

Summary
Human plasma-derived C1-esterase inhibitor (C1–INH) is an efficacious and safe treatment for hereditary angioedema. However, thrombotic events in subjects treated with C1–INH at recommended or offlabel, high doses have been reported. In this study, we addressed the potential prothrombotic risk of C1–INH treatment in high doses using a non-clinical rabbit model. Following intravenous infusion of C1–INH to rabbits at doses up to 800 IU/kg, the exposure and the pharmacodynamic efficacy of C1–INH in rabbits were confirmed by activity measurements of C1-esterase, and coagulation factors XIa and XIIa, respectively. Potential prothrombotic effects were assessed following induction of venous and arterial thrombosis using in vivo models of venous and arterial stasis, complemented by various in vitro assays of coagulation markers. Administration of C1–INH at doses up to 800 IU/ kg did not potentiate thrombus formation during venous stasis. In contrast, inhibition of arterial occlusion was observed upon C1–INH administration when compared with isotonic saline treatment, indicating antithrombotic rather than prothrombotic activity of high dose C1–INH treatment in vivo. This was further confirmed in vitro by decreased thrombin generation, increased activated partial thromboplastin time, clotting time and clot formation time, and inhibition of platelet aggregation. No relevant changes in fibrinolysis or in the levels of thrombin-antithrombin complexes, and prothrombin fragment 1+2 were observed upon high dose C1–INH treatment. The data suggest that treatment of healthy rabbits with high doses of C1–INH could potentially inhibit coagulation and thrombus formation rather than induce a prothrombotic risk.
Keywords: Animal model, C1-esterase inhibitor, coagulation, contact activation system, thrombosis
Introduction
C1-esterase inhibitor (C1–INH), a plasma glycoprotein with a molecular weight of 104 kDa, belongs to the protein family of serine protease inhibitors (serpins) which regulate the activity of serine proteases by inhibiting their catalytic activity ( 1 ). As the only known inhibitor of the activated serine proteases C1s and C1r, C1–INH inhibits the classical pathway of the complement system. Furthermore, C1–INH is a major inhibitor of the contact activation system due to its ability to inhibit the activated serine proteases factor XIIa (FXIIa), factor XIa (FXIa), and plasma kallikrein ( 2 , 3 ). Deficiency in C1–INH leads to the clinical manifestation of hereditary angioedema (HAE), which is characterised by episodes of acute angioedema attacks in subcutaneous or submucosal tissues such as the skin, larynx, or visceral organs ( 4 ). Subtypes of HAE are characterised by genetically reduced synthesis of C1–INH (type I), functional deficiency of C1–INH (type II), or FXII mutations (type III), resulting in increased bradykinin release upon stimulation of the contact activation system ( 5 ). HAE attacks can be treated effectively by administering exogenous C1–INH ( 4 , 5 ). Due to its inhibitory effects on the complement and the contact activation systems, C1–INH substitution restores normal haemostatic function and inhibits the excessive formation of vasoactive peptides such as bradykinin, which mediate the formation of angioedema.
Even though treatment with C1–INH purified from pooled human plasma has been demonstrated to be safe for many years ( 6 , 7 ), there have been recent reports suggesting increased risk of thromboembolic complications associated with the use of C1–INH ( 8 , 9 ), particularly beyond the approved clinical indications and doses. During off-label administration of C1–INH to neonates and infants undergoing cardiopulmonary bypass surgery at doses up to 500 IU/kg, i. e. 25 times higher than recommended for HAE patients, to prevent capillary leakage, occasionally lethal thromboembolic complications in the upper venous system were observed. These adverse events were believed to be at least partly associated with C1–INH treatment due to its inhibitory effects on the fibrinolytic system ( 6 , 10 ). However, it should be noted that the induction of thrombosis in neonates under surgery is highly complex. Central venous lines, surgically traumatised vessels, alteration of coagulation by extracorporeal circulation, and the immature haemostatic system of the newborn could considerably contribute to a thrombotic risk.
Horstick et al. ( 11 ) examined the cardioprotective effects of C1–INH using a pig model of coronary artery occlusion and reperfusion, and reported protective effects of C1–INH at low doses (< 100 IU/kg). However, adverse effects were observed at higher doses (≥100 IU/kg), including clot formation (inhibiting blood aspiration from non-heparinised intravenous [i. v.] catheters) and increased thrombin-antithrombin (TAT) levels. Another investigation into the potential thrombogenicity of a C1–INH product using a rabbit stasis model for venous thrombosis concluded that C1–INH presents a thrombogenic risk with a threshold of between 100 and 200 IU/kg and a non-observed adverse effect level (NOAEL) of ≤ 50 IU/kg ( 12 ). In contrast, Tassani et al. ( 13 ) demonstrated a clinical benefit of administering C1–INH at a dose of 100 IU/kg in neonates undergoing arterial switch operations.
A recent retrospective data analysis based on reports from the United States Food and Drug Administration’s Adverse Event Reporting System database resulted in 10 confirmed cases of C1–INH product-associated thrombotic events in HAE patients using recommended doses ( 14 ). However, the analysis did not allow any causal relationship between the investigated drug treatment and adverse events to be established. Despite the possibility that the thrombotic events associated with the specific C1–INH product may be related to the use of catheters or underlying risk factors ( 9 ), further studies on the potential risk associated with C1–INH treatment are warranted.
To support the safety of a recombinant C1–INH product, Relan et al. ( 15 ) evaluated the effects of C1–INH treatment on coagulation and fibrinolysis in a randomised clinical trial in symptomatic HAE patients. Infusion of the recombinant C1–INH did not affect parameters reflecting activation of the coagulation and fibrinolytic systems, such as levels of TAT, D-dimers, or plasmin-antiplasmin (PAP) complexes, with the exception of the activated partial thromboplastin time (aPTT), which was prolonged, and of prothrombin fragment 1+2 (F1+2) levels, which were decreased. However, the baseline levels of F1+2, TAT, D-dimers, and PAP complexes were already elevated, and aPTT was low to normal in the majority of these patients prior to recombinant C1–INH treatment, indicating activation of both coagulation and fibrinolysis in HAE patients. These data are supported by observations by Cugno et al. ( 16 ), who had previously suggested activation of the coagulation cascade in HAE patients during attacks. Overall, Relan et al. ( 15 ) observed no thromboembolic events in HAE patients following recombinant C1–INH treatment at doses up to 100 IU/kg.
Interestingly, antithrombotic effects were observed in a murine model of ischaemic stroke upon C1–INH treatment ( 17 ), as well as in several preclinical thrombosis models, upon inhibition ( 18 , 19 ) or genetic deletion ( 20 , 21 ) of C1–INH target molecules, i. e. FXII and FXI.
Against this background, we conducted a preclinical study designed to complement previous investigations and to provide further insight into the effects of C1–INH on the coagulation system, as well as to further assess the risk of thrombotic complications following administration of high doses of human plasma-derived C1–INH.
Materials and methods
Additional details of the methods are included in the Supplementary Material (available online at www.thrombosis-online.com ).
Animals and anaesthesia
Three to 4 months old Chinchilla Bastard (CHB) or New Zealand White (NZW) rabbits were used. Except for plasma sampling for pharmacokinetic analysis, all treatments were conducted upon anaesthesia induced by ketamine and xylazine.
Activity of C1-esterase and coagulation FXIa and FXIIa
Purified C1–INH obtained from pooled human plasma (Berinert ® , CSL Behring, Marburg, Germany) was injected i. v. at a dose of 200 IU/kg to CHB rabbits, and blood samples were collected at different time points. C1–INH activity levels were analysed based on the inhibition of human C1-esterase activity (Berichrom ® C1-In-hibitor, Siemens Healthcare Diagnostics Products, Marburg, Germany). Using the same method, C1–INH activity was determined following i. v. infusion of cumulative doses of 100, 400, and 800 IU/ kg C1–INH or equal volumes of isotonic saline (0.9 % sodium chloride) to CHB rabbits.
