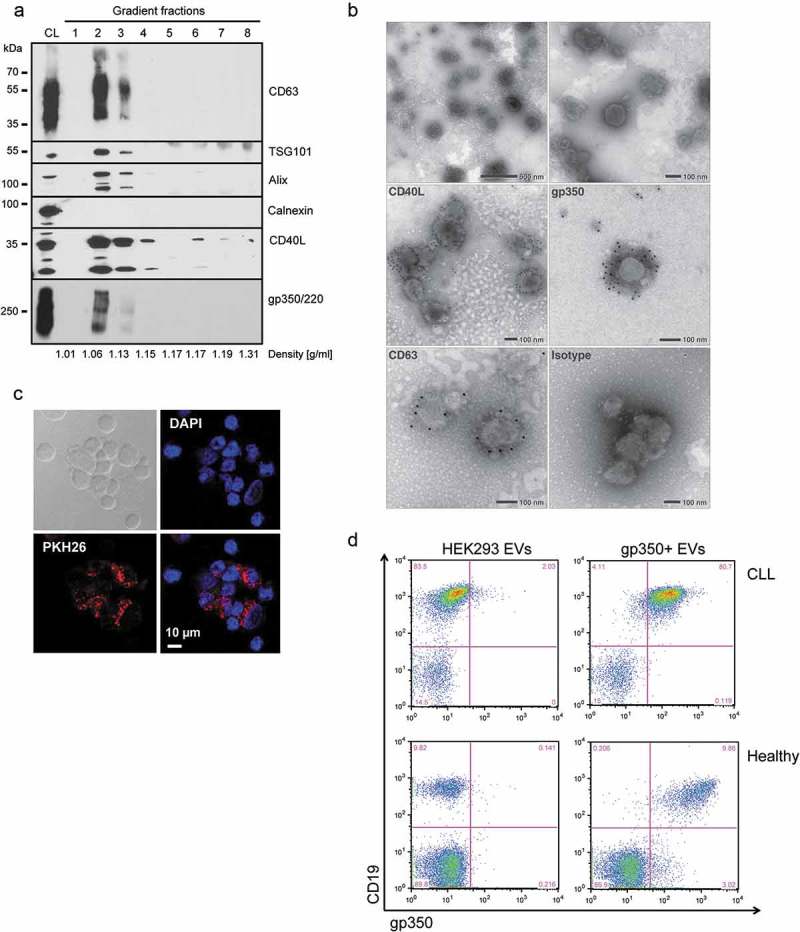
CD40L and gp350 are incorporated into EVs and confer B-cell tropism.
HEK293 cells were transfected with CD40L and gp350 expression plasmids and EVs were isolated from conditioned cell culture supernatant by serial (ultra-) centrifugation and density gradient centrifugation. (a) The fractions of a bottom-up iodixanol gradient were analysed by Western blotting. EVs carrying CD40L and gp350 and the EV-enriched proteins CD63, TSG101 and Alix were mainly detected in fractions two and three. CL: cell lysate. (b) Electron microscopy analysis of EV preparations. For immune electron microscopy antibodies against CD63, CD40L, gp350 or an isotype control were used and detected by a gold-conjugated secondary antibody. (c) Confocal microscopy of CLL cells incubated for 24 h with CD40L+/gp350+ EVs labelled with the fluorescent dye PKH26. Cell nuclei were counterstained with DAPI (40× magnification). (d) EVs from untransfected HEK293 cells or cells expressing gp350 were incubated with PBMCs from a healthy donor or primary CLL cells. After 48 h, cells were washed, stained with CD19- and gp350-specific antibodies and analysed by flow cytometry.
In order to demonstrate that engineered EVs interact with primary CLL cells, we labelled gp350+ EVs with the membrane dye PKH26 and incubated them with freshly isolated CLL cells. Separation of stained EVs from free excess dye via density gradient centrifugation ensured the specificity of the labelling (Supplemental Figure 3(a)). Flow cytometry analysis showed that CD40L+/gp350+ EVs interacted with CLL cells much more efficiently than CD40L+ EVs, corroborating the crucial role of gp350 for the interaction of EVs with B cells (Supplemental Figure 3(b)). Binding of labelled gp350+ EVs to CLL cells was further monitored by confocal microscopy after 24 h of incubation (Figure 1(c)). To demonstrate that gp350 confers B-cell tropism, we next incubated gp350+ EVs with CLL cells or PBMCs of healthy individuals and measured binding by flow cytometry two days later. As shown in Figure 1(d), gp350 was exclusively detectable on CD19+ B cells but not on CD19- cells.
