

Abstract
Background
This study investigated whether therapeutic hypercapnia (TH) ameliorated blood–brain barrier (BBB) damage and improved the neurologic outcome in a rat model of lateral fluid percussion injury (FPI), and explored the possible underlying mechanism.
Methods
Rats underwent lateral FPI and received inhalation of 30%O2–70%N2 or 30%O2–N2 plus CO2 to maintain arterial blood CO2 tension (PaCO2) between 80 and 100 mmHg for 3 h. To further explore the possible mechanisms for the protective effects of TH, a PKC inhibitor staurosporine or PKCαβ inhibitor GÖ6976 was administered via intracerebral ventricular injection.
Results
TH significantly improved neurological function 24 h, 48 h, 7 d, and 14 d after FPI. The wet/dry ratio, computed tomography values, Evans blue content, and histological lesion volume were significantly reduced by TH. Moreover, numbers of survived neurons and the expression of tight junction proteins (ZO-1, occludin, and claudin-5) were significantly elevated after TH treatment at 48-h post-FPI. TH significantly increased the expression of protein kinase Cε (PKCε) at 48-h post-FPI, but did not significantly change the expression of PKCα and PKCβII. PKC inhibitor staurosporine (but not the selective PKCαβ inhibitor-GÖ6976) inhibited the protective effect of TH.
Conclusions
Therapeutic hypercapnia is a promising candidate that should be further evaluated for clinical treatment. It not only protects the traumatic penumbra from secondary injury and improves histological structure but also maintains the integrity of BBB and reduces neurologic deficits after trauma in a rat model of FPI.
Keywords: Hypercapnia, Traumatic brain injury, Protein kinases, Blood–brain barrier
Background
Traumatic brain injury (TBI), a leading cause of disability and death worldwide, poses a substantial burden on health outcomes and expenditure. In the USA, 1.7 million people per year suffer from TBI and 50,000 of these people die [1, 2]. TBI basically consists of two phases: the initial injury that occurs at the time of the traumatic impact and the secondary injury that activates cascades of endogenous auto-destructive biochemical processes following initial injury. Secondary injury lasts over hours to days and involves many cellular processes such as blood–brain barrier (BBB) breakdown, apoptosis, oxidative stress, neuronal excitability, inflammation, and immune responses [3–5]. Secondary injury is regarded as a major cause for long-term post-TBI neurological dysfunction and death [6]. Furthermore, increasing size of injury around the contusion called “traumatic penumbra” is caused by the secondary injury [7]. Since secondary injury represents a region of tissue that is most affected by clinical management and neuroprotective interventions, it suggests a potential therapeutic window and a therapy target. Standard procedures for TBI therapy include hyperventilation, hyper-osmotic fluid treatment, and decompressive craniectomy. Unfortunately, to date, no effective therapies can be applied immediately after TBI to ameliorate the secondary damage and improve clinical outcomes.
Accumulating data show that the permeability of BBB serves as a key factor to mediate secondary brain damage, thus improving BBB integrity optimizes TBI treatment [6, 8, 9]. Protein kinase C (PKC) has been demonstrated to play a pivotal role in modulating microvascular permeability [10] and thus serves as a critical factor in BBB structural and functional integrity in pathological conditions [11–13]. Three important PKC isozymes—PKCα, PKCβ, and PKCε—directly modify components of the tight junction (TJ) complex to regulate microvascular permeability [14]. PKCα or PKCβ signaling may enhance TJ opening while PCKε has been shown to be associated with BBB maintenance [15–17].
Hypercapnia exerts neuroprotective effects following cerebral ischemic reperfusion, possibly via suppression of AQP4 and anti-apoptosis mechanisms [18–20]. Zhou et al. [21] reported that mild to moderate hypercapnia (PaCO2, 60–100 mmHg) does not significantly increase intracranial pressure (ICP) for the treatment of global cerebral ischemia in adult rats. Therefore, we hypothesize that hypercapnia may have a potential protective effect on BBB function against TBI.
In this study, we aimed to evaluate the therapeutic efficacy of hypercapnia on BBB function in rats with fluid percussion injury (FPI) and to explore whether hypercapnia regulated specific PKC isozymes against BBB disruption following TBI.
Methods
Animals and ethics approval
Adult male Sprague–Dawley rats (weighing 280–320 g) were obtained from the Second Affiliated Hospital of Harbin Medical University (Harbin, P. R. China). Animals were housed in standard cages, given free access to food and water, and kept in a 12-h day/night cycle. The experimental protocols were approved by the Institutional Animal Care Committee of Harbin Medical University, and all procedures were conducted in strict accordance with the guidelines for the care and use of laboratory animals of Harbin Medical University as well as the ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines for animal research.
Experiment procedure
Each rat was anesthetized via intraperitoneal injection of 30 mg/kg pentobarbital. All rats were intubated and received mechanical ventilation with a gas mixture of 30% O2–70% N2 using a Harvard small animal ventilator 683 (Natick, MA, USA). The tidal volume was 9 ml/kg, and the respiratory rate was 45 breaths/min with a 1:1 inspiratory to respiratory ratio. The left femoral artery was cannulated with a 24-gauge (G) catheter for recording the mean arterial pressure (MAP), heart rate (HR), and blood gas analysis (PaO2, PaCO2, pH). The left femoral vein was cannulated for injection of pentobarbital (3 mg/kg per hour) and Evans blue dye. ICP was continuously measured via a 4-mm-long catheter (22 G) placed into the lateral ventricle (1 mm posterior to the bregma, 1.5 mm right of the sagittal suture).
Experimental groups
Rats were randomly assigned (by drawing lots) to five groups (n = 24 per group):
Sham group rats received drilled craniotomy and were placed on the FPI device without injury. During the following 3 h period, rats were mechanically ventilated with a gas mixture of 30% O2–70% N2.
Trauma group (group T): the FPI model was established followed by mechanical ventilation with a gas mixture of 30% O2–70% N2 for 3 h.
Hypercapnia group (group T+H): the FPI model was established followed by mechanical ventilation with a gas mixture of 30%O2–N2–CO2 for 3 h. The gas flow of N2 and CO2 was continuously adjusted to maintain PaCO2 between 80 and 100 mmHg based on the arterial blood gas analysis.
PKC inhibitor staurosporine group (group T+H+PKCi): rats received the same treatment procedure as rats in group T+H, except that they were given intraventricular injection of 5 μL staurosporine (0.1 μM) (Santa Cruz, CA, USA) immediately after the FPI was established.
PKCαβ inhibitor group (group T+H+PKCαβi): rats were treated with the same procedure as rats in group T+H, except that they were given intraventricular injection of 5 μL PKCαβ inhibitor GÖ6976 (1 μM) (ab141413; Abcam, Cambridge, UK) immediately after the FPI was established.
In each group, six rats were used for the Evans blue test, six for Nissl staining, six for wet/dry ratio calculation, six for Western blot, six for PCR test, six for CT scanning, and six for behavior test scoring.