The activity of FXIa or FXIIa was assessed in plasma from CHB rabbits treated with cumulative doses of C1–INH or equal volumes of isotonic saline, based on aPTT measurement using the BCS ® XP system (Siemens Healthcare Diagnostics Products).
Comparison of effects of C1-INH on coagulation in rabbit and human plasma in vitro
C1–INH efficacy was assessed, following addition of 10, 15, 20, 40, 60, and 80 IU/ml C1–INH or equal volumes of saline into rabbit plasma or standard human plasma (SHP), based on measurements of FXIa and FXIIa activity, aPTT and PT using the BCS ® XP system (Siemens Healthcare Diagnostics Products).
Venous and arterial thrombosis
In vivo models of venous thrombosis
Venous thrombosis was investigated in NZW rabbits using the Wessler model ( 22 ) modified by Giles et al. ( 23 ). Venous stasis was induced for 30 min following single i. v. administrations of isotonic saline, Feiba ® NF 500 E (Factor Eight Inhibitor Bypassing Activity, Baxter, Unterschleissheim, Germany) at doses of 5, 10, 25, 50, or 100 IU/kg and C1–INH at doses of 200, 400, and 800 IU/kg. Thrombus score and thrombus wet weight were assessed, as described in the Supplementary Material (available online at www.thrombosis-online.com ).
Thrombus wet weight was also assessed after 3 hours (h) of venous stasis which was induced at the end of the administration of C1–INH cumulative doses (100, 400, and 800 IU/kg) or equal volumes of isotonic saline. Prior to each infusion and after the last infusion, blood samples were drawn for analysis of coagulation markers (see “ In vitro coagulation assays”).
In vivo model of arterial thrombosis
C1–INH (800 IU/kg) or an equal volume of isotonic saline were infused to CHB rabbits. Thereafter, arterial thrombosis was induced using a modification of the method originally described by Reimann-Hunziger ( 24 ) and Kurz et al. ( 25 ). Shortly, both femoral arteries were treated with 70 % ferric chloride (FeCl 3 ) for 60 minutes (min) while arterial occlusion was assessed for up to 90 min, using a flow module TS420 and perivascular flow probes (Transonic Systems Inc., Ithaca, NY, USA).
In vitro coagulation assays
The assessment of coagulation by in vitro assays was performed following either i. v. treatment of CHB rabbits at cumulative doses of C1–INH (100, 400, and 800 IU/kg) or equal volumes of isotonic saline, as described in “ In vivo models of venous thrombosis” or addition of C1–INH to rabbit plasma, SHP, or washed platelets derived from rabbit whole blood.
Thrombus formation and fibrinolysis using thromboelastography
Overall clot formation was assessed by determination of throm-boelastographic parameters (clot formation time [CFT], clotting time [CT], maximum clot firmness [MCF]) using the Rotem ® 05 system (Tem International, Munich, Germany). Coagulation of citrated whole blood from C1–INH or isotonic saline treated CHB rabbits was activated by use of INTEM, EXTEM, or NATEM.
Fibrinolysis was induced by addition of tissue plasminogen activator (tPA) (0.1 µg/ml; Actilyse ® , Boehringer Ingelheim, Biberach, Germany) to rabbit whole blood from C1–INH or isotonic saline treated CHB rabbits, followed by addition of EXTEM; the lysis index 60 (LI60) was measured.
Platelet aggregation
Platelets were activated within heparinised whole blood (5 IU/ml heparin; Ratiopharm, Ulm, Germany) from CHB rabbits treated with C1–INH or isotonic saline, by addition of either collagen (COLtest, 100 µg/ml) or adenosine diphosphate (ADP) (ADPtest, 0.2 mM). Platelet aggregation was assessed using impedance aggregometry (Multiplate Analyzer 5.0, Instrumentation Laboratory, Munich, Germany).
In addition, washed platelets were spiked with C1–INH at concentrations of 3.125 IU/ml to 50 IU/ml (reflecting C1–INH plasma levels following i. v. doses of approximately 100 to 1,600 IU/kg) (n=2). Platelet aggregation was then measured using the APACT 4 system (Rolf Greiner BioChemica, Flacht, Germany) following activation with rabbit thrombin (0.1 IU/ml; Sigma Aldrich, Munich, Germany).
Activated partial thromboplastin time and prothrombint ime
Blood samples were drawn from CHB rabbits treated with C1–INH or isotonic saline. APTT was determined following intrinsic activation, and prothrombin time (PT) was determined, following extrinsic activation, using the BCS ® XP analyser (Siemens Healthcare Diagnostics Products).
Thrombin generation
The thrombin generation assay (TGA) was conducted using calibrated thrombinography (Thrombinoscope, Maastricht, the Netherlands) in plasma samples from the C1–INH or isotonic saline treated animals, upon intrinsic or extrinsic activation. The thrombin calibrated automated thrombograms (dF/dt vs time) were used to determine the TGA parameters: peak thrombin levels, lag-time, and endogenous thrombin potential (ETP).
Thrombin-antithrombin complexes
TAT complexes were determined in blood samples drawn from CHB rabbits treated with C1–INH or isotonic saline, using a TAT enzyme-linked immunosorbent assay (ELISA) kit (Cusabio Biotech, Wuhan, China) according to the manufacturer’s instructions.
In addition, TAT complexes were determined in plasma samples obtained during the assessment of arterial thrombosis as described in “ In vivo model of arterial thrombosis”.
Prothrombin fragment 1+2
F1+2 levels were determined in plasma samples obtained during the assessment of arterial thrombosis as described in “ In vivo model of arterial thrombosis”, using a F1+2 ELISA Kit (Cusabio), according to the manufacturer’s instructions.
Statistical analysis and data presentation
Data from each dose of C1–INH were compared with corresponding data from the control group, using test statistics for two independent samples. Pre-dose data were accounted for by using analysis of covariance models, if possible. In that case, the estimated treatment effects were derived from the statistical model. P–values below 0.05 were termed statistically significant. Details on the statistical analysis can be found in the Supplementary Material (available online at www.thrombosis-online.com ).
Results
Evaluation of the pharmacokinetic and pharmaco-dynamic properties of C1–INH in rabbits
The pharmacological activity of plasma-derived human C1–INH was compared between rabbit and human, based on the inhibition of FXIa and FXIIa activities, as well as aPTT and PT measurements in vitro . To this end, rabbit plasma or SHP were spiked with increasing concentrations of C1–INH, which correspond to in vivo exposures of 400 to 800 IU/kg and above. Similar concentration-dependent effects of C1–INH on FXIIa inhibition between rabbit-and human-derived plasma were observed (Suppl. Figure 1A , available online at www.thrombosis-online.com ), while C1-INH mediated FXIa inhibition was stronger in human plasma compared to rabbit plasma (Suppl. Figure 1B , available online at www. thrombosis-online.com), indicating that the pharmacodynamics of C1–INH were at least partially comparable between rabbits and humans. Interestingly, in this assay, in rabbit plasma a 1.49-fold higher FXII- and a 3.8-fold higher baseline FXI activity compared to human plasma were found (data not shown) which is in line with Karges et al. ( 26 ). Additionally, aPTT and PT were analysed in rabbit- and human-derived plasma. As expected, no influence of C1-INH on PT was found whereas rabbit aPTT was even stronger affected by increasing concentrations of C1-INH compared to human aPTT values (Suppl. Figure 1C–D , available online at www.thrombosis-online.com ). Furthermore, efficacy of C1-INH regarding the inhibition of human and rabbit contact system activation was demonstrated via Western blotting of high-molecular weight kininogen (HMWK) cleavage (Suppl. Figure 2 , available online at www.thrombosis-online.com ). Here, a similar dose-response in the inhibition of HMWK cleavage upon activation of the kallikrein-kinin system was found in both rabbit and human plasma following addition of C1-INH in vitro . These data clearly demonstrate the pharmacological activity of C1–INH in rabbit plasma.