Functional CD40L, transferred via engineered EVs, restores the antigen-presenting capacity of CLL cells
Engagement of CD40 on B cells regulates a wide spectrum of molecular and cellular processes and downstream signalling pathways, including activation of the noncanonical and canonical NF-κB pathways and the mitogen-activated protein (MAP) kinases ERK and JNK. To investigate whether engineered EVs also activate the CD40 pathway upon binding to B cells, we performed Western blot analysis of lysates from CLL cells, which had been pre-incubated or not with CD40L+/gp350+ EVs. It became clear that treatment of cells with CD40L+/gp350+ EVs induced ERK and JNK phosphorylation as early as 20 min of incubation (Figure 2(a)), indicative for CD40 activation and functionality of EV-tethered CD40L.
Figure 2.
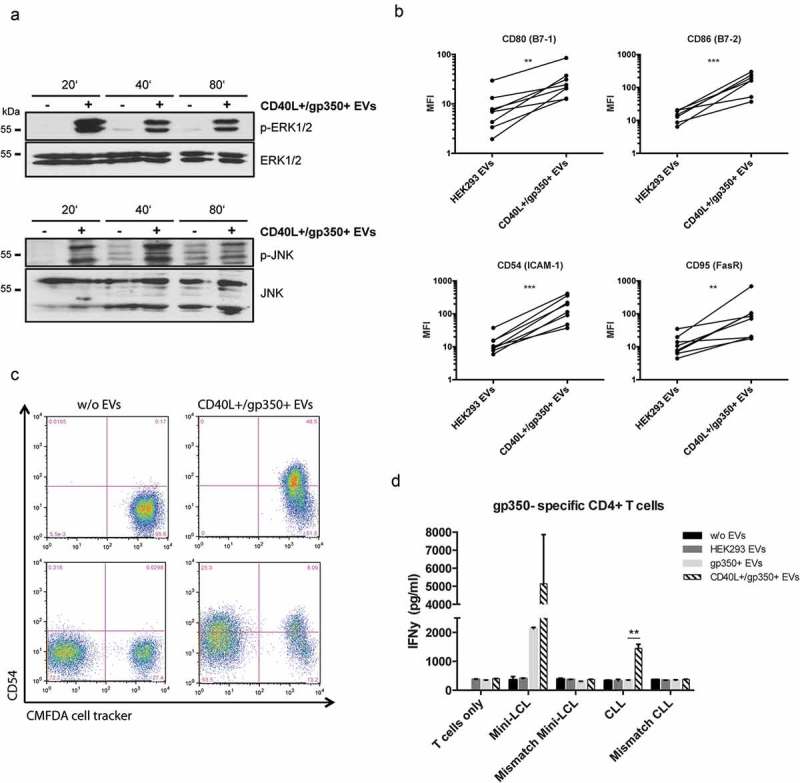
CLL cells treated with CD40L+/gp350+ EVs upregulate costimulatory surface molecules and become immunogenic for gp350-specific CD4+ T cells.
(a) Western Blot of ERK1/2 (upper panel) and JNK (lower panel) phosphorylation in untreated CLL cells or cells treated with gp350+/CD40L+ EVs. Cell lysates were prepared after 20, 40 and 80 min of incubation. (b) PBMCs were isolated from seven patients diagnosed with CLL and stimulated with EVs from untransfected HEK293 cells and cells expressing CD40L and gp350 for 48 h. The upregulation of the immune accessory molecules CD80, CD86, CD54 and CD95 was studied by measuring the mean fluorescence intensity (MFI) by flow cytometry. Asterisks represent statistically significant different responses to EV treatment (Mann–Whitney–Wilcoxon test, **p < 0.01, ***p < 0.001). (c) CLL cells were labelled with CFMDA cell tracker dye and incubated with CD40L+/gp350+ EVs (upper right panel) or left untreated (upper left panel) overnight. The cells were mixed with untreated CFMDA-negative cells and CD54 expression was analysed by flow cytometry after 24 h (lower panel). (d) HLA-DR13+ mini-LCLs and primary CLL cells, as well as mismatched control cells, were used as antigen-presenting cells and incubated with 500 ng of different EVs, as indicated. After coincubation for 24 h with HLA-DR13-restricted gp350-specific CD4+ T cells, IFN-γ secretion was measured by ELISA. The results are shown as mean and SD of triplicates. P values were calculated with an unpaired t-test.
To further investigate the transfer of EV-associated functional CD40L to CLL cells, PMBCs were isolated from seven CLL patients (≥80% of CD5+/CD19+ leukaemic cells) and incubated in the presence of 500 ng CD40L+/gp350+ EVs or control EVs for 48 h. We found that the expression of the accessory molecule CD54 (ICAM-1), the costimulatory molecules B7-1 (CD80) and B7-2 (CD86), and the death receptor CD95 (Fas) on CD19+ CLL cells was significantly upregulated upon incubation with CD40L+/gp350+ EVs but not upon incubation with control EVs (Figure 2(b)).