Establishment of lateral fluid percussion injury model
FPI was established using the fluid percussion device (FP 302, AmScien Instruments, Richmond, VA, USA) as previously described [22–24]. Rats were fixed into a stereotaxic frame (Narishige, Scientific Instrument Lab, Tokyo, Japan). A 4-mm-diameter craniotomy was made 2 mm posterior to the bregma and 2.5 mm left to the sagittal suture, using a dental drill. The dura was exposed intact, and care was taken to avoid any damage to the blood vessels in the dura. The dura was connected to a stainless-steel female Leur-Lot fitting. Dental acrylate was used to fill the space between the dura and the fitting. The pendulum bob was placed at 17° and was later released to produce an impact pressure of between 1.5 and 2 atm. This pressure range induced a moderate brain injury [23]. Rats were then removed from the device and placed on a warming pad to maintain normal body temperature. The scalp was sutured, and animals were ventilated for 3 h. The intubation tube was removed after the rats recovered spontaneous breathing.
Administration of PKC inhibitor
After the FPI model was established, rats were fixed into a stereotaxic frame. Injection of 5 μL staurosporine (0.1 μM) or GÖ6976 (1 μM) into the lateral ventricle (0.8 mm posterior to the bregma and 1.5 mm lateral to the sagittal suture, 4 mm deep) was performed.
Measurement of brain edema
Forty-eight hours after FPI, each rat was anesthetized and then decapitated; the brain was removed. The injured hemisphere was weighed and recorded as wet weight (W). Next, the brain was dried at 80 °C for 48 h and then remeasured as dry weight. The W/D ratio was calculated by an investigator who was blinded to the grouping according to the following equation:
W/D ratio = [(wet weight – dry weight)/wet weight] × 100% [25].
Computed tomography scanning
Forty-eight hours after FPI, the rats were anesthetized. For brain computed tomography, CT images were acquired using a whole-body CT scanner (GE LightSpeed VCT, GE Healthcare, Chicago, IL, USA). The rats’ brains were scanned using 140 kVp, 400 mA, and an exposure time of 2 s. Data were reconstructed in real time using a Dose system (GE Healthcare). Edema was defined as CT values less than 20 HU, while the CT values in the normal brain tissue varied between 25 and 40 HU. Scanning was started from the border of cerebral lesion, and CT images were acquired with a thickness of 0.625 mm. CT values of six sequential slices (with a lesion diameter of 3.75 mm) were required and compared by an investigator who was blinded to the protocol.
Evans blue detection
Evans blue (EB) dye was used to assess the permeability of BBB and was performed and analyzed by a grouping-blinded researcher. EB dye (2%, 5 ml/kg) was injected through the left femoral vein 3 h after FPI establishment. One hour after EB injection, rats were transcardially perfused with normal saline for 10 min. The brain of each rat was then removed, and the left prefrontal cortex was separated, weighed, and homogenized in 50% trichloroacetic acid (TCA). Homogenates were centrifuged for 10 min, and the supernatant was collected. EB dye was measured using an absorbance spectrophotometer at 620 nm. EB dye content was calculated and expressed as nanogram/milligram tissue as previously described [26].
Nissl staining and measurement of lesion volume
Forty-eight hours after FPI, rats were euthanized by overdose anesthesia. Rats were then transcardially perfused with normal saline followed by 4% paraformaldehyde. Brains were removed, fixed in 4% paraformaldehyde for 24 h, and dehydrated in 30% sucrose in PBS at room temperature. Cerebral lesion was sequentially cut at 25 um thick, and 167 ± 16 slices were obtained for each rat. The slices were mounted and then stained with Nissl stain as previously described [27]. Nissl-stained sections were then photographed. At 40× magnification, five fields in each section of the peri-lesion were randomly chosen to count the number of neurons.
The Nissl-stained images of the whole brain section were obtained using a scanner (LaserJet Pro MFP m128fn). The lesion volumes were measured by a researcher blinded to the experimental conditions using Image-Pro Plus version 6.0 software (Media Cybernetics, Bethesda, MD, USA). The lesion area was obtained by bordering damaged or abnormal tissues in the ipsilateral cortex. The lower boundary of the lesion was outlined by the area in which the neuron density was less than normal. Edema area was calculated by ipsilateral hemisphere area minus contralateral hemisphere area. To eliminate the effects of edema, the percentage of lesion volume was calculated according to the following equation: [measured lesion area – edema area] / [(ipsilateral hemisphere area + contralateral hemisphere area) – edema area]. The lesion of each slice was calculated, and the average of the whole brain was recorded and compared by a researcher who was blinded to the experiment protocol.
Western blot analysis
The peri-lesion part of the ipsilateral hemisphere, defined as “penumbra,” was removed and immediately stored at − 80 °C and later used for Western blot analysis as previously described [28] at 48 h post-FPI. Protein samples from the left prefrontal cortex were dissolved in lysis buffer and protease inhibitor cocktail (Sigma) at 4 °C for 1 h. After centrifugation for 10 min at 12,000×g/min, the supernatant was collected and the protein concentration was calculated by BCA protein measurement kit (Bio-Rad). Equal amount of proteins were loaded in SDS-PAGE gel, and the products of the electrophoresis were transferred to PVDF membrane (Invitrogen) following the wet transfer procedure (transfer buffer: 25 mM Tris, 192 mM glycine, 20% methanol). The membrane was blocked with 5% nonfat milk in TBST buffer and then incubated with primary antibodies overnight at 4 °C. The primary antibodies (1:1000 dilution) against PKCα (sc-8393; Santa Cruz Biotechnology, Dallas, TX, USA); PKCβII (sc-13,149; Santa Cruz Biotechnology); PKCε (sc-1681, Santa Cruz Biotechnology); ZO-1 (sc-33,725; Santa Cruz Biotechnology); occludin (sc-133,256; Santa Cruz Biotechnology); and claudin-5 (sc-28,670; Santa Cruz Biotechnology) were used. After extensive rinsing in TBST buffer, the membranes were incubated with HRP-conjugated secondary antibody (diluted at 1:5000, Rockland Inc., Gilbertsville, PA, USA) at 37 °C for 1 h. The final product was viewed using chemiluminescent substrates by a grouping-blinded researcher. The expression level of β-actin was used as loading control.
Reverse transcription-quantitative polymerase chain reaction
Total RNA was extracted from peri-lesion tissues using TRIzol reagents (Invitrogen, Carlsbad, CA, USA). According to the previously published description of the reverse transcription-quantitative polymerase chain reaction (RT-qPCR) [29], 1 μg of total RNA was used for reverse transcription with PrimeScript RT Enzyme Mix I (Takara Bio Inc., Otsu, Shiga, Japan) and a reverse transcription primer.
cDNA solution (1 μL) was mixed with SYBR PrimeScript Ex Taq II (Takara Bio Inc.) and the primers in a total volume of 20 μl. The result was analyzed with a LightCycler 2.0 (Roche, Basel, Switzerland) and an investigator who was blinded to grouping applied the 2△△−Ct method to calculate the relative quantity of PKCε mRNA. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was used as internal control (forward primer, 5′-CTCTGCTCCTCCTGTTCGAC-3′, and reverse primer, 5′-TTAAAAGCAGCCCTGGTGAC-3′). The primers for qPCR were designed on the basis of previous data: forward primer 5′-TACGAAGTGCGCTGGGCTAA-3′ and reverse primer 5′-GGAGCCACAGTGGTCACAGAA-3′.