Figure 1: Pharmacokinetic and pharmacodynamic properties of C1-esterase inhibitor (C1–INH) in rabbits.

. C1–INH activity was assessed by measuring the inhibition of C1-esterase activity (A), at various time points upon administration of 200 IU/kg C1–INH and (B) upon administration of cumulative doses of C1–INH or equal volumes of isotonic saline. Values are expressed as percentage of C1–INH activity in standard human plasma (norm). C) Coagulation factors XIa (FXIa) and XIIa (FXIIa) activity levels were measured in rabbit plasma following administration of cumulative doses of C1–INH or equal volumes of isotonic saline. C1–INH values were normalised to pre-dose and corresponding isotonic saline values. For A-C: Dots and bars show means ± SD. **P=0.0024, ***P< 0.0001.
Figure 2: Effects of C1-esterase inhibitor (C1–INH) on venous thrombosis in rabbits in vivo.

. Thrombus score (A) and thrombus wet weight (B) were measured following single administrations of isotonic saline, Feiba ® , or C1–INH and 30 min of venous stasis. Bars show means ± SD. *P< 0.05. C) Thrombus wet weight was measured following 3 h of venous stasis in rabbits treated with cumulative doses of C1–INH up to 800 IU/kg or equal volumes of isotonic saline. Bars show means ± SD.
The assessment of the in vivo exposure and pharmacokinetic behaviour of plasma-derived human C1–INH in rabbits was based on measurements of C1-esterase inhibition.
Following i. v. administration of a single dose of 200 IU/kg, an initial rise in C1–INH activity to 737 % of endogenous normal (SHP) levels was measured, corresponding to a 14-fold increase from baseline (► Figure 1A ). Maximum C1–INH activity levels dropped by 8 % and 24 %, at 2 h and 3 h post-dose, respectively, and reached baseline levels after 168 h (Day 7). In addition, a terminal half-life of 1.4 ± 0.2 days, an overall exposure (area under the curve [AUC]) of 6.3 ± 0.8 day × IU/ml and clearance of 39 ± 4 ml/kg/day were determined (means ± standard deviation [SD]).
Cumulative doses of 100 IU/kg (0 to 5 min), 400 IU/kg (5 to 15 min), and 800 IU/kg (15 to 30 min) of C1–INH resulted in a dose-dependent increase in C1–INH activity ranging between 238 ± 31 % and 1587 ± 184 % of endogenous normal levels at 100 and 800 IU/kg compared with 32 ± 7 % at pre-dose (means ± SD; ► Figure 1 B). At the highest dose level tested, this translates to a 49.6-fold increase in C1-INH activity. In comparison, volume-adjusted treatment with isotonic saline resulted in a dilution effect observed as a reduction of C1–INH activity levels from 32 % to 21 % of endogenous normal levels.
Measurements of FXIa and FXIIa activities demonstrated the pharmacodynamic activity of human plasma-derived C1–INH in rabbits. Administration of cumulative doses of C1–INH to CHB rabbits resulted in a dose-dependent inhibition of FXIa and FXIIa activity compared with isotonic saline treatment (► Figure 1C ). The inhibition was statistically significant, reaching levels of 78 % (p< 0.0001) of isotonic saline for FXIa and 66 % (p=0.0024) of isotonic saline for FXIIa at a dose of 400 IU/kg, as well as 62 % (p< 0.0001) for FXIa and 59 % (p< 0.0001) for FXIIa at a dose of 800 IU/kg (analysis of covariance of log-transformed data).
Assessment of the prothrombotic potential of C1-INH in vivo
As shown in ► Figure 2 A and B, treatment of NZW rabbits with isotonic saline or C1–INH at doses up to 800 IU/kg following 30 min of venous stasis, did not induce any thrombus formation. In contrast, i. v. administration of Feiba ® , which was used as a positive control, resulted in a dose-dependent increase in thrombus formation reaching statistical significance (p< 0.05) at doses of 25 to 100 IU/kg for thrombus score and at 100 IU/kg for thrombus wet weight.
Prolongation of venous stasis time to 3 h led to non-occlusive thrombus formation, even in animals treated with isotonic saline (23.3–106.1 mg thrombus wet weight). Even under these experimental conditions, administration of C1–INH at a high dose (i. e. 800 IU/kg) did not result in any potentiation of venous thrombus formation (► Figure 2C ). The mean thrombus wet weight was slightly but not statistically significantly lower after C1–INH administration (47.2 mg) compared with isotonic saline (66.0 mg).
Since the modified Wessler models applied above only address potential prothrombotic activity of C1–INH during venous stasis, its effect was further assessed using a rabbit model of FeCl 3 -induced arterial thrombosis. Interestingly, treatment of CHB rabbits with 800 IU/kg C1–INH led to an inhibition of arterial thrombus formation. Occlusion rates were up to 2.2-fold lower upon C1–INH treatment compared with isotonic saline, suggesting a rather antithrombotic role for C1–INH under these experimental conditions (► Figure 3 ; p=0.08 for the log-rank test applied to mean time until occlusion of both arteries per animal).
Figure 3: Effects of C1-esterase inhibitor (C1–INH) on arterial thrombosis in rabbits in vivo.
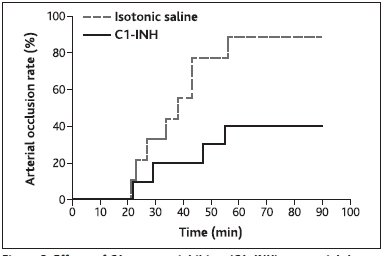
. A single dose of 800 IU/kg C1–INH or an equal volume of isotonic saline was administered to rabbits followed by induction of arterial occlusion via FeCl3 in each arteria femoralis. Occlusion rate was calculated as percentage distribution of arteries showing full occlusion over time.
In vitro assessment of C1-INH effects on coagulation
Effects of C1-INH on activated partial thromboplastin time and prothrombin time
Plasma from rabbits treated with cumulative C1–INH doses up to 800 IU/kg showed an increased aPTT from 21 seconds (sec) at pre-dose to 84 sec at 400 IU/kg C1–INH. At 800 IU/kg, an aPTT of 92 sec could only be measured in one animal, while the aPTTs for the remaining animals were above the cut-off value of 160 sec. Therefore, the aPTT was set to 160 sec for these animals (► Figure 4A ). The prolongation of aPTT following C1–INH treatment was statistically significant compared with isotonic saline for all cumulative doses: 100 IU/kg (p=0.035), 400 IU/kg (p< 0.001), and 800 IU/kg (p< 0.001) (Wilcoxon test) and corresponding to the initial in vitro spike experiments (Suppl. Figure 1C , available online at www.thrombosis-online.com ). No relevant effect of C1–INH treatment was observed on PT (data not shown) which ranged between 9.9 sec and 10.3 sec (mean values), with differences between C1–INH and isotonic saline treatments smaller than 0.15 sec at all doses (p>0.5; analysis of covariance). These values were also in line with results obtained in the in vitro spike assay (Suppl. Figure 1D , available online at www.thrombosis-online.com ).
Figure 4: Effects of C1-esterase inhibitor (C1–INH) on activated partial thromboplastin time (aPTT) and thrombin generation in rabbits.