The so-called bystander effect describes the transfer of a particular phenotype from manipulated cells to untreated naïve cells in close vicinity. Because not each and every tumour cell can be isolated from a patient and thus is not amenable to a directed ex vivo manipulation, the efficacy of immunotherapeutic approaches also depends on this effect to occur after re-infusion of manipulated cells. We, therefore, wished to elucidate whether CLL cells, pre-incubated with engineered EVs, transfer their activated immunophenotype to naïve bystander CLL cells. For this, we stained CLL cells with the fluorescent CellTracker Green CMFDA dye and then incubated them with CD40L+/gp350+ EVs. As expected, the activation of CLL cells became evident by the induction of CD54 as measured by flow cytometry 24 h later (Figure 2(c), upper right panel). Next, we co-incubated the EV-activated, CFMDA-stained CLL cells with untreated, unstained CLL cells from the same donor for another 24 h. A flow cytometric analysis performed thereafter revealed a clear induction of ICAM-1 also on the hitherto untreated CLLs, thus confirming the activation of naïve bystander cells by EV-activated CLL cells (Figure 2(c), lower right panel).
As a next step, we investigated whether CLL cells reactivated by CD40L+ EVs become functional antigen-presenting cells (APCs) and consequently are able to reactivate specific T cells. To address this question, primary CLL cells as well as mini-LCLs, a B-cell line generated by immortalization with an EBV-derived vector [30], were used as APCs. Cells were incubated overnight with different EVs, as indicated in Figure 2(d), and thereafter co-incubated with a gp350-specific HLA-DR13-restricted CD4+ T-cell clone at a 1:1 ratio. HLA-mismatched LCLs and CLL cells alone were used as negative controls. Next, the concentration of IFN-γ in the cell culture supernatants after 24 h of incubation was quantified by ELISA. CLL cells alone and cells incubated with gp350+ EVs did not induce detectable release of IFN-γ. This is mainly because CLL cells, in contrast to LCLs, display a reduced expression of important costimulatory molecules and consequently efficient interaction with T cells is severely impaired. However, CLL cells, which had been pre-incubated with CD40L+/gp350+ EVs, induced a significant secretion of IFN-γ from co-cultured T cells, pointing out to the crucial role of CD40L for the antigen-presenting capacity of CLL cells.
B cells loaded with CD40L+/gp350+/pp65+ EVs efficiently stimulate pp65-specific CD4+ and CD8+ T cells
Co-opting the robust cellular T-cell immunity against EBV and, in particular, CMV, is an attractive strategy for immunotherapeutic approaches against CLL [29,34], but malignant cells normally are not infected with either virus, and thus do not express, and present, EBV- or CMV-derived proteins. The described strong CMV-specific immunity in CMV-seropositive CLL patients prompted us to investigate whether engineered EVs could be harnessed as conveyors of anti-viral immunity to malignant CLL cells. For this, we generated CD40L+/gp350+ EVs that additionally carried pp65 (=CD40L+/gp350+/pp65+), which is the immunodominant tegument protein of CMV known to elicit both CD4+ and CD8+ T-cell immune responses in CLL patients [27,28].
CD40L+/gp350+/pp65+ EVs were generated by overexpressing the proteins in HEK293 cells and EVs were isolated from conditioned cell cultured media by differential centrifugation and subsequent density gradient fractionation 3 days later, as described. Like CD40L and gp350, also pp65 was detected by immunoblotting mainly in fractions 2, 3 and 4 of the gradient (Figure 3(a)). To analyse the immunogenicity of EV-incorporated pp65, EVs were incubated with EBV-infected mini-LCLs overnight and then co-cultivated with HLA-matched, pp65-specific CD4+ and CD8+ T-cell clones for another 24 h. As expected, CD40L+/gp350+/pp65+ EVs efficiently induced IFN-γ release from the CD4+ T-cell clone (Figure 3(c), left diagram), while pp65-carrying EVs negative for gp350 were less effective in this assay, most likely due to reduced binding to mini-LCLs. Remarkably, CD40L+/gp350+/pp65+ EVs also reactivated pp65-specific CD8+ T cells to release IFN-γ (Figure 3(c), middle) and granzyme B indicative for cytolytic activity (Figure 3(c), right).
Figure 3.