Behavioral testing
The Modified Neurological Severity Score (mNSS) was used to evaluate neurologic function as previously described by Chen et al. [30]. mNSS tests were performed before FPI; and at 24 h, 48 h, 7 d, 14 d, and 28 d after FPI, by an investigator blinded to the experimental condition. This 18-score system consisted of motor, sensory, reflex, and beam balance tests. A higher score indicates a more severe neurologic impairment.
Statistical analysis
Statistical analyses were performed using SAS version 9.3 software (SAS Institute Inc., Cary, NC, USA). Data are expressed as mean ± standard error of Measurement (SEM). The results from physiological variables and biochemical and animal behavior studies were examined by repeated measures analysis of variance (ANOVA). We selected the mixed effect model to analyze it: after the interaction effect was statistically significant, single-effect comparison was performed by the “contrast program” in SAS software. Using statistician programming and setting of the error, we corrected the p value using code. A p value < 0.05 was considered significant for all statistical analyses in this study.
Results
Physiologic parameters
We first examined the physiological parameters in the sham group, group T, and group T+H. Table 1 showed the changes in ICP, MAP, PaO2, PaCO2, and pH after FPI with hypercapnia. ICP in the sham group did not change significantly during the experiment. FPI resulted in a significant increase in ICP, peaked at 68.00 ± 0.76 mmHg in group T and 67.00 ± 0.65 mmHg in group T+H rats and gradually dropped to a normal level within 30 min. ICP did not increase significantly during 3 h of CO2 inhalation and was kept within physiological values in the three groups.
Table 1.
Physiologic parameters
| Group | Time | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | Trauma | CO2 inhalation | ||||||||
| 15 min | 30 min | 60 min | 90 min | 120 min | 150 min | 180 min | ||||
| ICP (mmHg) | Sham | 14.50 ± 0.46 | 14.25 ± 0.34 | 15.75 ± 0.60 | 15.00 ± 0.29 | 15.25 ± 0.45 | 14.50 ± 0.46 | 14.75 ± 0.34 | 14.50 ± 0.46 | 14.50 ± 0.20 |
| T | 14.75 ± 0.53 | 68.00 ± 0.76# | 17.25 ± 0.53 | 17.00 ± 1.26 | 17.25 ± 0.93 | 18.25 ± 1.36# | 19.50 ± 1.93# | 18.75 ± 1.17# | 19.25 ± 0.97# | |
| T+H | 13.50 ± 0.61 | 67.00 ± 0.65# | 21.25 ± 0.60*# | 18.50 ± 1.06# | 16.00 ± 1.61 | 14.50 ± 0.94* | 14.50 ± 1.10* | 15.50 ± 1.02* | 15.00 ± 0.76* | |
| MAP (mmHg) | Sham | 106.89 ± 2.81 | 106.89 ± 2.76 | 104.09 ± 3.32 | 104.78 ± 3.61 | 106.33 ± 2.29 | 106.50 ± 1.76 | 110.61 ± 2.30 | 105.72 ± 3.29 | 106.48 ± 4.48 |
| T | 99.56 ± 3.57 | 163.76 ± 5.15# | 90.61 ± 3.18# | 91.22 ± 4.0# | 94.83 ± 2.05# | 97.00 ± 1.96 | 102.39 ± 4.72 | 99.89 ± 5.92 | 97.06 ± 5.06 | |
| T+H | 102.89 ± 2.51 | 163.38 ± 5.22# | 87.22 ± 3.74# | 89.33 ± 2.12# | 97.83 ± 4.08 | 100.28 ± 3.37 | 100.42 ± 3.49 | 101.94 ± 2.66 | 100.74 ± 2.64 | |
| PaO2 (mmHg) | Sham | 103.42 ± 2.6 | 110.75 ± 2.42 | 113.22 ± 2.20 | 114.5 ± 2.42 | 112.62 ± 2.78 | 112.82 ± 1.64 | 113.85 ± 2.12 | 113.67 ± 2.11 | |
| T | 108.87 ± 3.69 | 104.58 ± 5.00 | 111.78 ± 4.16 | 110.72 ± 3.55 | 109.10 ± 5.0 | 111.37 ± 2.93 | 109.38 ± 2.44 | 108.55 ± 3.68 | ||
| T+H | 104.83 ± 3.32 | 118.82 ± 5.23* | 130.28 ± 3.08*# | 138.78 ± 4.10*# | 138.30 ± 6.28*# | 141.40 ± 7.00*# | 138.65 ± 3.36*# | 146.87 ± 3.65*# | ||
| PaCO2 (mmHg) | Sham | 37.10 ± 1.18 | 39.97 ± 1.67 | 38.38 ± 1.52 | 37.20 ± 0.73 | 39.07 ± 1.07 | 39.58 ± 1.47 | 42.45 ± 1.51 | 39.93 ± 1.31 | |
| T | 37.33 ± 2.01 | 41.90 ± 2.85 | 39.83 ± 2.06 | 39.93 ± 1.72 | 40.67 ± 2.97 | 37.92 ± 2.44 | 38.48 ± 2.73 | 39.20 ± 2.47 | ||
| T+H | 34.05 ± 1.83 | 72.53 ± 8.15*# | 84.25 ± 4.21*# | 89.97 ± 4.07*# | 97.78 ± 2.29*# | 94.60 ± 4.43*# | 92.48 ± 3.98*# | 91.58 ± 3.56*# | ||
| pH | Sham | 7.38 ± 0.02 | 7.38 ± 0.02 | 7.40 ± 0.01 | 7.39 ± 0.02 | 7.38 ± 0.01 | 7.43 ± 0.01 | 7.40 ± 0.02 | 7.38 ± 0.01 | |
| T | 7.41 ± 0.02 | 7.36 ± 0.01 | 7.37 ± 0.02 | 7.39 ± 0.02 | 7.35 ± 0.02 | 7.34 ± 0.02 | 7.33 ± 0.02 | 7.37 ± 0.02 | ||
| T+H | 7.39 ± 0.01 | 7.15 ± 0.01*# | 7.06 ± 0.04*# | 7.07 ± 0.02*# | 7.05 ± 0.02*# | 7.06 ± 0.01*# | 7.08 ± 0.02*# | 7.09 ± 0.02*# | ||
ICP, MAP, PaO2, PaCO2 and pH in the sham group, group T, and group T+H. Data were expressed as means ± SEM. n = 6
#p < 0.05 vs. sham, *p < 0.05 vs. group T
MAP significantly elevated immediately after trauma in both group T and group T+H, compared with the control group (p < 0.05), MAP elevation lasted for 60 min in group T and 30 min in group T+H. The fluctuation of MAP was believed to be in response to ICP changes.
PaCO2 in group T+H rats was maintained within the range of 80–100 mmHg during 3-h CO2 inhalation, and was significantly higher compared with the sham group (p < 0.05). For PaO2, compared with group T, hypercapnia significantly increased PaO2 as early as 15 min post-PFI, which lasted for 3 h (p < 0.05). pH values in group T+H were significantly lower compared with the other two groups (p < 0.05).
