

Abstract
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC) is a rare autosomal recessive disorder that is caused by mutation in genes coding for tight junction proteins claudin-16 and claudin-19. It is characterized by renal wasting of magnesium and calcium associated with the development of nephrocalcinosis and renal stones by early childhood. Most of them progress to end-stage renal failure by the second or third decade. Here, we report two siblings with FHHNC, who presented with nephrocalcinosis without any extrarenal manifestations, one of them having novel homozygous nonsense mutation in claudin-16 (CLDN16) (c.620G>A, p. Trp207Ter). Both were treated with dietary changes, hydrochlorothiazide, potassium citrate, and magnesium supplementation. FHHNC is a rare cause of nephrocalcinosis, and we believe that it should be considered in the presence of nephrocalcinosis with hypercalciuria and hypomagnesemia.
Keywords: Chronic kidney disease, claudin, familial nephrocalcinosis, hypercalciuria, hypomagnesemia
Introduction
The term nephrocalcinosis refers to generalized deposition of calcium oxalate or calcium phosphate in the kidney. It usually involves the renal medulla (97%) and rarely the renal cortex.[1,2] The common causes are hyperparathyroidism, distal renal tubular acidosis, idiopathic hypercalciuria, and hyperoxaluria.[1,2] Rarely an autosomal recessive disorder, familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC) caused by mutation in tight junction proteins claudin-16 and claudin-19 characterizes hypomagnesemia and hypercalciuria that can result in nephrocalcinosis and renal failure.[3] We herein report this rare syndrome in two siblings. To the best of our knowledge, this is the third case report from India, the second involving siblings and the first one to be proved by genetic analysis.
Case Report
Two siblings, a 10-year-old girl and 3-year-old boy, born out of nonconsanguineous marriage presented to us with complaints of pain in abdomen, recurrent urinary tract infection, and history of passing gravels in urine for past 1 year. There was no history of similar complaints or any metabolic disorder in other family members. Pedigree chart is shown in Figure 1. On examination, both siblings were malnourished and stunted with both weight and height for age between 25th and 3rd percentile of their corresponding age group. Their systemic examination was normal. Basic anthropometric, clinical examination and laboratory investigation details are given in Table 1. Arterial blood gas analysis was normal in both the siblings.
Figure 1.
Pedigree chart
Table 1.
Clinical and laboratory details
| Parameter | Sibling 1 (S1) | Sibling 2 (S2) |
|---|---|---|
| Age | 10 years | 3 years |
| Sex | Female | Male |
| Height | 132 cm | 85 cm |
| Weight | 23.1 kg | 10.5 kg |
| BMI | 13.3 kg/m2 | 14.5 kg/m2 |
| Blood pressure | 104/68 mmHg | 84/54 mmHg |
| Fundus | Normal | Normal |
| ENT-audiogram | Normal | Normal |
| Systemic examination | Within normal limits | Within normal limits |
| Serum creatinine | 1.2 mg/dl | 0.3 mg/dl |
| eGFR (Schwartz formula) | 45.4 ml/min/1.73m2 | 117 ml/min/1.73m2 |
| Urine routine | Occ. RBC and plenty of pus cells/hpf | Plenty of RBCs and pus cells/hpf |
| Urine spot protein creatinine ratio | 0.43 | 0.24 |
| Serum sodium | 141 mEq/L | 137 mEq/L |
| Serum potassium | 4.0 mEq/L | 4.0 mEq/L |
| Serum bicarbonate | 22 mEq/L | 21 mEq/L |
| Serum calcium | 9.2 mg/dl | 8.2 mg/dl |
| Serum phosphorus | 5.3 mg/dl | 3.9 mg/dl |
| Serum magnesium | 1.0 mg/dl | 1.2 mg/dl |
| Serum uric acid | 6.3 mg/dl | 5.1 mg/dl |
| Vitamin D | 39.6 ng/ml | 32.5 ng/ml |
| Serum PTH | 125.5 pg/ml | 480 pg/ml |
BMI: Body mass index, RBC: Red blood cell, ENT: Ear Nose and Throat examination, eGFR: Estimated Glomerular filtration rate, PTH: Parathyroid Hormone
X-ray kidney–ureter–bladder showed medullary nephrocalcinosis in both siblings. Ultrasound abdomen of both showed normal-size kidneys with increased echoes and calcified foci in the bilateral medulla. Bladder showed calcareous deposits over wall [Figure 2]. In view of increased parathyroid hormone (PTH) in the female child (S1), ultrasound neck was done which showed small hypoechoic lesion near the inferior and lateral margin of the right lobe of thyroid gland, but parathyroid scintigraphy did not show any uptake, and the lesion was thought to be anatomical variant. Ultrasound neck of male child (S2) did not reveal any lesion.