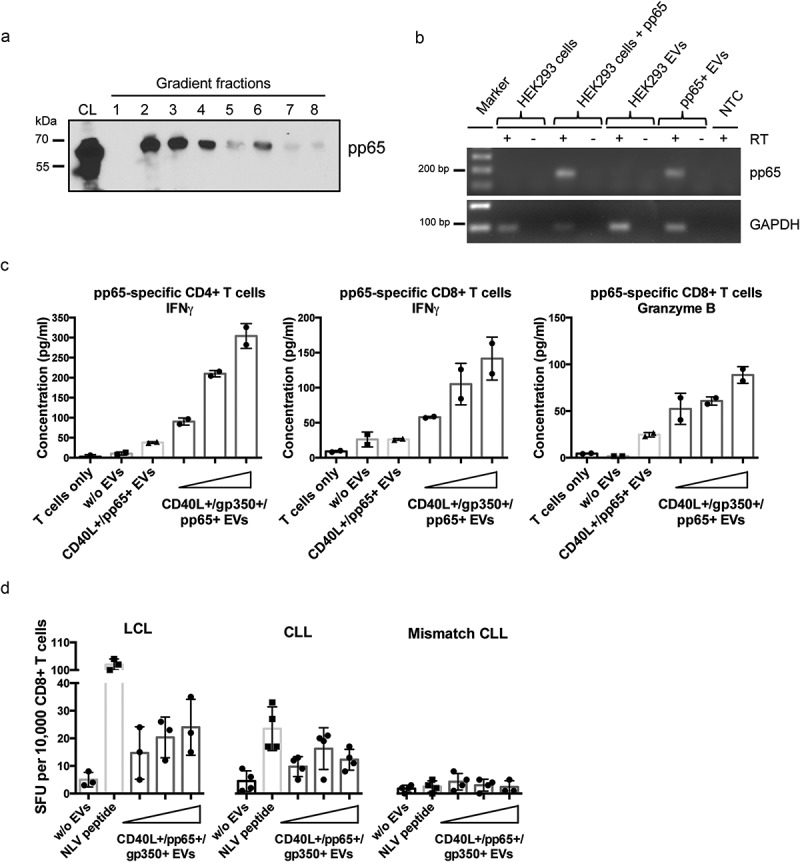
CMV pp65 is incorporated into HEK293-derived EVs and induces specific CD4+ and CD8+ T-cell responses.
(a) HEK293 cells were transiently transfected with a pp65 expression plasmid and EVs were isolated 3 days later by serial centrifugation and density gradient fractionation. Eight fractions were collected and analysed for pp65 by Western blotting. CL: cell lysate. (b) EVs derived from HEK293 cells expressing pp65 contain pp65 mRNA, as confirmed by reverse transcriptase (RT-) PCR using pp65-specific primers and cDNA isolated from HEK293 cells and the corresponding EVs. GAPDH mRNA was detected in both EVs from untransfected and transfected HEK293 cells. RT: reverse transcriptase; NTC: no template control. (c) 10,000 HLA-DQB1+ mini-LCLs were incubated overnight with 1000 ng of different types of EVs or left untreated, as indicated. CD40L+/gp350+/pp65+ EVs were applied in three different concentrations (250 ng, 500 ng, 1000 ng). EV-loaded mini-LCLs were co-cultured with an HLA-DQB1-restricted pp65-specific CD4+ T-cell clone at a 1:1 ratio. After 24 h, IFN-γ secretion was measured by ELISA. Middle and right diagram: HLA-A2+ LCLs were used to stimulate an HLA-A2-restricted pp65-specific CD8+ T-cell clone. The same settings as for CD4+ T cells were used. The concentrations of IFN-γ (middle) and granzyme B (right) in the supernatants were measured by ELISA. (d) LCLs or primary CLL cells isolated from an HLA-A2+ CMV-negative donors were loaded with increasing amounts of CD40L+/gp350+/pp65+ EVs and co-cultured on IFN-γ ELISpot filter plates with a HLA-A2-restricted pp65-specific CD8+ T-cell clone (10,000 cells/well) at a 1:1 ratio for 24 h. Wells containing T cells co-cultured with unloaded APCs or HLA-mismatched CLL cells were used as controls to confirm the specificity of the assay. Mean +SD of IFN-γ spot-forming units (SFU) per 10,000 T cells was enumerated.
We obtained similar results when we treated primary CLL cells with CD40L+/gp350+/pp65+ EVs and used them as stimulators for pp65-specific T cells. For this, CLL cells from a HLA-A2+ patient were loaded with either pp65-derived NLV peptide or increasing amounts of CD40L+/gp350+/pp65+ EVs, washed and then incubated with allogeneic HLA-A2-restricted pp65-specific CD8+ T cells, as described above. T-cell activity was measured with an IFN-γ ELISpot assay. As depicted in Figure 3(d), EV-treated CLL cells reactivated pp65-specific CD8+ T cells almost as efficiently as EBV-infected mini-LCLs, which are known as potent APCs for foreign antigens [35]. These results demonstrated that EV-loaded CLL cells are either capable of cross-presenting exogenous EV-derived proteins in association with MHC class I molecules or that pp65 mRNAs which are transferred to CLL cells by CD40L+/gp350+/pp65+ EVs are de novo translated and, upon degradation, presented via the MHC class I pathway (Figure 3(b); Supplemental Figure 4).
CLL cells loaded with CD40L+/gp350+/pp65+ EVs efficiently reactive and expand autologous pp65-specific CD4+ and CD8+ T cells
The previous experiments underlined the potential of normal and malignant B cells, loaded with CD40L+/gp350+/pp65+ EVs, to efficiently reactivate allogeneic pp65-specific CD4+ and CD8+ T-cell clones. In order to investigate a clinically much more relevant situation, we wished to elucidate whether EV-loaded CLL cells also have the capacity to reactivate autologous CMV-specific T cells. For this, we incubated primary PBMCs from CMV-seropositive HLA-A2+ CLL patients with CD40L+/gp350+/pp65+ EVs on an IFN-γ ELISpot plate overnight and then counted the number of IFN-γ-producing T cells. Figure 4(a) shows that the incubation of CLL cells with CD40L+/gp350+/pp65+ EVs indeed reactivates pp65-specific autologous T cells.