Hypercapnia ameliorates cerebral edema after FPI
At 48-h post-FPI, the W/D ratio in group T rats was significantly increased compared with the sham group (p < 0.05, Fig. 1a). The W/D ratio was significantly reduced in group T+H compared with the group T rats (p < 0.05, Fig. 1a), suggesting that hypercapnia ameliorated cerebral edema. PKCi (but not PKCαβi) significantly inhibited hypercapnia-induced decrease in cerebral edema (p > 0.05) (Fig. 1a).
Fig. 1.

Hypercapnia reduces brain edema. a The wet/dry (W/D) ratio of the injured brain of rats in the sham group, group T, group T+H, group T+H+PKCi, and group T+H+PKCαβi rats at 48-h post-FPI. b Representative computed tomography images of coronal section of rats in the sham group, group T, group T+H, group T+H+PKCi, and group T+H+PKCαβi rats at 48-h post-FPI. The darker areas with lower CT values represented cerebral edema. c CT values. The red arrow indicates post-traumatic brain cerebral edema. Data were expressed as means ± SEM. n = 6. #p < 0.05 vs. sham, *p < 0.05 vs. group T, &p < 0.05 vs. group T+H
Similarly, CT values was significantly decreased in group T compared with the sham group, suggesting that PFI induced cerebral edema, The CT values in group T+H rats were significantly higher than those in group T rats. PKCi (but not PKCαβi) significantly inhibited hypercapnia-induced increase in CT values (p < 0.05) (Fig. 1b, c).
The permeability of the blood–brain barrier was ameliorated by hypercapnia
EB extravasation in the injured prefrontal cortex was significantly increased 3 h after FPI in group T rats. Hypercapnia significantly ameliorated EB extravasation (p < 0.05). PKCi (but not PKCαβi) significantly blocked the effect of hypercapnia (Fig. 2).
Fig. 2.
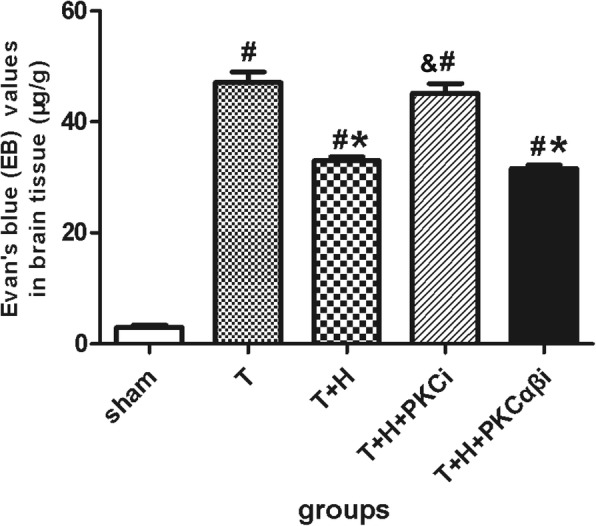
Evans blue values of the injured brain from rats in the sham group, group T, group T+H, group T+H+PKCi, and group T+H+PKCαβi rats at 3-h post-FPI. Data were expressed as means ± SEM. n = 4. #p < 0.05 vs. sham, *p < 0.05 vs. group T, &p < 0.05 vs. group T+H.
Hypercapnia reduced lesion volume and improved neuronal survival
Hypercapnia resulted in a significant decrease in lesion volume and a significant increase in survived neurons at 48 h post-FPI (p < 0.05, Fig. 3). A significant increase in lesion volume and a significant decrease in survived neurons was observed in group T+H+PKCi compared with those in group T+H (p < 0.05) rats. However, PKCαβ treatment did not result in significant changes in lesion volume and neuronal survival compared with group T+H rats (p < 0.05) (Fig. 3).
Fig. 3.

Hypercapnia reduced lesion volume and improved neuronal survival. A, B The whole brain (A) and coronal section of the injured brain (B) in the sham group, group T, group T+H, group T+H+PKCi, and group T+H+PKCαβi rats at 48-h post-FPI. a, the peri-lesion part (“penumbra”). C, D Representative images showing outlined lesion area (C) and Nissl staining (×4 (D) and ×40 (E)). Scale bars: D = 2000 μm, E = 200 μm. F Lesion volume was calculated and analyzed. G Survived neurons in the penumbra. n = 4. #p < 0.05 vs. sham, *p < 0.05 vs. group T, &p < 0.05 vs. group T+H
Hypercapnia resulted in an increase in the expression of tight junction proteins and a significant increase in the expression of PKCε
The expression of TJ proteins and PKC isozymes were shown in the Fig. 4a and b. Hypercapnia increases the expression of ZO-1 (Fig. 4c), occludin (Fig. 4d), and claudin-5 (Fig. 4e). PKCi (but not PKCαβi) blocked hypercapnia-induced upregulation of ZO-1, occludin, and claudin-5. After FPI, the expression levels of PKCα (Fig. 4f), PKCβII (Fig. 4g), and PKCε (Fig. 4h) were significantly increased, while hypercapnia only significantly elevated the expression of PKCε (Fig. 4h). PKCi decreased the expression of all the three PKCs and PKCαβi inhibited the expression of PKCαβ.
Fig. 4.

Hypercapnia increased the expression of TJ proteins and PKCε. a, b Representative Western blot results showing the expression of the tight junction proteins ZO-1, occludin, and claudin-5 (a) and PKCα, PKCβII, and PKCε (b) in the sham group, group T, group T+H, group T+H+PKCi, and group T+H+PKCαβ rats at 48-h post-FPI. c–h Quantification of the expression of ZO-1 (c), occludin (d), claudin-5 (e), PKCα (f), PKCβII (g), and PKCε (h) in the sham group, group T, group T+H, group T+H+PKCi, and group T+H+PKCαβi rats. i RT-PCR results showing the mRNA expression of PKCε in the sham group, group T, group T+H, group T+H+PKCi, and group T+H+PKCαβI rats. Data were expressed as means ± SEM. n = 4. #p < 0.05 vs. sham, *p < 0.05 vs. group T, &p < 0.05 vs. group T+H
mRNA of PKCε was detected by RT-PCR. Hypercapnia significantly increased the mRNA expression of PKCε. However, only PKCi (but not PKCαβi) inhibited the mRNA expression of PKCε (p < 0.05) (Fig. 4i).
Hypercapnia improved mNSS scores
At day 1, day 2, day 7, and day 14 after FPI, mNSS scores in group T rats were elevated compared with those in the sham group rats (p < 0.05). The neurological dysfunction of group T rats recovered within 28 days after FPI. mNSS scores in group T+H rats were significantly reduced at days 1, 2, 7, and 14 after FPI compared with group T rats (p < 0.05), and there were no significant differences between the two groups at day 28 (p > 0.05) (Fig. 5).
Fig. 5.