Figure 2.
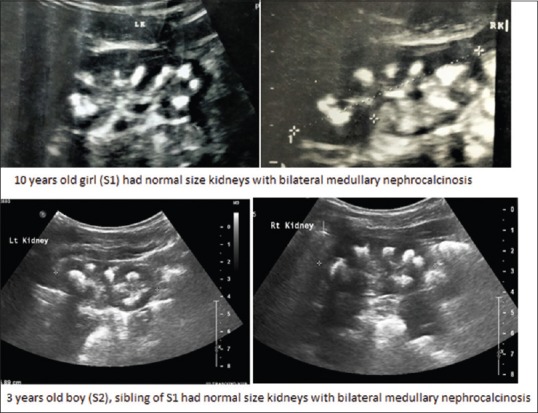
Ultrasonography images of two siblings
On further investigating the cause of nephrocalcinosis, both siblings (S1 and S2) had hypomagnesemia (serum magnesium - 1.0 mg/dl and 1.2 mg/dl, respectively) and hyperuricemia (serum uric acid - 6.3 mg/dl and 5.1, respectively). Hence, 24 h urinary testing was carried out, which showed increased urinary calcium and magnesium loss [Table 2]. Hence, these siblings with medullary nephrocalcinosis and normal calcium and phosphorus levels, low magnesium levels with increased urinary excretion of calcium and magnesium, high uric acid, and PTH levels were thought to have familial hypomagnesemic hypercalciuric nephrocalcinosis (FHHNC).
Table 2.
24 h urinary analysis
| 24-h urinary tests | Sibling 1 | Sibling 2 | Normal values |
|---|---|---|---|
| Volume | 2610 ml | 1080 ml | |
| Creatinine | 390 mg | 189 mg | 15-18 mg/kg |
| Calcium | 154 mg (6.7 mg/kg) | 104 mg (9.9 mg/kg) | <4 mg/kg |
| Spot urine Ca/Cr-0.75 | <0.1 | ||
| Spot urine Ca/Cr-0.36 | |||
| FeMg | 29% | 36.9% | Normally <2% |
| Oxalate | 37 mg | 25 mg | <45 mg/1.73 m2 |
| Citrate | 596 mg | 482 mg | >310 mg/1.73 m2 in females >365 mg/1.73 m2 in males |
| Phosphorus | 240 mg | 280 mg | <1000 mg/1.73 m2 |
| Uric acid | 181 mg | 204 mg | <750 mg/1.73 m2 |
FeMg: Fractional excretion of magnesium
Genetic testing further showed homozygous nonsense mutation of claudin-16 gene (CLDN16) (c.620G>A, p. Trp207Ter) in the female child (S1). Hence, she was diagnosed to have FHHNC Type I due to claudin-16 mutation with autosomal recessive inheritance. For the younger boy (S2), genetic analysis was not carried out as he had the similar features like his sister and the diagnosis was already confirmed. Both were managed plenty of oral fluids, low salt, and normal calcium intake in diet and were supplemented with potassium citrate, magnesium, and hydrochlorothiazide.