Figure 4.
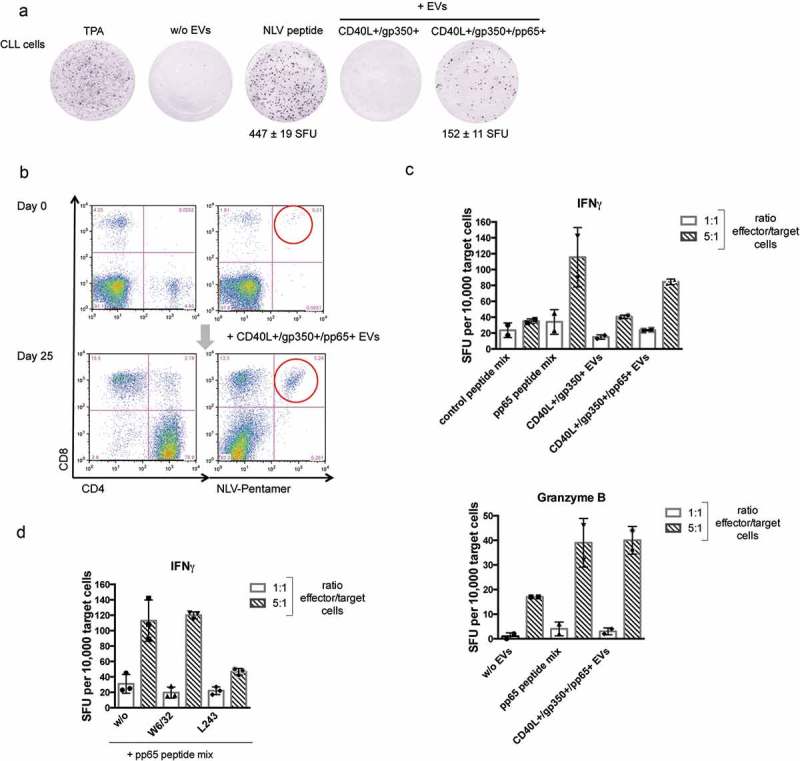
Autologous pp65-specific T cells from CLL patients are stimulated by pp65+ engineered EVs and are functional effector cells.
(a) CLL cells were seeded in IFN-γ ELISpot microwells (500,000 cells/well) and challenged with TPA/ionomycin, NLV peptide or different EVs, as indicated. SFU: spot-forming unit. (b) PBMCs were isolated from three CMV-positive CLL donors. Repetitive stimulation with CD40L+/gp350+/pp65+ EVs and IL-2 promoted cell survival. The cells were stained with CD4- and CD8- specific antibodies and the NLV pentamer on day 0 and day 25. One representative donor out of three is shown. (c) After 25 days of incubation, autologous CLL cells were thawed and loaded with EVs or the indicated peptide pools (0.5 µg/ml final concentration of each peptide) overnight. These target cells were mixed with the enriched effector cells at 1:1 and 1:5 ratios. Secretion of IFN-γ (c, d) and granzyme B (c, lower panel) was measured by ELISpots. Mean +SD of spot-forming units per 10,000 target cells were enumerated in duplicates. (d) The CLL target cells were incubated with the MHC class I blocking antibody W6/32 or the MHC class II blocking antibody L243 for 30 min prior to pp65 peptide loading.
In the next series of experiments, we addressed the question whether CD40L+/gp350+/pp65+ EVs not only reactivate, but even can expand pp65-specific T cells from CLL patients. For this, PBMCs isolated from three HLA-A2+, CMV+ CLL patients were treated with different EVs in the presence of IL-2, as described earlier. Cells were restimulated 7 and 14 days later by adding new EVs and IL-2. Cells were harvested and analysed by flow cytometry on day 25 of incubation. At that time point, no viable cells could be detected in cultures left unstimulated or stimulated with control EVs (not shown). In contrast, cultures stimulated three times with CD40L+/gp350+/pp65+ EVs contained a large fraction of vital cells, almost exclusively consisting of a predominant population of CD4+ T cells and a minor population of CD8+ T cells (Figure 4(b)). Staining with the pp65-specific HLA-A2 pentamer NLV revealed that the number of pp65-specific CD8+ T cells had increased several folds.