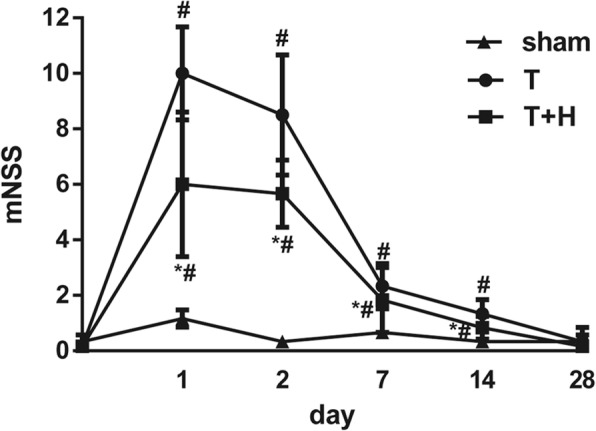
The Modified Neurological Severity Score (mNSS) in the sham group, group T, and group T+H rats at 1, 2, 7, 14, and 28 days post-FPI. Data were expressed as means ± SEM. n = 6. #p < 0.05vs.sham, *p < 0.05 vs. group T
Discussion
Hypercapnia was first found to produce neuroprotection against hypoxic ischemic injury in immature animal models [31, 32]. Later, hypercapnia has been reported to reduce neurologic deficit and ameliorate histopathologic damage and cerebral apoptosis in global cerebral ischemia [33]. In addition, hypercapnia improves functional recovery in focal cerebral ischemia and increases the 28-day survival rate [18, 34].
Penumbra is defined as a region of intermediate cerebral blood flow depression surrounding the densely ischemic core in stroke [35]. Traumatic penumbra may need to be defined physiologically rather than confined in terms of perilesional anatomy, is most vulnerable to secondary ischemic injury, and can be treated by therapeutic interventions [7, 35, 36]. TBI has similar pathophysiology to ischemic injury, including BBB interruption, edema, neuronal loss, apoptosis, and cognitive dysfunction. This study served as the first study to use therapeutic hypercapnia (TH) for the treatment of TBI in rats and showed for the first time how hypercapnia exerted neuroprotective effects on TBI in rats—by reducing brain edema, improving BBB function, decreasing lesion volume, and improving neurological outcome.
PKC family is widely expressed in tissues and consists of at least 12 isozymes, including conventional (a, βΙ, βII, and γ); novel (δ, ε, η, θ, μ, ѵ); and atypical (ζ, ɩ) isoforms with distinct distribution and different sensitivity to Ca2+, and regulation of diverse signal transduction and cellular activities [37, 38].
Among these isozymes, three important PKCs—PKCα, PKCβII, and PKCε—were shown to be most revolved in the regulation of the expression of TJ proteins. There are several possible mechanisms by which PKC may be altering BBB permeability. One possible mechanism is direct regulation (phosphorylation) of the TJ proteins by PKC. PKCα and PKCβ have both been implicated in regulating cell permeability after ischemia and inflammatory stimuli [10]. PKCε was found to play a beneficial role in the regulation of TJ proteins, while PKCβII aggravated the disruption of TJ proteins during aglycemic hypoxia [15]. In addition, increased PKCα activity exacerbates neuronal damage after poststroke brain injury [39].
We found that FPI increased the expression of injurious PKCα and PKCβII in groups T and T+H compared with sham, as well as the beneficial PKCε. Although our results do not show an increase in PKCα and β-expression by hypercapnia treatment compared to group T, our results do not exclude the possibility that PKCα and β and other isozymes may play a role in altering BBB endothelial cell integrity during FPI. In addition, our data, which show an increase in PKCε expression in group T+H, suggest that this may be this one isozyme that is important in regulating endothelial barrier integrity by hypercapnia. In addition, hypercapnia significantly increased the mRNA expression of PKCε at 48-hpost-FPI. This was accompanied by augmented expression of TJ proteins ZO-1, occludin, and claudin-5 as well as decreased brain edema. As the BBB is formed by endothelial cell TJs, the increased TJ proteins after hypercapnia suggests that BBB function were improved [14].
Furthermore, the non-selective PKC inhibitor staurosporine significantly decreases the expression of PKCα, PKCβII, and PKCε, and this blocked the protective effects of hypercapnia. GÖ6976 (an inhibitor of PKCα, PKCβ, and PKCγ but not PKCε) had no effect on hypercapnia-induced neuroprotection. These results suggest that hypercapnia may exert neuroprotective effects via upregulation of PKCε expression.
As noted, animal model of cerebral ischemic and TBI share certain comparable pathobiology. It has been reported that PKCε increased BBB integrity by upregulation of the expression of TJ proteins [11, 16, 40, 41]. In addition, PKCε activation protects BBB acutely and improves neuronal function chronically [28]. In cerebral ischemia models, PKCε activation improves neuronal protection and increases cerebral blood flow (CBF) after ischemia [42, 43]. In addition, PKCε activation plays a protective role in ischemic pre-conditioning (IPC) conditions [34, 44]. PKCε exerts its neuroprotective effect by activating adenosine-induced ATP-sensitive mitochondrial potassium (mKATP) channels, decreasing the production of ROS and calcium overload [45], preserving mitochondrial membrane potential, maintaining energy, and reducing calcium influx [46]. PKCε has also been found to be closely associated with IPC and N-methyl-d-aspartate (NMDA)-induced ischemic tolerance [47, 48]. We believe that this evidence suggests that PKCε serves as a key factor in protective effects against TBI.
Although we did not measure CBF in this study, our previous study found that hypercapnia treatment resulted in CBF recovery in rats treated with mild to moderate hypoxia (PaO2 > 50 mmHg) [20]. In addition, though in combination with acidosis and hyperoxia, carbon dioxide inhalation has been reported to cause vasodilation and increase CBF [49]. We speculate that in this rat model of FPI, hypercapnic acidosis appears to increase PaO2 through two mechanisms: increased PaCO2 and decreased pH leads to right shift of the O2-hemoglobin deviation curve and augmented oxygen delivery through increased cerebral blood flow (CBF). Hypercapnic acidosis also decreases the shunt of the lung and increases arterial oxygenation, which increases the cardiac output and decreases the oxygen consumption. These systemic effects may all explain better cerebral oxygenation and improved neurological outcome. Since PKCε modulates CBF in animals after ischemia [42], we could not exclude the possibility that hypercapnia produces cerebral protection via increasing CBF by upregulation of PKCε. In addition, PKCε upregulation by hypercapnia reduced neuronal loss and decreased apoptosis cells in this rat model of FPI (data not shown). This is consistent with a previous report showing that PKCε activation is responsible for anti-apoptosis by regulation of BCL-2 [50–52].
Together, these studies suggest that PKCε may be the key factor for hypercapnia-induced neuroprotection via different mechanisms including preserving the BBB, increasing CBF regulation, and inhibiting apoptosis. However, it remains unclear which PKC downstream signaling pathways (such as PKCε-CREB-Nrf2, PKCε/MAPK, PKCε/MERK, or PKC-mKATP) are responsible for the beneficial effects of hypercapnia following FPI.