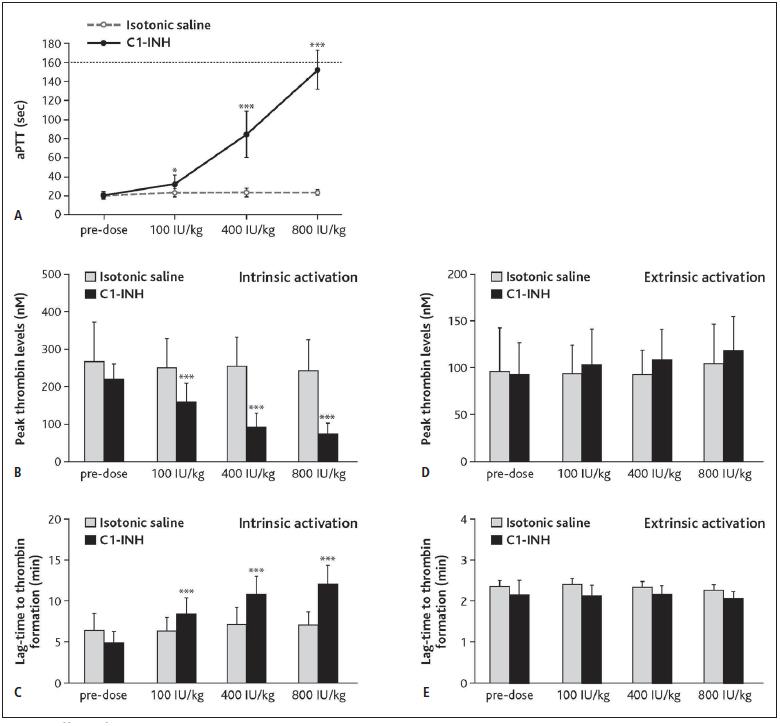
. Rabbits were injected with cumulative doses of C1–INH or equal volumes of isotonic saline and blood samples were drawn. A) APTT was measured following intrinsic activation (cut-off at 160 sec). Dots show means ± SD. B-C) Peak thrombin levels (B) and lag-time to thrombin formation (C) were measured by calibrated thrombography upon intrinsic activation. For A-C: *P=0.035, ***P< 0.001. Peak thrombin levels (D) and lag-time to thrombin formation (E) were measured by calibrated thrombography upon extrinsic activation. All data are statistically not significant with P≥ 0.05. For B-D: Bars show means ± SD.
Effects of C1-INH on thrombin generation
Administration of up to 800 IU/kg C1–INH to rabbits revealed inhibitory effects of C1–INH on thrombin generation after intrinsic activation, reflected by changes in peak thrombin levels and lagtime (► Figure 4B and C ). Compared with isotonic saline, peak thrombin levels were significantly reduced (p< 0.001) after treatment with C1–INH by 29 % (100 IU/kg), 61 % (400 IU/kg), and 66 % (800 IU/kg) (analysis of covariance of log-transformed data for all TGA parameters; ► Figure 4B ). Lag-time was significantly prolonged (p< 0.001) by 1.5-fold (100 IU/kg C1–INH), 1.8-fold (400 IU/kg C1–INH), and 2.0-fold (800 IU/kg C1–INH) compared with isotonic saline treatment (► Figure 4C ). In contrast, a very mild effect in the opposite direction was seen on peak thrombin levels and lag-time upon C1–INH treatment following extrinsic activation. The effect had borderline significance at some doses (peak thrombin levels increased by 16 % at 800 IU/kg C1–INH and lag-time decreased by 6 % at 100 IU/kg and 800 IU/kg C1–INH, compared with isotonic saline; p=0.05 in each case; ► Figure 4D and E ). ETP (measured as AUC) was not significantly affected by C1–INH treatment after intrinsic or extrinsic activation (differences between C1–INH and isotonic saline treatment were within ± 8 %; p>0.08 for all doses; data not shown).
Effects of C1-INH on platelet aggregation
To elucidate C1–INH-mediated effects on platelet function, ADP or collagen was applied to blood samples drawn from C1–INH or isotonic saline treated rabbits in order to induce platelet aggregation. C1–INH administration of doses up to 800 IU/kg showed only a slight trend towards inhibitory effects on maximum platelet aggregation in the presence of collagen, although this was in a dose-independent manner and statistically not significant (► Figure 5A ). Platelet aggregation was not significantly affected by C1–INH treatment upon ADP induction (increase of AUC between 1 % and 23 % compared with isotonic saline; p>0.3; data not shown).
Figure 5: Effects of C1-esterase inhibitor (C1–INH) on platelet aggregation in rabbits.
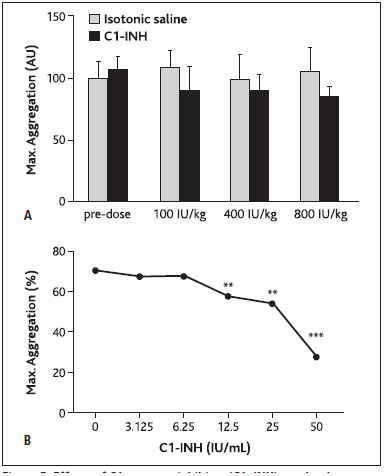
. A) Rabbits were injected with cumulative doses of C1–INH or equal volumes of isotonic saline and blood samples were drawn. Maximum aggregation was measured upon collagen addition, using impedance aggregometry. Bars show means ± SD. AU=arbitrary units. B) Washed platelets from rabbit blood were spiked with C1–INH and platelet aggregation was induced by thrombin. Maximum aggregation was expressed as percentage of aggregated sample volume. Dots show means.
In a thrombin-induced in vitro assay of platelet aggregation (► Figure 5B ), platelet concentrate was spiked with C1–INH at doses up to 50 IU/ml (translating to an in vivo dose of 1600 IU/kg) and a dose-dependent decline in thrombin-induced maximum platelet aggregation was observed (p=0.0031, p=0.0024, and p< 0.0001 at 12.5 IU/ml, 25 IU/ml, and 50 IU/ml, respectively, compared to isotonic saline; analysis of variance).
Effects of C1-INH on thrombus formation and fibrinolysis using thromboelastography
Upon intrinsic activation of the coagulation cascade (INTEM test), CT and CFT increased in a dose-dependent manner between 10 % and 23 % (CT), and 13 % and 29 % (CFT) compared with isotonic saline, at C1–INH doses of 100 IU/kg and 800 IU/kg, respectively (data not shown). The effects at 400 IU/kg C1–INH for CT (p=0.037) and at 800 IU/kg C1–INH for CT and CFT (p=0.013 and p=0.034, respectively) were statistically significant (analysis of covariance of log-transformed data). MCF was not affected by C1–INH treatment and differed within a 2.3 % range from isotonic saline levels at all doses tested (data not shown).
After extrinsic activation (EXTEM test), CFT increased by 7 % at 400 IU/kg (p=0.46) and 17 % at 800 IU/kg (p=0.08) C1-INH administration compared with isotonic saline, while CT and MCF remained unaffected, showing differences in the range of 3.0 % (CT) and 0.6 % (MCF) compared with isotonic saline levels at all doses (p>0.5) (data not shown).
In addition, the NATEM test was used as one of the most sensitive methods assessing effects on the coagulation cascade. Here, in the absence of extrinsic or intrinsic activation, recalcification resulted in a prolongation of CT and CFT upon C1–INH administration. CT was prolonged by 21 % at 100 IU/kg (p=0.13), 42 % at 400 IU/kg (p=0.03), and 92 % at 800 IU/kg (p< 0.0001) compared with isotonic saline administration (analysis of covariance of log-transformed data; ► Figure 6A ). Median CFT was prolonged by 40 % at 100 IU/kg (p=0.03), 100 % at 400 IU/kg (p< 0.001), and more than 100 % at 800 IU/kg (p=0.006; log-rank test) (► Figure 6B ).