To further analyse the specificity of EV-enriched effector cells, freshly thawed autologous CLL target cells were loaded with CD40L+/gp350+/pp65+ EVs, a pp65 peptide pool or an adenovirus-derived control peptide pool and were added to the cultures. The number of IFN-γ secreting cells was enumerated 24 h later with an ELISpot assay (Figure 4(c)). Enumeration of spot-forming units (SFU) showed that CLL cells loaded with a pp65 peptide mix induced IFN-γ secretion from a larger number of cells than CLL cells loaded with the control peptide pool. Also, treatment with CD40L+/gp350+/pp65+ EVs resulted in a much higher number of SFUs as compared to incubation with pp65-negative EVs, indicating that pp65-specific T cells are present in the effector cell population (Figure 4(c)) and that EV-loaded CLL cells were targets for pp65-specific autologous T cells. Also, measurement of granzyme B secretion showed clearly that target cells loaded with either pp65 peptides or CD40L+/gp350+/pp65+ EVs amplified granzyme B secretion of functional pp65-specific T-effector cells (Figure 4(c), lower panel). Finally, we wished to elucidate the composition of the activated effector cells. Treatment of target cells with the MHC class II blocking antibody L243 prior to loading with pp65 peptides reduced the number of IFN-γ-secreting effector cells (Figure 4(d)). In contrast, an effect of the MHC class I blocking antibody W6/32 on IFN-γ-secreting cells was not observed, thus identifying CD4+ T cells as the major effector T-cell population.
Discussion
Since it is known that EVs are important conveyors of information between nearby or distant cells, their role in both physiological and pathological conditions has been studied extensively. Yet, EV research is still in its infancy and many mechanisms underlying EV biogenesis and EV-mediated cell–cell communication remain to be elucidated. Though, it is beyond doubt that EVs constitute attractive tools for novel therapeutic interventions as they can be engineered to efficiently transfer functional proteins of interest to target cells (reviewed in [9]). The aim of our work was to generate and evaluate the potential of engineered EVs as immunotherapeutic agents for the adjuvant treatment of CLL.
Several characteristics qualify CLL as an attractive indication for the development of such new experimental approaches. Firstly, expression of CD40L is reduced on T lymphocytes from CLL patients causing improper stimulation, and thus dysfunction, of CD40 on malignant CLL cells. In the same line, a number of experimental therapeutic strategies have demonstrated that the ectopic expression of CD40L can rescue CD40 signalling, resulting in an increased immunogenicity of CLL cells [36–38]. Secondly, in CLL patients the overall seroprevalence for EBV and CMV is high [39] and the cellular herpesviral immunity, in particular against CMV, remains intact even at advanced stages of the disease [28,40]. Thirdly, B-CLL cells express high levels of CD21, which is the physiological receptor for EBV gp350.
Here, we took advantage of these combined characteristics for an innovative EV-based immunotherapeutic approach, for which we equipped EVs with (i) gp350, the major glycoprotein of EBV, which confers B-cell tropism to these engineered EV and, at the same time, serves as a viral neo-antigen for CD4+ T cells in EV-treated CLL cells, with (ii) functional CD40L, which engages the CD40 receptor on CLL cells resulting in the induction of immune accessory molecules and enhanced immunogenicity and, finally, with (iii) pp65, the immunodominant tegument protein of CMV, which is known to elicit strong CD4+ and CD8+ cellular immune responses in infected hosts. Accordingly, our in vitro results demonstrate that engineered EVs strongly and specifically interact with CD21+ normal and malignant B cells and that the EV-mediated transfer of CD40L induced a robust induction of immune accessory cells like CD54 and CD80. Intriguingly, we observed that EV-treated and -activated CLL cells transfer their immunogenic phenotype to untreated cells, a process known as bystander effect, which is regarded as relevant for the efficacy of immunotherapies.
Even more important, CLL cells treated with CD40L+/gp350+/pp65+ engineered EVs became potent stimulators of CD4+ and CD8+ T-cell clones specific for gp350 and pp65. This is important to mention since the capacity to cross-present exogenous proteins on MHC class I molecules is mainly ascribed to dendritic cells, and the published literature is controversial about whether B lymphocytes share this capacity [41,42]. Since we could demonstrate that EVs can be engineered to contain both pp65 protein and pp65 mRNA, it is therefore tempting to speculate that the EV-mediated transfer of mRNAs to recipient B cells results in de novo translation and subsequent presentation of pp65-derived peptide in association with MHC class I molecules. In our experiments, autologous T cells were efficiently expanded by co-incubation with EV-loaded CLL cells and IL-2, so that after 25 days of incubation, cultures almost exclusively consisted of antigen-specific CD4+ and CD8+ T cells. Thus, stimulation of T cells with EV-loaded CLL cells is an attractive and powerful option to selectively expand specific T cells from CLL cells in vitro.
Clearly, further in vivo studies are necessary to further validate the potential of CD40L+/gp350+/pp65+ EVs as effective immunotherapeutic agents in a clinical setting. However, our in vitro studies strongly suggest the use of engineered EVs as promising tools for the development of new immunotherapeutic modalities. Above all, their potential to equip malignant cells with viral neo-antigens and to induce cytolytic CD4+ and CD8+ cellular immune responses against these cells should be further investigated.
Supplementary Material
Funding Statement
This work was supported by the Marga und Walter Boll-Stiftung [210-05-13] and by institutional grants.