We used staurosporine and GÖ6976 to identify the specific PKC isozyme involved in the beneficial effect of hypercapnia following FPI. Staurosporin is a nonspecific PKC inhibitor that inhibits various PKC isozymes, including PKCα, PKCβ, PKCγ, PKCδ, and PKCε [33, 53]. GÖ6976 is a relatively selective PKC inhibitor, which inhibits PKCa, PKCβ, and PKCγ. Although our study may indicate that PKCε is important for the beneficial effect of hypercapnia following FPI, a specific PKCε inhibitor εV1–2 [54] should have been used in this study, which we could not obtain in this experiment.
The model of FPI is one of the most frequently used models to mimic the clinic condition of TBI without skull fracture and has the similar histopathological changes as TBI. The severity of FPI is classified according to the values of impact force, which were controlled between 1.5 and 2 atm (classified as moderate FPI) in our study to reduce the mortality to 19.97% (data not shown).
Zhou et al. reported that in a rat model of global cerebral ischemia reperfusion injury (CIRI), PaCO2 of 80–100 mmHg produced a better neuroprotective effect than PaCO2 of 60–80 mmHg [21]. Therefore, we used PaCO2 of 80–100 mmHg in this experiment. Future studies will be performed to explore the beneficial effects of TH at a sub-acute time window at different duration and levels and to identify the underlying neuroprotective mechanism.
TH has been used in various brain injury animal models, and the data indicates neuroprotective effects of elevated CO2. Though still in the experimental period, promising laboratory studies and accumulating clinical studies suggest potential roles for clinical application of CO2 for patients with brain disease, preferably in avoiding the coning crisis period when first applied in the clinic. In a clinical study [55], it is reported that in patients with subarachnoid hemorrhage, hypercapnia (PCO2 50–60 mmHg) was not associated with increasing of the ICP and hypercapnia could possibly be safely performed in these patients. In a clinical study from our department, continuous inhalation of CO2 during one-lung ventilation (OLV) improved respiratory function and mitigated the OLV-related lung and systemic inflammation in patients undergoing a lobectomy [56]. Moreover, according to the literature, patients can tolerate PaCO2 elevation (more than 150 mmHg according to several case reports) more than previously assumed. Such findings are in line with our results. CO2-related ICP proved to be reversible without permanent damage to the health of patients in these clinical reports [55, 57]. Considering that TH does not significantly alter ICP and is associated with less adverse effects in these studies, TH is now available for further study and clinical use.
Conclusions
In summary, we found that hypercapnia (for 3 h with PaCO2 levels 80–100 mmHg) after TBI reduced brain edema, improved BBB function, inhibited lesion volume, and improved neurological outcome in a rat model of FPI. Hypercapnia improves neuronal function following FPI, possibly via upregulation of PKCε.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural Science Foundation of China (grant no. 81400989) and China Postdoctoral Science Foundation Grant (grant no. 2015 M581474).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- BBB
Blood–brain barrier
- CBF
Cerebral blood flow
- FPI
Fluid percussion injury
- HR
Heart rate
- ICP
Intracranial pressure
- MAP
Mean arterial pressure
- OLV
One-lung ventilation
- TBI
Traumatic brain injury
- TH
Therapeutic hypercapnia
- TJ
Tight junction
Authors’ contributions
WCY and WZL designed the study. QW and LTC collected and analyzed the data. YZW and HLC advised on the histological staining and analysis. LTC and YZW contributed in the sample collection and intellectual input. WCY and QW drafted and wrote the manuscript. WZL revised the manuscript critically for intellectual content. All authors gave intellectual input to the study and approved the final version of the manuscript.
Ethics approval and consent to participate
The experimental protocols were approved by the Institutional Animal Care Committee of Harbin Medical University, and all procedures were conducted in strict accordance with the guidelines for the care and use of laboratory animals of Harbin Medical University as well as the ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines for animal research.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Wan-Chao Yang, Email: Dachao_1980@126.com.
Qi Wang, Email: Qiqiwang2004@126.com.
Lai-Ting Chi, Email: Clt525290@163.com.
Yue-Zhen Wang, Email: 117410297@qq.com.
Hong-Ling Cao, Email: 851263331@qq.com.
Wen-Zhi Li, Email: Wenzhili9@126.com.
References
- 1.Hoge CW, McGurk D, Thomas JL, Cox AL, Engel CC, Castro CA. Mild traumatic brain injury in U.S. soldiers returning from Iraq. N Engl J Med. 2008;358:453–463. doi: 10.1056/NEJMoa072972. [DOI] [PubMed] [Google Scholar]
- 2.Kou Z, VandeVord PJ. Traumatic white matter injury and glial activation: from basic science to clinics. Glia. 2014;62:1831–1855. doi: 10.1002/glia.22690. [DOI] [PubMed] [Google Scholar]
- 3.Graham DI, McIntosh TK, Maxwell WL, Nicoll JA. Recent advances in neurotrauma. J Neuropathol Exp Neurol. 2000;59:641–651. doi: 10.1093/jnen/59.8.641. [DOI] [PubMed] [Google Scholar]
- 4.Albensi BC. Models of brain injury and alterations in synaptic plasticity. J Neurosci Res. 2001;65:279–283. doi: 10.1002/jnr.1151. [DOI] [PubMed] [Google Scholar]
- 5.Gorrie C, Oakes S, Duflou J, Blumbergs P, Waite PM. Axonal injury in children after motor vehicle crashes: extent, distribution, and size of axonal swellings using beta-APP immunohistochemistry. J Neurotrauma. 2002;19:1171–1182. doi: 10.1089/08977150260337976. [DOI] [PubMed] [Google Scholar]
- 6.Shlosberg D, Benifla M, Kaufer D, Friedman A. Blood–brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol. 2010;6:393–403. doi: 10.1038/nrneurol.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menon DK. Procrustes, the traumatic penumbra, and perfusion pressure targets in closed head injury. Anesthesiology. 2003;98:805–807. doi: 10.1097/00000542-200304000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Jungner M, Siemund R, Venturoli D, Reinstrup P, Schalén W, Bentzer P. Blood-brain barrier permeability following traumatic brain injury. Minerva Anestesiol. 2016;82:525–533. [PubMed] [Google Scholar]