Discussion and Review of Literature
Incidence rates and etiology of children with urolithiasis or nephrocalcinosis vary due to differences in geographic, genetic, and socioeconomic background.[4] While the exact rates for nephrocalcinosis are unknown, the incidence of urolithiasis in childhood is believed to be approximately 10% of that in adults, which is around 5% in industrialized countries.[5,6] There is scarcity of data from developing country like India. Urolithiasis or nephrocalcinosis in childhood differs considerably from that of adults with regard to etiology, clinical manifestations, and treatment. Most of the children have a metabolic background of stone disease, and nearly 40% of children with urolithiasis have a positive family history of kidney stones.[1,2] The most common heritable causes of nephrocalcinosis are due to (1) hypercalciuria induced: autosomal dominant hypocalcemic hypercalciuria, FHHNC, Bartter's syndrome, Dent's disease, Lowe syndrome, and others; (2) hyperoxaluria induced: primary hyperoxaluria; (3) cystinuria; (4) Purine/pyrimidine induced: hyperuricosuria- and hypouricosuria-associated urolithiasis; and (5) distal renal tubular acidosis.[1,2,7] Detailed medical history, imaging studies, and specific urine analysis lead way to the determination of the pathophysiological background of stone disease, to initiate early and appropriate treatment and to prevent recurrence and morbidity of stone disease.
The two siblings with nephrocalcinosis were evaluated methodically and diagnosed to have FHHNC by genetic testing. Nephrocalcinosis in FHHNC is characterized by the deposition of calcium oxalate/phosphate in the kidney parenchyma and tubules.[3] It was described in 1972 by Michelis et al.[8] (historically known as Michelis–Gutmo syndrome) and designated as FHHNC by Praga et al. in 1995[9] and is a rare inherited renal tubular disease with nephrocalcinosis frequently complicated by progressive renal failure during childhood or adolescence.
Earlier, Simon et al.[10] and Weber et al.,[11] respectively, analyzed group of families with FHHNC and identified a gene Paracellin-1 (PCLN1) [now described as Claudin-16 (CLDN16)] which was mutated in this disorder. Later in 2006, Konrad et al.[12] studied other group of FHHNC patients who lack mutations in CLDN16 and found to have severe ocular defect and identified a second gene, Claudin-19 (CLDN19) linked to the disease. Claudins or paracellins are tight junction membrane proteins that function as paracellular pathway in the epithelial cells.[13] In the thick ascending limb of Henle's loop, claudins have an important role in the absorption of calcium and magnesium.[13] Hence, defective claudin leads to calcium and magnesium leak in the urine that forms the main pathogenesis of this disease. Two types of FHHNC exist, Type I due to CLDN16 defect and Type II due to CLDN19 defect.[3] Extrarenal manifestations such as ocular abnormalities and hearing impairment are observed in nearly 50% and 10% of cases, respectively, in patients with Type II FHHNC.[14]
The mode of inheritance is autosomal recessive.[3] The usual presenting features are polyuria, polydipsia, renal stones, abdominal colic, recurrent urinary tract infections, failure to thrive, growth retardation, and rickets.[3] Our patients presented to us with pain in abdomen, recurrent urinary tract infection, and history of passing gravels in urine. Additional manifestations related to magnesium deficiency such as tetanic convulsions, muscle cramps, and muscle weakness are reported,[3] and some patients have incomplete distal tubular acidosis.[15] Increased urinary calcium excretion, magnesium deficiency, and urinary acidification defects lead to nephrocalcinosis and nephrolithiasis in FHHNC.[3] Despite marked calcium excretion in urine, these patients usually do not develop hypocalcemia as it is compensated by increased intestinal absorption of calcium and enhanced release of calcium from bone.