Acknowledgements
We are indebted to all CLL patients for blood donation. We thank Josef Mautner and Andreas Moosmann, Research Unit Gene Vectors, Helmholtz Centre Munich, for the preparation of gp350- and pp65-specific T-cell clones and helpful discussions.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Théry C, Ostrowski M, Segura E.. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–13. [DOI] [PubMed] [Google Scholar]
- [2].Théry C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569–579. [DOI] [PubMed] [Google Scholar]
- [3].Ruiss R, Jochum S, Mocikat R, et al. EBV-gp350 confers B-cell tropism to tailored exosomes and is a neo-antigen in normal and malignant B cells - a new option for the treatment of B-CLL. PLoS One. 2011;6:e25294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Valadi H, Ekström K, Bossios A, et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659. [DOI] [PubMed] [Google Scholar]
- [5].Ruiss R, Jochum S, Wanner G, et al. A virus-like particle-based Epstein-Barr virus vaccine. J Virol. 2011;85:13105–13113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Beauvillain C, Juste MO, Dion S, et al. Exosomes are an effective vaccine against congenital toxoplasmosis in mice. Vaccine. 2009;27:1750–1757. [DOI] [PubMed] [Google Scholar]
- [7].Delcayre A, Estelles A, Sperinde J, et al. Exosome Display technology: applications to the development of new diagnostics and therapeutics. Blood Cells Mol Dis. 2005;35:158–168. [DOI] [PubMed] [Google Scholar]
- [8].Rountree RB, Mandl SJ, Nachtwey JM, et al. Exosome targeting of tumor antigens expressed by cancer vaccines can improve antigen immunogenicity and therapeutic efficacy. Cancer Res. 2011;71:5235–5244. [DOI] [PubMed] [Google Scholar]
- [9].Yang Y, Hong Y, Cho E, et al. Extracellular vesicles as a platform for membrane- associated therapeutic protein delivery. J Extracell Vesicles. 2018;7: 1440131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cramer P, Hallek M, Eichhorst B. State-of-the-art treatment and novel agents in chronic lymphocytic leukemia. Oncol Res Treat. 2016;39:25–32. [DOI] [PubMed] [Google Scholar]
- [11].Morabito F, Recchia AG, Vigna E, et al. Promising therapies for the treatment of chronic lymphocytic leukemia. Expert Opin Investig Drugs. 2015;24:795–807. [DOI] [PubMed] [Google Scholar]
- [12].Fischer K, Bahlo J, Fink AM, et al. Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood. 2018;127:208–216. [DOI] [PubMed] [Google Scholar]
- [13].Wolos JA, Davey FR. B lymphocyte function in B cell chronic lymphocytic leukaemia. Br J Haematol. 1981;49:395–403. [DOI] [PubMed] [Google Scholar]
- [14].Cantwell M, Hua T, Pappas J, et al. Acquired CD40-ligand deficiency in chronic lymphocytic leukemia. Nat Med. 1997;3:984–989. [DOI] [PubMed] [Google Scholar]
- [15].Battle TE, Wierda WG, Rassenti LZ, et al. In vivo activation of signal transducer and activator of transcription 1 after CD154 gene therapy for chronic lymphocytic leukemia is associated with clinical and immunologic response. Clin Cancer Res. 2003;9:2166–2172. [PubMed] [Google Scholar]
- [16].Takahashi S, Rousseau RF, Yotnda P, et al. Autologous antileukemic immune response induced by chronic lymphocytic leukemia B cells expressing the CD40 ligand and interleukin 2 transgenes. Hum Gene Ther. 2001;12:659–670. [DOI] [PubMed] [Google Scholar]
- [17].Wierda WG, Cantwell MJ, Woods SJ, et al. CD40-ligand (CD154) gene therapy for chronic lymphocytic leukemia. Blood. 2000;96:2917–2924. [PubMed] [Google Scholar]
- [18].Wendtner CM, Kofler DM, Mayr C, et al. The potential of gene transfer into primary B-CLL cells using recombinant virus vectors. Leuk Lymphoma. 2004;45:897–904. [DOI] [PubMed] [Google Scholar]
- [19].Mayr C, Kofler DM, Büning H, et al. Transduction of CLL cells by CD40 ligand enhances an antigen-specific immune recognition by autologous T cells. Blood. 2005;106:3223–3226. [DOI] [PubMed] [Google Scholar]
- [20].Stanfield BA, Luftig MA. Recent advances in understanding Epstein-Barr virus. F1000Res. 2017;6:386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tarrand JJ, Keating MJ, Tsimberidou AM, et al. Epstein-Barr virus latent membrane protein 1 mRNA is expressed in a significant proportion of patients with chronic lymphocytic leukemia. Cancer. 2010;116:880–887. [DOI] [PubMed] [Google Scholar]