- 9.Donkin JJ, Vink R. Mechanisms of cerebral edema in traumatic brain injury: therapeutic developments. Curr Opin Neurol. 2010;23:293–299. doi: 10.1097/WCO.0b013e328337f451. [DOI] [PubMed] [Google Scholar]
- 10.Yuan SY. Protein kinase signaling in the modulation of microvascular permeability. Vasc Pharmacol. 2002;39:213–223. doi: 10.1016/S1537-1891(03)00010-7. [DOI] [PubMed] [Google Scholar]
- 11.Fleegal MA, Hom S, Borg LK, Davis TP. Activation of PKC modulates blood-brain barrier endothelial cell permeability changes induced by hypoxia and posthypoxic reoxygenation. Am J Physiol Heart Circ Physiol. 2005;289:H2012–H2019. doi: 10.1152/ajpheart.00495.2005. [DOI] [PubMed] [Google Scholar]
- 12.Yang T, Roder KE, Bhat GJ, Thekkumkara TJ, Abbruscato TJ. Protein kinase C family members as a target for regulation of blood-brain barrier Na,K,2Cl-cotransporter during in vitro stroke conditions and nicotine exposure. Pharm Res. 2006;23:291–302. doi: 10.1007/s11095-005-9143-2. [DOI] [PubMed] [Google Scholar]
- 13.Padmaperuma B, Mark R, Dhillon HS, Mattson MP, Prasad MR. Alterations in brain protein kinase C after experimental brain injury. Brain Res. 1996;714:19–26. doi: 10.1016/0006-8993(95)01579-5. [DOI] [PubMed] [Google Scholar]
- 14.Harhaj NS, Antonetti DA. Regulation of tight junctions and loss of barrier function in pathophysiology. Int J Biochem Cell Biol. 2004;36:1206–1237. doi: 10.1016/j.biocel.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 15.Kim YA, Park SL, Kim MY, Lee SH, Baik EJ, Moon CH, et al. Role of PKCbetaII and PKCdelta in blood-brain barrier permeability during aglycemic hypoxia. Neurosci Lett. 2010;468:254–258. doi: 10.1016/j.neulet.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 16.Sonobe Y, Takeuchi H, Kataoka K, Li H, Jin S, Mimuro M, et al. Interleukin-25 expressed by brain capillary endothelial cells maintains blood-brain barrier function in a protein kinase C-dependent manner. J Biol Chem. 2016;291:12573. doi: 10.1074/jbc.A109.025940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abdullah Z, Bayraktutan U. Suppression of PKC-alpha attenuates TNF-alpha-evoked cerebral barrier breakdown via regulations of MMP-2 and plasminogen-plasmin system. Biochim Biophys Acta. 1862;2016:1354–1366. doi: 10.1016/j.bbadis.2016.03.014. [DOI] [PubMed] [Google Scholar]
- 18.Tao T, Liu Y, Zhang J, Xu Y, Li W, Zhao M. Therapeutic hypercapnia improves functional recovery and attenuates injury via antiapoptotic mechanisms in a rat focal cerebral ischemia/reperfusion model. Brain Res. 2013;1533:52–62. doi: 10.1016/j.brainres.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 19.Tregub P, Kulikov V, Motin Y, Bespalov A, Osipov I. Combined exposure to hypercapnia and hypoxia provides its maximum neuroprotective effect during focal ischemic injury in the brain. J Stroke Cerebrovasc Dis. 2015;24:381–387. doi: 10.1016/j.jstrokecerebrovasdis.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 20.Yang W, Zhang X, Wang N, Tan J, Fang X, Wang Q, et al. Effects of acute systemic hypoxia and hypercapnia on brain damage in a rat model of hypoxia-ischemia. PLoS One. 2016;11:e0167359. doi: 10.1371/journal.pone.0167359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou Q, Cao B, Niu L, Cui X, Yu H, Liu J, et al. Effects of permissive hypercapnia on transient global cerebral ischemia-reperfusion injury in rats. Anesthesiology. 2010;112:288–297. doi: 10.1097/ALN.0b013e3181ca8257. [DOI] [PubMed] [Google Scholar]
- 22.Kharatishvili I, Nissinen JP, McIntosh TK, Pitkanen A. A model of posttraumatic epilepsy induced by lateral fluid-percussion brain injury in rats. Neuroscience. 2006;140:685–697. doi: 10.1016/j.neuroscience.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 23.McIntosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Soares H, et al. Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience. 1989;28:233–244. doi: 10.1016/0306-4522(89)90247-9. [DOI] [PubMed] [Google Scholar]
- 24.Alder J, Fujioka W, Lifshitz J, Crockett DP, Thakker-Varia S. Lateral fluid percussion: model of traumatic brain injury in mice. J Vis Exp. 2011;54:3063. PMID 21876530. [DOI] [PMC free article] [PubMed]
- 25.Zweckberger K, Eros C, Zimmermann R, Kim SW, Engel D, Plesnila N. Effect of early and delayed decompressive craniectomy on secondary brain damage after controlled cortical impact in mice. J Neurotrauma. 2006;23:1083–1093. doi: 10.1089/neu.2006.23.1083. [DOI] [PubMed] [Google Scholar]
- 26.Dehghan F, Khaksari Hadad M, Asadikram G, Najafipour H, Shahrokhi N. Effect of melatonin on intracranial pressure and brain edema following traumatic brain injury: role of oxidative stresses. Arch Med Res. 2013;44:251–258. doi: 10.1016/j.arcmed.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 27.Wang G, Jiang X, Pu H, Zhang W, An C, Hu X, et al. Scriptaid, a novel histone deacetylase inhibitor, protects against traumatic brain injury via modulation of PTEN and AKT pathway : Scriptaid protects against TBI via AKT. Neurotherapeutics. 2013;10:124–142. doi: 10.1007/s13311-012-0157-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lucke-Wold BP, Logsdon AF, Smith KE, Turner RC, Alkon DL, Tan Z, et al. Bryostatin-1 restores blood brain barrier integrity following blast-induced traumatic brain injury. Mol Neurobiol. 2015;52:1119–1134. doi: 10.1007/s12035-014-8902-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang L, Qin Y, Tong L, Wu S, Wang Q, Jiao Q, et al. MiR-342-5p suppresses coxsackievirus B3 biosynthesis by targeting the 2C-coding region. Antivir Res. 2012;93:270–279. doi: 10.1016/j.antiviral.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 30.Chen J, Li Y, Wang L, Zhang Z, Lu D, Lu M, et al. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke. 2001;32:1005–1011. doi: 10.1161/01.STR.32.4.1005. [DOI] [PubMed] [Google Scholar]
- 31.Vannucci RC, Towfighi J, Heitjan DF, Brucklacher RM. Carbon dioxide protects the perinatal brain from hypoxic-ischemic damage: an experimental study in the immature rat. Pediatrics. 1995;95:868–874. [PubMed] [Google Scholar]
- 32.Vannucci RC, Brucklacher RM, Vannucci SJ. Effect of carbon dioxide on cerebral metabolism during hypoxia-ischemia in the immature rat. Pediatr Res. 1997;42:24–29. doi: 10.1203/00006450-199707000-00005. [DOI] [PubMed] [Google Scholar]