Figure 6: Effects of C1-esterase inhibitor (C1–INH) on thrombus formation in rabbits using thromboelastography.

. Clotting time (CT) (A) and clot formation time (CFT) (B) were measured after recalcification only (NATEM) of citrated whole blood from rabbits treated with cumulative doses of C1–INH or equal volumes of isotonic saline. Horizontal lines correspond to medians.
Measurements of LI60 following induction of clot lysis by addition of tPA, in order to assess potential inhibitory effects of C1–INH on fibrinolysis, revealed no indication of antifibrinolytic activity of C1–INH (► Figure 7 ; p>0.2 at all doses; analysis of covariance of log-transformed data).
Figure 7: Effects of C1-esterase inhibitor (C1–INH) on fibrinolysis in rabbits.
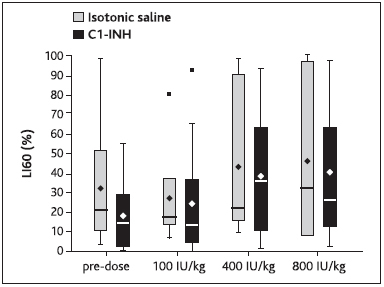
. CHB rabbits were injected with cumulative doses of C1–INH or equal volumes of isotonic saline and blood samples were drawn. Fibrinolysis was induced by addition of tPA and upon extrinsic activation. Lysis index 60 (LI60) was measured using thromboelastography. Bottom and top of the box represent the 25th and 75th percentile, respectively, the band within the box represents the median, the diamond symbol represents the mean, and the low and high ends of the whiskers represent the lowest datum still within 1.5 interquartile range (IQR) of the lower quartile, and the highest datum still within 1.5 IQR of the upper quartile, respectively.
Effects of C1-INH on TAT and F1+2 levels
After the administration of 800 IU/kg C1-INH to rabbits, no relevant changes in the TAT and F1+2 levels were observed compared to isotonic saline treatment (data not shown).
Discussion
Overall, C1–INH replacement therapy has been recognized as a well-established, effective, and safe treatment for HAE acute attacks for over 20 years as well as in prophylaxis. However, adverse events reported in association with C1–INH treatment include risk of thromboembolic complications upon administration at both high and recommended doses ( 8 , 9 ). Recently, few clinical and preclinical reports have provided contradictory evidence on the possible prothrombotic potential of C1–INH treatment ( 11 , 13 , 14 ). However, no causative mechanism has been demonstrated so far and remains elusive. Therefore, our study aimed to provide further insight into the involvement of C1–INH in coagulatory functions and its role in thromboembolic events.
To this end, the rabbit was chosen as a suitable experimental model, providing high sensitivity due to its high endogenous levels of coagulation factors compared with humans or other experimental species ( 26 ). In our study, the pharmacokinetic behaviour of C1–INH in rabbits was in agreement with previous observations in rabbits ( 27 ), and C1–INH showed pharmacody-namic efficacy, as demonstrated by a dose-dependent inhibition of FXIa and FXIIa activities (► Figure 1 ). Based on additional in vitro experiments, the dose-response of this effect was similar between human- and rabbit-derived plasma although more pronounced for human plasma in the case of FXIa inhibition (Suppl. Figure 1 , available online at www.thrombosis-online.com ). On the other hand, the increased effect on aPTT seen in rabbit plasma (► Figure 1C ) may point to a stronger inhibitory effect of of C1-INH on rabbit plasma kallikrein compared to human kallikrein. Via Western blotting using C1-INH it was found that inhibition of contact system activation was similar between human and rabbit plasma (Suppl. Figure 2 , available online at www.thrombosis-online.com ). Furthermore, literature is available suggesting that human C1-INH has not just strong inhibitory potency on human complement proteases C1r, C1s and MASP-1, but also on rabbit C1s, further supporting the rabbit as a pharmacologically relevant animal model to study the effects of human C1-INH ( 28 ). However, it remains to be elucidated whether human C1-INH is equally potent in rabbits as it is in humans. Therefore, the potential differences in the contact system between human and rabbit, including the differences between the effects of C1-INH on FXIa inhibition and aPTT or discrepancies between FXIa and PT baseline levels as seen in this study, have to be kept in mind when interpreting and translating data from rabbit models to humans.
At present, a potential procoagulatory role of C1-INH is being discussed. An underlying mechanism leading to this class of adverse events observed has not been demonstrated; however, potential antifibrinolytic effects of C1-INH have been considered. Due to its ability to inhibit the function of FXIa, FXIIa, and kallikrein, C1–INH could potentially elicit those procoagulatory events by interfering with downstream cascades such as the fibrinolytic pathway (► Figure 8A ). During fibrinolysis, plasmin is generated by plasminogen upon the action of tPA and urokinase-like plasminogen activator (uPA); plasmin subsequently catalyses the degradation of fibrin clots ( 29 ). C1–INH could interfere with plasmin production in multiple ways: directly, by inhibiting the activity of tPA ( 30 ) or the cleavage of plasminogen by FXIIa ( 32 ), or indirectly, by inhibiting the activity of kallikrein. The latter could lead to decreased urokinase and subsequently reduced plasminogen activation ( 31 ), as well as to decreased tPA levels due to reduced bradykinin production downstream of kallikrein ( 32 ). The contribution of C1–INH to tPA inhibition seems to depend on the active levels of plasminogen activator inhibitor-1 (PAI-1), and C1–INH may inhibit as little as 5 % of plasma tPA under certain active PAI-1 concentrations ( 30 ). Since active PAI-1 levels can vary between healthy individuals and certain patient populations ( 30 ), different subjects may exhibit varying sensitivities to the potential antifibrinolytic effects of C1–INH. However, C1–INH seems to play only a limited role in tPA inhibition. Clinical data has demonstrated that the levels of tPA and uPA remained normal during both remission and acute attacks of HAE patients ( 33 ) and increased C1–INH-tPA complexes were only observed in presence of high tPA levels in the circulation ( 3 , 34 ).
Figure 8: Potential pro (A) and anticoagulatory (B) effects of C1-esterase inhibitor (C1–INH) function.
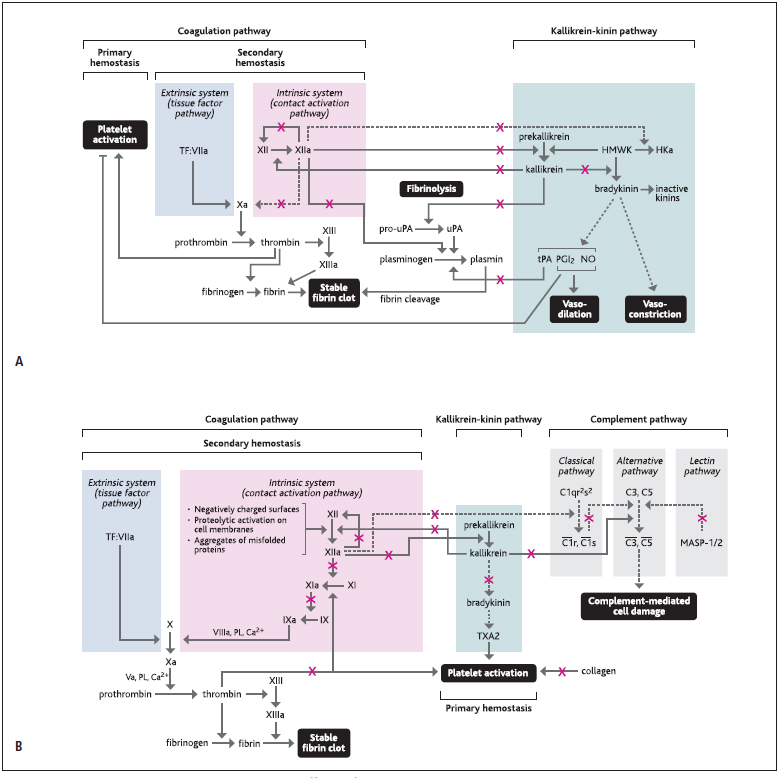
. C1–INH can potentially elicit procoagulatory effects (A), by inhibiting fibrinolysis or increasing platelet activation, but also anticoagulatory effects (B), by inhibiting thrombin formation, platelet activation, or the function of complement pathway. Dotted arrows indicate indirect actions and continuous arrows indicate direct actions. Red Xs indicate inhibition by C1–INH. Ca=calcium; coagulation factors Va, VIIIa, IX/IXa, X/Xa, XI/XIa, XII/XIIa, XIII/XIIIa; HKa=cleaved kinin-free kininogen; HMWK=high-molecular weight kininogen; MASP=mannose-binding lectin-associated serine protease; NO=nitric oxide; PGI2=prostaglandin I2; PL=phospholipids; pro-uPA=pro-urokinase-type plasminogen activator; TF:VIIa=TF complex with factor VIIa; tPA=tissue plasminogen activator; TXA2=thromboxane A2; uPA=urokinase-type plasminogen activator.