- [22].Dolcetti R, Carbone A. Epstein-Barr virus infection and chronic lymphocytic leukemia: a possible progression factor? Infect Agent Cancer. 2010;5:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ferrajoli A, Ivan C, Ciccone M, et al. Epstein-Barr virus microRNAs are expressed in patients with chronic lymphocytic leukemia and correlate with overall survival. Ebiom. 2015;2:572–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Nemerow GR, Wolfert R, Mcnaughton ME, et al. Identification and characterization of the Epstein-Barr virus receptor on human B lymphocytes and its relationship to the C3d complement receptor (CR2). J Virol. 1985;55:347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Vallhov H, Gutzeit C, Johansson S, et al. Exosomes containing glycoprotein 350 released by EBV-transformed B cells selectively target B cells through CD21 and block EBV infection in vitro. J Immunol. 2011;186:73–82. [DOI] [PubMed] [Google Scholar]
- [26].Khan N, Hislop A, Gudgeon N, et al. Herpesvirus-specific CD8 T cell immunity in old age: cytomegalovirus impairs the response to a coresident EBV infection. J Immunol. 2004;173:7481–7489. [DOI] [PubMed] [Google Scholar]
- [27].Mackus WJM, Frakking FNJ, Grummels A, et al. Expansion of CMV-specific CD8+CD45RA+CD27- T cells in B-cell chronic lymphocytic leukemia. Blood. 2003;102:1057–1063. [DOI] [PubMed] [Google Scholar]
- [28].Pourgheysari B, Bruton R, Parry H, et al. The number of cytomegalovirus-specific CD4+ T cells is markedly expanded in patients with B-cell chronic lymphocytic leukemia and determines the total CD4+ T-cell repertoire. Blood. 2010;116:2968–2974. [DOI] [PubMed] [Google Scholar]
- [29].Te Raa GD, Pascutti MF, García-Vallejo JJ, et al. CMV-specific CD8+ T-cell function is not impaired in chronic lymphocytic leukemia. Blood. 2014;123:717–724. [DOI] [PubMed] [Google Scholar]
- [30].Van Deun J, Mestdagh P, Agostinis P, et al. EV-TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nat Methods. 2017;14:228–232. [DOI] [PubMed] [Google Scholar]
- [31].Graham FL, Smiley J, Russell WC, et al. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol. 1977;36:59–74. [DOI] [PubMed] [Google Scholar]
- [32].Kempkes B, Pich D, Zeidler R, et al. Immortalization of human primary B lymphocytes in vitro with DNA. Proc Natl Acad Sci U S A. 1995;92:5875–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Théry C, Clayton A, Amigorena S, et al. Isolation and characterization of exosomes from cell culture supernatants. Curr Protoc Cell Biol. 2006;Chapter 3:Unit3.22. [DOI] [PubMed] [Google Scholar]
- [34].Kater AP, Remmerswaal EBM, Nolte MA, et al. Autologous cytomegalovirus-specific T cells as effector cells in immunotherapy of B cell chronic lymphocytic leukaemia. Br J Haematol. 2004;126:512–516. [DOI] [PubMed] [Google Scholar]
- [35].Moosmann A, Khan N, Cobbold M, et al. B cells immortalized by a mini-Epstein-Barr virus encoding a foreign antigen efficiently reactivate specific cytotoxic T cells. Blood. 2002;100:1755–1764. [PubMed] [Google Scholar]
- [36].Biagi E, Rousseau R, Yvon E, et al. Responses to human CD40 ligand/human interleukin-2 autologous cell vaccine in patients with B-cell chronic lymphocytic leukemia. Clin Cancer Res. 2005;11:6916–6923. [DOI] [PubMed] [Google Scholar]
- [37].Messmer D, Kipps TJ. CD154 gene therapy for human B-cell malignancies. Ann N Y Acad Sci. 2005;1062:51–60. [DOI] [PubMed] [Google Scholar]
- [38].Wendtner C, Kofler DM, Theiss HD, et al. Efficient gene transfer of CD40 ligand into primary B-CLL cells using recombinant adeno-associated virus (rAAV) vectors. Blood. 2013;100:1655–1661. [PubMed] [Google Scholar]
- [39].Steininger C, Rassenti LZ, Vanura K, et al. Relative seroprevalence of human herpes viruses in patients with chronic lymphocytic leukaemia. Eur J Clin Invest. 2009;39:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Vanura K, Rieder F, Kastner M, et al. Chronic lymphocytic leukemia patients have a preserved cytomegalovirus-specific antibody response despite progressive hypogammaglobulinemia. PLoS One. 2013;8:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Keller SA, von Allmen CE, Heather J, et al. Follicular and marginal zone B cells fail to cross-present MHC class I-restricted epitopes derived from viral particles. J Immunol. 2009;182:6261–6266. [DOI] [PubMed] [Google Scholar]
- [42].Hon H, Oran A, Brocker T, et al. B lymphocytes participate in cross-presentation of antigen following gene gun vaccination. J Immunol. 2005;174:5233–5242. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.