- 33.Mochly-Rosen D, Das K, Grimes KV. Protein kinase C, an elusive therapeutic target? Nat Rev Drug Discov. 2012;11:937–957. doi: 10.1038/nrd3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lange-Asschenfeldt C, Raval AP, Dave KR, Mochly-Rosen D, Sick TJ, Perez-Pinzon MA. Epsilon protein kinase C mediated ischemic tolerance requires activation of the extracellular regulated kinase pathway in the organotypic hippocampal slice. J Cereb Blood Flow Metab. 2004;24:636–645. doi: 10.1097/01.WCB.0000121235.42748.BF. [DOI] [PubMed] [Google Scholar]
- 35.Coles JP, Fryer TD, Smielewski P, Chatfield DA, Steiner LA, Johnston AJ, et al. Incidence and mechanisms of cerebral ischemia in early clinical head injury. J Cereb Blood Flow Metab. 2004;24:202–211. doi: 10.1097/01.WCB.0000103022.98348.24. [DOI] [PubMed] [Google Scholar]
- 36.Coles JP. Regional ischemia after head injury. Curr Opin Crit Care. 2004;10:120–125. doi: 10.1097/00075198-200404000-00008. [DOI] [PubMed] [Google Scholar]
- 37.Newton AC. Regulation of protein kinase C. Curr Opin Cell Biol. 1997;9:161–167. doi: 10.1016/S0955-0674(97)80058-0. [DOI] [PubMed] [Google Scholar]
- 38.Dempsey EC, Newton AC, Mochly-Rosen D, Fields AP, Reyland ME, Insel PA, et al. Protein kinase C isozymes and the regulation of diverse cell responses. Am J Physiol Lung Cell Mol Physiol. 2000;279:L429–L438. doi: 10.1152/ajplung.2000.279.3.L429. [DOI] [PubMed] [Google Scholar]
- 39.Shirai Y, Adachi N, Saito N. Protein kinase Cepsilon: function in neurons. FEBS J. 2008;275:3988–3994. doi: 10.1111/j.1742-4658.2008.06556.x. [DOI] [PubMed] [Google Scholar]
- 40.Krizbai I, Szabo G, Deli M, Maderspach K, Lehel C, Olah Z, et al. Expression of protein kinase C family members in the cerebral endothelial cells. J Neurochem. 1995;65:459–462. doi: 10.1046/j.1471-4159.1995.65010459.x. [DOI] [PubMed] [Google Scholar]
- 41.Sonobe Y, Takeuchi H, Kataoka K, Li H, Jin S, Mimuro M, et al. Interleukin-25 expressed by brain capillary endothelial cells maintains blood-brain barrier function in a protein kinase Cepsilon-dependent manner. J Biol Chem. 2009;284:31834–31842. doi: 10.1074/jbc.M109.025940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Della-Morte D, Raval AP, Dave KR, Lin HW, Perez-Pinzon MA. Post-ischemic activation of protein kinase C epsilon protects the hippocampus from cerebral ischemic injury via alterations in cerebral blood flow. Neurosci Lett. 2011;487:158–162. doi: 10.1016/j.neulet.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bright R, Sun GH, Yenari MA, Steinberg GK, Mochly-Rosen D. epsilonPKC confers acute tolerance to cerebral ischemic reperfusion injury. Neurosci Lett. 2008;441:120–124. doi: 10.1016/j.neulet.2008.05.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang Z, Sun W, Hu K. Molecular mechanism underlying adenosine receptor-mediated mitochondrial targeting of protein kinase C. Biochim Biophys Acta. 1823;2012:950–958. doi: 10.1016/j.bbamcr.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 45.Capuani B, Pacifici F, Pastore D, Palmirotta R, Donadel G, Arriga R, et al. The role of epsilon PKC in acute and chronic diseases: possible pharmacological implications of its modulators. Pharmacol Res. 2016;111:659–667. doi: 10.1016/j.phrs.2016.07.029. [DOI] [PubMed] [Google Scholar]
- 46.Teshima Y, Akao M, Li RA, Chong TH, Baumgartner WA, Johnston MV, et al. Mitochondrial ATP-sensitive potassium channel activation protects cerebellar granule neurons from apoptosis induced by oxidative stress. Stroke. 2003;34:1796–1802. doi: 10.1161/01.STR.0000077017.60947.AE. [DOI] [PubMed] [Google Scholar]
- 47.Raval AP, Dave KR, Mochly-Rosen D, Sick TJ, Perez-Pinzon MA. Epsilon PKC is required for the induction of tolerance by ischemic and NMDA-mediated preconditioning in the organotypic hippocampal slice. J Neurosci. 2003;23:384–391. doi: 10.1523/JNEUROSCI.23-02-00384.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jia J, Wang X, Li H, Han S, Zu P, Li J. Activations of nPKCepsilon and ERK1/2 were involved in oxygen-glucose deprivation-induced neuroprotection via NMDA receptors in hippocampal slices of mice. J Neurosurg Anesthesiol. 2007;19:18–24. doi: 10.1097/01.ana.0000211020.88431.e2. [DOI] [PubMed] [Google Scholar]
- 49.Duong TQ, Iadecola C, Kim SG. Effect of hyperoxia, hypercapnia, and hypoxia on cerebral interstitial oxygen tension and cerebral blood flow. Magn Reson Med. 2001;45:61–70. doi: 10.1002/1522-2594(200101)45:1<61::AID-MRM1010>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 50.Steinberg R, Harari OA, Lidington EA, Boyle JJ, Nohadani M, Samarel AM, et al. A protein kinase Cepsilon-anti-apoptotic kinase signaling complex protects human vascular endothelial cells against apoptosis through induction of Bcl-2. J Biol Chem. 2007;282:32288–32297. doi: 10.1074/jbc.M704001200. [DOI] [PubMed] [Google Scholar]
- 51.Okhrimenko H, Lu W, Xiang C, Hamburger N, Kazimirsky G, Brodie C. Protein kinase C-epsilon regulates the apoptosis and survival of glioma cells. Cancer Res. 2005;65:7301–7309. doi: 10.1158/0008-5472.CAN-05-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mylroie H, Dumont O, Bauer A, Thornton CC, Mackey J, Calay D, et al. PKCepsilon-CREB-Nrf2 signalling induces HO-1 in the vascular endothelium and enhances resistance to inflammation and apoptosis. Cardiovasc Res. 2015;106:509–519. doi: 10.1093/cvr/cvv131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Swannie HC, Kaye SB. Protein kinase C inhibitors. Curr Oncol Rep. 2002;4:37–46. doi: 10.1007/s11912-002-0046-7. [DOI] [PubMed] [Google Scholar]
- 54.Gray MO, Karliner JS, Mochly-Rosen D. A selective epsilon-protein kinase C antagonist inhibits protection of cardiac myocytes from hypoxia-induced cell death. J Biol Chem. 1997;272:30945–30951. doi: 10.1074/jbc.272.49.30945. [DOI] [PubMed] [Google Scholar]
- 55.Petridis AK, Doukas A, Kienke S, Maslehaty H, Mahvash M, Barth H, et al. The effect of lung-protective permissive hypercapnia in intracerebral pressure in patients with subarachnoid haemorrhage and ARDS. A retrospective study. Acta Neurochir. 2010;152:2143–2145. doi: 10.1007/s00701-010-0761-z. [DOI] [PubMed] [Google Scholar]
- 56.Gao W, Liu DD, Li D, Cui GX. Effect of therapeutic hypercapnia on inflammatory responses to one-lung ventilation in lobectomy patients. Anesthesiology. 2015;122:1235–1252. doi: 10.1097/ALN.0000000000000627. [DOI] [PubMed] [Google Scholar]
- 57.Urwin L, Murphy R, Robertson C, Pollok A. A case of extreme hypercapnia: implications for the prehospital and accident and emergency department management of acutely dyspnoeic patients. Emerg Med J. 2004;21:119–120. doi: 10.1136/emj.2003.005009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.