Furthermore, it could be speculated from the literature that C1–INH could potentially elicit additional procoagulatory effects by indirectly increasing platelet activation and/or fibrin clot formation (► Figure 8A ). Reduced bradykinin levels, due to C1–INH-mediated inhibition of kallikrein, could likely decrease the release of prostaglandin I2 and nitric oxide from endothelial cells, which usually exert platelet inhibitory effects, thus decreasing the barrier for platelet activation ( 35 , 36 ). However, this hypothesis was not further investigated or proven as part of the present study. In addition, cleaved kinin-free kininogen was shown to compete with fibrinogen for deposition on negatively-charged surfaces, neutrophils, and platelets ( 31 ). Therefore, reduced cleaved kinin-free kininogen levels, due to inhibition of FXIIa by C1–INH, may potentially lead to reduced displacement of fibrinogen and increased fibrin formation.
Nevertheless, despite the theoretical impact of the potential procoagulatory effects of C1–INH described above, in the present work no such findings underlining this hypothesis were observed. Thus, our study clearly suggests no prothrombotic risk associated with C1–INH treatment in rabbits, even at supratherapeutic doses. In the current study, in vivo models of thrombogenicity were employed to assess the prothrombotic potential of C1–INH, as a sensitive readout of the interplay between blood cells, cell membranes, vascular wall, blood flow conditions, and local or systemic conditions such as hypoxia or acidosis. The modified Wessler test in rabbits is a well-established model of acute and prolonged venous stasis, mimicking major aspects of deep-vein thrombosis in humans. Following venous stasis, C1–INH doses up to 800 IU/kg did not result in any potentiation of thrombus formation (► Figure 2 ). Interestingly, the additional application of a chemically-induced arterial thrombosis model in rabbits revealed anticoagulatory properties for C1–INH, with doses of 800 IU/kg C1–INH resulting in a 2.2-fold reduction of total arterial occlusion compared with isotonic saline treatment (► Figure 3 ).
The in vivo observations were further supported by a variety of in vitro assays assessing the impact of C1–INH treatment on the contact activation system, thrombin generation, platelet aggregation, and fibrinolysis. In contrast to the theoretical potential of C1–INH to enhance clot formation as described above, a prolongation of CT, CFT, aPTT and lag-time of thrombin formation, and a decrease in peak thrombin levels were observed in the presence of C1–INH upon intrinsic activation of thrombin generation, pointing towards an anticoagulatory effect for this treatment (► Figure 4A–C and ► Figure 6 ). No effect of C1-INH was observed on PT (Suppl. Figure 1D , available online at www.thrombosis-online.com ) or on thrombin generation following extrinsic activation (► Figure 4D–E ).
Our findings showing a potential anticoagulatory role for C1–INH are not surprising considering the multiple functions of this protein within the contact activation and complement cascades and the complex interplay among these pathways.
Firstly, any interference of C1–INH with the contact activation system, i. e. through inhibition of FXIIa, FXIa, or kallikrein, could modulate vascular biology by decreasing thrombin generation (► 3 ) (► Figure 8B ), a finding that was observed in the present study following intrinsic activation of thrombin generation as demonstrated in Figure 4B–C .
In addition, the observed inhibition of platelet aggregation when thrombin was used as platelet agonist (► Figure 5 ) in the presence of C1–INH further argues against a prothrombotic role for this treatment. These findings are partly in line with results from Copolla et al. who hypothesised a negative regulating role of C1-INH in platelet aggregation ( 37 , 38 ). In general, platelet activation is positively regulated by thrombin, collagen, and thromb-oxane A2 (TXA2) ( 37 ). C1–INH could interfere with this process either directly, by binding to thrombin ( 39 , 40 ) and collagen ( 41 ), or indirectly by downregulating bradykinin levels, leading to reduced TXA2 production via the arachidonic acid pathway ( 42 – 44 ). In addition, reduced FXIIa and kallikrein activity, due to C1–INH inhibition, may potentially affect neutrophil-platelet interactions ( 45 , 46 ).
The potential interaction of C1-INH with thrombin may consequently enhance the fibrinolytic system, considering the inhibitory effects of thrombin on fibrinolysis ( 47 ) and the decrease in thrombin generation upon C1–INH action. However, no relevant changes in fibrinolytic activity were observed in this study upon C1–INH treatment (► Figure 7 ).
Furthermore, C1–INH could potentially exert anticoagulatory effects via its action on the complement system (► Figure 8B ). C1–INH can interfere with the complement system directly, by inhibiting the activity of the serine proteases C1s and C1r, as well as of MASP-1 (mannose-binding lectin-associated serine protease) and MASP-2, but also indirectly, by blocking FXII autoactivation and kallikrein. Upon cleavage of FXIIa by kallikrein, the FXIIa â-fragment is produced which can transform the macromolecular C1qr 2 s 2 complex into enzymatically active C1r and C1s ( 48 , 49 ). Moreover, kallikrein can directly activate C3 and C5. Therefore, inhibition of FXII autoactivation and kallikrein by C1–INH could lead to the downregulation of the classical, lectin, and alternative pathways of the complement system and may consequently suppress the stimulation of downstream procoagulatory events triggered by complement fixation on cell surfaces ( 50 ) or anaphylatoxin (C3a and C5a)-mediated inflammatory responses ( 51 ).
Overall, C1–INH administration at supratherapeutic doses in rabbits revealed an antithrombotic effect for this treatment. However, it should be taken into consideration that the adverse thromboembolic events reported previously have not been observed under healthy conditions but against a background of capillary leakage syndrome or heart insufficiencies, which are off-label indications for C1–INH. In addition, it cannot be neglected that C1–INH activity can be affected by co-treatments or by the status or severity of HAE. In the first case it has been demonstrated that co-treatment with glycosaminoglycans such as Heparins can increase C1–INH activity ( 52 , 53 ) and the mean half-life of therapeutically administered C1–INH has been shown to vary from 33 h up to 48 h for patients with severe HAE and patients with moderate HAE, respectively (54).
What is known about this topic?
C1–INH is a potent inhibitor of the complement and contact activation systems, and its deficiency can lead to the manifestation of hereditary angioedema. Replacement therapy with human plasma-derived C1–INH is an efficacious and safe treatment for this disease.
However, thrombotic events have been associated with the use of C1–INH at recommended or off-label, high doses.
What does this paper add?
High-dose C1–INH administration to rabbits did not potentiate venous thrombosis, and interestingly inhibited arterial thrombosis.
Decreased thrombin generation and platelet aggregation, as well as prolonged activated partial thromboplastin time, clotting time, and clot formation time were observed upon high dose administration of C1–INH.