The two siblings (S1 and S2) had medullary nephrocalcinosis, normal calcium and phosphorus levels, low magnesium levels with increased urinary excretion of calcium and magnesium and thus raised the suspicious of FHHNC, which was confirmed by genetic test later. These two patients also had high uric acid and PTH levels. Most of FHHNC patients also have hyperuricemia, and the mechanism of abnormal paracellular secretory process for uric acid is not clear so far.[11] However, these patients generally do not have hyperuricosuria. Some authors report elevated serum PTH levels early during the course of disease (as seen in our patients), independent of reduced GFR, and therefore, probably reflecting chronic calcium depletion.[11]
The following genes – CLDN16, CLDN19, EGF, TRPM6, CNNM2, and FXYD2 – were screened as part of hypomagnesemia workup. In our patient (S1, 10-year-old girl), a homozygous nonsense variation in exon 4 of the CLDN16 gene, chromosome 3 (variant c.620G>A) that results in a stop codon and premature truncation of protein at codon 207 (p. Trp207Ter) was detected by targeted gene sequencing method. The CLDN16 variant Trp207Ter lies in a critical domain of the protein-PMP-22/EMP/MP20/claudin family (domain length-72-253aa), and it is considered as a hotspot for mutations. As the variant results in a truncated (shortened) protein, it may affect its interaction with other proteins. The reference codon is conserved across species. Based on this evidence, this CLDN16 variation is classified as a pathogenic variant. She was diagnosed to have Type I FHHNC. For the other sibling, genetic analysis was not carried out. This is a limitation of the study as it is required to confirm the same change in the homozygous state in other affected sibling (S2) and as heterozygous change in parents. However, it was not feasible due to financial constraints. Both of them did not have extrarenal manifestations. They lack ocular and hearing abnormalities consistent with claudin-16 mutation whereas claudin-19 defect does have extrarenal features.
Most patients with FHHNC progress to end-stage renal disease (ESRD) in the second or third decade.[3,16] One of our patient, elderly female child of 10 years (S1), was already in CKD stage 3 (eGFR-45.4 ml/min/1.73 m2) at the time of diagnosis. The other child, 3-year-old boy (S2), had normal renal function (117 ml/min/1.73 m2) at the time of presentation. At 12-month follow-up, both had significant decline in renal function despite medical correction of magnesium and other supplements [the elder sibling (S1) had eGFR of 40 ml/min/1.73 m2 and the younger sibling (S2) had 91 ml/min/1.73 m2 eGFR]. Like with other inherited rare disorders, modification of the primary genetic defects is not yet realistic. There is no specific treatment for this disease, and there have been no clinical trials that guide us toward a therapeutic option. Therefore, the current therapy is basically supportive. Renal transplantation is the definitive treatment if they reach ESRD. FHHNC patients are given oral magnesium (to supplement magnesium loss in urine), citrate (to alkalinize urine, to inhibit stone crystal formation, and to reduce calcium supersaturation in urine), and thiazide diuretics (to reduce calcium excretion in urine) to prevent the progression of nephrocalcinosis or nephrolithiasis, but none had considerable effect in slowing the deterioration of renal function.[3]
To date, >50 different CLDN16 and 17 different CLDN19 pathogenic mutations and only about 131 affected families (178 patients) subjected to molecular-genetic testing have been reported in the literature.[17] Only two cases are reported from India before this, one from southern India and other from northern India, but none were confirmed with genetic testing.[18,19]
It should be noted that FHHNC Type I and Type II are very rare disorders, and their population prevalence is not widely reported or unrecognized. To the best of our knowledge, our case is the first patient of FHHNC confirmed by genetic test in India. FHHNC is likely to have been underdiagnosed due to a lack of awareness of this disorder. Hence, the diagnosis of this rare but intriguing disease deserves consideration in any patient with nephrocalcinosis, hypercalciuria, and renal failure to explore the research for the treatment options in future.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Habbig S, Beck BB, Hoppe B. Nephrocalcinosis and urolithiasis in children. Kidney Int. 2011;80:1278–91. doi: 10.1038/ki.2011.336. [DOI] [PubMed] [Google Scholar]