C1–INH administration to healthy rabbits at high doses seems to have anticoagulatory and antithrombotic effects, rather than a prothrombotic potential.
In summary, assessment of the prothrombotic risk of C1–INH treatment in rabbits revealed a rather anticoagulatory role for C1–INH in this study. Venous thrombosis was not potentiated and arterial thrombosis was inhibited upon C1–INH administration at supratherapeutic doses. Inhibition of the contact activation system, reduced thrombin generation, and reduced platelet aggregation were observed in blood samples of C1–INH treated animals, further supporting the anticoagulatory potential of C1–INH. Furthermore, there was no apparent C1–INH-mediated inhibition of fibrinolysis. Nevertheless, as the present study was conducted in healthy rabbits, future investigations of the prothrombotic risk of C1–INH are warranted in various clinical backgrounds reflecting the potentially complex influences of underlying disease states in patients.
Acknowledgments
The authors would like to thank Franz Kaspereit, Wilfried Krege, Bärbel Dörr, Sabrina Schenk, and Patrick Letmade (all CSL Behring GmbH) for their excellent technical assistance, and Dr. Ioanna Bethani at Trilogy Writing & Consulting (Frankfurt am Main, Germany) for assistance in preparing the manuscript. Work on inhibition of HMWK cleavage was performed using funding provided by Vetenskapsrådet (K2013–65X-21462–04–5), German Research Society (SFBs 841 & 877) and European Research Council grant (ERC-StG-2012–311575_F-12). CSL Behring sponsored the Open Access fee for this manuscript.
Funding Statement
Financial support: This work was funded by CSL Behring GmbH.
Conflicts of interest D. Schürmann, E. Herzog, E. Raquet, M. W. Nolte, F. May, J. Müller-Cohrs, G. Dickneite, and I. Pragst are employees of CSL Behring GmbH. J. Björkqvist declares no conflicts of interest.
Equal contribution.
References
- 1.Bock SC, Skriver K, Nielsen E. Human C1 inhibitor: primary structure, cDNA cloning, and chromosomal localization. Biochemistry. 1986;25:4292–4301. doi: 10.1021/bi00363a018. [DOI] [PubMed] [Google Scholar]
- 2.Davis AE. The pathophysiology of hereditary angioedema. Clin Immunol. 2005;114:3–9. doi: 10.1016/j.clim.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 3.Caliezi C, Wuillemin WA, Zeerleder S. C1-Esterase Inhibitor: an anti-inflammatory agent and its potential use in the treatment of diseases other than hereditary angioedema. Pharmacol Rev. 2000;52:91–112. [PubMed] [Google Scholar]
- 4.Longhurst H, Cicardi M. Hereditary angio-oedema. Lancet. 2012;379:474–481. doi: 10.1016/S0140-6736(11)60935-5. [DOI] [PubMed] [Google Scholar]
- 5.Bork K. Diagnosis and treatment of hereditary angioedema with normal C1 inhibitor. Poster presented at the 2012 AAAAI Annual Meeting, Orlando, Florida. Allergy Asthma Clin Immunol. 2010;6:15. doi: 10.1186/1710-1492-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bork K, Korger G, Kreuz W.Review of the long-term safety of a human pasteurized C1 inhibitor concentrate J Allergy Clin Immunol 201212902SAB222..et al. [Google Scholar]
- 7.Berinert® US Prescribing Information Available at:http://labeling.cslbehring.com/PI/US/Berinert/EN/Berinert-Prescribing-Information.pdfAccessed May 22, 2013.
- 8.Cinryze® US Prescribing Information Available at:http://www.cinryze.com/pdfs/cinryze-prescribing-information.pdfAccessed May 22, 2013.
- 9.German Medical Profession’s Drugs Committee Severe thrombus formation of Berinert® HS. Deutsches Ärzteblatt2000; 97: A-1016/B-864/C-812.
- 10.Horstick G, Berg O, Heimann A. Application of C1-esterase inhibitor during reperfusion of ischemic myocardium: dose-related beneficial versus detrimental effects. Circulation. 2001;104:3125–3131. doi: 10.1161/hc5001.100835. [DOI] [PubMed] [Google Scholar]
- 11.Pharmacology / Toxicology Review Memorandum STN 125267 - C1 Esterase Inhibitor for the treatment of human angioedema (HAE). Buehler PWDepartment of Health and Human Services Food and Drug Administration Center for Biologics Evaluation and Research. August, 2007. Available at:www.fda.gov/downloads/BiologicsBloodVaccines/BloodBloodProducts/ApprovedProducts/LicensedProductsBLAs/FractionatedPlasmaProducts/UCM229783.pdfAccessed May 22, 2013.
- 12.Tassani P, Kunkel R, Richter JA. Effect of C1-esterase-inhibitor on capillary leak and inflammatory response syndrome during arterial switch operations in neonates. J Cardiothorac Vasc Anesth. 2001;15:469–473. doi: 10.1053/jcan.2001.24989. [DOI] [PubMed] [Google Scholar]
- 13.Ghandi PK, Gentry WM, Bottorff MB. Thrombotic events associated with C1 esterase inhibitor products in patients with hereditary angioedema: Investigation from the United States Food and Drug Administration Adverse Event Reporting System Database. Pharmacotherapy. 2012;32:902–909. doi: 10.1002/j.1875-9114.2012.01126. [DOI] [PubMed] [Google Scholar]
- 14.Relan A, Bakhtiari K, van Amersfoort ES. Recombinant C1-inhibitor. Effects on coagulation and fibrinolysis in patients with hereditary angioedema. Biodrugs. 2012;26:43–52. doi: 10.2165/11599490-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 15.Cugno M, Cicardi M, Bottasso B. Activation of the coagulation cascade in C1-inhibitor deficiencies. Blood. 1997;89:3213–3218. [PubMed] [Google Scholar]
- 16.Heydenreich N, Nolte MW, Göb E. C1-inhibitor protects from brain ischemia-reperfusion injury by combined antiinflammatory and antithrombotic mechanisms. Stroke. 2012;43:2457–2467. doi: 10.1161/STROKEAHA.112.660340. [DOI] [PubMed] [Google Scholar]
- 17.Hagedorn I, Schmidbauer S, Pleines I. Factor XIIa inhibitor recombinant human albumin Infestin-4 abolishes occlusive arterial thrombus formation without affecting bleeding. Circulation. 2010;121:1510–1517. doi: 10.1161/CIRCULATIONAHA.109.924761. [DOI] [PubMed] [Google Scholar]
- 18.Schumacher WA, Seiler SE, Steinbacher TE. Antithrombotic and hemostatic effects of a small molecule factor XIa inhibitor in rats. Eur J Pharmacol. 2007;570:167–174. doi: 10.1016/j.ejphar.2007.05.043. [DOI] [PubMed] [Google Scholar]
- 19.Renné T, Pozgajová M, Grüner S. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. 2005;202:271–281. doi: 10.1084/jem.20050664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosen ED, Gailani D, Castellino FJ. FXI is essential for thrombus formation following FeCl3-induced injury of the carotid artery in the mouse. Thromb Haemost. 2002;87:774–776. [PubMed] [Google Scholar]
- 21.Wessler S, Ward K, Ho C. Studies in intravascular coagulation. III. The pathogenesis of serum-induced venous thrombosis. J Clin Invest. 1955;34:647–651. doi: 10.1172/JCI103114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giles AR, Johnston M, Hoogendoorn H. The thrombogenicity of prothrombin complex concentrates: I. The relationship between in vitro characteristics and in vivo thrombogenicity in rabbits. Thromb Res. 1980;17:353–366. doi: 10.1016/0049-3848(80)90070-5. [DOI] [PubMed] [Google Scholar]