- 2.Hoppe B, Kemper MJ. Diagnostic examination of the child with urolithiasis or nephrocalcinosis. Pediatr Nephrol. 2010;25:403–13. doi: 10.1007/s00467-008-1073-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Claverie-Martin F. Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis: Clinical and molecular characteristics. Clin Kidney J. 2015;8:656–64. doi: 10.1093/ckj/sfv081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.López M, Hoppe B. History, epidemiology and regional diversities of urolithiasis. Pediatr Nephrol. 2010;25:49–59. doi: 10.1007/s00467-008-0960-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hesse A. Reliable data from diverse regions of the world exist to show that there has been a steady increase in the prevalence of urolithiasis. World J Urol. 2005;23:302–3. doi: 10.1007/s00345-005-0033-3. [DOI] [PubMed] [Google Scholar]
- 6.Sarica K. Pediatric urolithiasis: Etiology, specific pathogenesis and medical treatment. Urol Res. 2006;34:96–101. doi: 10.1007/s00240-005-0018-0. [DOI] [PubMed] [Google Scholar]
- 7.Edvardsson VO, Goldfarb DS, Lieske JC, Beara-Lasic L, Anglani F, Milliner DS, et al. Hereditary causes of kidney stones and chronic kidney disease. Pediatr Nephrol. 2013;28:1923–42. doi: 10.1007/s00467-012-2329-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michelis MF, Drash AL, Linarelli LG, De Rubertis FR, Davis BB. Decreased bicarbonate threshold and renal magnesium wasting in a sibship with distal renal tubular acidosis. (Evaluation of the pathophysiological role of parathyroid hormone) Metabolism. 1972;21:905–20. doi: 10.1016/0026-0495(72)90025-x. [DOI] [PubMed] [Google Scholar]
- 9.Praga M, Vara J, González-Parra E, Andrés A, Alamo C, Araque A, et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis. Kidney Int. 1995;47:1419–25. doi: 10.1038/ki.1995.199. [DOI] [PubMed] [Google Scholar]
- 10.Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, et al. Paracellin-1, a renal tight junction protein required for paracellular mg2+ resorption. Science. 1999;285:103–6. doi: 10.1126/science.285.5424.103. [DOI] [PubMed] [Google Scholar]
- 11.Weber S, Schneider L, Peters M, Misselwitz J, Rönnefarth G, Böswald M, et al. Novel paracellin-1 mutations in 25 families with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol. 2001;12:1872–81. doi: 10.1681/ASN.V1291872. [DOI] [PubMed] [Google Scholar]
- 12.Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet. 2006;79:949–57. doi: 10.1086/508617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hou J, Goodenough DA. Claudin-16 and claudin-19 function in the thick ascending limb. Curr Opin Nephrol Hypertens. 2010;19:483–8. doi: 10.1097/MNH.0b013e32833b7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Faguer S, Chauveau D, Cintas P, Tack I, Cointault O, Rostaing L, et al. Renal, ocular, and neuromuscular involvements in patients with CLDN19 mutations. Clin J Am Soc Nephrol. 2011;6:355–60. doi: 10.2215/CJN.02870310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Passer J. Incomplete distal renal tubular acidosis in hypomagnesemia-dependent hypocalcemia. Arch Intern Med. 1976;136:462–6. [PubMed] [Google Scholar]
- 16.Müller D, Kausalya PJ, Meij IC, Hunziker W. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: Blocking endocytosis restores surface expression of a novel claudin-16 mutant that lacks the entire C-terminal cytosolic tail. Hum Mol Genet. 2006;15:1049–58. doi: 10.1093/hmg/ddl020. [DOI] [PubMed] [Google Scholar]
- 17.Claverie-Martín F, Vargas-Poussou R, Müller D, García-Nieto V. Clinical utility gene card for: Familial hypomagnesemia with hypercalciuria and nephrocalcinosis with/without severe ocular involvement. Eur J Hum Genet. 2015;23:889. doi: 10.1038/ejhg.2014.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prabahar MR, Manorajan R, Fernando ME, Venkatraman R, Balaraman V, Jayakumar M, et al. Nephrocalcinosis in siblings – Familial hypomagnesemia, hypercalciuria with nephrocalcinosis (FHHNC syndrome) J Assoc Physicians India. 2006;54:497–500. [PubMed] [Google Scholar]
- 19.Lodha R, Hari P, Bagga A. Syndrome of renal magnesium wasting and nephrocalcinosis. Indian Pediatr. 1999;36:1046–8. [PubMed] [Google Scholar]