- 23.Reimann-Hunziger G. Über experimentelle Thrombose und ihre Behandlung mit Heparin. Schweiz Med Wschr. 1944;74:66–69. [Google Scholar]
- 24.Kurz KD, Main BW, Sandusky GE. Rat model of arterial thrombosis induced by ferric chloride. Thromb Res. 1990;60:269–280. doi: 10.1016/0049-3848(90)90106-m. [DOI] [PubMed] [Google Scholar]
- 25.Karges HE, Funk KA, Ronneberger H. Activity of coagulation and fibrinolysis parameters in animals. Arzneimittelforschung. 1994;44:793–797. [PubMed] [Google Scholar]
- 26.Minta JO. The role of sialic acid in the functional activity and the hepatic clearance of C1-INH. J Immunol. 1981;126:245–249. [PubMed] [Google Scholar]
- 27.Buerke M, Schwertz H, Seitz W. Novel small molecule inhibitor of C1s exerts cardioprotective effects in ischemia-reperfusion injury in rabbits. J Immunol. 2001;167:5375–5380. doi: 10.4049/jimmunol.167.9.5375. [DOI] [PubMed] [Google Scholar]
- 28.Schaller J, Gerber SS. The plasmin-antiplasmin system: structural and functional aspects. Cell Mol Life Sci. 2011;68:785–801. doi: 10.1007/s00018-010-0566-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chandler WL, Levy WC, Stratton JR. The circulatory regulation of TPA and UPA secretion, clearance, and inhibition during exercise and during the infusion of isoproterenol and phenylephrine. Circulation. 1995;92:2984–2994. doi: 10.1161/01.cir.92.10.2984. [DOI] [PubMed] [Google Scholar]
- 30.Colman RW, Schmaier AH. Contact System: A vascular biology modulator with anticoagulant, profibrinolytic, antiadhesive, and proinflammatory attributes. Blood. 1997;90:3819–3843. [PubMed] [Google Scholar]
- 31.Brown NJ, Gainer JV, Stein CM. Bradykinin stimulates tissue plasminogen activator release in human vasculature. Hypertension. 1999;33:1431–1435. doi: 10.1161/01.hyp.33.6.1431. [DOI] [PubMed] [Google Scholar]
- 32.Cugno M, Hack CE, de Boer JP. Generation of plasmin during acute attacks of hereditary angioedema. J Lab Clin Med. 1993;121:38–43. [PubMed] [Google Scholar]
- 33.Huisman LGM, van Griensven JMT, Kluft C. On the role of C1-inhibitor as inhibitor of tissue-type plasminogen activator in human plasma. Thromb Hae-most. 1995;73:466–471. [PubMed] [Google Scholar]
- 34.Sainz IM, Pixley RA, Colman RW. Fifty years of research on the plasma kallikrein-kinin system: From protein structure and function to cell biology and in-vivo pathophysiology. Thromb Haemost. 2007;98:77–83. [PubMed] [Google Scholar]
- 35.Brass LF. Thrombin and platelet activation. Chest. 2003;124:18S–25S.. doi: 10.1378/chest.124.3_suppl.18s. [DOI] [PubMed] [Google Scholar]
- 36.Coppola L, Tirelli A, Giunta R. C1-inhibitor and platelet aggregation. Haematologica. 1988;73:153–161. [PubMed] [Google Scholar]
- 37.Coppola L, Guastafierro S, Verrazzo G. C1 inhibitor infusion modifies platelet activity in hereditary angioedema patients. Arch Pathol Lab Med. 2002;126:842–845. doi: 10.5858/2002-126-0842-CIIMPA. [DOI] [PubMed] [Google Scholar]
- 38.Caccia S, Castelli R, Maiocchi D. Interaction of C1 inhibitor with thrombin on the endothelial surface. Blood Coagul Fibrinolysis. 2011;22:571–575. doi: 10.1097/MBC.0b013e3283494ba7. [DOI] [PubMed] [Google Scholar]
- 39.Cugno M, Bos I, Lubbers Y. In vitro interaction of C1-inhibitor with thrombin. Blood Coagul Fibrinolysis. 2001;12:253–260. doi: 10.1097/00001721-200106000-00005. [DOI] [PubMed] [Google Scholar]
- 40.Patston PA, Schapira M. Regulation of C1-inhibitor function by binding to type IV collagen and heparin. Biochem Biophys Res Commun. 1997;230:597–601. doi: 10.1006/bbrc.1996.6010. [DOI] [PubMed] [Google Scholar]
- 41.Gecse A, Kis B, Mezei Z. The effect of bradykinin and substance P on the arachidonate cascade of platelets. Immunopharmacology. 1996;33:167–170. doi: 10.1016/0162-3109(96)00035-5. [DOI] [PubMed] [Google Scholar]
- 42.Crutchley DJ, Ryan JW, Ryan US. Bradykinin-induced release of prostacyclin and thromboxanes from bovine pulmonary artery endothelial cells. Studies with lower homologs and calcium antagonists. Biochim Biophys Acta. 1983;751:99–107. doi: 10.1016/0005-2760(83)90261-8. [DOI] [PubMed] [Google Scholar]
- 43.Schrör K. Role of prostaglandins in the cardiovascular effects of bradykinin and angiotensin-converting enzyme inhibitors. J Cardiovasc Pharmacol. 1992;20:S68–73. [PubMed] [Google Scholar]
- 44.von Brühl ML, Stark K, Steinhart A. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819–835. doi: 10.1084/jem.20112322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zarbock A, Polanowska-Grabowska RK, Ley K. Platelet-neutrophil-interactions: linking hemostasis and inflammation. Blood Rev. 2007;21:99–111. doi: 10.1016/j.blre.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 46.Mosnier LO, Bouma BN. Regulation of fibrinolysis by thrombin activatable fibrinolysis inhibitor, an unstable carboxypeptidase B that unites the pathways of coagulation and fibrinolysis. Arterioscler Thromb Vasc Biol. 2006;26:2445–2453. doi: 10.1161/01.ATV.0000244680.14653.9a. [DOI] [PubMed] [Google Scholar]
- 47.Kaplan AP, Ghebrehiwet B. The plasma bradykinin-forming pathways and its interrelationships with complement. Mol Immunol. 2010;47:2161–2169. doi: 10.1016/j.molimm.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 48.Schmaier AH. The elusive physiologic role of Factor XII. J Clin. Invest. 2008;118:3006–3009. doi: 10.1172/JCI36617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carson SD, Johnson DR. Consecutive enzyme cascades: complement activation at the cell surface triggers increased tissue factor activity. Blood. 1990;76:361–367. [PubMed] [Google Scholar]
- 50.Hartmann K, Henz BM, Krüger-Krasagakes S. C3a and C5a stimulate chemotaxis of human mast cells. Blood. 1997;89:2863–2870. [PubMed] [Google Scholar]
- 51.Wuillemin WA, te Velthuis H, Lubbers YT. Potentiation of C1 inhibitor by glycosaminoglycans: dextran sulfate species are effective inhibitors of in vitro complement activation in plasma. J Immunol. 1997;159:1953–1960. [PubMed] [Google Scholar]
- 52.Wuillemin WA, Eldering E, Citarella F. Modulation of contact system pro-teases by glycosaminoglycans. Selective enhancement of the inhibition of factor XIa. J Biol Chem. 1996;271:12913–12918. doi: 10.1074/jbc.271.22.12913. [DOI] [PubMed] [Google Scholar]
- 53.Martinez-Saguer I, Rusicke E, Aygören-Pürsün E. Pharmacokinetic analysis of human plasma-derived pasteurized C1-inhibitor concentrate in adults and children with hereditary angioedema: a prospective study. Transfusion. 2010;50:354–360. doi: 10.1111/j.1537-2995.2009.02394.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.